Engineering Lipid Bilayer Membranes for Protein Studies

Abstract
:1. Introduction
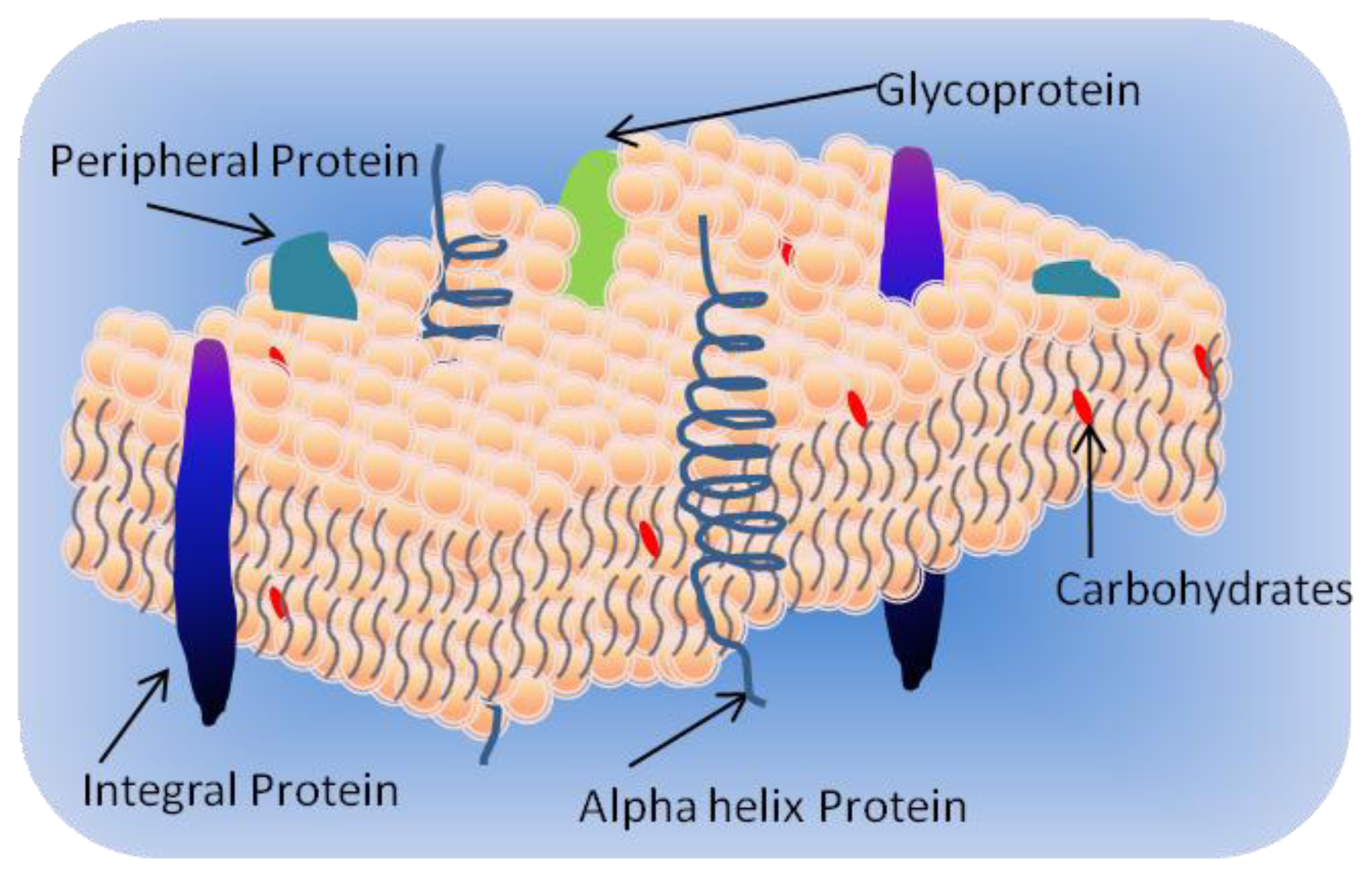
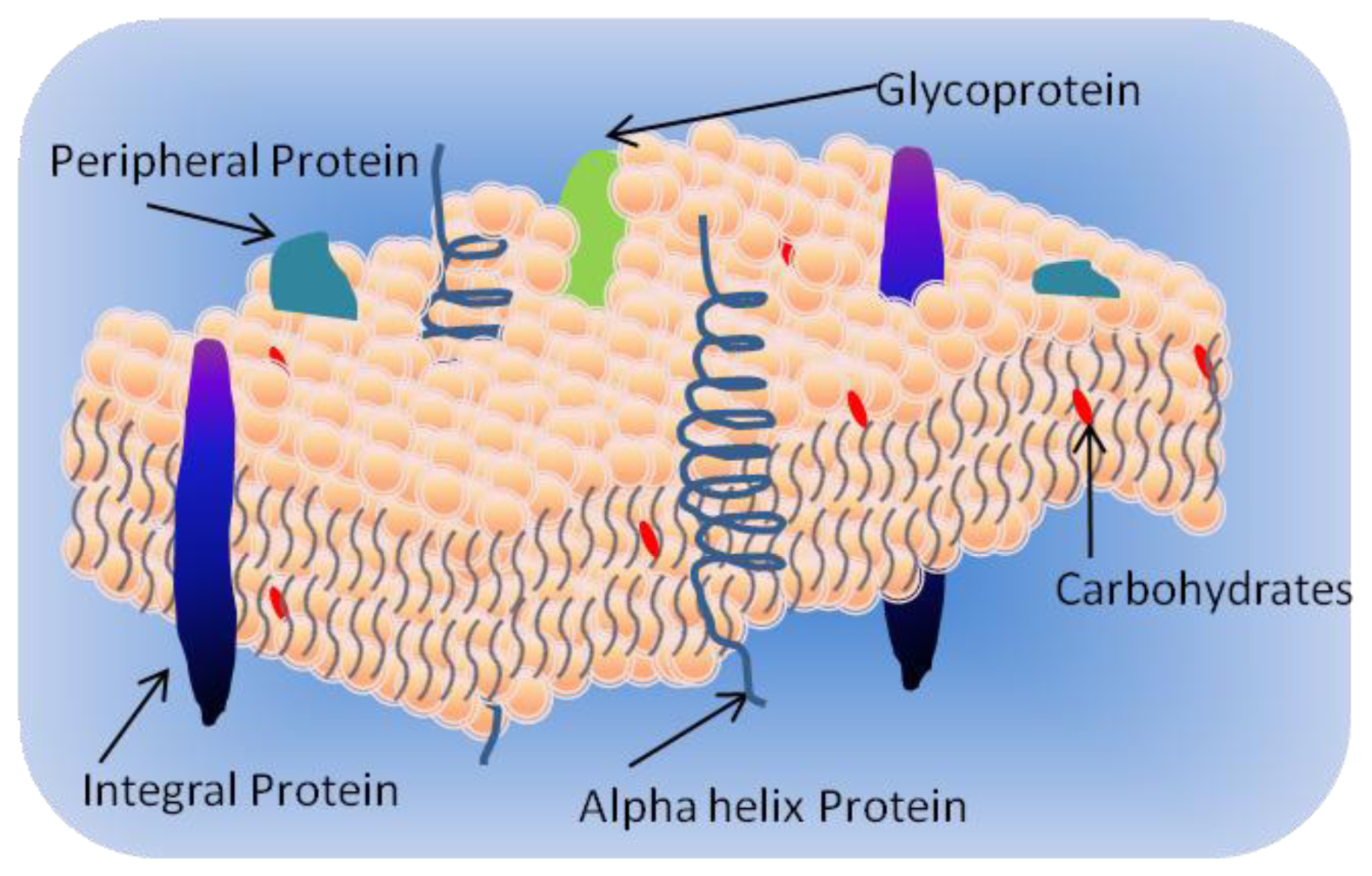
1.1. Cell Membrane
- Integral proteins which have one or more transmembrane domains;
- Peripheral proteins associated at the surface of the bilayer; and
- Lipid-anchored proteins covalently attached to the fatty acids in the membrane.
1.2. Classification of Membrane Lipids
1.2.1. Phospholipids
1.2.2. Sphingolipids
1.2.3. Sterols
1.3. Membrane Proteins
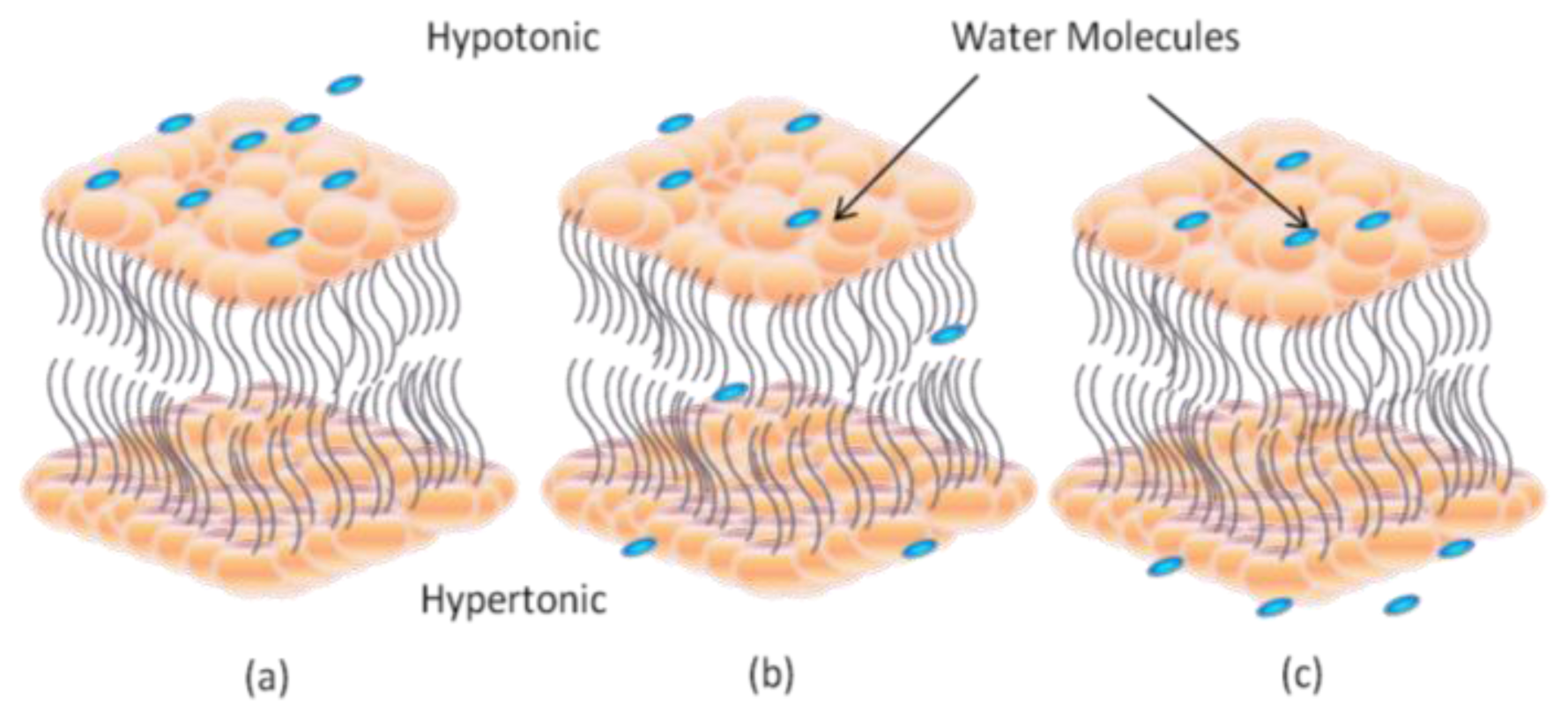
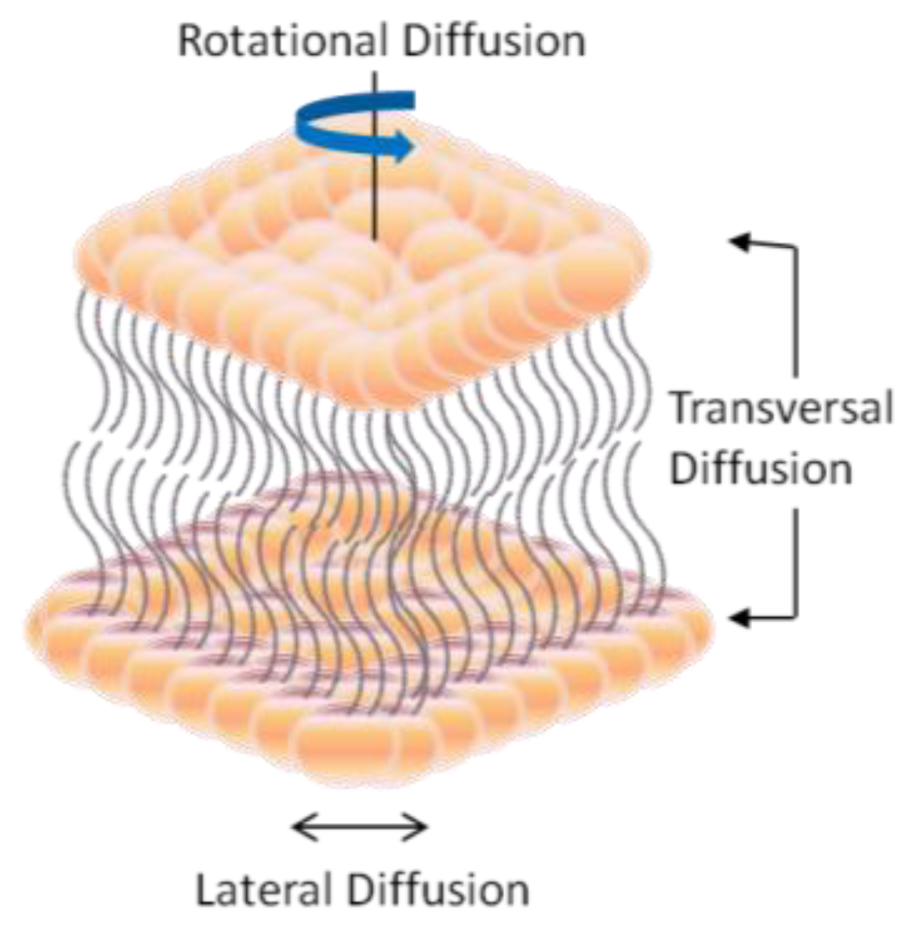
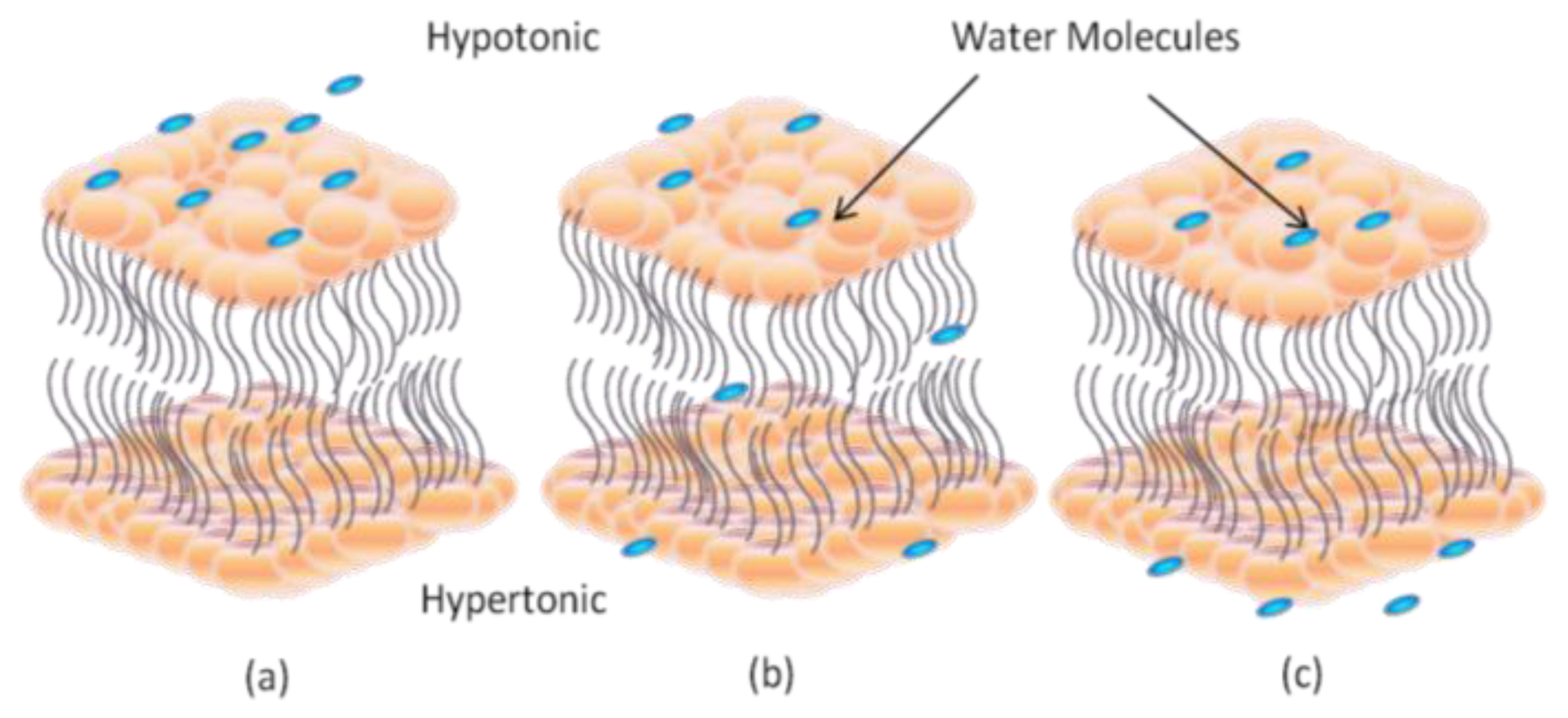
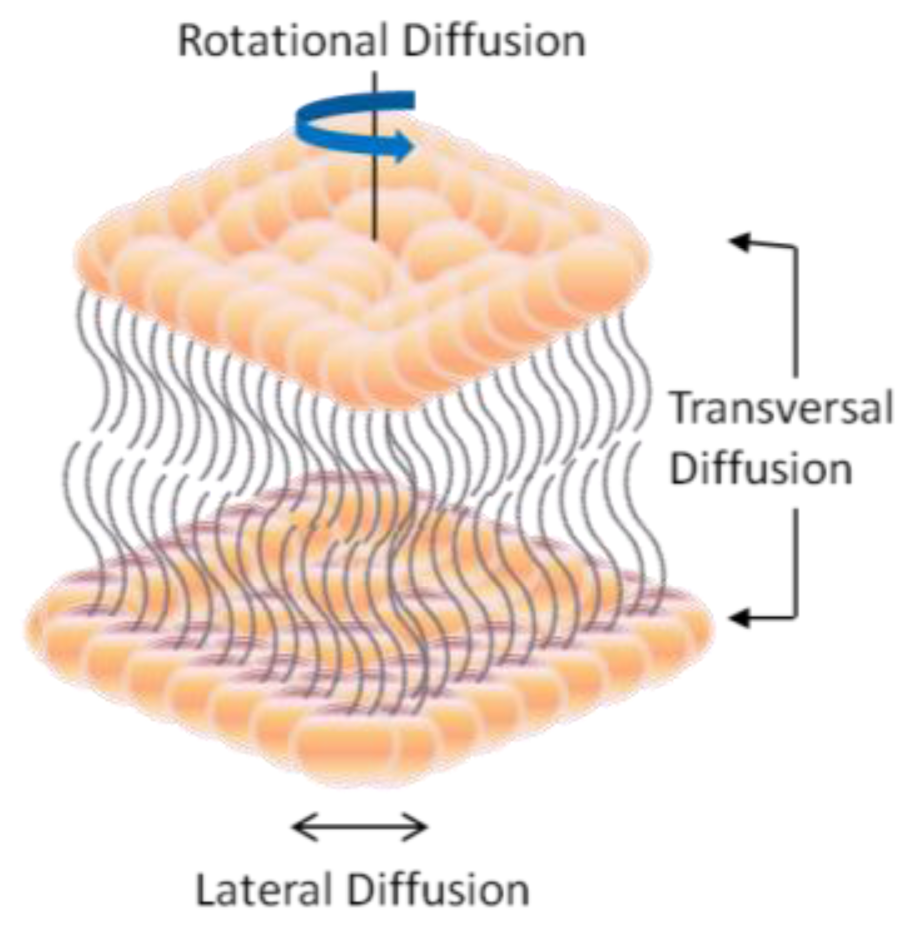
2. Dynamic Structure and Formation of Lipid Bilayer Membrane (LBM)
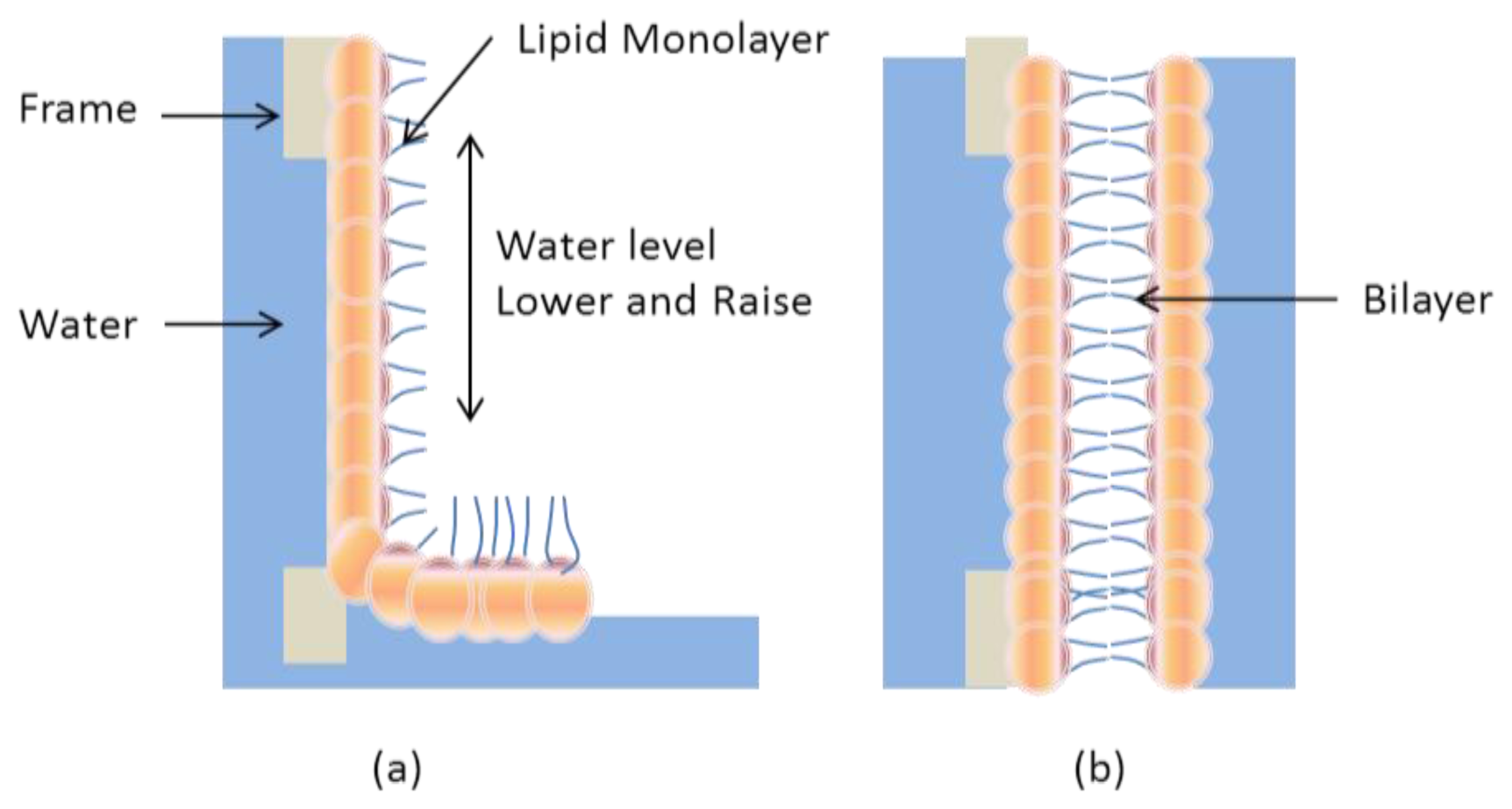
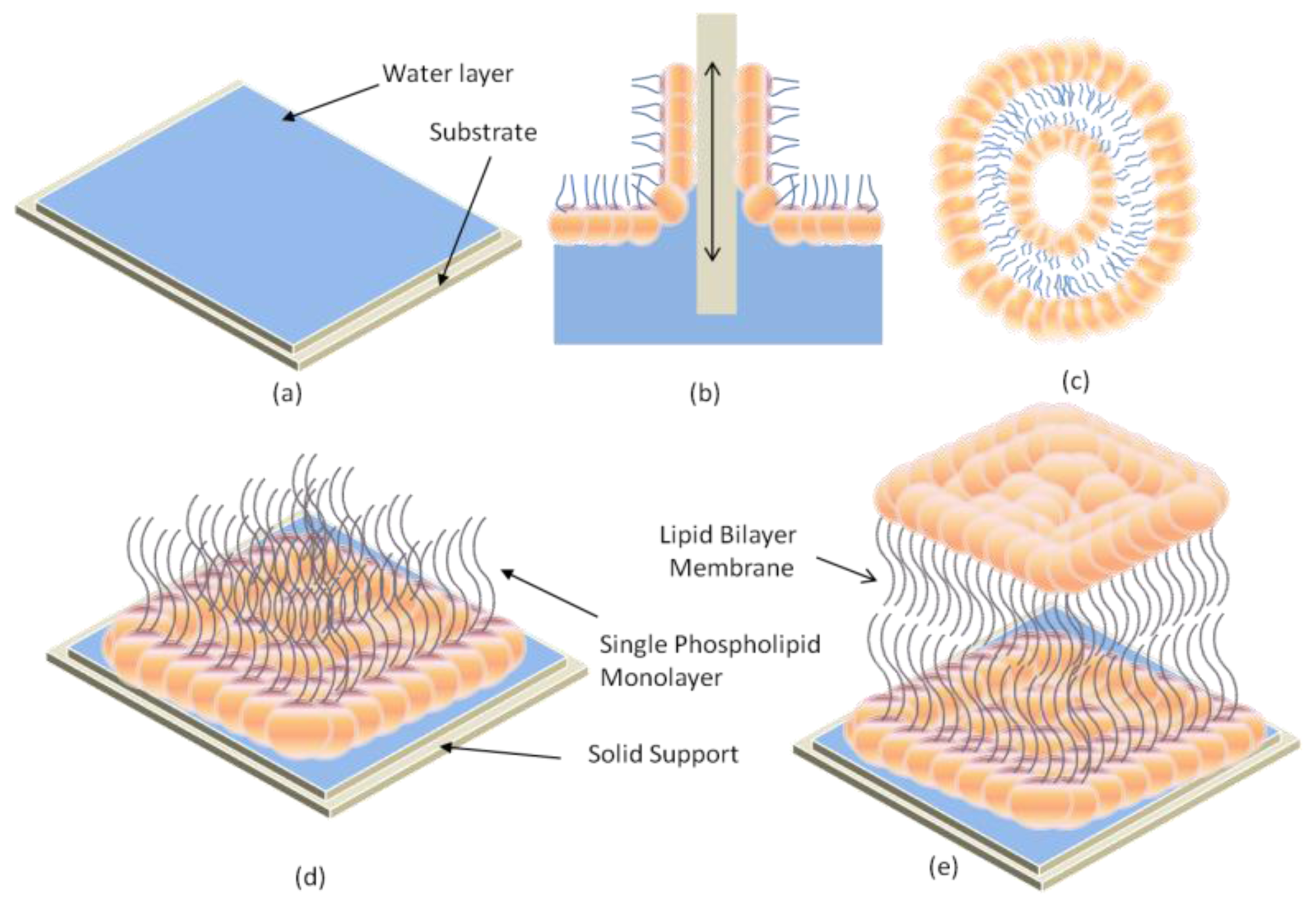
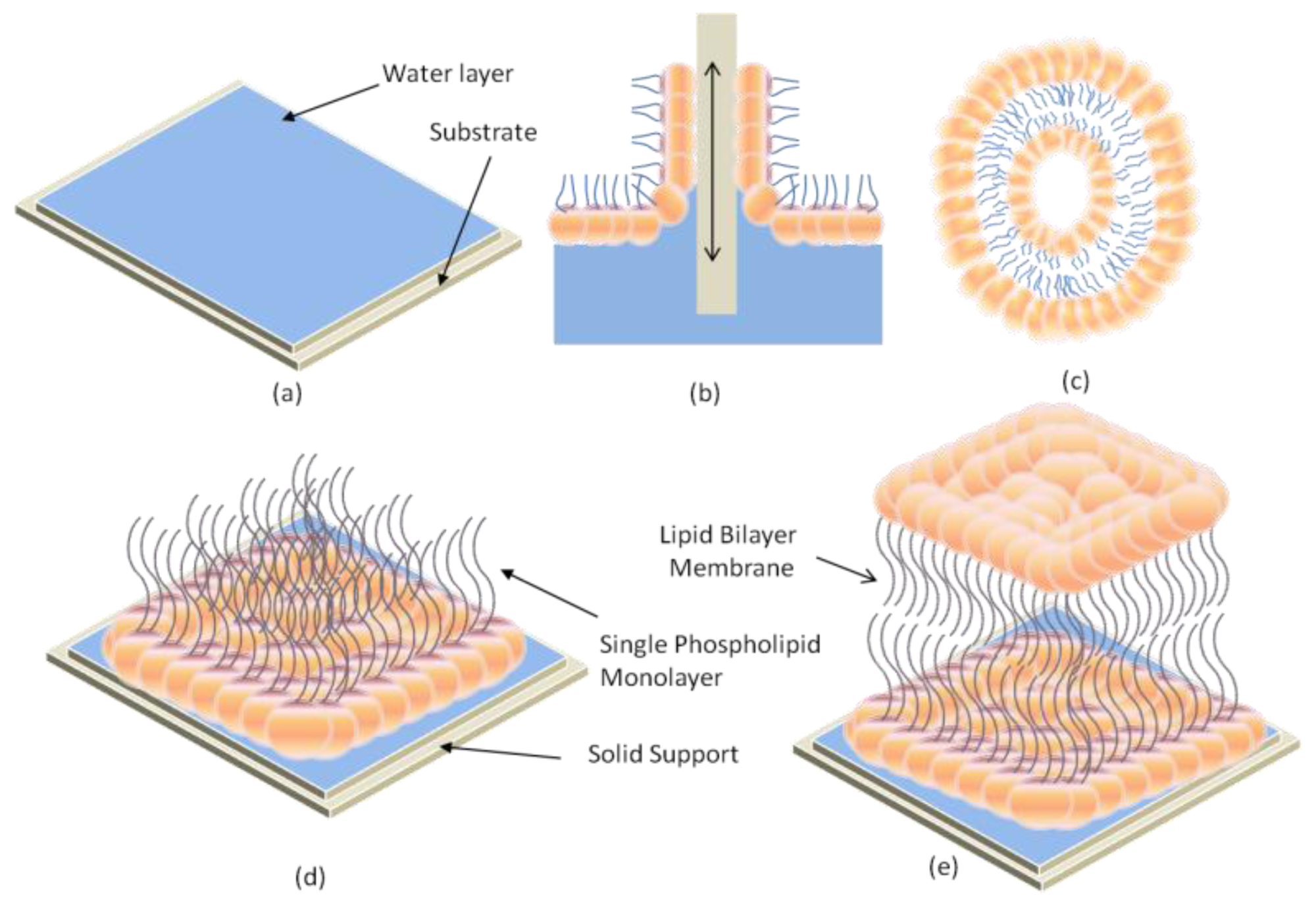
2.1. Black Lipid Membrane (BLM)
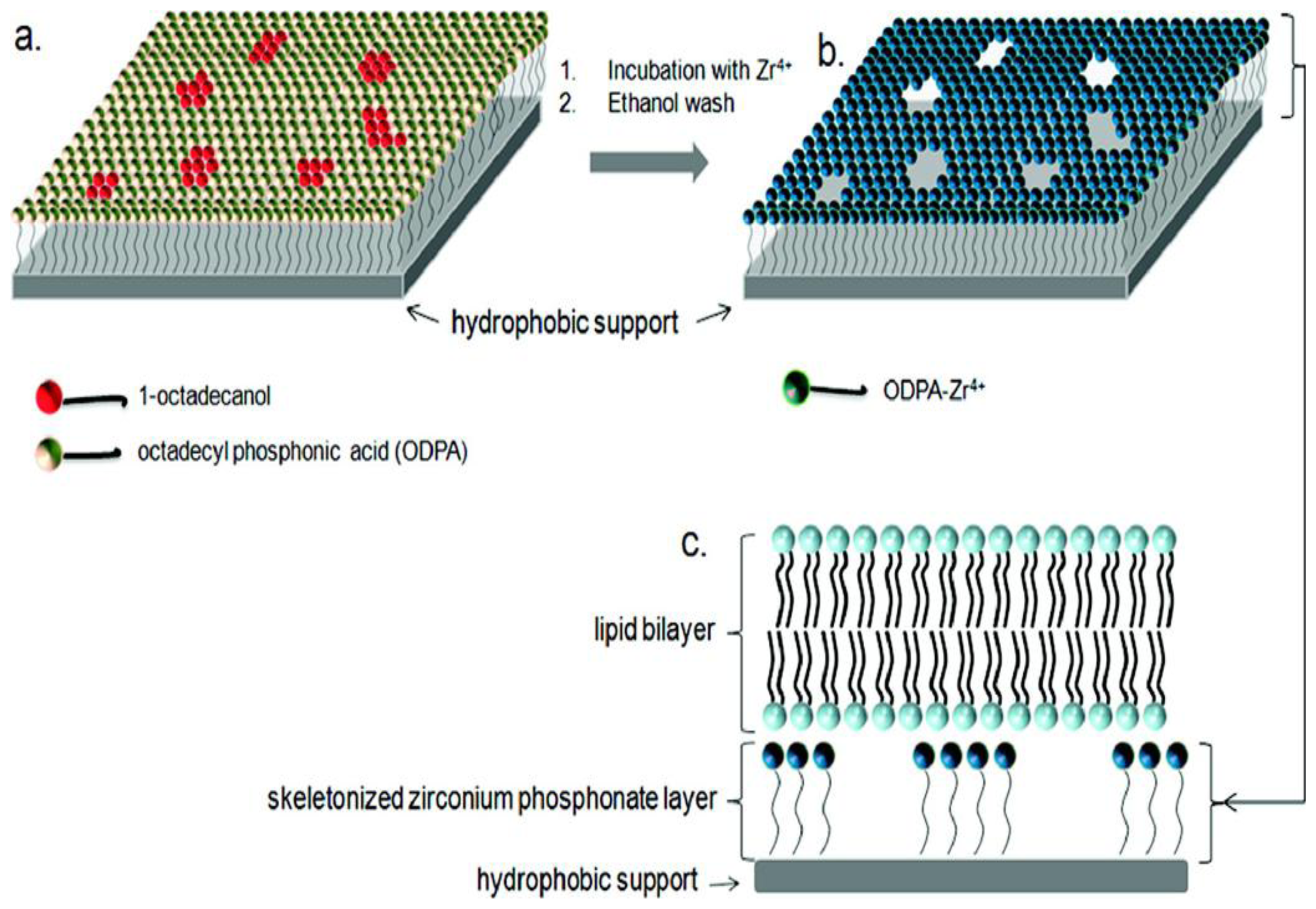
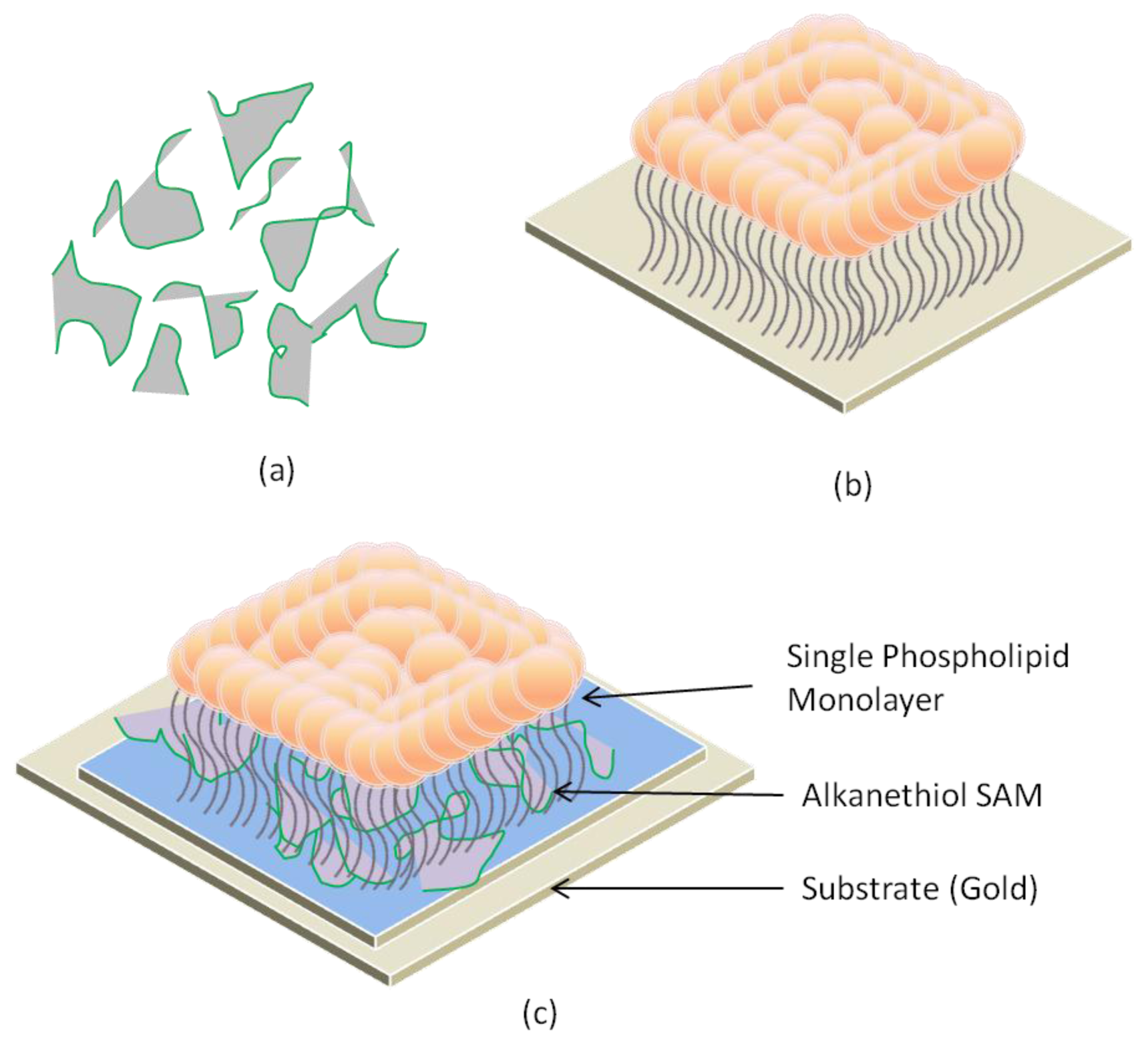
2.2. Supported Lipid Bilayer Membrane (s-LBM)
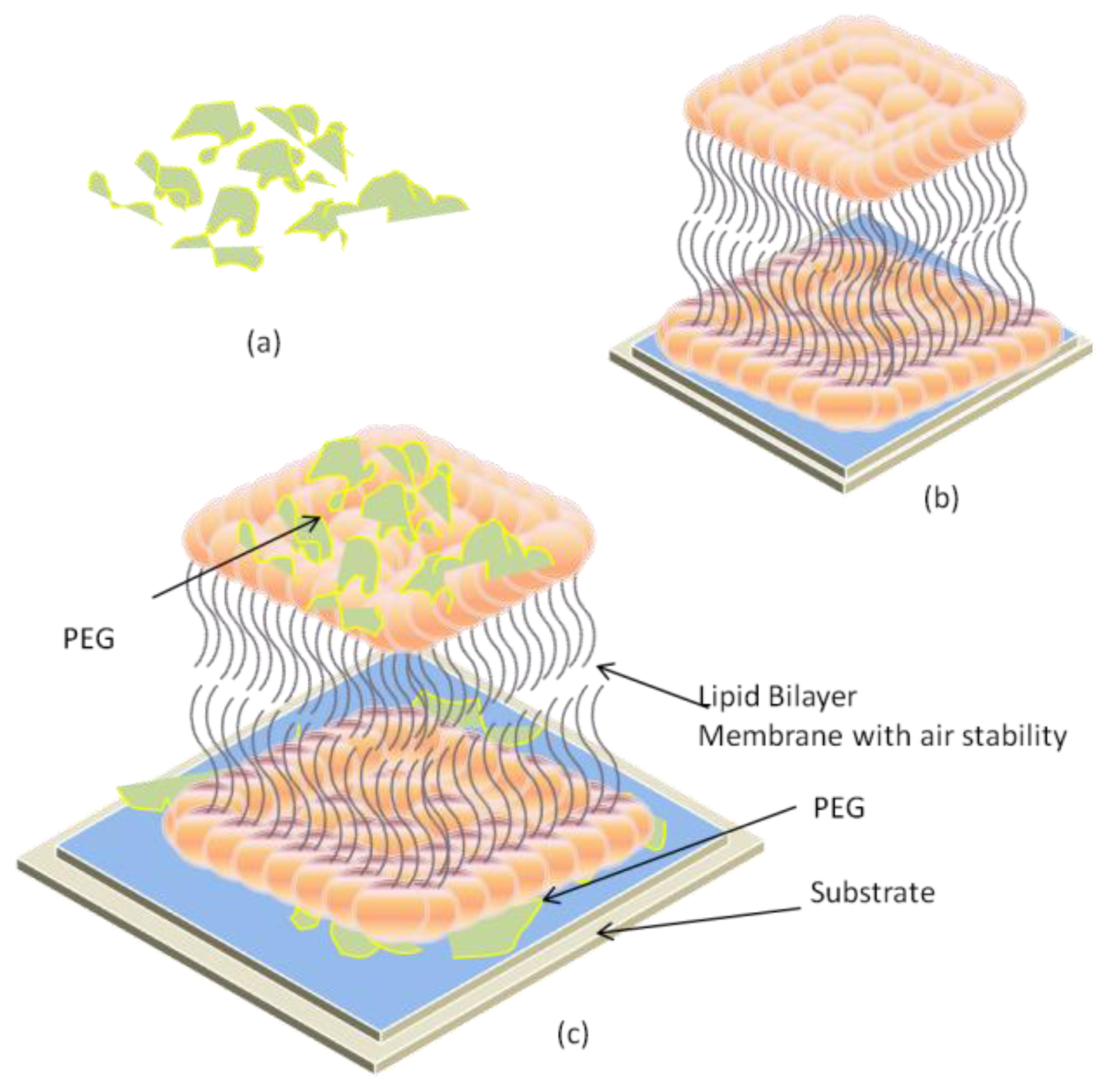
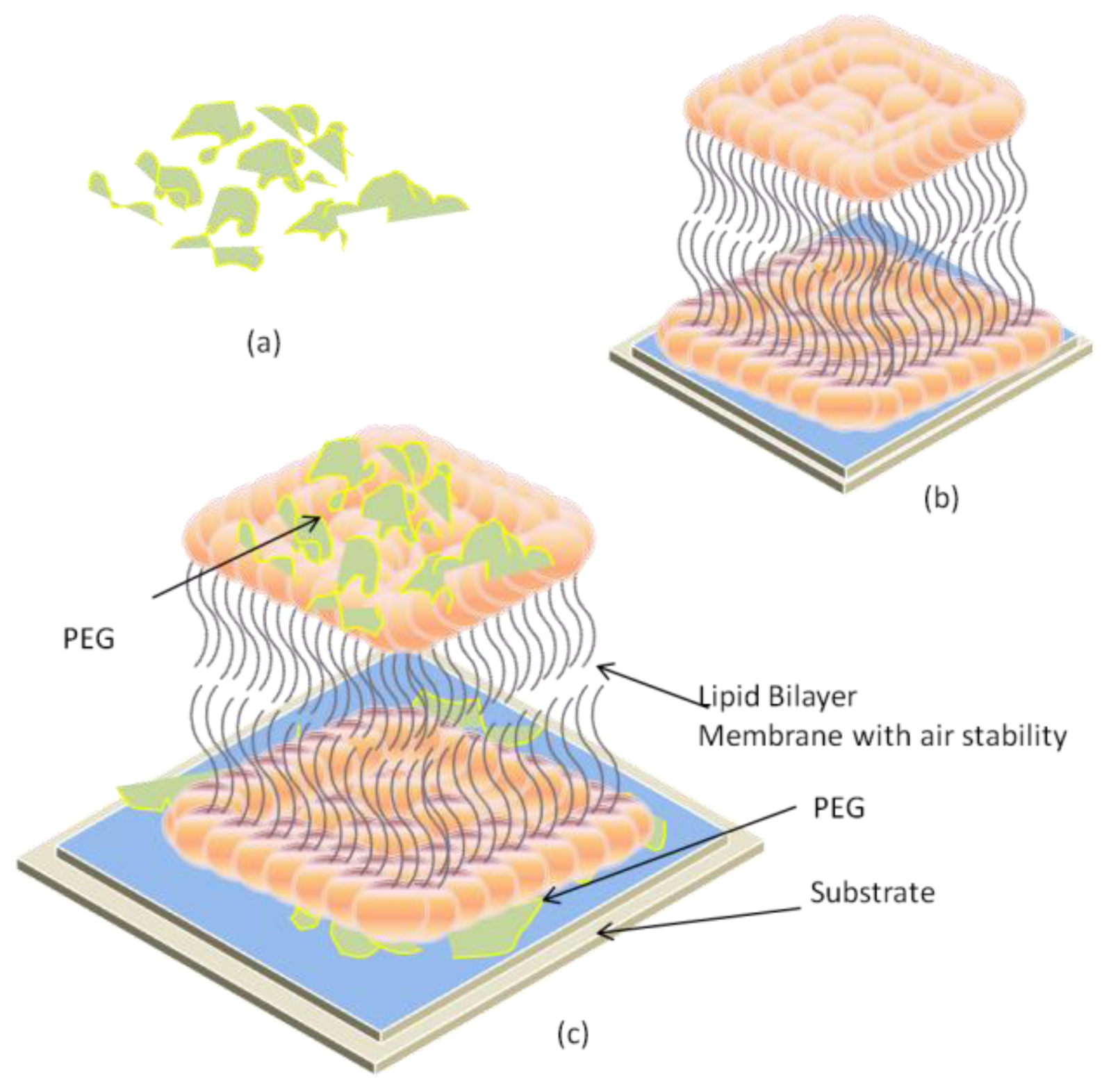
2.3. Air-Stable Lipid Bilayer Membrane (as-LBM)
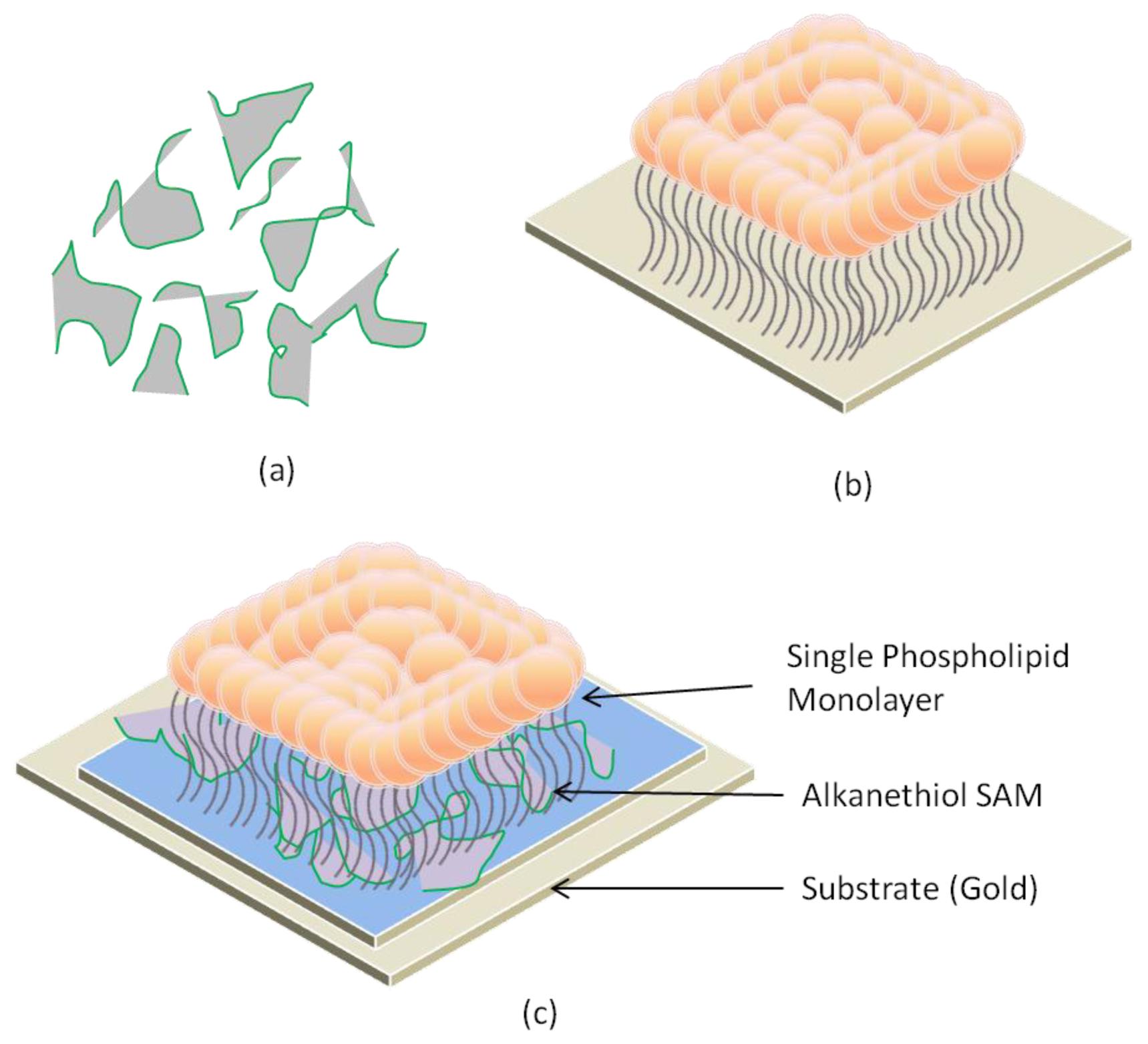
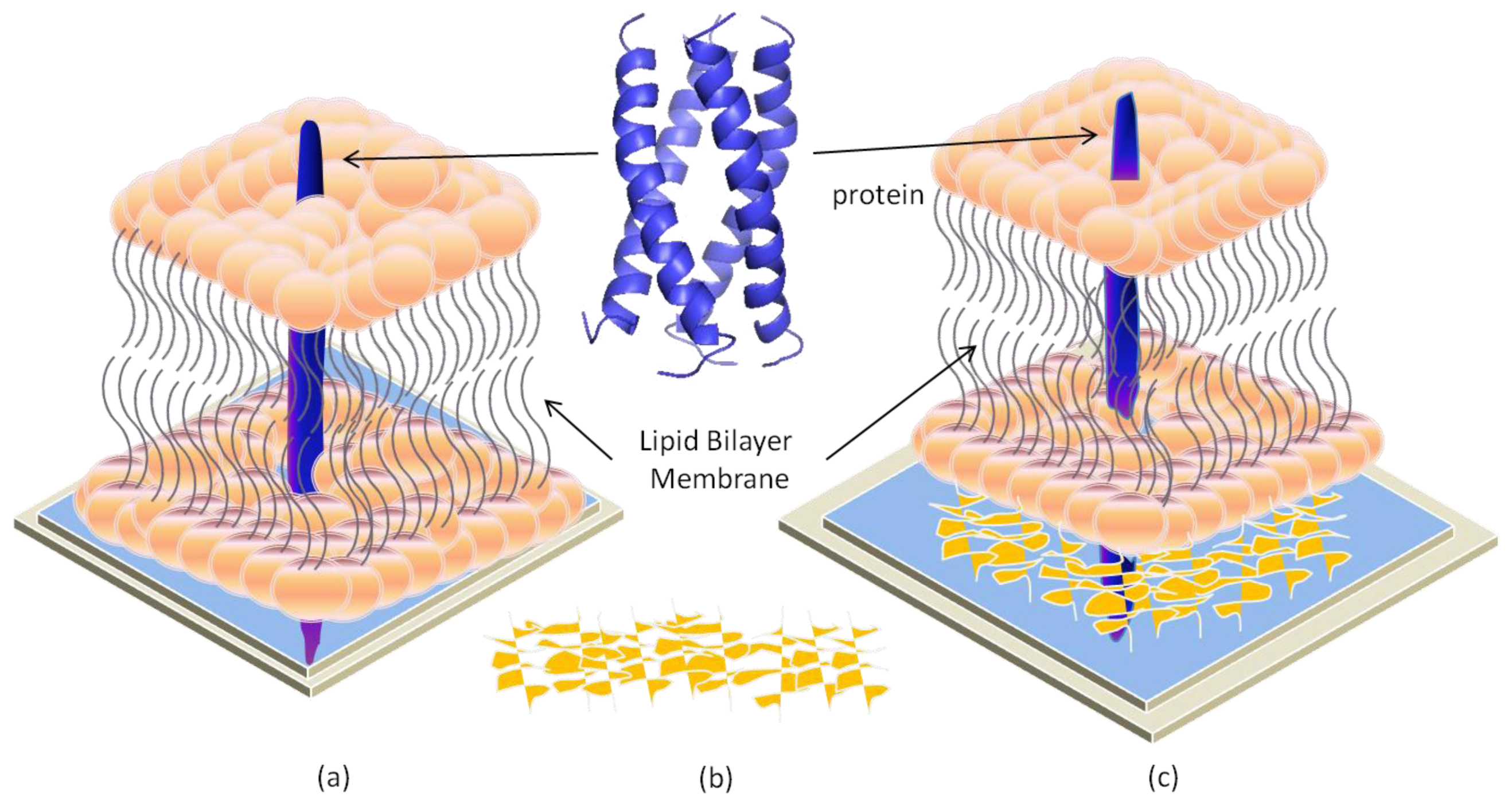
2.4. Hybrid Lipid Bilayer Membrane (h-LBM)
2.5. Polymer-Cushioned Lipid Bilayer Membrane (pc-LBM)
3. Probing Transmembrane Proteins on Solid Surfaces
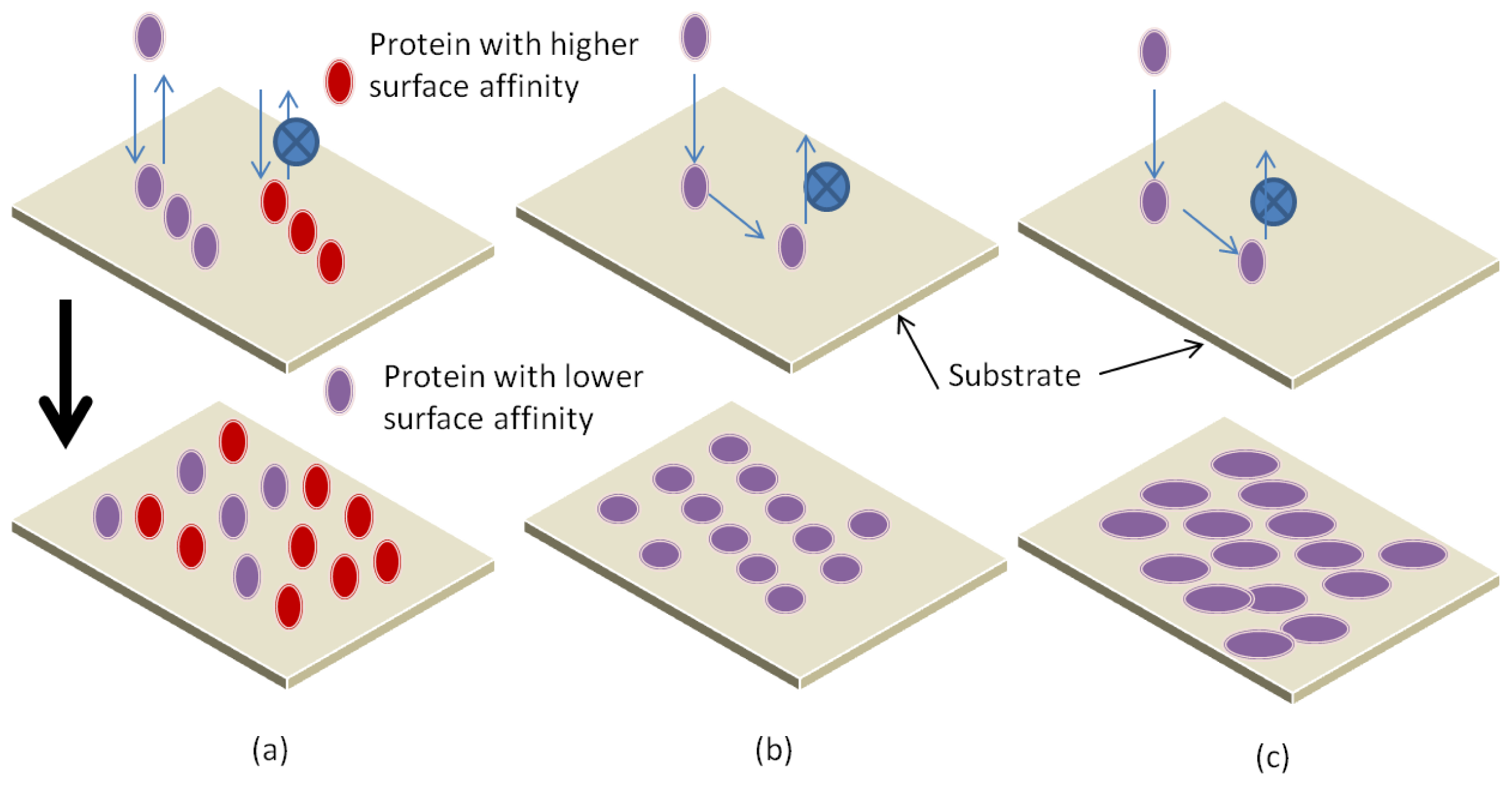
3.1. Parameters Affecting Protein Interaction with Substrate
3.2. Adsorption Mechanisms
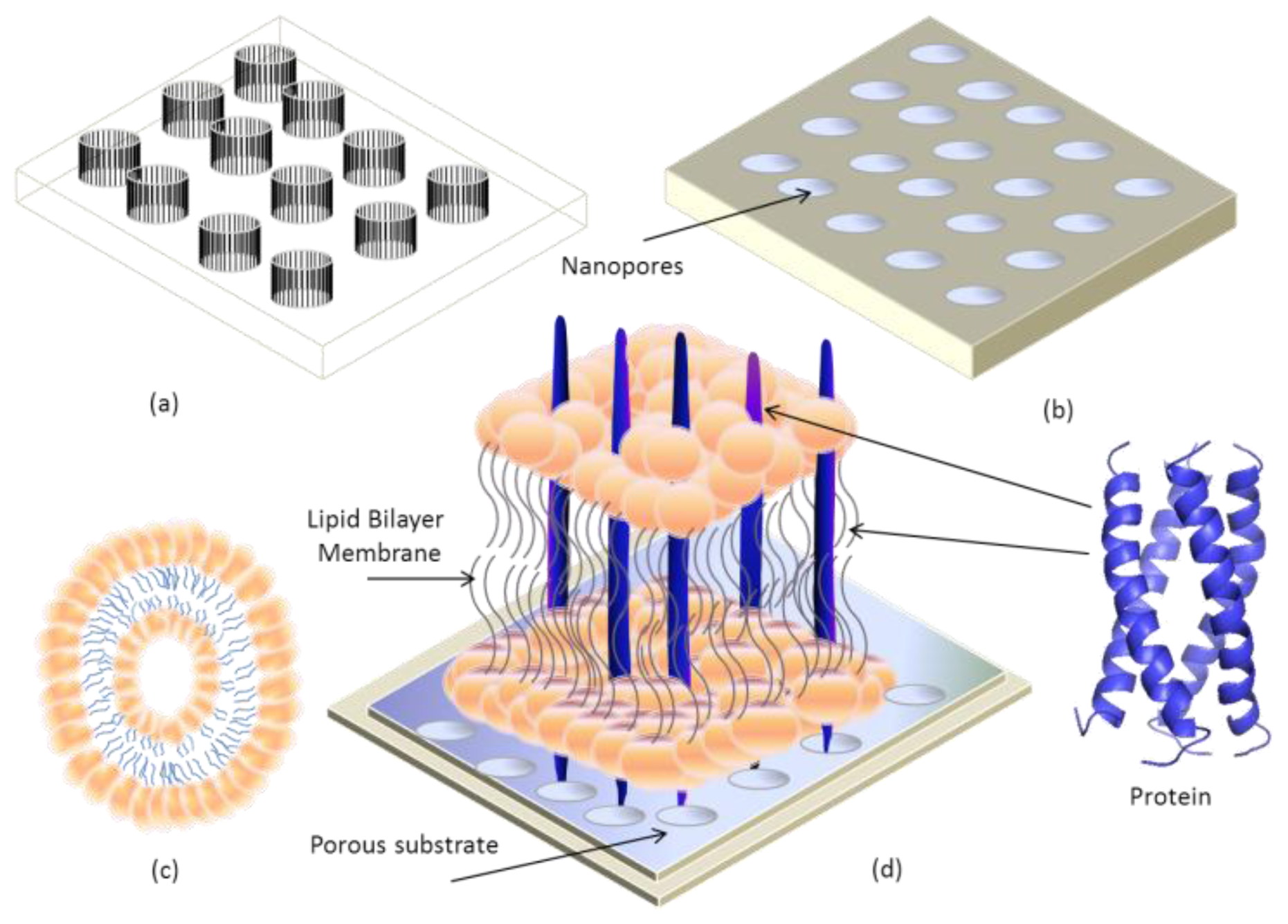
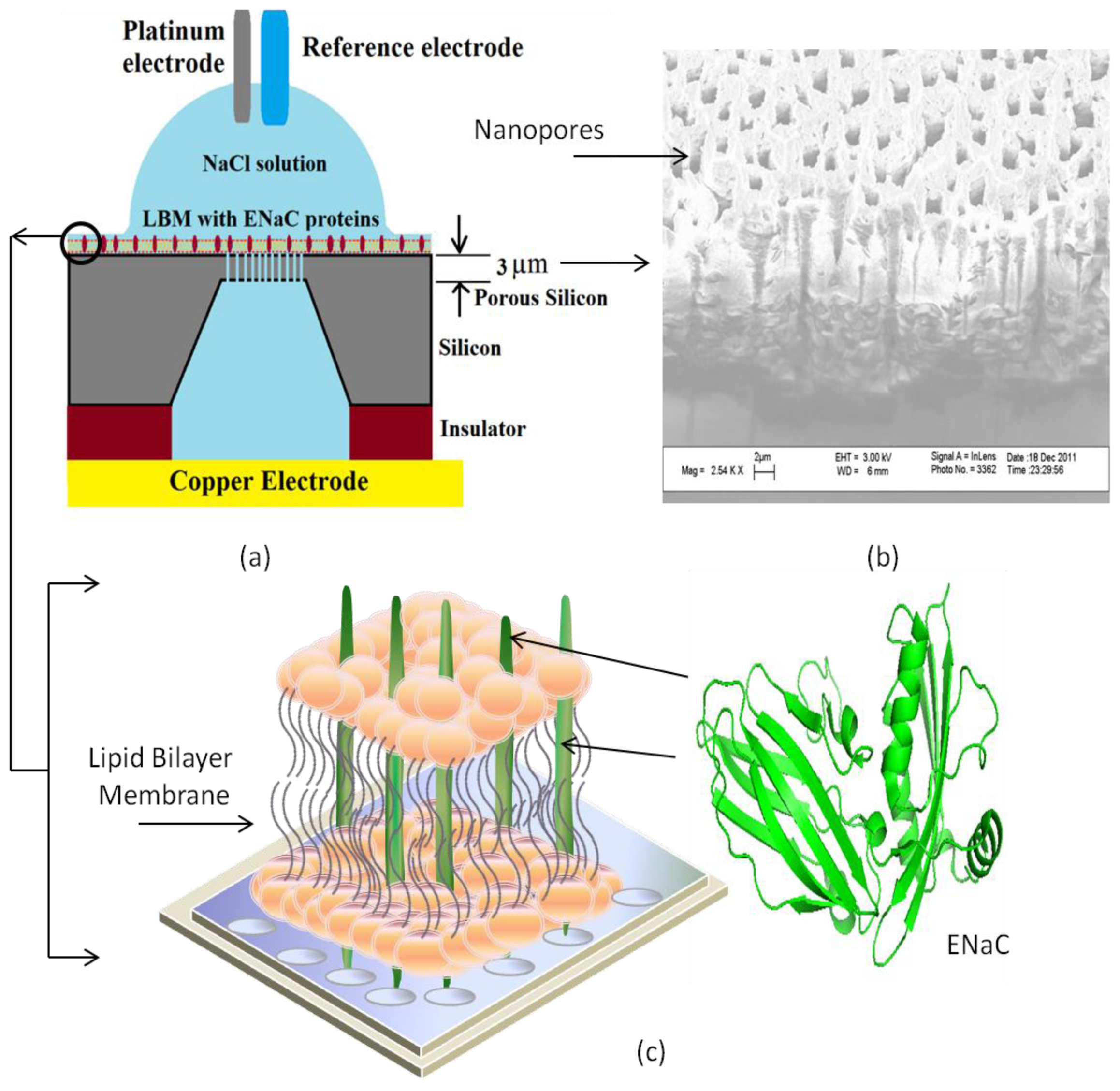
4. Transmembrane Protein Fused in Bilayer Lipid Membrane on Porous Silicon (PSi)
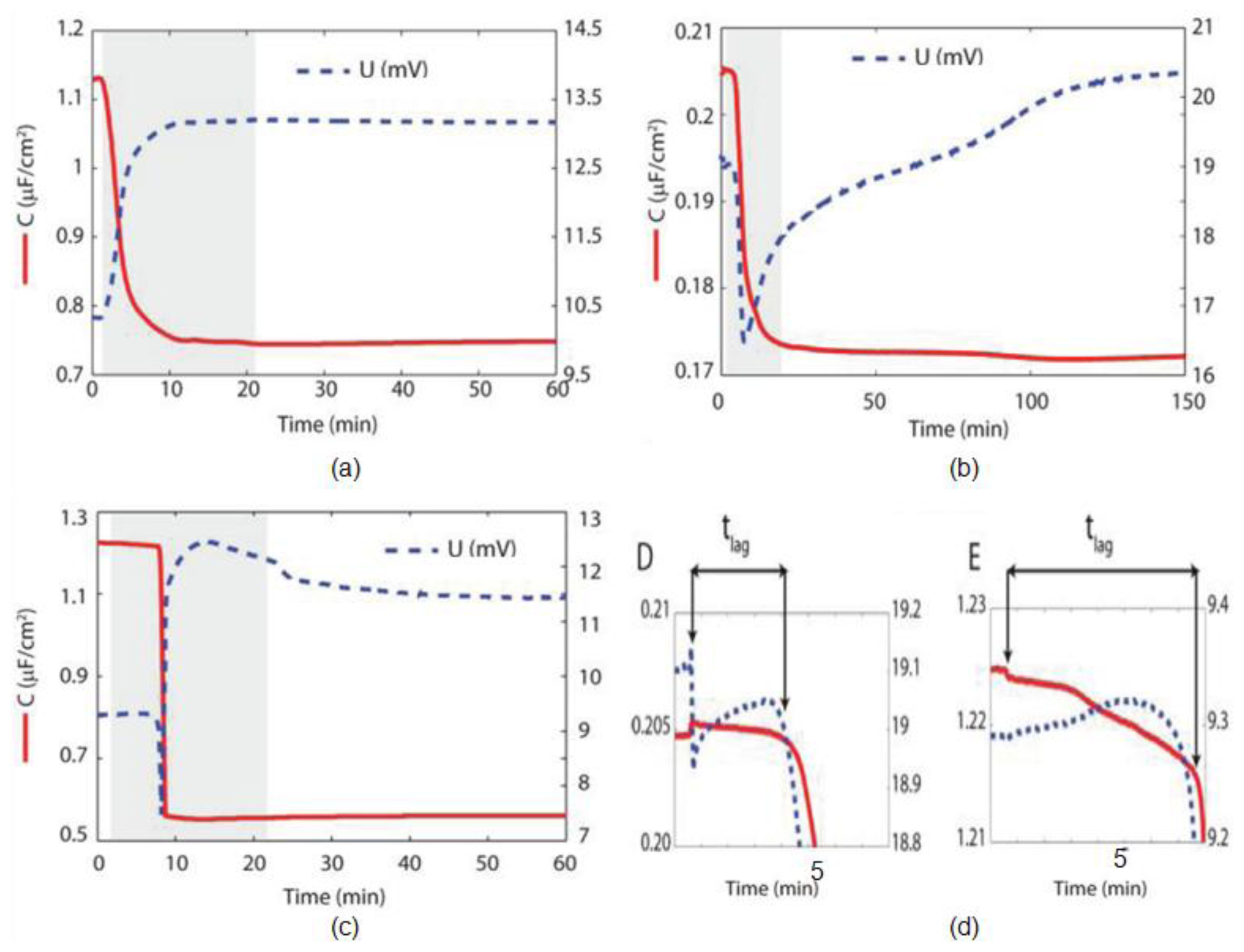
5. Electrochemical Impedance Spectroscopy
- DOPC: 1,2-di-sn-glycero-3-phosphocholine,
- TR-DHPE: Dihexadecanoyl-phosphoethanolamine,
- DPPE: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine,
- DPPTE: 1,2-dipalmitoyl-sn-glycero-3-phosphoethanol,
- DPPS: 1,2-dipalmitoyl-sn-glycero-3-phosphoserine,
- DPHPC: 1,2-diphytanoyl-sn-glycero-3-phosphocholin,
- DMPC: dimyristoylphosphatidylcholine,
- DHADAB: dihexadecyldimethylammonium bromide,
- POPG: 1-Palmitoyl-2-oleoyl-sn-glycero-3-[phosphor-rac-(1-glycerol)],
- POEPC: 1-palmitoyl-2-oleoyl-sn-glycero-3-ethylphosphocholine and
- POPC: 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine.
6. Conclusion and Future Directions






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Reference | Supporting surface | Pore size (nm) | Lipid membrane | Protein | Membrane capacitance (μFcm−2) | Membrane resistance (GΩcm2) |
|---|---|---|---|---|---|---|
| [25] | Porous alumina | 55–280 | Bilayer (DPPTE and DPHPC) | Gramicidin (A, B, C, D) | 0.65 ± 0.2 | 0.16 |
| [155] | Porous silicon nitride | 200–700 | Bilayer (DOPC) | Gramicidin (A, B, C, D) | 5.9 | 38.8–53.6 * |
| [80] | Glass slides coated with Indium-Tin-Oxide | - | Composed of 42 mol % DMPC, 49 mol % cholesterol, and 9 mol % DHADAB on regenerated cellulose by vesicle fusion | Gramicidin D | 0.57 | 44 × 10−5 |
| [150] | Porous alumina | 200 | Bilayer (DOPC and TR-DHPE) | α-hemolysin | - | 9.0 |
| [152] | Teflon with gold coated | 0.02 | Phosphatidylcholines | Gramicidin D & Proteoliposome | (1.8 ± 0.2) × 10−3 | 25.9 ± 4.1 |
| [48,159] | Porous silicon | 0.0002–0.002 | Bilayer (DPPS and DPPE) | ENaC | 0.63 | 4 × 10−6* |
| [179] | Alkanethiol (gold) | - | Monolayer (POEPC) | Gramicidin D | 2.1 ± 0.04 | - |
| SiO2 | - | Bilayer (POEPC and POPC) | 1.29 ± 0.2 | - | ||
| 1.16 ± 0.3 | - | |||||
| Bilayer (POPG) | - | 2.4 (ordered) | - | |||
| [180] | Porous alumina (ordered/non ordered) | 20–50 | Bilayer (DPPTE and DOPC) | - | 0.99 (non-ordered with gold covered alumina), 0.90 (non-ordered) | 4 × 10−4* |
Acknowledgments
Conflicts of Interest
References
- Reece, J.B.; Cain, M.L.; Urry, S.L.A. Campbell Biology, 9th ed; Benjamin Cummings: San Francisco, CA, USA, 2011; pp. 125–141. [Google Scholar]
- Luckey, M. Membrane Structural Biology: With Biochemical and Biophysical Foundations, 1st ed.; Cambridge University Press: Cambridge, UK, 2002; pp. 1–12. [Google Scholar]
- Yeagle, P. The Structure of Biological Membranes, 3rd ed.; CRC Press, Taylor & Francis Group: Boca Raton, FL, USA, 2012; pp. 1–8. [Google Scholar]
- Nikolov, A.D.; Wasan, D.T. Ordered micelle structuring in thin films formed from anionic surfactant solutions. I. Experimental. J. Colloid Interface Sci 1989, 133, 1–12. [Google Scholar]
- Voet, D.; Voet, J.G. Biochemistry, 4th ed; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2011; pp. 389–395. [Google Scholar]
- Singer, S.J.; Nicolson, G.L. The fluid mosaic model of the structure of cell membranes. Science 1972, 175, 720–731. [Google Scholar]
- Simons, K.; Ikonen, E. Functional rafts in cell membranes. Nature 1997, 397, 569–572. [Google Scholar]
- Op den Kamp, J.A.F. Lipid asymmetry in membranes. Annu. Rev. Biochem 1979, 48, 47–71. [Google Scholar]
- Zhang, Y.M.; Rock, C.O. Thematic review series: Glycerolipids, acyltransferases in bacterial glycerophospholipid synthesis. J. Lipid Res 2008, 49, 1867–1874. [Google Scholar]
- Brown, R.E. Sphingolipid organization in biomembranes: What physical studies of model membranes reveal. J. Cell Sci 1998, 111, 1–9. [Google Scholar]
- Dufourc, E.J. Sterols and membrane dynamics. J. Chem. Biol 2008, 1, 63–77. [Google Scholar]
- Meleard, P.; Gerbeaud, C.; Pott, T.; Fernandez-Puente, L.; Bivas, I.; Mitov, M.D.; Dufourcq, J.; Bothorel, P. Bending elasticities of model membranes: Influences of temperature and sterol content. Biophys. J 1997, 72, 2616–2629. [Google Scholar]
- Redondo-Morata, L.; Giannotti, M.I.; Sanz, F. Influence of cholesterol on the phase transition of lipid bilayers: A temperature-controlled force spectroscopy study. Langmuir 2012, 28, 12851–12860. [Google Scholar]
- Pucadyil, T.J.; Chattopadhyay, A. Effect of cholesterol on lateral diffusion of fluorescent lipid probes in native hippocampal membranes. Chem. Phys. Lipids 2006, 143, 11–21. [Google Scholar]
- Xu, X.; London, E. The effect of sterol structure on membrane lipid domains reveals how cholesterol can induce lipid domain formation. Biochemistry 2000, 39, 843–849. [Google Scholar]
- Smith, E.A.; Wang, W.; Dea, P.K. Effects of cholesterol on phospholipid membranes: Inhibition of the interdigitated gel phase of F-DPPC and F-DPPC/DPPC. Chem. Phys. Lipids 2012, 165, 151–159. [Google Scholar]
- De Pont, J.J.H.H.M. Molecular Aspects of Transport Proteins, 1st ed; Elsevier Science Pub Co: Amsterdam, The Netherlands, 1992; pp. 1–23. [Google Scholar]
- East, J.M.; Lee, A.G. Lipid selectivity of the calcium and magnesium ion dependent adenosine triphosphatase, studied with fluorescence quenching by a brominated phospholipid. Biochemistry 1982, 21, 6441–6446. [Google Scholar]
- Cui, H.; Mim, C.; Vázquez, F.X.; Lyman, E.; Unger, V.M.; Voth, G.A. Understanding the role of amphipathic helices in N-BAR domain driven membrane remodeling. Biophys. J 2013, 104, 404–411. [Google Scholar]
- McMahon, H.T.; Gallop, J.L. Membrane curvature and mechanisms of dynamic cell membrane remodelling. Nature 2005, 438, 590–596. [Google Scholar]
- Muller, P.; Herrmann, A. Rapid transbilayer movement of spin-labeled steroids in human erythrocytes and in liposomes. Biophys. J 2002, 82, 1418–1428. [Google Scholar]
- Mueller, P.; Rudin, D.O.; Tien, H.T.; Westcott, W.C. Reconstitution of excitable cell membrane structure in vitro. Circulation 1962, 26, 1167–1171. [Google Scholar]
- Shalaev, E.Y.; Steponkus, P.L. Phase diagram of 1,2-dioleoylphosphatidylethanolamine (DOPE): Water system at subzero temperatures and at low water contents. Biochim. Biophys. Acta 1999, 1419, 229–247. [Google Scholar]
- Montal, M.; Mueller, P. Formation of bimolecular membranes from lipid monolayers and a study of their electrical properties. Proc. Nat. Acad. Sci. USA 1972, 69, 3561–3566. [Google Scholar]
- Römer, W.; Steinem, C. Impedance analysis and single-channel recordings on nano-black lipid membranes based on porous alumina. Biophys. J 2004, 86, 955–965. [Google Scholar]
- Gomezlagunas, F.; Pena, A.; Lievano, A.; Darszon, A. Incorporation of ionic channels from yeast plasma membranes into black lipid membranes. Biophys. J 1989, 56, 115–119. [Google Scholar]
- Van Gelder, P.; Dumas, F.; Winterhalter, M. Understanding the function of bacterial outer membrane channels by reconstituting into black lipid membranes. Biophys. Chem 2000, 85, 153–167. [Google Scholar]
- Castellana, E.T.; Cremer, P.S. Solid supported lipid bilayers: From biophysical studies to sensor design. Surf. Sci. Rep 2006, 61, 429–444. [Google Scholar]
- Ryan, S.R.; Hyeon, C.; Rikard, B.; Francisco, B.; James, R.H. Black lipid membranes: Visualizing the structure, dynamics, and substrate dependence of membranes. J. Phys. Chem. B 2004, 108, 16040–16049. [Google Scholar]
- Tien, H.T. Black lipid membranes: Thickness determination and molecular organization by optical methods. J. Theor. Biol 1967, 16, 97–110. [Google Scholar]
- Starr, T.E.; Thompson, N.L. Total internal reflection with fluorescence correlation spectroscopy: Combined surface reaction and solution diffusion. Biophys. J 2001, 80, 1575–1584. [Google Scholar]
- Cremer, P.S.; Boxer, S.G. Formation and spreading of lipid bilayers on planar glass supports. J. Phys. Chem. B 1999, 103, 2554–2559. [Google Scholar]
- Egawa, H.; Furusawa, K. Liposome adhesion on mica surface studied by atomic force microscopy. Langmuir 1999, 15, 1660–1666. [Google Scholar]
- Starr, T.E.; Thompson, N.L. Formation and characterization of planar phospholipid bilayers supported on TiO2 and SrTiO3 single crystals. Langmuir 2000, 16, 10301–10308. [Google Scholar]
- Ajo-Franklin, C.M.; Kam, L.; Boxer, S.G. High refractive index substrates for fluorescence microscopy of biological interfaces with high z contrast. Proc. Nat. Acad. Sci. USA 2001, 98, 13643–13648. [Google Scholar]
- Rossetti, F.F.; Bally, M.; Michel, R.; Textor, M.; Reviakine, I. Interaction between titanium dioxide and phosphatidyl serine-containing liposomes: Formation and patterning of supported phospholipid bilayers on the surface of a medically relevant material. Langmuir 2005, 21, 6443–6450. [Google Scholar]
- Fabre, R.M.; Okeyo, G.O.; Talham, D.R. Supported lipid bilayers at skeletonized surfaces for the study of transmembrane proteins. Langmuir 2012, 28, 2835–2841. [Google Scholar]
- Petty, M.C. Langmuir-Blodgett Films: An Introduction; Cambridge University Press: Cambridge, UK, 1996. [Google Scholar]
- Jass, J.; Tjärnhage, T.; Puu, G. From liposomes to supported, planar bilayer structures on hydrophilic and hydrophobic surfaces: An atomic force microscopy study. Biophys. J 2000, 79, 3153–3163. [Google Scholar]
- Kalb, E.; Frey, S.; Tamm, L.K. Formation of supported planar bilayers by fusion of vesicles to supported phospholipid monolayers. Biochim. Biophys. Acta 1992, 1103, 307–316. [Google Scholar]
- Nollert, P.; Kiefer, H.; Jähnig, F. Lipid vesicle adsorption versus formation of planar bilayers on solid surfaces. Biophys. J 1995, 69, 1447–1455. [Google Scholar]
- Mimms, L.T.; Zampighi, G.; Nozaki, Y.; Tanford, C.; Reynolds, J.A. Phospholipid vesicle formation and transmembrane protein incorporation using octyl glucoside. Biochemistry 1981, 20, 833–840. [Google Scholar]
- Nayar, R.; Hope, M.J.; Cullis, P.R. Generation of large unilamellar vesicles from long-chain saturated phosphatidylcholines by extrusion technique. Biochim. Biophys. Acta 1989, 986, 200–206. [Google Scholar]
- Reimhult, E.; Hook, F.; Kasemo, B. Intact vesicle adsorption and supported biomembrane formation from vesicles in solution: Influence of surface chemistry, vesicle size, temperature, and osmotic pressure. Langmuir 2003, 19, 1681–1691. [Google Scholar]
- Stelzle, M.; Weissmüller, G.; Sackmann, E. On the application of supported bilayers as receptive layers for biosensors with electrical detection. J. Phys. Chem 1993, 97, 2974–2981. [Google Scholar]
- Gritsch, S.; Nollert, P.; Jahnig, F.; Sackmann, E. Impedance spectroscopy of porin and gramicidin pores reconstituted into supported lipid bilayers on indium-tin-oxide electrodes. Langmuir 1998, 14, 3118–3125. [Google Scholar]
- Berquand, A.; Mazeran, P.E.; Pantigny, J.; Proux-Delrouyre, V.; Laval, J.M.; Bourdillon, C. Two-step formation of streptavidin-supported lipid bilayers by PEG-triggered vesicle fusion. Fluorescence and atomic force microscopy characterization. Langmuir 2003, 19, 1700–1707. [Google Scholar]
- Tantawi, K.H.; Cerro, R.; Berdiev, B.; Williams, J. Investigations on transmembrane ion channels suspended over porous silicon membranes. Nanotechnology 2013, 3, 198–201. [Google Scholar]
- Tantawi, K.H.; Cerro, R.; Berdiev, B.; Martina-Diaz, M.; Montez, F.; Patel, D.; Williams, J. Investigation of transmembrane protein fused in lipid bilayer membranes supported on porous silicon. J. Med. Eng. Technol 2013, 37, 28–34. [Google Scholar]
- Plant, A.L. Supported hybrid bilayer membrane as rugged cell membrane mimics. Langmuir 1999, 15, 5128–5135. [Google Scholar]
- Holden, M.A.; Jung, S.Y.; Yang, T.L.; Castellana, E.T.; Cremer, P.S. Creating fluid and air-stable solid supported lipid bilayers. J. Am. Chem. Soc 2004, 126, 6512–6513. [Google Scholar]
- Conboy, J.C.; Liu, S.C.; O’Brien, D.F.; Saavedra, S.S. Planar supported bilayer polymers formed from bis-diene lipids by Langmuir-Blodgett deposition and UV irradiation. Biomacromolecules 2003, 4, 841–849. [Google Scholar]
- Morigaki, K.; Kiyosue, K.; Taguchi, T. Micropatterned composite membranes of polymerized and fluid lipid bilayers. Langmuir 2004, 20, 7729–7735. [Google Scholar]
- Albertorio, F.; Diaz, A.J.; Yang, T.L.; Chapa, V.A.; Kataoka, S.; Castellana, E.T.; Cremer, P.S. Fluid and air-stable lipopolymer membranes for biosensor applications. Langmuir 2005, 21, 7476–7482. [Google Scholar]
- Charrier, A.; Thibaudau, F. Main phase transitions in supported lipid single-bilayer. Biophys. J 2005, 89, 1094–1101. [Google Scholar]
- Tanaka, M.; Sackmann, E. Polymer-supported membranes as models of the cell surface. Nature 2005, 437, 656–663. [Google Scholar]
- Zhang, L.F.; Spurlin, T.A.; Gewirth, A.A.; Granick, S. Electrostatic stitching in gel-phase supported phospholipid bilayers. J. Phys. Chem. B 2006, 110, 33–35. [Google Scholar]
- Deng, Y.; Wang, Y.; Holtz, B.; Li, J.; Traaseth, N.; Veglia, G.; Stottrup, B.J.; Elde, R.; Pei, D.; Guo, A.; et al. Fluidic and air-stable supported lipid bilayer and cell-mimicking microarrays. J. Am. Chem. Soc 2008, 130, 6267–6271. [Google Scholar]
- Zhang, Y.; Chen, Y.; Jin, G. PEGylated phospholipid membrane on polymer cushion and its interaction with cholesterol. Langmuir 2010, 26, 11140–11144. [Google Scholar]
- Meuse, C.W.; Krueger, S.; Majkrzak, C.F.; Dura, J.A.; Fu, J.; Connor, J.T.; Plant, A.L. Hybrid bilayer membranes in air and water: Infrared spectroscopy and neutron reflectivity studies. Biophys. J 1998, 74, 1388–1398. [Google Scholar]
- Han, X.; Critchley, K.; Zhang, L.; Pradeep, S.N.D.; Bushby, R.J.; Evans, S.D. A novel method to fabricate patterned bilayer lipid membranes. Langmuir 2007, 23, 1354–1358. [Google Scholar]
- Glazier, S.A.; Vanderah, D.J.; Plant, A.L.; Bayley, H.; Valincius, G.; Kasianowicz, J.J. Reconstitution of the pore-forming toxin a-hemolysin in phospholipid/18-octadecyl-1-thiahexa (ethylene oxide) and phospholipid/n-octadecanethiol supported bilayer membranes. Langmuir 2000, 16, 10428–10435. [Google Scholar]
- Valincius, G.; Meškauskas, T.; Ivanauskas, F. Electrochemical impedance spectroscopy of tethered bilayer membranes. Langmuir 2012, 28, 977–990. [Google Scholar]
- Robertson, J.W.; Friedrich, M.G.; Kibrom, A.; Knoll, W.; Naumann, R.L.; Walz, D. Modeling ion transport in tethered bilayer lipid membranes. 1. Passive ion permeation. J. Phys. Chem. B 2008, 112, 10475–10482. [Google Scholar]
- Spencelayh, M.J.; Cheng, Y.L.; Bushby, R.J.; Bugg, T.D.H.; Li, J.J.; Henderson, P.L.F.; O’Reilly, J.; Evans, S.D. Antibiotic action and peptidoglycan formation on tethered lipid bilayer membranes. Angew. Chem. Int. Ed 2006, 45, 2111–2116. [Google Scholar]
- Jeuken, L.J.C.; Connell, S.D.; Nurnabi, M.; O’Reilly, J.; Henderson, P.J.F.; Evans, S.D.; Bushby, R.J. Direct electrochemical interaction between a modified gold electrode and a bacterial membrane extract. Langmuir 2005, 21, 1481–1488. [Google Scholar]
- Terrettaz, S.; Mayer, M.; Vogel, H. Highly electrically insulating tethered lipid bilayers for probing the function of ion channel proteins. Langmuir 2003, 19, 5567–5569. [Google Scholar]
- Terrettaz, S.; Vogel, H. Investigating the function of ion channels in tethered lipid membranes by impedance spectroscopy. MRS Bull 2005, 30, 207–210. [Google Scholar]
- Kalb, E.; Tamm, L.K. Incorporation of cytochrome b5 into supported phospholipid bilayers by vesicle fusion to supported monolayers. Thin Solid Films 1992, 210/211, 763–765. [Google Scholar]
- Rossi, C.; Chopineau, J. Biomimetic tethered lipid membranes designed for membrane-protein interaction studies. Eur. Biophys. J 2007, 36, 955–965. [Google Scholar]
- Sackmann, E.; Tanaka, M. Supported membranes on soft polymer cushions: Fabrication, characterization and applications. Trends Biotechnol 2000, 18, 58–64. [Google Scholar]
- Wagner, M.L.; Tamm, L.K. Tethered polymer-supported planar lipid bilayers for reconstitution of integral membrane proteins: Silane-polyethyleneglycol-lipid as a cushion and covalent linker. Biophys. J 2000, 79, 1400–1414. [Google Scholar]
- Renner, L.; Pompe, T.; Lemaitre, R.; Drechsel, D.; Werner, C. Controlled enhancement of transmembrane enzyme activity in polymer cushioned supported bilayer membranes. Soft Matter 2010, 6, 5382–5389. [Google Scholar]
- Kibrom, A.; Roskamp, R.F.; Jonas, U.; Menges, B.; Knoll, W.; Paulsen, H.; Naumann, R.L. Hydrogel-supported protein-tethered bilayer lipid membranes: A new approach toward polymer-supported lipid membranes. Soft Matter 2011, 7, 237–246. [Google Scholar]
- Zhang, L.Q.; Longo, M.L.; Stroeve, P. Mobile phospholipid bilayers supported on a polyion/alkylthiol layer pair. Langmuir 2000, 16, 5093–5099. [Google Scholar]
- Wong, J.Y.; Park, C.K.; Seitz, M.; Israelachvili, J. Polymer-cushioned bilayers. II. An investigation of interaction forces and fusion using the surface forces apparatus. Biophys. J 1999, 77, 1458–1468. [Google Scholar]
- Ma, C.; Srinivasan, M.P.; Waring, A.J.; Lehrer, R.I.; Longo, M.L.; Stroeve, P. Supported lipid bilayers lifted from the substrate by layer-by-layer polyion cushions on self-assembled monolayers. Colloids Surf. B 2003, 28, 319–329. [Google Scholar]
- Kugler, R.; Knoll, W. Polyelectrolyte-supported lipid membranes. Bioelectrochemistry 2002, 56, 175–178. [Google Scholar]
- Lee, H.; Dellatore, S.M.; Miller, W.M.; Messersmith, P.B. Mussel-inspired surface chemistry for multifunctional coatings. Science 2007, 318, 426–430. [Google Scholar]
- Hillebrandt, H.; Wiegand, G.; Tanaka, M.; Sackmann, E. High electric resistance polymer/lipid composite films on indium-tin-oxide electrodes. Langmuir 1999, 15, 8451–8459. [Google Scholar]
- Baumgart, T.; Offenhausser, A. Polysaccharide-supported planar bilayer lipid model membranes. Langmuir 2003, 19, 1730–1737. [Google Scholar]
- Wang, T.; Li, D.; Lu, X.; Khmaladze, A.; Han, X.; Ye, S.; Yang, P.; Xue, G.; He, N.; Chen, Z. Single lipid bilayers constructed on polymer cushion studied by sum frequency generation vibrational spectroscopy. J. Phys. Chem. C 2011, 115, 7613–7620. [Google Scholar]
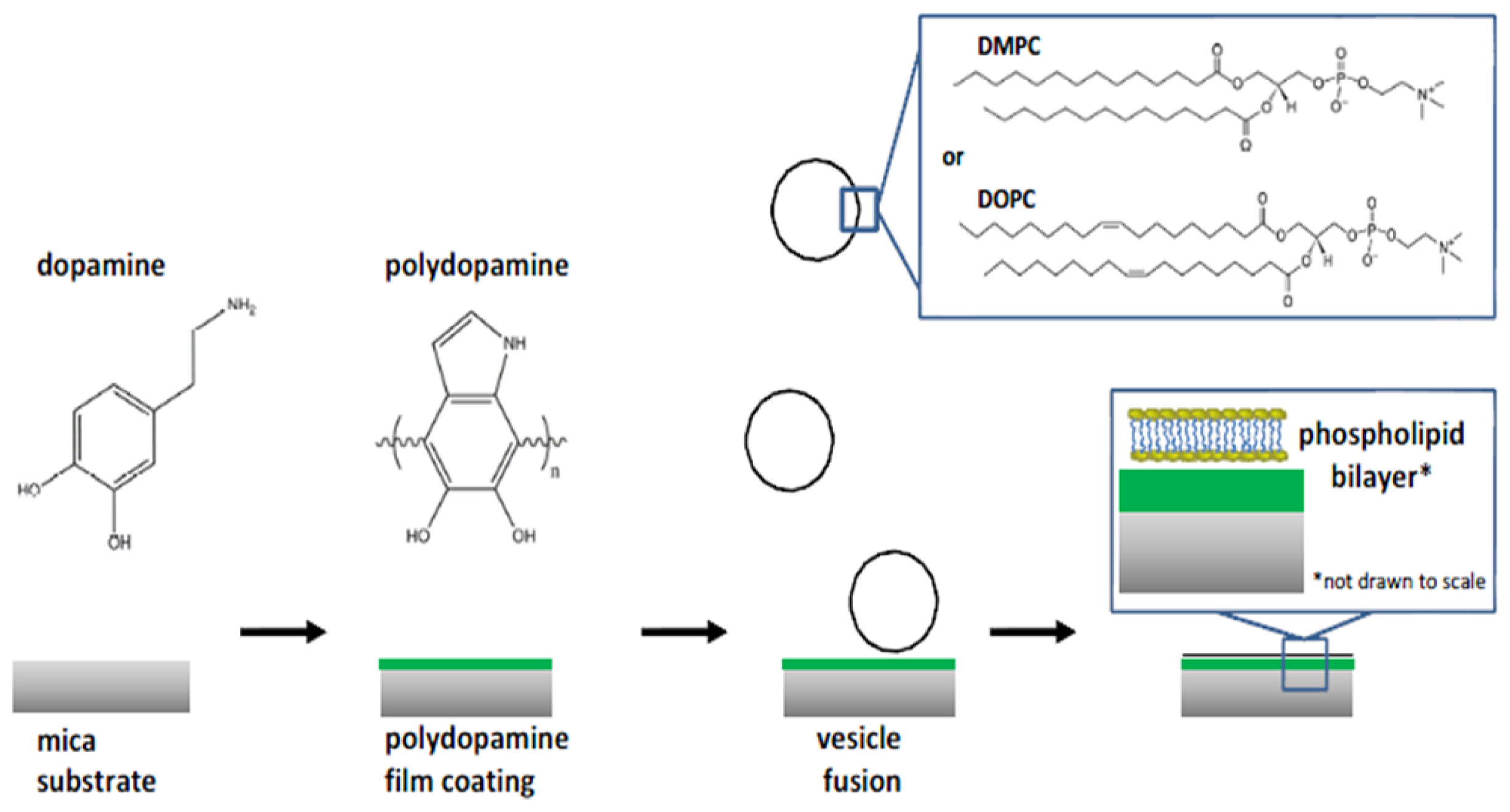
- Nirasay, S.; Badia, A.; Leclair, G.; Claverie, J.P.; Marcotte, I. Polydopamine-supported lipid bilayers. Materials 2012, 5, 2621–2636. [Google Scholar]
- Tsapikouni, T.S.; Missirlis, Y.F. Protein-material interactions: From micro-to-nano scale. Mater. Sci. Eng. B 2008, 152, 2–7. [Google Scholar]
- Hlady, V.; Buijs, J.; Jennissen, H.P. Methods to study protein adsorption. Methods Enzymol 1999, 309, 402–429. [Google Scholar]
- Rabe, M.; Verdes, D.; Seeger, S. Understanding protein adsorption phenomena at solid surfaces. Adv. Colloid Interface Sci 2011, 162, 87–106. [Google Scholar] [Green Version]
- Koehler, J.A.; Ulbricht, M.; Belfort, G. Intermolecular forces between a protein and a hydrophillic polysulfone film with relevance to filtration. Langmuir 2001, 16, 10419–10427. [Google Scholar]
- Dupont-Gillain, C.C.; Fauroux, C.M.J.; Gardner, D.C.J.; Leggett, G.J. Use of AFM to probe the adsorption strength and time-dependent changes of albumin on self-assembled monolayers. J. Biomed. Mater. Res 2003, 67A, 548–558. [Google Scholar]
- Anand, G.; Sharma, S.; Kumar, S.K.; Belfort, G. Stability of tethered proteins. Langmuir 2009, 25, 4998–55005. [Google Scholar]
- Kidoaki, S.; Nakayama, Y.; Matsuda, T. Measuerment of interaction forces between proteins and iniferter-based graft-polymerized surfaces with an atomic force microscope in an aqueous media. Langmuir 2001, 17, 1080–1087. [Google Scholar]
- McColl, J.; Yakubov, G.E.; Ramsden, J.J. Complex desorption of mucin from silica. Langmuir 2007, 23, 7096–7100. [Google Scholar]
- Reichhart, C.; Czeslik, C. Native-like structure of proteins at a planar poly(acrylic acid) brush. Langmuir 2009, 25, 1047–1053. [Google Scholar]
- Cha, T.-W.; Guo, A.; Zhu, X.-Y. Formation of supported phospholipid bilayers on molecular surfaces: Role of surface charge density and electrostatic interaction. Biophys. J 2006, 90, 1270–1274. [Google Scholar]
- Reisch, A.; Hemmerle, J.; Voegel, J.C.; Gonthier, E.; Decher, G.; Benkirane-Jessel, N.; Chassepot, A.; Mertz, D.; Lavalle, P.; Mesini, P.; et al. Polyelectrolyte multilayer coatings that resist protein adsorption at rest and under stretching. J. Mater. Chem 2008, 18, 4242–4245. [Google Scholar]
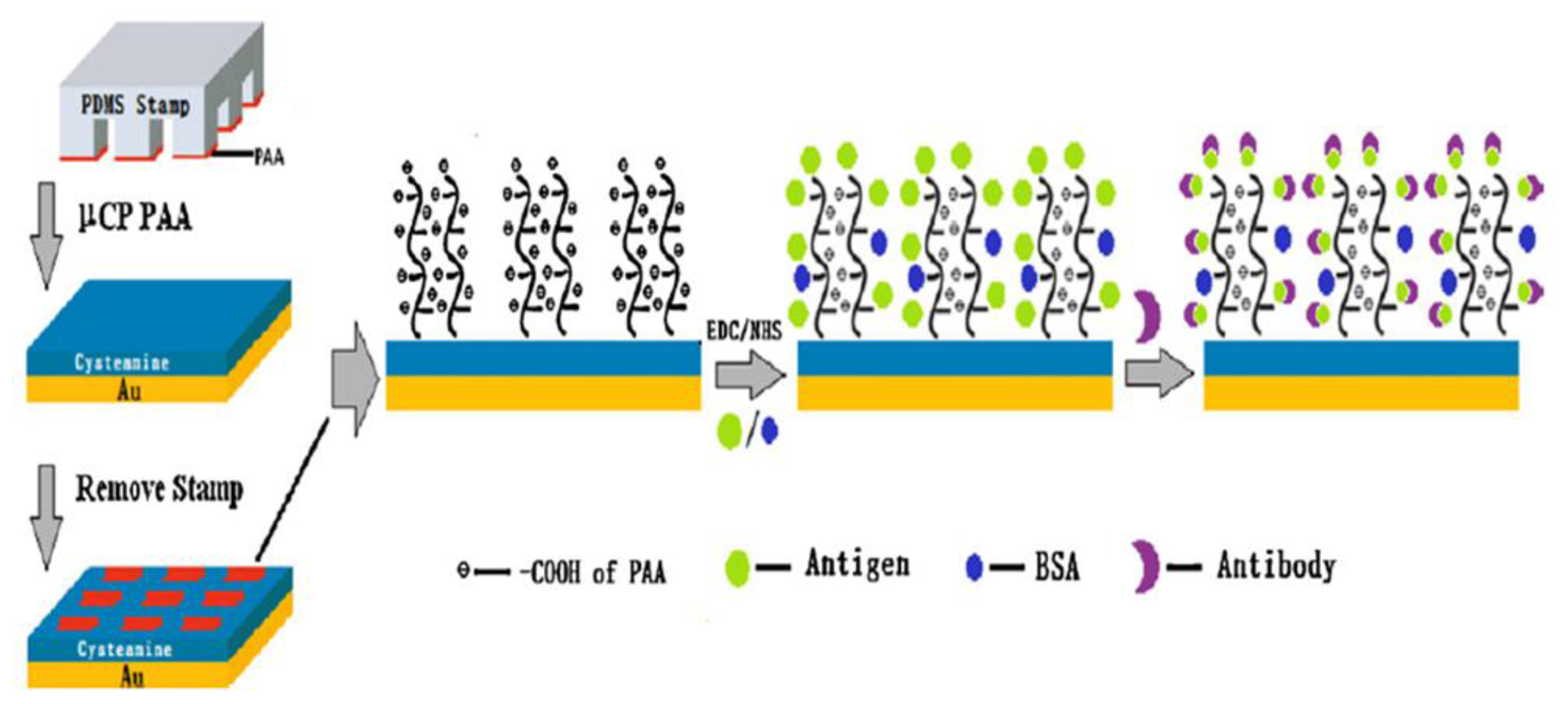
- Wang, Y.-M.; Cui, Y.; Cheng, Z.-Q.; Song, L.-S.; Wang, Z.-Y.; Han, B.-H.; Zhu, J.-S. Poly(acrylic acid) brushes pattern as a 3D functional biosensor surface for microchips. Appl. Surf. Sci 2013, 266, 313–318. [Google Scholar]
- Silva, R.A.; Urzua, M.D.; Petri, D.F.S.; Dubin, P.L. Protein adsorption onto polyelectrolyte layers: Effects of protein hydrophobicity and charge anisotropy. Langmuir 2010, 26, 14032–14038. [Google Scholar]
- Wang, C.; Yan, Q.; Liu, H.-B.; Zhou, X.-H.; Xiao, S.-J. Different EDC/NHS activation mechanisms between PAA and PMAA brushes and the following amidation reactions. Langmuir 2011, 27, 12058–12068. [Google Scholar]
- Evers, F.; Reichhart, C.; Steitz, R.; Tolan, M.; Czeslik, C. Probing adsorption and aggregation of insulin at a poly(acrylic acid) brush. Phys. Chem. Chem. Phys 2010, 12, 4375–4382. [Google Scholar]
- Czeslik, C.; Jackler, G.; Hazlett, T.; Gratton, E.; Steitz, R.; Wittemann, A.; Ballauff, M. Salt-induced protein resistance of polyelectrolyte brushes studied by using fluorescence correlation spectroscopy and neutron reflectometry. Phys. Chem. Chem. Phys 2004, 6, 5557–5563. [Google Scholar]
- Malmsten, M. Formation of adsorbed protein layers. J. Colloid Interface Sci 1998, 207, 186–199. [Google Scholar]
- Demanèche, S.; Chapel, J.P.; Monrozier, L.J.; Quiquampoix, H. Dissimilar pH-dependent adsorption features of bovine serum albumin and α-chymotrypsin on mica probed by AFM. Colloids Surf. B 2009, 70, 226–231. [Google Scholar]
- Rabe, M.; Verdes, D.; Zimmermann, J.; Seeger, S. Surface organization and cooperativity during nonspecific protein adsorption events. J. Phys. Chem. B 2008, 112, 13971–13980. [Google Scholar]
- Vendruscolo, M.; Dobson, C.M. Chemical biology: More charges against aggregation. Nature 2007, 449, 555. [Google Scholar]
- Norde, W. My voyage of discovery to proteins in flatland…and beyond. Colloids Surf. B 2008, 61, 1–9. [Google Scholar]
- Norde, W.; Giacomelli, C.E. BSA structural changes during homomolecular exchange between the adsorbed and the dissolved states. J. Biotechnol 2000, 79, 259–268. [Google Scholar]
- Andrade, J.D.; Hlady, V.; Wei, A.P. Adsorption of complex proteins at interfaces. Pure Appl. Chem 1992, 64, 1777–1781. [Google Scholar]
- Andrade, J.D.; Hlady, V. Plasma protein adsorption: The big twelve. Ann. N. Y. Acad. Sci 1987, 516, 158–172. [Google Scholar]
- Lu, J.R.; Zhao, X.B.; Yaseen, M. Protein adsorption studied by neutron reflection. Curr. Opin. Colloid Interface Sci 2007, 12, 9–16. [Google Scholar]
- Su, T.J.; Lu, J.R.; Thomas, R.K.; Cui, Z.F.; Penfold, J. The effect of pH on the adsorption of lysozyme at the hydrophilic silicon oxide-water interface, a neutron reflection study. Langmuir 1998, 14, 438–445. [Google Scholar]
- Wertz, C.F.; Santore, M.M. Adsorption and reorientation kinetics of lysozyme on hydrophobic surfaces. Langmuir 2002, 18, 1190–1199. [Google Scholar]
- Rabe, M.; Verdes, D.; Rankl, M.; Artus, G.R.J.; Seeger, S. A comprehensive study of concepts and phenomena of the non-specific adsorption of β-lactoglobulin. Chem. Phys. Chem 2007, 8, 862–872. [Google Scholar]
- Daly, S.M.; Przybycien, T.M.; Tilton, R.D. Coverage dependent orientation of lysozyme adsorbed on silica. Langmuir 2003, 19, 3848–3857. [Google Scholar]
- Andrade, J.D.; Hlady, V. Protein adsorption and materials biocompatibility: A tutorial review and suggested hypotheses. Adv. Polym. Sci 1986, 79, 1–63. [Google Scholar]
- Karlsson, M.; Carlsson, U. Protein adsorption orientation in the light of fluorescent probes: Mapping of the interaction between site-directly labeled human carbonic anhydrase II and silica nanoparticles. Biophys. J 2005, 88, 3536–3545. [Google Scholar]
- Zoungrana, T.; Findenegg, G.H.; Norde, W. Structure, stability and activity of adsorbed enzymes. J. Colloid Interface Sci 1997, 190, 437–448. [Google Scholar]
- Pancera, S.M.; Gliemann, H.; Schimmel, T.; Petri, D.F.S. Adsorption behavior and activity of hexokinase. J. Colloid Interface Sci 2006, 302, 417–423. [Google Scholar]
- Sonesson, A.W.; Callisen, T.H.; Brismar, H.; Elofsson, U.M. Adsorption and activity of Thermomyces lanuginosus lipase on hydrophobic and hydrophilic surfaces measured with dual polarization interferometry (DPI) and confocal microscopy. Colloids Surf. B 2008, 61, 208–215. [Google Scholar]
- Tie, Y.; Calonder, C.; van Tassel, P.R. Protein adsorption: Kinetics and history dependence. J. Colloid Interface Sci 2003, 268, 1–11. [Google Scholar]
- Leermakers, F.A.M.; Ballauff, M.; Borisov, O.V. On the mechanisms of interaction of globular proteins with polyelectrolyte brushes. Langmuir 2007, 23, 3937–3946. [Google Scholar]
- Wittemann, A.; Ballauff, M. Interaction of proteins with linear polyelectrolytes and spherical polyelectrolyte brushes in aqueous solution. Phys. Chem. Chem. Phys 2006, 8, 5269–5275. [Google Scholar]
- De Vos, W.M.; Leermakers, F.A.M.; de Keizer, A.; Stuart, M.A.C.; Kleijn, J.M. Field theoretical analysis of driving forces for the uptake of proteins by like-charged polyelectrolyte brushes: Effects of charge regulation and patchiness. Langmuir 2010, 26, 249–259. [Google Scholar]
- Hollmann, O.; Gutberlet, T.; Czeslik, C. Structure and protein binding capacity of a planar PAA brush. Langmuir 2007, 23, 1347–1353. [Google Scholar]
- Chelmowski, R.; Prekelt, A.; Grunwald, C.; Woll, C. A case study on biological activity in a surface-bound multicomponent system: The biotin-streptavidin-peroxidase system. J. Phys. Chem. A 2007, 111, 12295–12303. [Google Scholar]
- Chatelier, R.C.; Minton, A.P. Adsorption of globular proteins on locally planar surfaces: Models for the effect of excluded surface area and aggregation of adsorbed protein on adsorption equilibria. Biophys. J 1996, 71, 2367–2374. [Google Scholar]
- Minton, A.P. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins. Biophys. Chem 2000, 86, 239–247. [Google Scholar]
- Minton, A.P. Effects of excluded surface area and adsorbate clustering on surface adsorption of proteins II. Kinetic models. Biophys. J 2001, 80, 1641–1648. [Google Scholar]
- Vroman, L.; Adams, A.L.; Fischer, G.C.; Munoz, P.C. Interaction of high molecular weight kininogen, factor XII, and fibrinogen in plasma at interfaces. Blood 1980, 55, 156–159. [Google Scholar]
- Daly, S.M.; Przybycien, T.M.; Tilton, R.D. Aggregation of lysozyme and of poly(ethylene glycol)-modified lysozyme after adsorption to silica. Colloids Surf. B 2007, 57, 81–88. [Google Scholar]
- Menon, A.K.; Holowka, D.; Webb, W.W.; Baird, B. Clustering, mobility, and triggering activity of small oligomers of immunoglobulin E on rat basophilic leukemia cells. J. Cell Biol 1986, 102, 534–540. [Google Scholar]
- Weiss, A.; Schlessinger, J. Switching signals on or off by receptor dimerization. Cell 1998, 94, 277–280. [Google Scholar]
- Alberts, B. The cell as a collection of protein machines: Preparing the next generation of molecular biologists. Cell 1998, 92, 291–284. [Google Scholar]
- Friedenberger, M.; Bode, M.; Krusche, A.; Schubert, W. Fluorescence detection of protein clusters in individual cells and tissue sections by using toponome imaging system: Sample preparation and measuring procedures. Nat. Protoc 2007, 2, 2285–2294. [Google Scholar]
- Trusova, V.M.; Gorbenko, G.P. Electrostatically-controlled protein adsorption onto lipid bilayer: Modeling adsorbate aggregation behavior. Biophys. Chem 2008, 133, 90–103. [Google Scholar]
- Rabe, M.; Verdes, D.; Seeger, S. Surface-induced spreading phenomenon of protein clusters. Soft Matter 2009, 5, 1039–1047. [Google Scholar]
- Zhdanov, V.P.; Kasemo, B. Protein adsorption and desorption on lipid bilayers. Biophys. Chem 2010, 146, 60–64. [Google Scholar]
- Junghans, A.; Champagne, C.; Cayot, P.; Loupiac, C.; Koper, I. Probing protein-membrane interactions using solid supported membranes. Langmuir 2011, 27, 2709–2716. [Google Scholar]
- Owicki, J.C.; Springate, M.W.; McConnell, H.M. Theoretical study of protein-lipid interactions in bilayer membranes. Proc. Natl. Acad. Sci. USA 1978, 75, 1616–1619. [Google Scholar]
- Owicki, J.C.; McConnell, H.M. A theoretical study of protein-protein and lipid-protein interactions. Proc. Natl. Acad. Sci. USA 1979, 76, 4570–4574. [Google Scholar]
- Jihing, F. Critical effects from lipid-protein interactions in membranes. I. Theoretical description. Biophys. J 1981, 36, 329–345. [Google Scholar]
- Scott, J.R.H.L.; Coe, T.J. A theoretical study of lipid-protein interactions in bilayers. Biophys. J 1983, 42, 219–224. [Google Scholar]
- Scotto, A.W.; Zakim, D. Reconstitution of membrane proteins: Spontaneous incorportaion of integral membrane protein into preformed bilayers of pure phospholipid. J. Biol. Chem 1998, 263, 18500–18506. [Google Scholar]
- Naumann, R.; Jonczyk, A.; Kopp, R.; van Esch, J.; Ringsdorf, H.; Knoll, W.; Griiber, P. Incorporation of membrane proteins in solid-supported lipid layers. Angew. Chem. Int. Ed. Engl 1995, 34, 2056–2058. [Google Scholar]
- Wiggins, P.A.; Phillips, R. Analytic models for mechanotransduction: Gating a mechanosensitive channel. Proc. Natl. Acad. Sci. USA 2004, 101, 4071–4076. [Google Scholar]
- Wiggins, P.; Phillips, R. Membrane-protein interactions in mechanosensitive channels. Biophys. J 2005, 88, 880–902. [Google Scholar]
- Lahiri, J.; Kalal, P.; Frutos, A.G.; Jonas, S.J.; Schaeffler, R. Method for fabricating supported bilayer lipid membranes on gold. Langmuir 2000, 16, 7805–7810. [Google Scholar]
- Naumann, R.; Schiller, S.M.; Giess, F.; Grohe, B.; Hartman, K.B.; Karcher, I.; Koper, I.; Lubben, J.; Vasilev, K.; Knoll, W. Tethered lipid bilayers on ultraflat gold surfaces. Langmuir 2003, 19, 5435–5443. [Google Scholar]
- Wang, X.; Shindel, M.M.; Wang, S.-W.; Ragan, R. A facile approach for assembling lipid bilayer membranes on template-stripped gold. Langmuir 2010, 26, 18239–18245. [Google Scholar]
- Dahlin, A.; Zach, M.; Rindzevicius, T.; Ka, M.; Sutherland, D.S.; Hook, F. Localized surface plasmon resonance sensing of lipid-membrane-mediated biorecognition events. J. Am. Chem. Soc 2005, 127, 5043–5048. [Google Scholar]
- Hennesthal, C.; Drexler, J.; Steinem, C. Membrane suspended nanocompartments based on ordered pores in alumina. Chem. Phys. Chem 2002, 10, 885–889. [Google Scholar]
- Venkatesan, B.M.; Polans, J.; Comer, J.; Sridhar, S.; Wendell, D.; Aksimentiev, A.; Bashir, R. Lipid bilayer coated Al2O3 nanopore sensors: Towards a hybrid biological solid-state nanopore. Biomed. Microdevices 2011, 13, 671–682. [Google Scholar]
- Mayer, M.; Kriebel, J.K.; Tosteson, M.T.; Whitesides, G.M. Microfabricated teflon membranes for low-noise recordings of ion channels in planar lipid bilayers. Biophys. J 2003, 85, 2684–2695. [Google Scholar]
- Phung, T.; Zhang, Y.; Dunlop, J.; Dalziel, J. Bilayer lipid membranes supported on Teflon filters: A functional environment for ion channels. Biosens. Bioelectron 2011, 26, 3127–3135. [Google Scholar]
- Gonçalves, R.P.; Agnus, G.; Sens, P.; Houssin, C.; Bartenlian, B.; Scheuring, S.; Two-chamber, AFM. Probing membrane proteins separating two aqueous compartments. Nat. Methods 2006, 3, 1007–1012. [Google Scholar]
- Worsfold, O.; Voelcker, N.H.; Nishiya, T. Biosensing using lipid bilayers suspended on porous silicon. Langmuir 2006, 22, 7078–7083. [Google Scholar]
- Zhu, Z.W.; Wang, Y.; Zhang, X.; Sun, C.F.; Li, M.G.; Yan, J.W.; Mao, B.W. Electrochemical impedance spectroscopy and atomic force microscopic studies of electrical and mechanical properties of nano-black lipid membranes and size dependence. Langmuir 2012, 28, 14739–14746. [Google Scholar]
- Simion, M.; Ruta, L.; Mihailescu, C.; Kleps, I.; Bragaru, A.; Miu, M.; Ignat, T.; Baciu, I. Porous silicon used as support for protein microarray. Superlattices Microstruct 2009, 46, 69–76. [Google Scholar]
- Thibault, C.; Carcenac, F.; Dague, E.; Chalmeau, J.; Vieu, C. Porous silicon membrane, with an integrated aqueous supply, for two chamber AFM. Microelectron. Eng 2009, 86, 1393–1395. [Google Scholar]
- Fertig, N.; Klau, M.; George, M.; Blick, R.H.; Behrends, J.C. Activity of single ion channel proteins detected with a planar microstructure. Appl. Phys. Lett 2002, 81, 4865–4867. [Google Scholar]
- Tantawi, K.H.; Berdiev, B.; Cerro, R.; Williams, J. Porous silicon membrane structure for investigation of transmembrane proteins. Superlattices Microstruct 2013, 58, 72–80. [Google Scholar]
- Martin-Palma, R.J.; Manso-Silvan, M.; Torres-Costa, V. Biomedical applications of nanostructured porous silicon: A review. J. Nanophoton 2010, 4, 042502. [Google Scholar]
- Claesson, M.; Frost, R.; Svedhem, S.; Andersson, M. Pore spanning lipid bilayers on mesoporous silica having varying pore size. Langmuir 2011, 27, 8974–8982. [Google Scholar]
- Torres-Costa, V.; Martín-Palma, R.J. Application of nanostructured porous silicon in the field of optics, a review. J. Mater. Sci 2010, 45, 2823–2838. [Google Scholar]
- Jane, A.; Dronov, R.; Hodges, A.; Voelcker, N.H. Porous silicon biosensors on the advance. Trends Biotechnol 2009, 27, 230–239. [Google Scholar]
- Phung, T.; Zhang, Y.; Dunlop, J.; Dalziel, J.E. Porous materials to support bilayer lipid membranes for ion channel biosensors. Int. J. Electrochem 2011. [Google Scholar] [CrossRef]
- Mulero, R.; Prabhu, A.S.; Freedman, K.J.; Kim, M.J. Nanopore-based devices for bioanalytical applications. J. Lab. Autom 2010, 15, 243–252. [Google Scholar]
- Pradeepa, M.; Venkatesan, P.; Menaka, E.; Kumaran, S. Fabrication of porous silicon nanoparticles to attach clorgyline for drug delivery. Inter. Proc. Chem. Biol. Environ. Eng 2011, 5, 327–330. [Google Scholar]
- Nussio, M.R.; Oncins, G.; Ridelis, I.; Szili, E.; Shapter, J.G.; Sanz, F.; Voelcker, N.H. Nanomechanical characterization of phospholipid bilayer islands on flat and porous substrates: A force spectroscopy study. J. Phys. Chem. B 2009, 113, 10339–10347. [Google Scholar]
- Gornall, J.L.; Mahendran, K.R.; Pambos, O.J.; Steinbock, L.J.; Otto, O.; Chimerel, C.; Winterhalter, M.; Keyser, U.F. Simple reconstitution of protein pores in nano lipid bilayers. Nano Lett 2011, 11, 3334–3340. [Google Scholar]
- Kowalczyk, S.W.; Blosser, T.R.; Dekker, C. Biomimetic nanopores: Learning from and about nature. Trends Biotechnol 2011, 12, 607–614. [Google Scholar]
- Wang, Y.; Tang, Z.; Kotov, N.A. Bioapplication of nanosemiconductors. Mater. Today 2005, 8, 20–31. [Google Scholar]
- Anglin, E.J.; Cheng, L.; Freeman, W.R.; Sailor, M.J. Porous silicon in drug delivery devices and materials. Adv. Drug Deliv. Rev 2008, 60, 1266–1277. [Google Scholar]
- Kumar, D.S.; Banji, D.; Madhavi, B.B.; Bodanapu, V.; Dondapati, S.; Sri, A.P. Nanostructured porous silicon—A novel biomaterial for drug delivery. Int. J. Pharm. Pharm. Sci 2009, 2, 8–16. [Google Scholar]
- Martín-Palma, R.J.; Manso, M.; Torres-Costa, V. Optical biosensors based on semiconductor nanostructures. Sensors 2009, 9, 5149–5172. [Google Scholar]
- Haidary, S.M.; Córcoles, E.P.; Ali, N.K. Nanoporous silicon as drug delivery systems for cancer therapies. J. Nanomater 2012, 12, 1–15. [Google Scholar]
- Giess, F.; Friedrich, M.G.; Heberle, J.; Naumann, R.L.; Knoll, W. The protein-tethered lipid bilayer: A novel mimic of the biological membrane. Biophys. J 2004, 87, 3213–3220. [Google Scholar]
- Chernomordik, L.; Sukharev, S.; Popov, S.; Pastushenko, V.; Sokirko, A.; Abidor, I.; Chizmadzhev, Y. The electrical breakdown of cell and lipid membranes: The similarity of phenomenologies. Biochim. Biophys. Acta 1987, 902, 360–373. [Google Scholar]
- Chernomordik, L.; Sukharev, S.; Abidor, I.; Chizmadzhev, Y. Breakdown of lipid bilayer membranes in an electric field. Biochim. Biophys. Acta 1983, 736, 203–213. [Google Scholar]
- Benz, R.; Beckers, F.; Zimmermann, U. Reversible electrical breakdown of lipid bilayer membranes: A charge-pulse relaxation study. J. Membr. Biol 1979, 48, 181–204. [Google Scholar]
- Lundgren, A.; Hedlund, J.; Andersson, O.; Branden, M.; Kunze, A.; Elwing, H.; Hook, F. Resonance-mode electrochemical impedance measurements of silicon dioxide supported lipid bilayer formation and ion channel mediated charge transport. Anal. Chem 2011, 83, 7800–7806. [Google Scholar]
- Drexler, J.; Steinem, C. Pore-suspending lipid bilayers on porous alumina investigated by electrical impedance spectroscopy. J. Phys. Chem. B 2003, 107, 11245–11254. [Google Scholar]
- Awayda, M.S. Specific and nonspecific effects of protein kinase C on the epithelial Na (+) channel. J. Gen. Physiol 2000, 115, 559–570. [Google Scholar]
- Berdiev, B.; Benos, D. Epithelial sodium channel in planar lipid bilayers. Methods Mol. Biol 2006, 337, 89–99. [Google Scholar]
- Naumowicz, M.; Petelska, A.D.; Figaszewski, Z.A. Impedance analysis of a phosphatidylcholine-phosphatidylethanolamine system in bilayer lipid membranes. Electrochem. Acta 2006, 51, 5024–5028. [Google Scholar]
- Naumowicz, M.; Kotynska, J. Impedance analysis of phosphatidylcholine membranes modified with valinomycin. Eur. Biophys. J 2006, 35, 239–246. [Google Scholar]
- Naumowicz, M.; Petelska, A.D.; Figaszewski, Z.A. Impedance analysis of complex formation equilibria in phosphatidylcholine bilayers containing decanoic acid or decylamine. Cell Biochem. Biophys 2011, 61, 145–155. [Google Scholar]
- Otera, H.; Ishihara, N.; Mihara, K. New insights into the function and regulation of mitochondrial fission. Biochim. Biophys. Acta 2013, 1833, 1256–1268. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Khan, M.S.; Dosoky, N.S.; Williams, J.D. Engineering Lipid Bilayer Membranes for Protein Studies. Int. J. Mol. Sci. 2013, 14, 21561-21597. https://doi.org/10.3390/ijms141121561
Khan MS, Dosoky NS, Williams JD. Engineering Lipid Bilayer Membranes for Protein Studies. International Journal of Molecular Sciences. 2013; 14(11):21561-21597. https://doi.org/10.3390/ijms141121561
Chicago/Turabian StyleKhan, Muhammad Shuja, Noura Sayed Dosoky, and John Dalton Williams. 2013. "Engineering Lipid Bilayer Membranes for Protein Studies" International Journal of Molecular Sciences 14, no. 11: 21561-21597. https://doi.org/10.3390/ijms141121561
APA StyleKhan, M. S., Dosoky, N. S., & Williams, J. D. (2013). Engineering Lipid Bilayer Membranes for Protein Studies. International Journal of Molecular Sciences, 14(11), 21561-21597. https://doi.org/10.3390/ijms141121561