Non-Coding RNAs: The “Dark Matter” of Cardiovascular Pathophysiology

Abstract
:
1. Introduction
2. An Overview on the Main Methods to Analyze the ncRNAs Expression
3. Functions of Non Coding RNAs
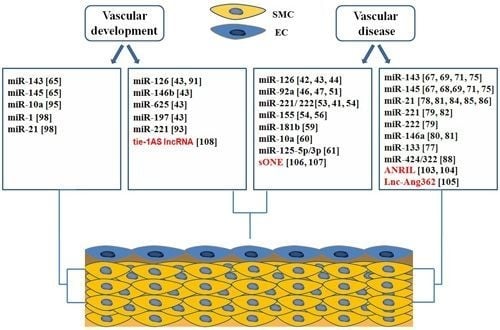
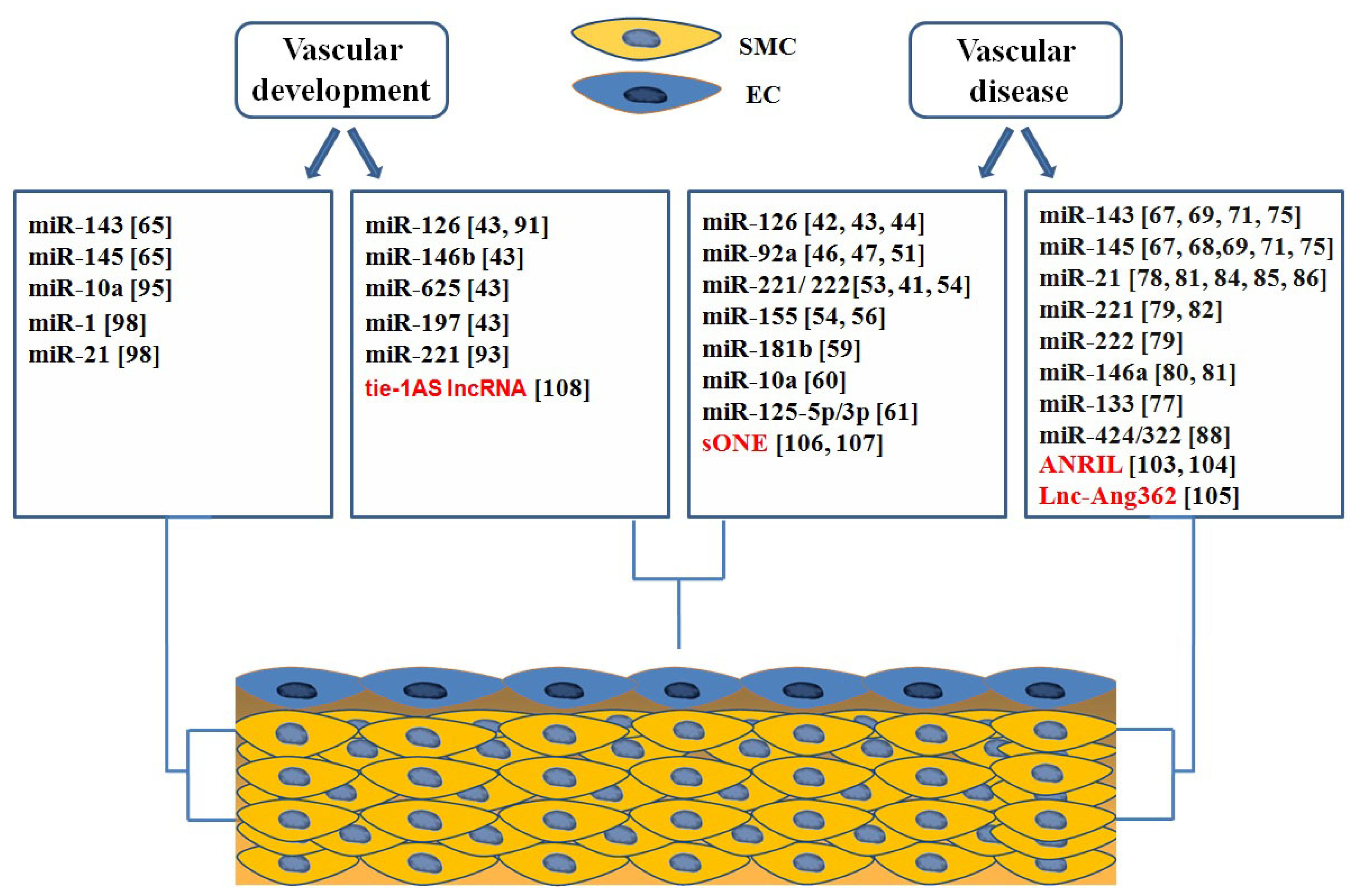
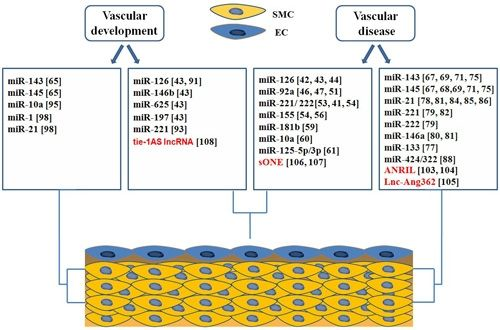
4. Roles of ncRNAs in Vascular Biology and Disease
4.1. microRNAs in Endothelial Biology and Dysfunction
4.2. microRNAs in Phenotypic Switching of VSMCs
4.2.1. miR-143 and miR-145 Play a Role in the Regulation of Phenotype of VSMCs in Response to Injury
4.2.2. miR-133 Is a Negative Regulator of SP-1 and Promotes Contractile Phenotype of VSMCs
4.2.3. microRNAs that Promote De-Differentiated Phenotype of VSMCs
4.3. Roles of microRNAs in Vascular Development
4.3.1. miRNAs Involved in Endothelial Development
4.3.2. microRNAs in Regulation of Vascular Smooth Muscle Cell Differentiation
4.4. Long Non Coding RNAs in Vascular Development and Disease
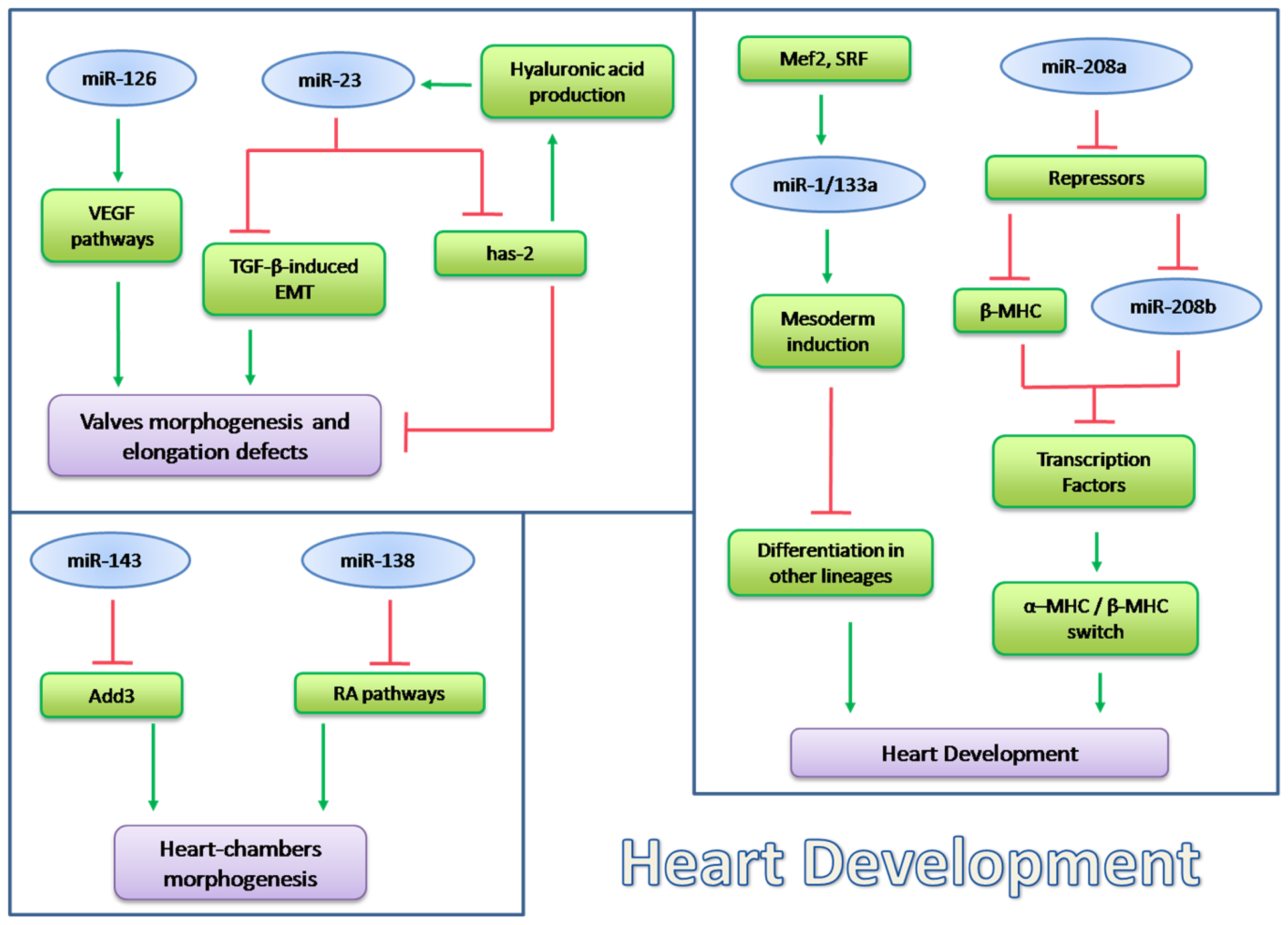
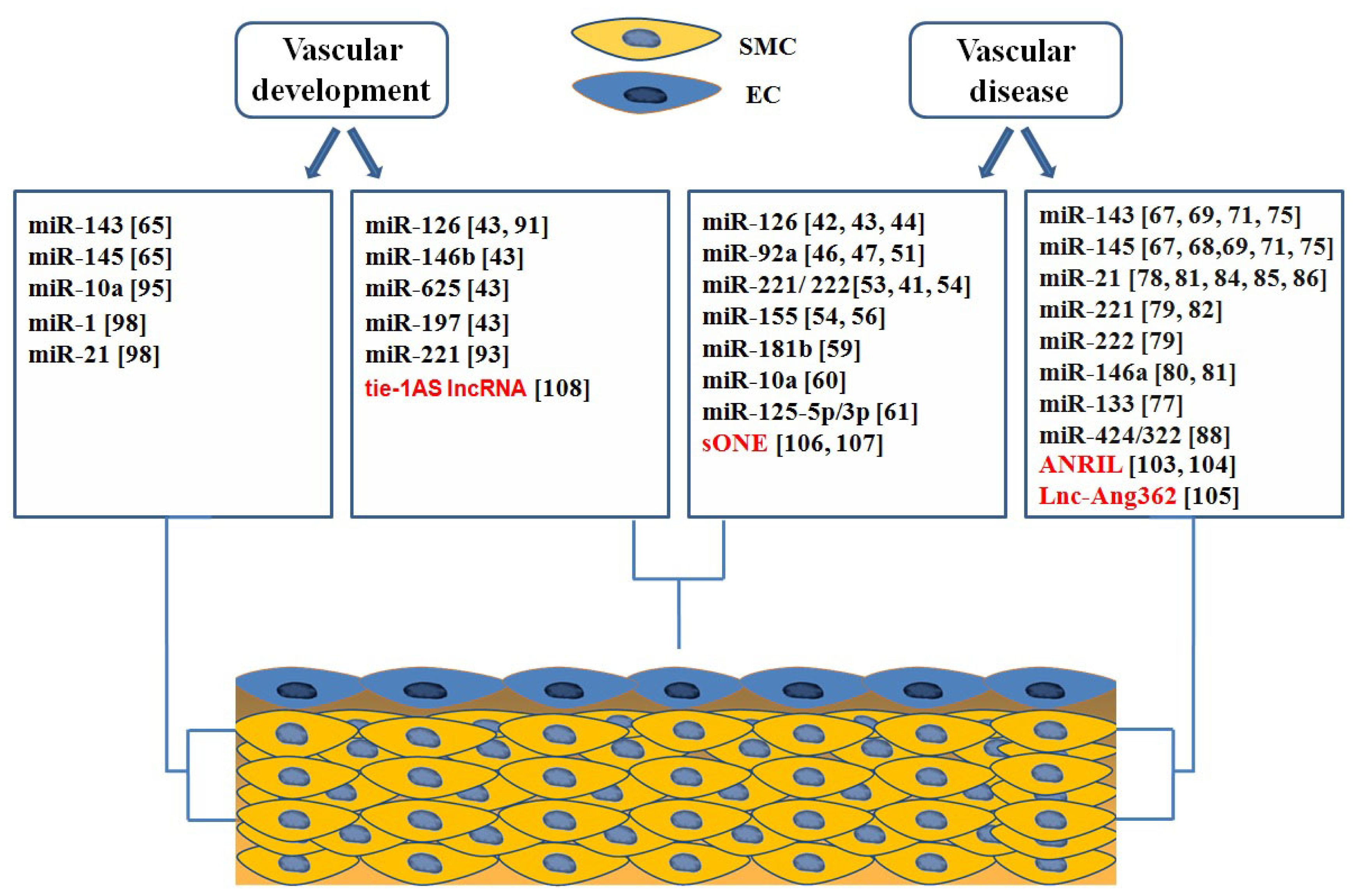
5. ncRNA in Heart Development and Pathophysiology
5.1. microRNA in Heart Development
5.1.1. miRNAs Encoded by MHC Genes
5.1.2. microRNAs in Heart Chamber Morphogenesis
5.1.3. microRNAs in Valves Development
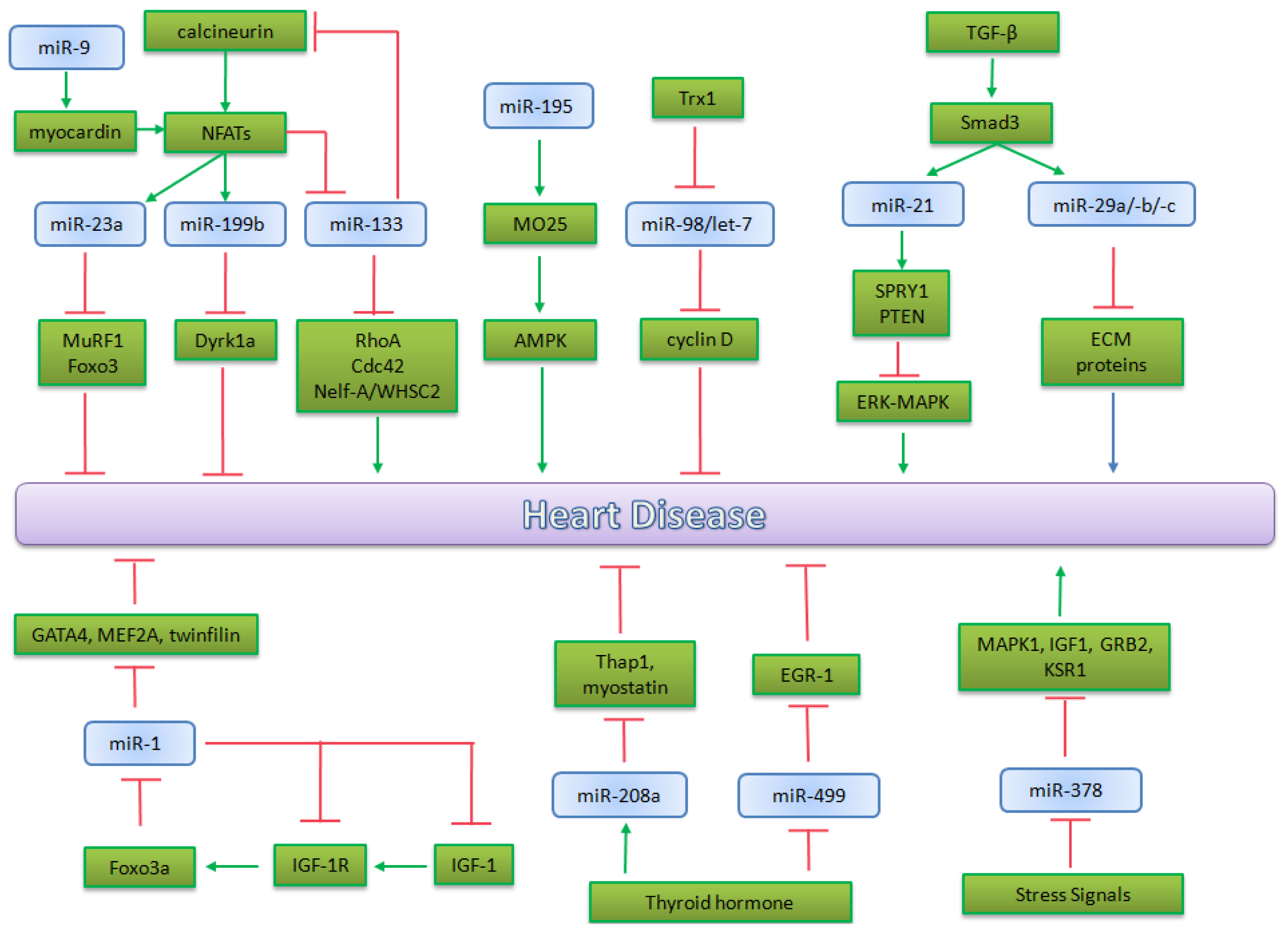
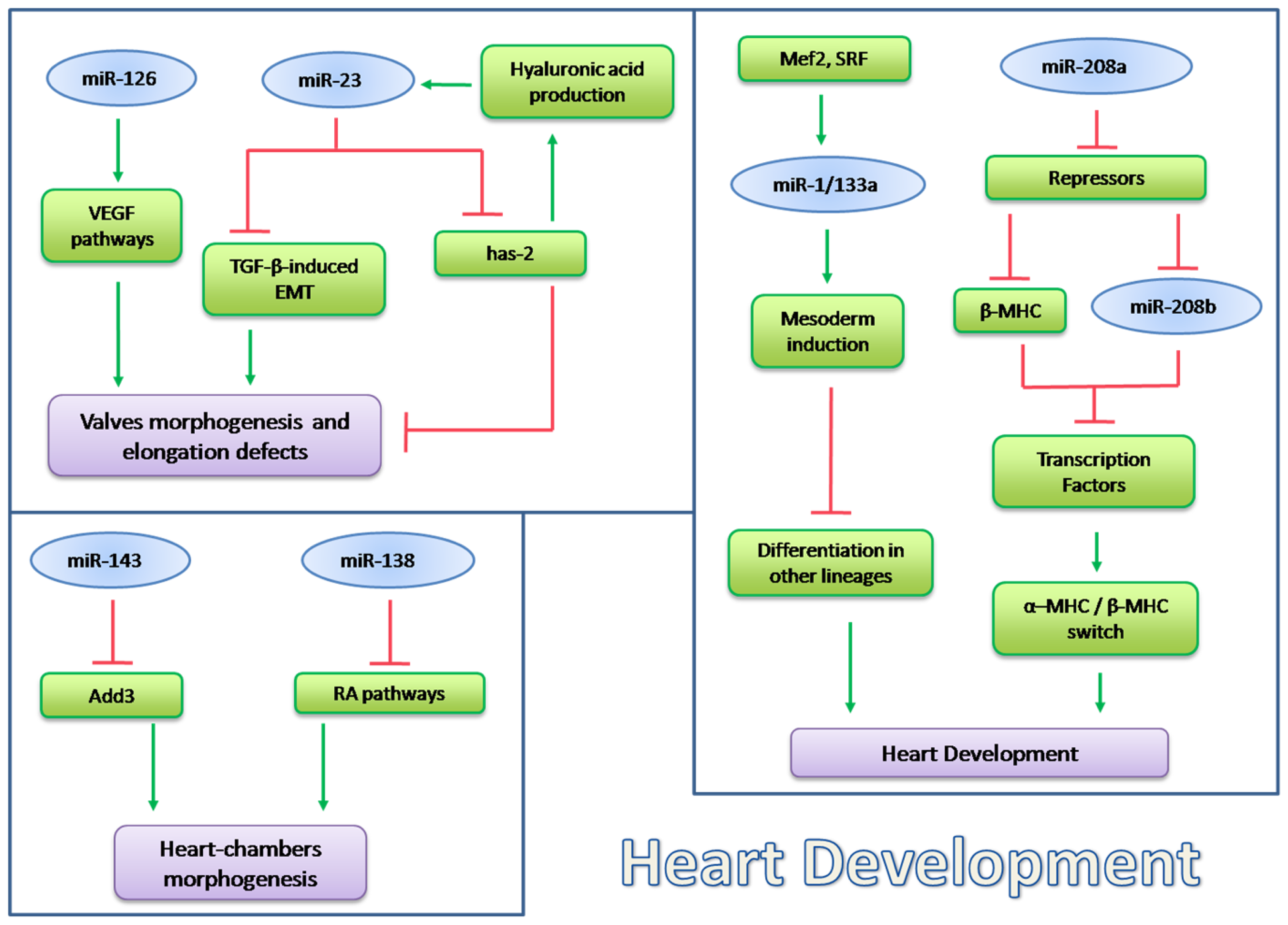
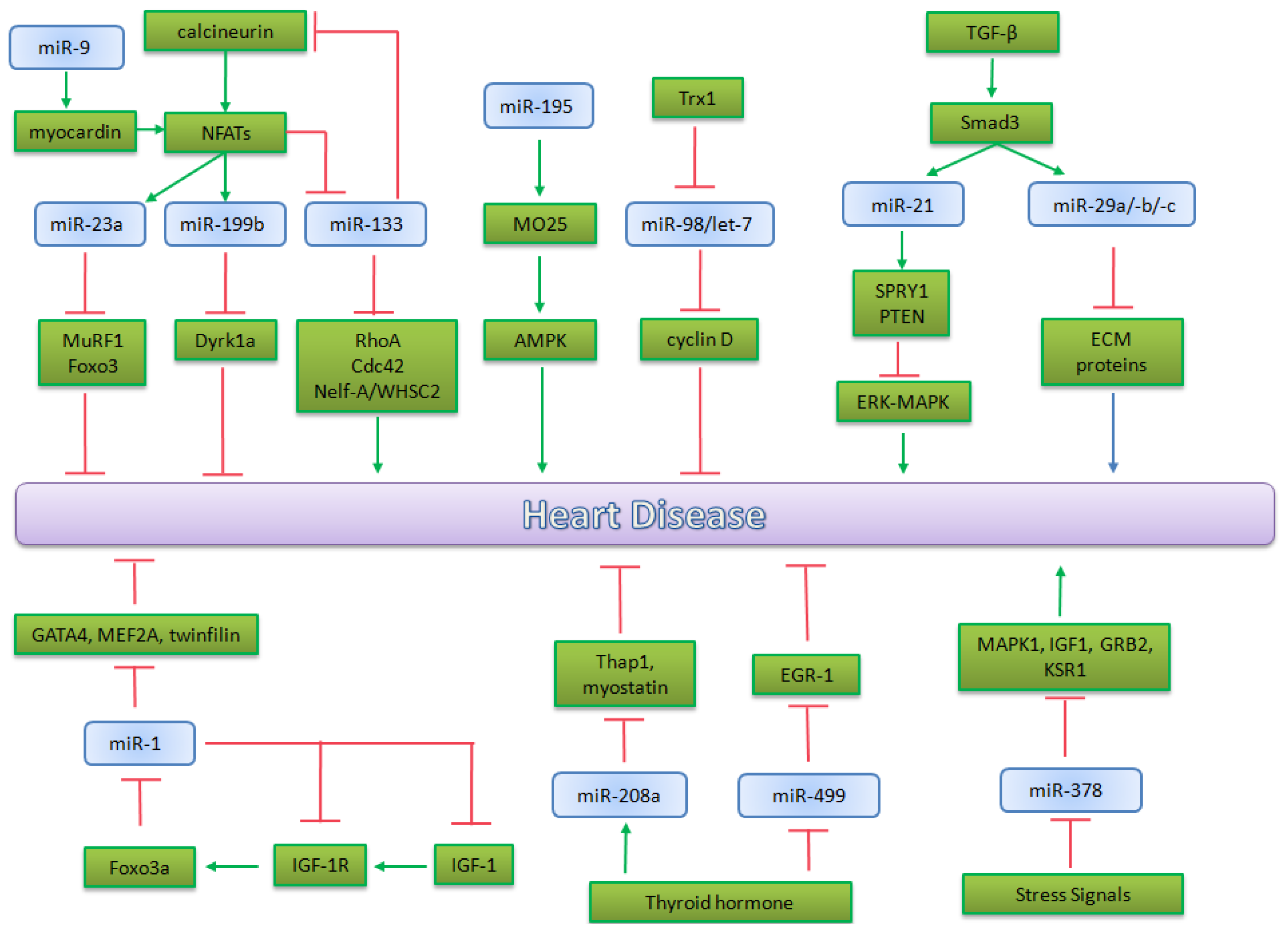
5.2. microRNA in Heart Pathophysiology
5.2.1. miR-195 and miR-98/let-7b in Cardiac Hypertrophy
5.2.2. miR-1 in Heart Physiopathology
5.2.3. miRNA Implicated in Calicineurin/NFATs Pathway
5.2.4. miRNAs Regulated by Thyroid Hormone
5.2.5. miRNAs Regulated by TGF-β in Heart Physiopathology
5.2.6. The Role of miR-378 in MAPK Signaling
5.3. lncRNA in Heart Pathophysiology
6. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Non-coding RNAs | Symbol | Functions |
|---|---|---|
| Structural ncRNAs | ||
| Transfer RNA | tRNA | mRNA translation |
| Ribosomal RNA | rRNA | mRNA translation |
| Regulatory ncRNA | ||
| Short ncRNA | ||
| Micro RNAs | miRNA | post-transcriptional regulators |
| PIWI-interacting RNA | piRNA | DNA methylation, transposon repression |
| Short interfering RNA | siRNA | RNA interference |
| Medium ncRNA | ||
| Small nucleolar RNAs | snoRNA | RNA modification, rRNA processing |
| Promoter upstream transcripts | PROMPTs | Associated with chromatin changes |
| Transcription initiation RNAs | tiRNAs | Epigenetic regulation |
| Long ncRNAs | ||
| Long intergenic ncRNA | lincRNAs | Epigenetic regulators of transcription |
| Enhancer-like ncRNA | eRNA | Transcriptional gene activation |
| Transcribed ultraconserved regions | T-UCRs | Regulation of miRNA and mRNA levels |
| Natural antisense transcripts | NATs | mRNA stability |
| Promoter-associated long RNAs | PALRs | chromatin changes |
| Pseudogenes | None | microRNA decoys |
| Long-ncRNA | Function | Reference |
|---|---|---|
| BraveHeart (AK143260) | Cardiomyocytes differentiation | [15,151] |
| ANRIL | Epigenetic modulation in cardiac development. | [152,153] |
| Fendrr | Heart development | [154] |
| MIAT | Susceptibility to myocardial infarction. | [155] |
| NATs | Regulation of gene expression | [157] |
| Kcnq1ot1 | Regulation of gene expression | [16] |
Acknowledgments
Conflicts of Interest
References
- Birney, E.; Stamatoyannopoulos, J.A.; Dutta, A.; Guigó, R.; Gingeras, T.R.; Margulies, E.H.; Weng, Z.; Snyder, M.; Dermitzakis, E.T.; Thurman, R.E.; et al. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 2007, 447, 799–816. [Google Scholar]
- Alexander, R.P.; Fang, G.; Rozowsky, J.; Snyder, M.; Gerstein, M.B. Annotating non-coding regions of the genome. Nat. Rev. Genet 2010, 11, 559–571. [Google Scholar]
- He, L.; Hannon, G.J. MicroRNAs: Small RNAs with a big role in gene regulation. Nat. Rev. Genet 2004, 5, 522–531. [Google Scholar]
- Mercer, T.R.; Dinger, M.E.; Mattick, J.S. Long non-coding RNAs insight into functions. Nat. Rev. Genet 2009, 10, 155–159. [Google Scholar]
- Mercer, T.R.; Mattick, J.S. Structure and function of long noncoding RNAs in epigenetic regulation. Nat. Struct. Mol. Biol 2013, 20, 300–307. [Google Scholar]
- Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet 2011, 12, 861–874. [Google Scholar]
- World Health Organization, The Global Burden of Disease 2004 Update; WHO: Geneva, Switzerland, 2008.
- Small, E.M.; Olson, E.N. Pervasive roles of microRNAs in cardiovascular biology. Nature 2011, 469, 336–342. [Google Scholar]
- Fiedler, J.; Thum, T. MicroRNAs in myocardial infarction. Arterioscler. Thromb. Vasc. Biol 2013, 33, 201–205. [Google Scholar]
- Holdt, L.M.; Teupser, D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler. Thromb. Vasc. Biol 2012, 32, 196–206. [Google Scholar]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Small, E.M.; Frost, R.J.; Olson, E.N. MicroRNAs add a new dimension to cardiovascular disease. Circulation 2010, 121, 1022–1032. [Google Scholar]
- Batista, P.J.; Chang, H.Y. Long noncoding RNAs: Cellular address codes in development and disease. Cell 2013, 152, 1298–1307. [Google Scholar]
- Hung, T.; Chang, H.Y. Long noncoding RNA in genome regulation: Prospects and mechanisms. RNA Biol 2010, 7, 582–585. [Google Scholar]
- Klattenhoff, C.A.; Scheuermann, J.C.; Surface, L.E.; Bradley, R.K.; Fields, P.A.; Steinhauser, M.L.; Ding, H.; Butty, V.L.; Torrey, L.; Haas, S.; et al. Braveheart, a long noncoding RNA required for cardiovascular lineage commitment. Cell 2013, 152, 570–583. [Google Scholar]
- Korostowski, L.; Sedlak, N.; Engel, N. The Kcnq1ot1 long non-coding RNA affects chromatin conformation and expression of Kcnq1, but does not regulate its imprinting in the developing heart. PLoS Genet 2012, 8, e1002956. [Google Scholar]
- Chen, C.; Ridzon, D.A.; Broomer, A.J.; Zhou, Z.; Lee, D.H.; Nguyen, J.T.; Barbisin, M.; Xu, N.L.; Mahuvakar, V.R.; Andersen, M.R.; et al. Real-time quantification of microRNAs by stem-loop RT-PCR. Nucleic Acids Res 2005, 33. [Google Scholar] [CrossRef]
- Ule, J.; Jensen, K.; Mele, A.; Darnell, R.B. CLIP: A method for identifying protein-RNA interaction sites in living cells. Methods 2005, 37, 376–386. [Google Scholar]
- Zhao, J.; Ohsumi, T.K.; Kung, J.T.; Ogawa, Y.; Grau, D.J.; Sarma, K.; Song, J.J.; Kingston, R.E.; Borowsky, M.; Lee, J.T. Genome-wide identification of polycomb-associated RNAs by RIP-seq. Mol. Cell 2010, 40, 939–953. [Google Scholar]
- Bittencourt, D.; Auboeuf, D. Analysis of co-transcriptional RNA processing by RNA-ChIP assay. Methods Mol. Biol 2012, 809, 563–577. [Google Scholar]
- Hu, W.; Yuan, B.; Flygare, J.; Lodish, H.F. Long noncoding RNA-mediated anti-apoptotic activity in murine erythroid terminal differentiation. Genes Dev 2011, 25, 2573–2578. [Google Scholar]
- Michelhaugh, S.K.; Lipovich, L.; Blythe, J.; Jia, H.; Kapatos, G.; Bannon, M.J. Mining Affymetrix microarray data for long non-coding RNAs: Altered expression in the nucleus accumbens of heroin abusers. In J. Neurochem; 2011; Volume 116, pp. 459–466. [Google Scholar]
- Bentwich, I.; Avniel, A.; Karov, Y.; Aharonov, R.; Gilad, S.; Barad, O.; Barzilai, A.; Einat, P.; Einav, U.; Meiri, E.; et al. Identification of hundreds of conserved and nonconserved human microRNAs. Nat. Genet 2005, 37, 766–770. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA MALAT1 regulates alternative splicing by modulating SR splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar]
- Yoon, J.H.; Abdelmohsen, K.; Srikantan, S.; Yang, X.; Martindale, J.L.; De, S.; Huarte, M.; Zhan, M.; Becker, K.G.; Gorospe, M. LincRNA-p21 suppresses target mRNA translation. Mol. Cell 2012, 47, 648–655. [Google Scholar]
- Guttman, M.; Donaghey, J.; Carey, B.W.; Garber, M.; Grenier, J.K.; Munson, G.; Young, G.; Lucas, A.B.; Ach, R.; Bruhn, L.; et al. lincRNAs act in the circuitry controlling pluripotency and differentiation. Nature 2011, 477, 295–300. [Google Scholar] [Green Version]
- Pandey, R.R.; Mondal, T.; Mohammad, F.; Enroth, S.; Redrup, L.; Komorowski, J.; Nagano, T.; Mancini-Dinardo, D.; Kanduri, C. Kcnq1ot1 antisense noncodingRNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell 2008, 32, 232–246. [Google Scholar]
- Wamstad, J.A.; Alexander, J.M.; Truty, R.M.; Shrikumar, A.; Li, F.; Eilertson, K.E.; Ding, H.; Wylie, J.N.; Pico, A.R.; Capra, J.A.; et al. Dynamic and coordinated epigenetic regulation of developmental transitions in the cardiac lineage. Cell 2012, 151, 206–220. [Google Scholar]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffold of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar]
- Hung, T.; Wang, Y.; Lin, M.F.; Koegel, A.K.; Kotake, Y.; Grant, G.D.; Horlings, H.M.; Shah, N.; Umbricht, C.; Wang, P.; et al. Extensive and coordinated transcription of noncoding RNAs within cell-cycle promoters. Nat. Genet 2011, 43, 621–629. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar]
- Poliseno, L.; Salmena, L.; Zhang, J.; Carver, B.; Haveman, W.J.; Pandolfi, P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature 2010, 465, 1033–1038. [Google Scholar]
- Memczak, S.; Jens, M.; Elefsinioti, A.; Torti, F.; Krueger, J.; Rybak, A.; Maier, L.; Mackowiak, S.D.; Gregersen, L.H.; Munschauer, M.; et al. Circular RNAs are a large class of animal RNAs with regulatory potency. Nature 2013, 495, 333–338. [Google Scholar]
- Hansen, T.B.; Jensen, T.I.; Clausen, B.H.; Bramsen, J.B.; Finsen, B.; Damgaard, C.K.; Kjems, J. Natural RNA circles function as efficient microRNA sponges. Nature 2013, 495, 384–388. [Google Scholar]
- Hansson, G.K. Inflammation, atherosclerosis, and coronary artery disease. N. Engl. J. Med 2005, 352, 1685–1695. [Google Scholar]
- Polimeni, A.; de Rosa, S.; Indolfi, C. Vascular miRNAs after balloon angioplasty. Trends Cardiovasc. Med 2013, 23, 9–14. [Google Scholar]
- Curcio, A.; Torella, D.; Indolfi, C. Mechanisms of smooth muscle cell proliferation and endothelial regeneration after vascular injury and stenting: Approach to therapy. Circ. J 2011, 75, 1287–1296. [Google Scholar]
- Kuehbacher, A.; Urbich, C.; Zeiher, A.M.; Dimmeler, S. Role of Dicer and Drosha for endothelial microRNA expression and angiogenesis. Circ. Res 2007, 101, 59–68. [Google Scholar]
- Suárez, Y.; Fernández-Hernando, C.; Pober, J.S.; Sessa, W.C. Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res 2007, 100, 1164–1173. [Google Scholar]
- Urbich, C.; Kuehbacher, A.; Dimmeler, S. Role of microRNAs in vascular diseases, inflammation, and angiogenesis. Cardiovasc. Res 2008, 79, 581–588. [Google Scholar]
- Fish, J.E.; Santoro, M.M.; Morton, S.U.; Yu, S.; Yeh, R.F.; Wythe, J.D.; Ivey, K.N.; Bruneau, B.G.; Stainier, D.Y.; Srivastava, D. miR-126 regulates angiogenic signaling and vascular integrity. Dev. Cell 2008, 15, 272–284. [Google Scholar]
- Harris, T.A.; Yamakuchi, M.; Ferlito, M.; Mendell, J.T.; Lowenstein, C.J. MicroRNA-126 regulates endothelial expression of vascular cell adhesion molecule 1. Proc. Natl. Acad. Sci. USA 2008, 105, 1516–1521. [Google Scholar]
- Bonauer, A.; Dimmeler, S. The microRNA-17–92cluster: Still a miRacle? Cell Cycle 2009, 8, 3866–3873. [Google Scholar]
- Bonauer, A.; Carmona, G.; Iwasaki, M.; Mione, M.; Koyanagi, M.; Fischer, A.; Burchfield, J.; Fox, H.; Doebele, C.; Ohtani, K.; et al. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in Mice. Science 2009, 324, 1710–1713. [Google Scholar]
- Fang, Y.; Davies, P.F. Site-specific microRNA-92a regulation of Kruppel-like factors 4 and 2 in atherosusceptible endothelium. Arterioscler. Thromb. Vasc. Biol 2012, 32, 979–987. [Google Scholar]
- Hamik, A.; Lin, Z.; Kumar, A.; Balcells, M.; Sinha, S.; Katz, J.; Feinberg, M.W.; Gerzsten, R.E.; Edelman, E.R.; Jain, M.K. Kruppel-like factor 4 regulates endothelial inflammation. J. Biol. Chem 2007, 282, 13769–13779. [Google Scholar]
- SenBanerjee, S.; Lin, Z.; Atkins, G.B.; Greif, D.M.; Rao, R.M.; Kumar, A.; Feinberg, M.W.; Chen, Z.; Simon, D.I.; Luscinskas, F.W.; et al. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med 2004, 199, 1305–1315. [Google Scholar]
- Lin, Z.; Kumar, A.; SenBanerjee, S.; Staniszewski, K.; Parmar, K.; Vaughan, D.E.; Gimbrone, M.A., Jr; Balasubramanian, V.; García-Cardeña, G.; Jain, M.K. KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med 2004, 199, 1305–1315. [Google Scholar]
- Iaconetti, C.; Polimeni, A.; Sorrentino, S.; Sabatino, J.; Pironti, G.; Esposito, G.; Curcio, A.; Indolfi, C. Inhibition of mir-92a increases endothelial proliferation and migration in vitro as well as reduces neointimal proliferation in vivo after vascular injury. Basic Res. Cardiol 2012, 107. [Google Scholar] [CrossRef]
- Indolfi, C.; Torella, D.; Coppola, C.; Curcio, A.; Rodriguez, F.; Bilancio, A.; Leccia, A.; Arcucci, O.; Falco, M.; Leosco, D.; et al. Physical training increases eNOS vascular expression and activity and reduces restenosis after balloon angioplasty or arterial stenting in rats. Circ. Res 2002, 91, 1190–1197. [Google Scholar]
- Poliseno, L.; Tuccoli, A.; Mariani, L.; Evangelista, M.; Citti, L.; Woods, K.; Mercatanti, A.; Hammond, S.; Rainaldi, G. MicroRNAs modulate the angiogenic properties of HUVECs. Blood 2006, 108, 3068–3071. [Google Scholar]
- Zhu, N.; Zhang, D.; Chen, S.; Liu, X.; Lin, L.; Huang, X.; Guo, Z.; Liu, J.; Wang, Y.; Yuan, W.; et al. Endothelial enriched microRNAs regulate angiotensin II-induced endothelial inflammation and migration. Atherosclerosis 2011, 215, 286–293. [Google Scholar]
- Zhan, Y.; Brown, C.; Maynard, E.; Anshelevich, A.; Ni, W.; Ho, I.C.; Oettgen, P. Ets-1 is a critical regulator of Ang IImediated vascular inflammation and remodeling. J. Clin. Investig 2005, 115, 2508–2516. [Google Scholar]
- Sun, H.X.; Zeng, D.Y.; Li, R.T.; Pang, R.P.; Yang, H.; Hu, Y.L.; Zhang, Q.; Jiang, Y.; Huang, L.Y.; Tang, Y.B.; et al. Essential role of microRNA-155 in regulating endothelium-dependent vasorelaxation by targeting endothelial nitric oxide synthase. Hypertension 2012, 60, 1407–1414. [Google Scholar]
- Hansson, G.K.; Libby, P. The immune response in atherosclerosis: A double-edged sword. Nat. Rev. Immunol 2006, 6, 508–519. [Google Scholar]
- Gareus, R.; Kotsaki, E.; Xanthoulea, S.; van der Made, I.; Gijbels, M.J.; Kardakaris, R.; Polykratis, A.; Kollias, G.; de Winther, M.P.; Pasparakis, M. Endothelial cell-specific NF-kappaB inhibition protects mice from atherosclerosis. Cell Metab 2008, 8, 372–383. [Google Scholar]
- Blackwell, T.S.; et al. MICU Registry. MicroRNA-181b regulates NF-κB-mediated vascular inflammation. J. Clin. Investig 2012, 122, 1973–1990. [Google Scholar]
- Fang, Y.; Shi, C.; Manduchi, E.; Civelek, M.; Davies, P.F. MicroRNA-10a regulation of proinflammatory phenotype in athero-susceptible endothelium in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2010, 107, 13450–13455. [Google Scholar]
- Li, D.; Yang, P.; Xiong, Q.; Song, X.; Yang, X.; Liu, L.; Yuan, W.; Rui, Y.C. MicroRNA-125a/b-5p inhibits endothelin-1 expression in vascular endothelial cells. J. Hypertens 2010, 28, 1646–1654. [Google Scholar]
- Ohkita, M.; Tawa, M.; Kitada, K.; Matsumura, Y. Pathophysiological roles of endothelin receptors in cardiovascular diseases. J. Pharmacol. Sci 2012, 119, 302–313. [Google Scholar]
- Alexander, M.R.; Owens, G.K. Epigenetic control of smooth muscle cell differentiation and phenotypic switching in vascular development and disease. Annu. Rev. Physiol 2012, 74, 13–40. [Google Scholar]
- Gomez, D.; Owens, G.K. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc. Res 2012, 95, 156–164. [Google Scholar]
- Indolfi, C.; Stabile, E.; Coppola, C.; Gallo, A.; Perrino, C.; Allevato, G.; Cavuto, L.; Torella, D.; Di Lorenzo, E.; Troncone, G.; et al. Membrane-bound protein kinase A inhibits smooth muscle cell proliferation in vitro and in vivo by amplifying cAMP-protein kinase A signals. Circ. Res 2001, 88, 319–324. [Google Scholar]
- Indolfi, C.; Avvedimento, E.V.; Rapacciuolo, A.; Di Lorenzo, E.; Esposito, G.; Stabile, E.; Feliciello, A.; Mele, E.; Giuliano, P.; Condorelli, G.; et al. Inhibition of cellular ras prevents smooth muscle cell proliferation after vascular injury in vivo. Nat. Med. 1995, 1, 541–545. [Google Scholar]
- Cordes, K.R.; Sheehy, N.T.; White, M.P.; Berry, E.C.; Morton, S.U.; Muth, A.N.; Lee, T.H.; Miano, J.M.; Ivey, K.N.; Srivastava, D. miR-145 and miR-143 regulate smooth muscle cell fate and plasticity. Nature 2009, 460, 705–710. [Google Scholar]
- Cheng, Y.; Liu, X.; Yang, J.; Lin, Y.; Xu, D.Z.; Lu, Q.; Deitch, E.A.; Huo, Y.; Delphin, E.S.; Zhang, C. MicroRNA-145, a novel smooth muscle cell phenotypic marker and modulator, controls vascular neointimal lesion formation. Circ. Res 2009, 105, 158–166. [Google Scholar]
- Elia, L.; Quintavalle, M.; Zhang, J.; Contu, R.; Cossu, L.; Latronico, M.V.; Peterson, K.L.; Indolfi, C.; Catalucci, D.; Chen, J.; et al. The knockout of miR-143 and -145 alters smooth muscle cell maintenance and vascular homeostasis in mice: Correlates with human disease. Cell Death Differ 2009, 16, 1590–1598. [Google Scholar]
- Owens, G.K. Molecular control of vascular smooth muscle cell differentiation and phenotypic plasticity. Novartis Found Symp 2007, 283, 174–191. [Google Scholar]
- Quintavalle, M.; Elia, L.; Condorelli, G.; Courtneidge, S.A. MicroRNA control of podosome formation in vascular smooth muscle cells in vivo and in vitro. J. Cell Biol. 2010, 189, 13–22. [Google Scholar]
- Lagna, G.; Ku, M.M.; Nguyen, P.H.; Neuman, N.A.; Davis, B.N.; Hata, A. Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors. J. Biol. Chem 2007, 282, 37244–37255. [Google Scholar]
- Ten Dijke, P.; Arthur, H.M. Extracellular control of TGFβ signalling in vascular development and disease. Nat. Rev. Mol. Cell Biol 2007, 8, 857–869. [Google Scholar]
- King, K.E.; Iyemere, V.P.; Weissberg, P.L.; Shanahan, C.M. Krüppel-like factor 4 (KLF4/GKLF) is a target of bone morphogenetic proteins and transforming growth factor beta 1 in the regulation of vascular smooth muscle cell phenotype. J. Biol. Chem 2003, 278, 11661–11669. [Google Scholar]
- Davis-Dusenbery, B.N.; Chan, M.C.; Reno, K.E.; Weisman, A.S.; Layne, M.D.; Lagna, G.; Hata, A. Down-regulation of Kruppel-like factor-4 (KLF4) by microRNA-143/145 is critical for modulation of vascular smooth muscle cell phenotype by transforming growth factor-beta and bone morphogenetic protein 4. J. Biol. Chem 2011, 286, 28097–28110. [Google Scholar]
- Deaton, R.A.; Gan, Q.; Owens, G.K. Sp1-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am. J. Physiol. Heart Circ. Physiol 2009, 296, 1027–1037. [Google Scholar]
- Torella, D.; Iaconetti, C.; Catalucci, D.; Ellison, G.M.; Leone, A.; Waring, C.D.; Bochicchio, A.; Vicinanza, C.; Aquila, I.; Curcio, A.; et al. MicroRNA-133 controls vascular smooth muscle cell phenotypic switch in vitro and vascular remodeling in vivo. Circ. Res. 2011, 109, 880–893. [Google Scholar]
- Ji, R.; Cheng, Y.; Yue, J.; Yang, J.; Liu, X.; Chen, H.; Dean, D.B.; Zhang, C. MicroRNA expression signa-ture and antisense-mediated depletion reveal an essential role of microRNA in vascular neointimal lesion formation. Circ. Res 2007, 100, 1579–1588. [Google Scholar]
- Liu, X.; Cheng, Y.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. A necessary role of miR-221and miR-222 in vascular smooth muscle cell proliferation and neointimal hyperplasia. Circ. Res 2009, 104, 476–487. [Google Scholar]
- Sun, S.G.; Zheng, B.; Han, M.; Fang, X.M.; Li, H.X.; Miao, S.B.; Su, M.; Han, Y.; Shi, H.J.; Wen, J.K. miR-146a and Kruppel-like factor 4 form a feedback loop to participate in vascular smooth musclecell proliferation. EMBO Reports 2011, 12, 56–62. [Google Scholar]
- Raitoharju, E.; Lyytikäinen, L.P.; Levula, M.; Oksala, N.; Mennander, A.; Tarkka, M.; Klopp, N.; Illig, T.; Kähönen, M.; Karhunen, P.J.; et al. miR-21, miR-210, miR-34a, and miR-146a/b are up-regulated in human atherosclerotic plaques in the Tampere Vascular Study. Atherosclerosis 2011, 219, 211–217. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Nguyen, P.H.; Lagna, G.; Hata, A. Induction of microRNA-221 by platelet-derived growth factor signaling is critical for modulation of vascular smooth muscle phenotype. J. Biol. Chem 2009, 284, 3728–3738. [Google Scholar]
- Zheng, B.; Han, M.; Wen, J.K. Role of Krüppel-like factor 4 in phenotypic switching and proliferation of vascular smooth muscle cells. IUBMB Life 2010, 62, 132–139. [Google Scholar]
- Horita, H.N.; Simpson, P.A.; Ostriker, A.; Furgeson, S.; van Putten, V.; Weiser-Evans, M.C.; Nemenoff, R.A. Serum response factor regulates expression of phosphatase and tensin homolog through a microRNA network in vascular smooth muscle cells. Arterioscler. Thromb. Vasc. Biol 2011, 31, 2909–2919. [Google Scholar]
- Maegdefessel, L.; Azuma, J.; Toh, R.; Deng, A.; Merk, D.R.; Raiesdana, A.; Leeper, N.J.; Raaz, U.; Schoelmerich, A.M.; McConnell, M.V.; et al. MicroRNA-21 blocks abdominal aortic aneurysm development and nicotine-augmented expansion. Sci. Transl. Med 2012, 22. [Google Scholar] [CrossRef]
- Cheng, Y.; Liu, X.; Zhang, S.; Lin, Y.; Yang, J.; Zhang, C. MicroRNA-21 protects against the H(2)O(2)-induced injury on cardiac myocytes via its target gene PDCD4. J. Mol. Cell. Cardiol 2009, 47, 5–14. [Google Scholar]
- Wang, M.; Li, W.; Chang, G.Q.; Ye, C.S.; Ou, J.S.; Li, X.X.; Liu, Y.; Cheang, T.Y.; Huang, X.L.; Wang, S.M. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler. Thromb. Vasc. Biol 2011, 31, 2044–2053. [Google Scholar]
- Merlet, E.; Atassi, F.; Motiani, R.K.; Mougenot, N.; Jacquet, A.; Nadaud, S.; Capiod, T.; Trebak, M.; Lompré, A.M.; Marchand, A. miR-424/322 regulates vascular smooth muscle cell phenotype and neointimal formation in the rat. Cardiovasc. Res 2013, 98, 458–468. [Google Scholar]
- Suárez, Y.; Fernández-Hernando, C.; Yu, J.; Gerber, S.A.; Harrison, K.D.; Pober, J.S.; Iruela-Arispe, M.L.; Merkenschlager, M.; Sessa, W.C. Dicer-dependent endothelial microRNAs are necessary for postnatal angiogenesis. Proc. Natl. Acad. Sci. USA 2008, 105, 14082–14087. [Google Scholar]
- Fish, J.E.; Srivastava, D. MicroRNAs: Opening a new vein in angiogenesis research. Sci. Signal 2009. [Google Scholar] [CrossRef]
- Wang, S.; Aurora, A.B.; Johnson, B.A.; Qi, X.; McAnally, J.; Hill, J.A.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. The endothelial-specific microRNA miR-126 governs vascular integrity and angiogenesis. Dev. Cell 2008, 15, 261–271. [Google Scholar]
- Kane, N.M.; Meloni, M.; Spencer, H.L.; Craig, M.A.; Strehl, R.; Milligan, G.; Houslay, M.D.; Mountford, J.C.; Emanueli, C.; Baker, A.H. Derivation of endothelial cells from human embryonic stem cells by directed differentiation: Analysis of microRNA and angiogenesis in vitro and in vivo. Arterioscler. Thromb. Vasc. Biol 2010, 30, 1389–1397. [Google Scholar]
- Nicoli, S.; Knyphausen, C.P.; Zhu, L.J.; Lakshmanan, A.; Lawson, N.D. miR-221 is required for endothelial tip cell behaviors during vascular development. Dev. Cell 2012, 22, 418–429. [Google Scholar]
- Albinsson, S.; Suarez, Y.; Skoura, A.; Offermanns, S.; Miano, J.M.; Sessa, W.C. MicroRNAs are necessary for vascular smooth muscle growth, differentiation, and function. Arterioscler. Thromb. Vasc. Biol 2010, 30, 1118–1126. [Google Scholar]
- Huang, H.; Xie, C.; Sun, X.; Ritchie, R.P.; Zhang, J.; Chen, Y.E. miR-10a contributes to retinoid acid-induced smooth muscle cell differentiation. J. Biol. Chem 2010, 285, 9383–9389. [Google Scholar]
- Xie, C.; Ritchie, R.P.; Huang, H.; Zhang, J.; Chen, Y.E. Smooth muscle cell differentiation: Models and underlying molecular mechanisms. Arterioscler. Thromb. Vasc. Biol 2011, 31, 1485–1494. [Google Scholar]
- Qiu, P.; Li, L. Histone acetylation and recruitment of serum responsive factor and CREB-binding protein onto SM22 promoter during SM22 gene expression. Circ. Res 2002, 90, 858–865. [Google Scholar]
- Xie, C.; Huang, H.; Sun, X.; Guo, Y.; Hamblin, M.; Ritchie, R.P.; Garcia-Barrio, M.T.; Zhang, J.; Chen, Y.E. MicroRNA-1 regulates smooth muscle cell differentiation by repressing Kruppel-like factor 4. Stem Cells Dev 2011, 20, 205–210. [Google Scholar]
- Davis, B.N.; Hilyard, A.C.; Lagna, G.; Hata, A. SMAD proteins control DROSHA-mediated microRNA maturation. Nature 2008, 454, 56–61. [Google Scholar]
- Samani, N.J.; Erdmann, J.; Hall, A.S.; Hengstenberg, C.; Mangino, M.; Mayer, B.; Dixon, R.J.; Meitinger, T.; Braund, P.; Wichmann, H.E.; et al. WTCCC and the Cardiogenics Consortium. Genome wide association analysis of coronary artery disease. N. Engl. J. Med 2007, 357, 443–453. [Google Scholar]
- McPherson, R.; Pertsemlidis, A.; Kavaslar, N.; Stewart, A.; Roberts, R.; Cox, D.R.; Hinds, D.A.; Pennacchio, L.A.; Tybjaerg-Hansen, A.; Folsom, A.R.; et al. A common allele on chromosome 9 associated with coronary heart disease. Science 2007, 316, 1488–1491. [Google Scholar]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. PROCARDIS consortium. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet 2008, 17, 806–814. [Google Scholar]
- Holdt, L.M.; Beutner, F.; Scholz, M.; Gielen, S.; Gäbel, G.; Bergert, H.; Schuler, G.; Thiery, J.; Teupser, D. ANRIL expression is associated with atherosclerosis risk at chromosome 9p21. Arterioscler. Thromb. Vasc. Biol 2010, 30, 620–627. [Google Scholar]
- Yap, K.L.; Li, S.; Muñoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar]
- Leung, A.; Trac, C.; Jin, W.; Lanting, L.; Akbany, A.; Sætrom, P.; Schones, D.E.; Natarajan, R. Novel long non-coding RNAs are regulated by angiotensin II in vascular smooth muscle cells. Circ. Res 2013, 113, 266–278. [Google Scholar]
- Fish, J.E.; Matouk, C.C.; Yeboah, E.; Bevan, S.C.; Khan, M.; Patil, K.; Ohh, M.; Marsden, P.A. Hypoxia-inducible expression of a natural cis-antisense transcript inhibits endothelial nitric-oxide synthase. J. Biol. Chem 2007, 282, 15652–15666. [Google Scholar]
- Robb, G.B.; Carson, A.R.; Tai, S.C.; Fish, J.E.; Singh, S.; Yamada, T.; Scherer, S.W.; Nakabayashi, K.; Marsden, P.A. Post-transcriptional regulation of endothelial nitric-oxide synthase by an overlapping antisense mRNA transcript. J. Biol. Chem 2004, 279, 37982–37996. [Google Scholar]
- Li, K.; Blum, Y.; Verma, A.; Liu, Z.; Pramanik, K.; Leigh, N.R.; Chun, C.Z.; Samant, G.V.; Zhao, B.; Garnaas, M.K.; et al. A noncoding antisense RNA in tie-1 locus regulates tie-1 function in vivo. Blood 2010, 115, 133–139. [Google Scholar]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar]
- Carè, A.; Catalucci, D.; Felicetti, F.; Bonci, D.; Addario, A.; Gallo, P.; Bang, M.L.; Segnalini, P.; Gu, Y.; Dalton, N.D.; et al. MicroRNA-133 controls cardiac hypertrophy. Nat. Med 2007, 13, 613–618. [Google Scholar]
- Chen, J.F.; Mandel, E.M.; Thomson, J.M.; Wu, Q.; Callis, T.E.; Hammond, S.M.; Conlon, F.L.; Wang, D.Z. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nat. Genet 2006, 38, 228–233. [Google Scholar]
- Liu, N.; Bezprozvannaya, S.; Williams, A.H.; Qi, X.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes Dev 2008, 22, 3242–3254. [Google Scholar]
- Ikeda, S.; He, A.; Kong, S.W.; Lu, J.; Bejar, R.; Bodyak, N.; Lee, K.H.; Ma, Q.; Kang, P.M.; Golub, T.R.; et al. MicroRNA-1 negatively regulates expression of the hypertrophy-associated calmodulin and Mef2a genes. Mol. Cell Biol 2009, 29, 2193–2204. [Google Scholar]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar]
- Gupta, M.P. Factors controlling cardiac myosin-isoform shift during hypertrophy and heart failure. J. Mol. Cell. Cardiol 2007, 43, 388–403. [Google Scholar]
- Morton, S.U.; Scherz, P.J.; Cordes, K.R.; Ivey, K.N.; Stainier, D.Y.; Srivastava, D. microRNA-138 modulates cardiac patterning during embryonic development. Proc. Natl. Acad. Sci. USA 2008, 105, 17830–17835. [Google Scholar]
- Taber, L.A. Biophysical mechanisms of cardiac looping. Int. J. Dev. Biol 2006, 50, 323–332. [Google Scholar]
- Deacon, D.C.; Nevis, K.R.; Cashman, T.J.; Zhou, Y.; Zhao, L.; Washko, D.; Guner-Ataman, B.; Burns, C.G.; Burns, C.E. The miR-143-adducin3 pathway is essential for cardiac chamber morphogenesis. Development 2010, 137, 1887–1896. [Google Scholar]
- Lagendijk, A.K.; Goumans, M.J.; Burkhard, S.B.; Bakkers, J. MicroRNA-23 restricts cardiac valve formation by inhibiting Has2 and extracellular hyaluronic acid production. Circ. Res 2011, 109, 649–657. [Google Scholar]
- Camenisch, T.D.; Spicer, A.P.; Brehm-Gibson, T.; Biesterfeldt, J.; Augustine, M.L.; Calabro, A., Jr.; Kubalak, S.; Klewer, S.E.; McDonald, J.A. Disruption of hyaluronan synthase-2 abrogates normal cardiac morphogenesis and hyaluronan-mediated transformation of epithelium to mesenchyme. J. Clin. Investig 2000, 106, 349–360. [Google Scholar]
- Stankunas, K.; Ma, G.K.; Kuhnert, F.J.; Kuo, C.J.; Chang, C.P. VEGF signaling has distinct spatiotemporal roles during heart valve development. Dev. Biol 2010, 347, 325–336. [Google Scholar]
- Kuhnert, F.; Mancuso, M.R.; Hampton, J.; Stankunas, K.; Asano, T.; Chen, C.Z.; Kuo, C.J. Attribution of vascular phenotypes of the murine Egfl7 locus to the microRNA miR-126. Development 2008, 135, 3989–3993. [Google Scholar]
- Li, Q.; Lin, X.; Yang, X.; Chang, J. NFATc4 is negatively regulated in miR-133a-mediated cardiomyocyte hypertrophic repression. Am. J. Physiol. Heart Circ. Physiol 2010, 298, H1340–H1347. [Google Scholar]
- Rao, P.K.; Toyama, Y.; Chiang, H.R.; Gupta, S.; Bauer, M.; Medvid, R.; Reinhardt, F.; Liao, R.; Krieger, M.; Jaenisch, R.; et al. Loss of cardiac microRNA-mediated regulation leads to dilated cardiomyopathy and heart failure. Circ. Res 2009, 105, 585–594. [Google Scholar]
- Chen, H.; Untiveros, G.M.; McKee, L.A.; Perez, J.; Li, J.; Antin, P.B.; Konhilas, J.P. Micro-RNA-195 and -451 regulate the LKB1/AMPK signaling axis by targeting MO25. PLoS One 2012, 7, e41574. [Google Scholar]
- Yang, Y.; Ago, T.; Zhai, P.; Abdellatif, M.; Sadoshima, J. Thioredoxin 1 negatively regulates angiotensin II-induced cardiac hypertrophy through upregulation of miR-98/let-7. Circ. Res 2011, 108, 305–313. [Google Scholar]
- Elia, L.; Contu, R.; Quintavalle, M.; Varrone, F.; Chimenti, C.; Russo, M.A.; Cimino, V.; de Marinis, L.; Frustaci, A.; Catalucci, D.; et al. Reciprocal regulation of microRNA-1 and insulin-like growth factor-1 signal transduction cascade in cardiac and skeletal muscle in physiological and pathological conditions. Circulation 2009, 120, 2377–2385. [Google Scholar]
- Obata, K.; Nagata, K.; Iwase, M.; Odashima, M.; Nagasaka, T.; Izawa, H.; Murohara, T.; Yamada, Y.; Yokota, M. Overexpression of calmodulin induces cardiac hypertrophy by a calcineurin-dependent pathway. Biochem. Biophys. Res. Commun 2005, 338, 1299–1305. [Google Scholar]
- Li, Q.; Song, X.W.; Zou, J.; Wang, G.K.; Kremneva, E.; Li, X.Q.; Zhu, N.; Sun, T.; Lappalainen, P.; Yuan, W.J.; et al. Attenuation of microRNA-1 derepresses the cytoskeleton regulatory protein twinfilin-1 to provoke cardiac hypertrophy. J. Cell Sci 2010, 123, 2444–2452. [Google Scholar]
- Tang, Y.; Zheng, J.; Sun, Y.; Wu, Z.; Liu, Z.; Huang, G. MicroRNA-1 regulates cardiomyocyte apoptosis by targeting bcl-2. Int. Heart J 2009, 50, 377–387. [Google Scholar]
- Sayed, D.; Hong, C.; Chen, I.Y.; Lypowy, J.; Abdellatif, M. MicroRNA play an essential role in development of cardiac hypertrophy. Circ. Res 2007, 100, 416–424. [Google Scholar]
- Curcio, A.; Torella, D.; Iaconetti, C.; Pasceri, E.; Sabatino, J.; Sorrentino, S.; Giampà, S.; Micieli, M.; Polimeni, A.; Henning, B.J.; et al. MicroRNA-1 downregulation increases connexin 43 displacement and induces ventricular tachyarrhythmias in rodent hypertrophic hearts. PLoS One 2013, 8, e70158. [Google Scholar]
- Dong, D.L.; Chen, C.; Huo, R.; Wang, N.; Li, Z.; Tu, Y.J.; Hu, J.T.; Chu, X.; Huang, W.; Yang, B.F. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: A novel mechanism for progressive cardiac hypertrophy. Hypertension 2010, 55, 946–952. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Liu, N.; Williams, A.H.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc. Natl. Acad. Sci. USA 2006, 103, 18255–18260. [Google Scholar]
- Matkovich, S.J.; Wang, W.; Tu, Y.; Eschenbacher, W.H.; Dorn, L.E.; Condorelli, G.; Diwan, A.; Nerbonne, J.M.; Dorn, G.W., 2nd. MicroRNA-133a protects against myocardial fibrosis and modulates electrical repolarization without affecting hypertrophy in pressure-overloaded adult hearts. Circ. Res 2010, 106, 166–175. [Google Scholar]
- Krützfeldt, J.; Rajewsky, N.; Braich, R.; Rajeev, K.G.; Tuschl, T.; Manoharan, M.; Stoffel, M. Silencing of microRNAs in vivo with “antagomirs”. Nature 2005, 438, 685–689. [Google Scholar]
- Wang, K.; Long, B.; Zhou, J.; Li, P.F. miR-9 and NFATc3 regulate myocardin in cardiac hypertrophy. J. Biol. Chem 2010, 285, 11903–11912. [Google Scholar]
- Lin, Z.; Murtaza, I.; Wang, K.; Jiao, J.; Gao, J.; Li, P.F. miR-23a functions downstream of NFATc3 to regulate cardiac hypertrophy. Proc. Natl. Acad. Sci. USA 2009, 106, 12103–12108. [Google Scholar]
- Da, Costa; Martins, P.A.; Salic, K.; Gladka, M.M.; Armand, A.S.; Leptidis, S.; El Azzouzi, H.; Hansen, A.; Coenen-de Roo, C.J.; Bierhuizen, M.F.; van der Nagel, R.; et al. MicroRNA-199b targets the nuclear kinase Dyrk1a in an auto-amplification loop promoting calcineurin/NFAT signalling. Nat. Cell Biol 2010, 12, 1220–1227. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Hill, J.; Olson, E.N. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science 2007, 316, 575–579. [Google Scholar]
- Callis, T.E.; Pandya, K.; Seok, H.Y.; Tang, R.H.; Tatsuguchi, M.; Huang, Z.P.; Chen, J.F.; Deng, Z.; Gunn, B.; Shumate, J.; et al. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. J. Clin. Investig 2009, 119, 2772–2786. [Google Scholar]
- Shieh, J.T.; Huang, Y.; Gilmore, J.; Srivastava, D. Elevated miR-499 levels blunt the cardiac stress response. PLoS One 2011, 6, e19481. [Google Scholar]
- Van Rooij, E.; Sutherland, L.B.; Thatcher, J.E.; DiMaio, J.M.; Naseem, R.H.; Marshall, W.S.; Hill, J.A.; Olson, E.N. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc. Natl. Acad. Sci. USA 2008, 105, 13027–13032. [Google Scholar]
- Thum, T.; Gross, C.; Fiedler, J.; Fischer, T.; Kissler, S.; Bussen, M.; Galuppo, P.; Just, S.; Rottbauer, W.; Frantz, S.; et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature 2008, 456, 980–984. [Google Scholar]
- Zavadil, J.; Narasimhan, M.; Blumenberg, M.; Schneider, R.J. Transforming growth factor-beta and microRNA:mRNA regulatory networks in epithelial plasticity. Cells Tissues Organs 2007, 185, 157–161. [Google Scholar]
- Roy, S.; Khanna, S.; Hussain, S.R.; Biswas, S.; Azad, A.; Rink, C.; Gnyawali, S.; Shilo, S.; Nuovo, G.J.; Sen, C.K. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc. Res 2009, 82, 21–29. [Google Scholar]
- Patrick, D.M.; Montgomery, R.L.; Qi, X.; Obad, S.; Kauppinen, S.; Hill, J.A.; van Rooij, E.; Olson, E.N. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J. Clin. Investig 2010, 120, 3912–3916. [Google Scholar]
- Zhu, S.M.; Si, M.L.; Wu, H.L.; Mo, Y.Y. MicroRNA-21 targets the tumor suppressor gene tropomyosin I (TPM1). J. Biol. Chem 2007, 282, 14328–14336. [Google Scholar]
- Ganesan, J.; Ramanujam, D.; Sassi, Y.; Ahles, A.; Jentzsch, C.; Werfel, S.; Leierseder, S.; Loyer, X.; Giacca, M.; Zentilin, L.; et al. MiR-378 controls cardiac hypertrophy by combined repression of mitogen-activated protein kinase pathway factors. Circulation 2013, 127, 2097–2106. [Google Scholar]
- Van Rooij, E.; Marshall, W.S.; Olson, E.N. Toward microRNA-based therapeutics for heart disease: The sense in antisense. Circ. Res 2008, 103, 919–928. [Google Scholar]
- Schonrock, N.; Harvey, R.P.; Mattick, J.S. Long noncoding RNAs in cardiac development and pathophysiology. Circ. Res 2012, 111, 1349–1362. [Google Scholar]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar]
- He, A.; Ma, Q.; Cao, J.; von Gise, A.; Zhou, P.; Xie, H.; Zhang, B.; Hsing, M.; Christodoulou, D.C.; Cahan, P.; et al. Polycomb repressive complex 2 regulates normal development of the mouse heart. Circ. Res 2012, 110, 406–415. [Google Scholar]
- Grote, P.; Wittler, L.; Hendrix, D.; Koch, F.; Währisch, S.; Beisaw, A.; Macura, K.; Bläss, G.; Kellis, M.; Werber, M.; et al. The tissue-specific lncRNA Fendrr is an essential regulator of heart and body wall development in the mouse. Dev. Cell 2013, 24, 206–214. [Google Scholar]
- Ishii, N.; Ozaki, K.; Sato, H.; Mizuno, H.; Saito, S.; Takahashi, A.; Miyamoto, Y.; Ikegawa, S.; Kamatani, N.; Hori, M.; et al. Identification of a novel non-coding RNA, MIAT, that confers risk of myocardial infarction. J. Hum. Genet 2006, 51, 1087–1099. [Google Scholar]
- Guttman, M.; Garber, M.; Levin, J.Z.; Donaghey, J.; Robinson, J.; Adiconis, X.; Fan, L.; Koziol, M.J.; Gnirke, A.; Nusbaum, C.; et al. Ab initio reconstruction of cell type-specific transcriptomes in mouse reveals the conserved multi-exonic structure of lincRNAs. Nat. Biotechnol 2010, 28, 503–510. [Google Scholar]
- Luther, H.P. Role of endogenous antisense RNA in cardiac gene regulation. J. Mol. Med 2005, 83, 26–32. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Iaconetti, C.; Gareri, C.; Polimeni, A.; Indolfi, C. Non-Coding RNAs: The “Dark Matter” of Cardiovascular Pathophysiology. Int. J. Mol. Sci. 2013, 14, 19987-20018. https://doi.org/10.3390/ijms141019987
Iaconetti C, Gareri C, Polimeni A, Indolfi C. Non-Coding RNAs: The “Dark Matter” of Cardiovascular Pathophysiology. International Journal of Molecular Sciences. 2013; 14(10):19987-20018. https://doi.org/10.3390/ijms141019987
Chicago/Turabian StyleIaconetti, Claudio, Clarice Gareri, Alberto Polimeni, and Ciro Indolfi. 2013. "Non-Coding RNAs: The “Dark Matter” of Cardiovascular Pathophysiology" International Journal of Molecular Sciences 14, no. 10: 19987-20018. https://doi.org/10.3390/ijms141019987