Spatial Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis

{kind=link}
Abstract
:1. Introduction
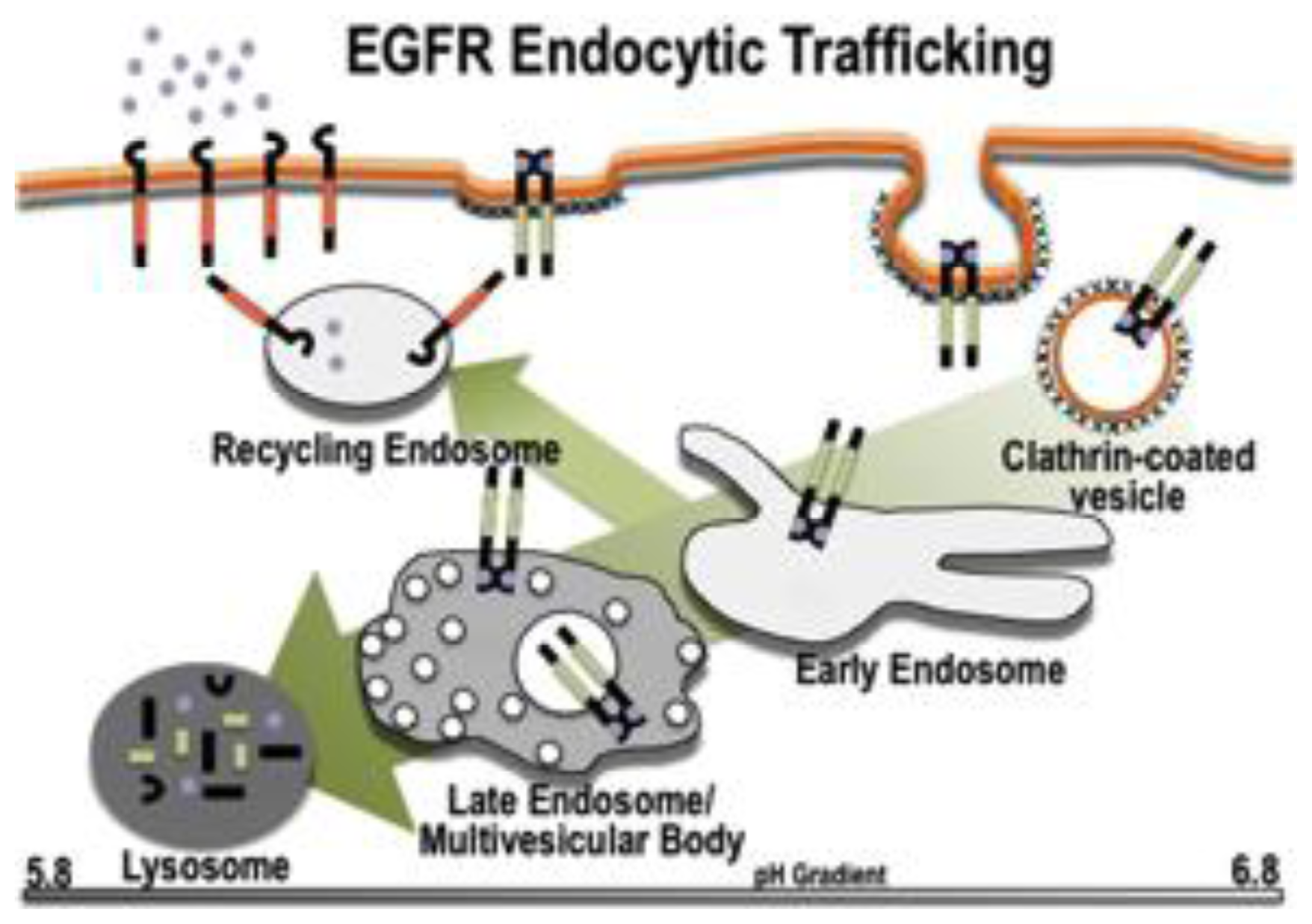
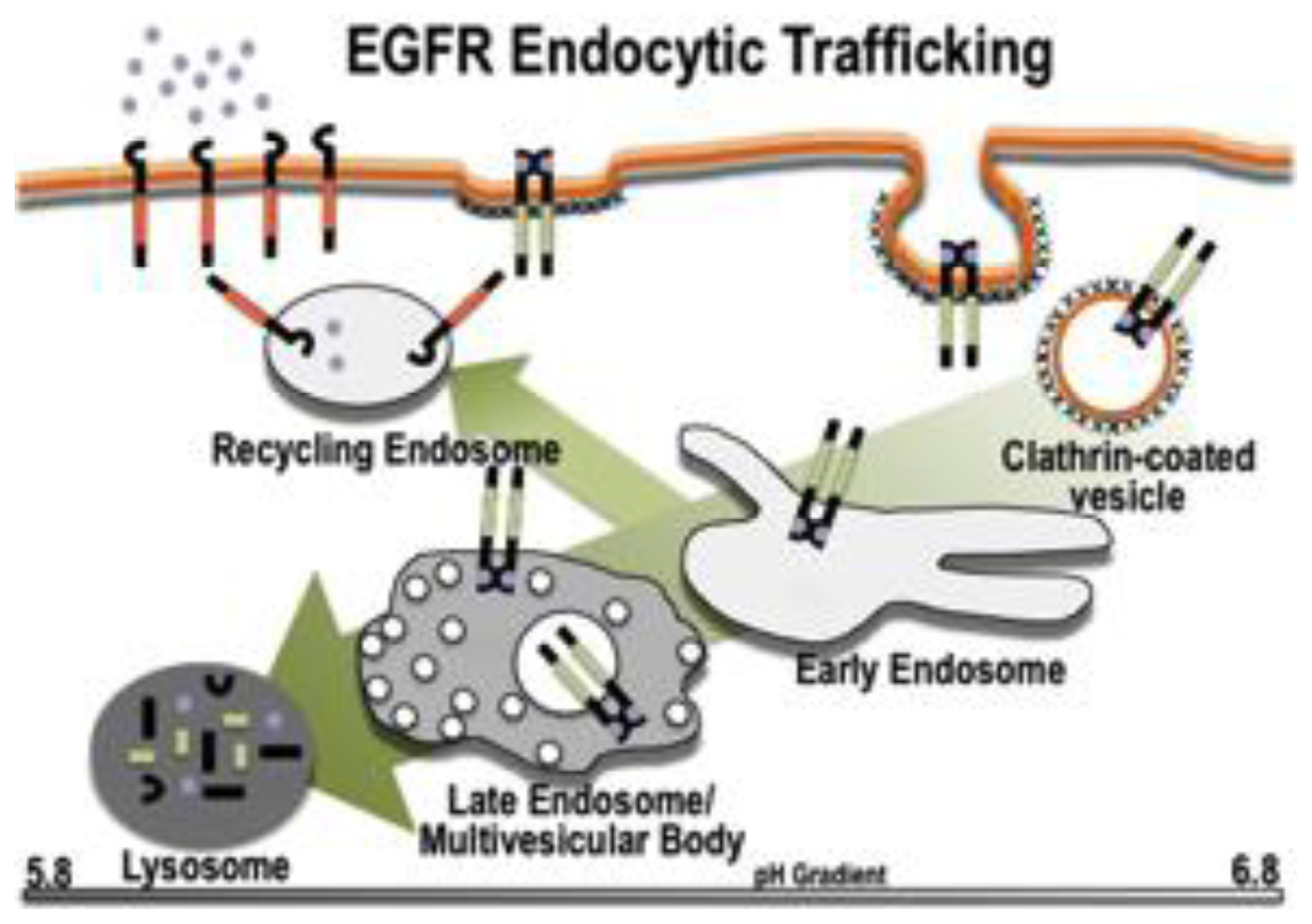
2. The Endocytic Pathway
2.1. Components of the Pathway
2.2. Regulators of EGFR Endocytosis
3. Clinical Relevance of EGFR Endocytic Trafficking
4. Models for Studying EGFRs Deficient in Endocytic Trafficking
4.1. Receptor Mutants
4.2. Inhibitors of the Endocytic Pathway
4.3. Immobilized Ligands
5. Recent Findings
5.1. Proteomic Approach
5.2. Effector Focused Approach
5.3. Transcription Factors
5.4. Cell Biology Approach
6. Conclusions
Acknowledgments
- Conflict of InterestThe author declares no conflict of interest.
References
- Zambruno, G.; Girolomoni, G.; Manca, V.; Segre, A.; Giannetti, A. Epidermal growth factor and transferrin receptor expression in human embryonic and fetal epidermal cells. Arch. Dermatol. Res 1990, 282, 544–548. [Google Scholar]
- Schneider, M.R.; Werner, S.; Paus, R.; Wolf, E. Beyond wavy hairs: The epidermal growth factor receptor and its ligands in skin biology and pathology. Am. J. Pathol 2008, 173, 14–24. [Google Scholar]
- Yu, F.S.; Yin, J.; Xu, K.; Huang, J. Growth factors and corneal epithelial wound healing. Brain Res. Bull 2010, 81, 229–235. [Google Scholar]
- Threadgill, D.W.; Dlugosz, A.A.; Hansen, L.A.; Tennenbaum, T.; Lichti, U.; Yee, D.; LaMantia, C.; Mourton, T.; Herrup, K.; Harris, R.C.; et al. Targeted disruption of mouse EGF receptor: Effect of genetic background on mutant phenotype. Science 1995, 269, 230–234. [Google Scholar]
- Luetteke, N.C.; Qiu, T.H.; Fenton, S.E.; Troyer, K.L.; Riedel, R.F.; Chang, A.; Lee, D.C. Targeted inactivation of the EGF and amphiregulin genes reveals distinct roles for EGF receptor ligands in mouse mammary gland development. Development 1999, 126, 2739–2750. [Google Scholar]
- Rowinsky, E.K. The erbB Family: Targets for therapeutic development against cancer and therapeutic strategies using monoclonal antibodies and tyrosine kinase inhibitors. Ann. Rev. Med 2004, 55, 433–457. [Google Scholar]
- Mayor, S.; Pagano, R.E. Pathways of clathrin-independent endocytosis. Nat. Rev. Mol. Cell Biol 2007, 8, 603–612. [Google Scholar]
- Sigismund, S.; Woelk, T.; Puri, C.; Maspero, E.; Tacchetti, C.; Transidico, P.; DiFiore, P.P.; Polo, S. Clathrin-independent endocytosis of ubiquinated cargos. Proc. Natl. Acad. Sci. USA 2005, 102, 2760–2765. [Google Scholar]
- French, A.R.; Sudlow, G.P.; Wiley, H.S.; Lauffenburger, D.A. Postendocytic trafficking of epidermal growth factor-receptor complexes is mediated through saturable and specific endosomal interactions. J. Biol. Chem 1994, 269, 15749–15755. [Google Scholar]
- Stoscheck, C.M.; Carpenter, G. Characterization of the metabolic turnover of epidermal growth factor receptor protein in A431 cells. J. Cell Phys 1984, 120, 296–302. [Google Scholar]
- McMahon, H.T.; Boucrot, E. Molecular mechanism and physiological functions of clathrin-mediated endocytosis. Nat. Rev. Mol. Cell Biol 2011, 12, 517–533. [Google Scholar]
- Carpenter, G.; Cohen, S. 125I-labeled human epidermal growth factor. Binding, internalization, and degradation in human fibroblasts. J. Cell Biol 1976, 71, 159–171. [Google Scholar]
- Felder, S.; LaVin, J.; Ullrich, A.; Schlessinger, J. Kinetics of binding, endocytosis, and recycling of EGF receptor mutants. J. Cell Biol 1992, 117, 203–212. [Google Scholar]
- Demory, M.L.; Boerner, J.L.; Davidson, R.; Faust, W.; Miyake, T.; Lee, I.; Huttemann, M.; Douglas, R.; Haddad, G.; Parsons, S.J. Epidermal growth factor receptor translocation to the mitochondria: Regulation and effect. J. Biol. Chem 2009, 284, 36592–36604. [Google Scholar]
- Progida, C.; Cogli, L.; Piro, F.; De Luca, A.; Bakke, O.; Bucci, C. Rab7b controls trafficking from endosomes to the TGN. J. Cell Sci 2010, 123, 1480–1491. [Google Scholar]
- Haj, F.G.; Verveer, P.J.; Squire, A.; Neel, B.G.; Bastiaens, P.I. Imaging sites of receptor dephosphorylation by PTP1B on the surface of the endoplasmic reticulum. Science 2002, 295, 1708–1711. [Google Scholar]
- Liao, H.J.; Carpenter, G. Role of the Sec61 translocon in EGF receptor trafficking to the nucleus and gene expression. Mol. Biol. Cell 2007, 18, 1064–1072. [Google Scholar]
- Lin, S.Y.; Makino, K.; Xia, W.; Matin, A.; Wen, Y.; Kwong, K.Y.; Bourguignon, L.; Hung, M.C. Nuclear localization of EGF receptor and its potential new role as a transcription factor. Nat. Cell Biol 2001, 3, 802–808. [Google Scholar]
- Vieira, A.V.; Lamaze, C.; Schmid, S.L. Control of EGF receptor signaling by clathrin-mediated endocytosis. Science 1996, 274, 2086–2089. [Google Scholar]
- Burke, P.; Schooler, K.; Wiley, H.S. Regulation of epidermal growth factor receptor signaling by endocytosis and intracellular trafficking. Mol. Biol. Cell 2001, 12, 1897–1910. [Google Scholar]
- Ceresa, B.P. Regulation of EGFR endocytic trafficking by rab proteins. Histol. Histopathol 2006, 21, 987–993. [Google Scholar]
- Kelly, E.E.; Horgan, C.P.; Goud, B.; McCaffrey, M.W. The Rab family of proteins: 25 years on. Biochem. Soc. Trans 2012, 40, 1337–1347. [Google Scholar]
- Ferguson, S.M.; De Camilli, P. Dynamin, a membrane-remodelling GTPase. Nat. Rev. Mol. Cell Biol 2012, 13, 75–88. [Google Scholar]
- Levkowitz, G.; Waterman, H.; Zamir, E.; Kam, Z.; Oved, S.; Langdon, W.Y.; Beguinot, L.; Geiger, B.; Yarden, Y. c-Cbl/Sli-1 regulates endocytic sorting and ubiquitination of the epidermal growth factor receptor. Genes Dev 1998, 12, 3663–3674. [Google Scholar]
- Waterman, H.; Katz, M.; Rubin, C.; Shtiegman, K.; Lavi, S.; Elson, A.; Jovin, T.M.; Yarden, Y. A mutant EGF-receptor defective in ubiquitylation and endocytosis unveils a role for Grb2 in negative signaling. EMBO J 2002, 21, 303–313. [Google Scholar]
- Grovdal, L.M.; Stang, E.; Sorkin, A.; Madshus, I.H. Direct interaction of Cbl with pTyr 1045 of the EGF receptor (EGFR) is required to sort the EGFR to lysosomes for degradation. Exp. Cell Res 2004, 300, 388–395. [Google Scholar]
- Oksvold, M.P.; Thien, C.B.; Widerberg, J.; Chantry, A.; Huitfeldt, H.S.; Langdon, W.Y. Serine mutations that abrogate ligand-induced ubiquitination and internalization of the EGF receptor do not affect c-Cbl association with the receptor. Oncogene 2003, 22, 8509–8518. [Google Scholar]
- Fallon, L.; Belanger, C.M.; Corera, A.T.; Kontogiannea, M.; Regan-Klapisz, E.; Moreau, F.; Voortman, J.; Haber, M.; Rouleau, G.; Thorarinsdottir, T.; et al. A regulated interaction with the UIM protein Eps15 implicates parkin in EGF receptor trafficking and PI(3)K-Akt signalling. Nat. Cell Biol 2006, 8, 834–842. [Google Scholar]
- Stang, E.; Blystad, F.D.; Kazazic, M.; Bertelsen, V.; Brodahl, T.; Raiborg, C.; Stenmark, H.; Madshus, I.H. Cbl-dependent ubituination is required for progression of EGF receptors into clathrin-coated pits. Mol. Biol. Cell 2006, 15, 3591–3604. [Google Scholar]
- Eden, E.R.; Huang, F.; Sorkin, A.; Futter, C.E. The role of EGF receptor ubiquitination in regulating its intracellular traffic. Traffic 2012, 13, 329–337. [Google Scholar]
- Longva, K.E.; Blystad, F.D.; Stang, E.; Larsen, A.M.; Johannessen, L.E.; Madshus, I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol 2002, 156, 843–854. [Google Scholar]
- Haglund, K.; Sigismund, S.; Polo, S.; Szymkiewicz, I.; Di Fiore, P.P.; Dikic, I. Multiple monoubiquitination of RTKs is sufficient for their endocytosis and degradation. Nat. Cell Biol 2003, 5, 461–466. [Google Scholar]
- Huang, F.; Goh, L.K.; Sorkin, A. EGF receptor ubiquitination is not necessary for its internalization. Proc. Natl. Acad. Sci. USA 2007, 104, 16904–16909. [Google Scholar]
- Bertelsen, V.; Sak, M.M.; Breen, K.; Rodland, M.S.; Johannessen, L.E.; Traub, L.M.; Stang, E.; Madshus, I.H. A chimeric pre-ubiquitinated EGF receptor is constitutively endocytosed in a clathrin-dependent, but kinase-independent manner. Traffic 2011, 12, 507–520. [Google Scholar]
- Mosesson, Y.; Mills, G.B.; Yarden, Y. Derailed endocytosis: An emerging feature of cancer. Nat. Rev. Cancer 2008, 8, 835–850. [Google Scholar]
- Chung, B.M.; Raja, S.M.; Clubb, R.J.; Tu, C.; George, M.; Band, V.; Band, H. Aberrant trafficking of NSCLC-associated EGFR mutants through the endocytic recycling pathway promotes interaction with Src. BMC Cell Biol 2009, 10, 84. [Google Scholar]
- McClintock, J.L.; Ceresa, B.P. Transforming growth factor-α (TGF-α) enhances corneal epithelial cell migration by promoting EGFR recycling. Invest. Opththalmol. Vis. Sci 2010, 51, 3455–3461. [Google Scholar]
- Assaker, G.; Ramel, D.; Wculek, S.K.; Gonzalez-Gaitan, M.; Emery, G. Spatial restriction of receptor tyrosine kinase activity through a polarized endocytic cycle controls border cell migration. Proc. Natl. Acad. Sci. USA 2010, 107, 22558–22563. [Google Scholar]
- Skorobogata, O.; Rocheleau, C.E. RAB-7 antagonizes LET-23 EGFR signaling during vulva development in Caenorhabditis elegans. PLoS One 2012, 7, e36489. [Google Scholar]
- Opresko, L.K.; Chang, C.P.; Will, B.H.; Burke, P.M.; Gill, G.N.; Wiley, H.S. Endocytosis and lysosomal targeting of epidermal growth factor receptors are mediated by distinct sequences independent of the tyrosine kinase domain. J. Biol. Chem 1995, 270, 4325–4333. [Google Scholar]
- Wells, A.; Welsh, J.B.; Lazar, C.S.; Wiley, H.S.; Gill, G.N.; Rosenfeld, M.G. Ligand-induced transformation by a noninternalizing epidermal growth factor receptor. Science 1990, 247, 962–964. [Google Scholar]
- Anderson, R.G.; Falck, J.R.; Goldstein, J.L.; Brown, M.S. Visualization of acidic organelles in intact cells by electron microscopy. Proc. Natl. Acad. Sci. USA 1984, 81, 4838–4842. [Google Scholar]
- King, A.C. Monensin, like methylamine, prevents degradation of 125I-epidermal growth factor, causes intracellular accumulation of receptors and blocks the mitogenic response. Biochem. Biophys. Res. Commun 1984, 124, 585–591. [Google Scholar]
- Presly, J.F.; Mayor, S.; McGraw, T.E.; Dunn, K.W.; Maxfield, F.R. Bafilomycin A1 treatment retards transferrin receptor recycling more than bulk membrane recycling. J. Biol. Chem 1997, 272, 13929–13936. [Google Scholar]
- Barbieri, M.A.; Fernandez-Pol, S.; Hunker, C.; Horazodovsky, B.F.; Stahl, P.D. Role of rab5 in EGF receptor-mediated signal transduction. Eur. J. Cell Biol 2004, 83, 305–314. [Google Scholar]
- Damke, H.; Baba, T.; Warnock, D.E.; Schmid, S.L. Induction of mutant dynamin specifically blocks endocytic coated vesicle formation. J. Cell Biol 1994, 127, 915–934. [Google Scholar]
- Dinneen, J.L.; Ceresa, B.P. Expression of dominant negative rab5 in HeLa cells regulates EGFR endocytic trafficking distal from the plasma membrane. Exp. Cell Res 2004, 294, 509–522. [Google Scholar]
- Chen, P.I.; Kong, C.; Su, X.; Stahl, P.D. Rab5 isoforms differentially regulate the trafficking and degradation of epidermal growth factor receptors. J. Biol. Chem 2009, 284, 30328–30338. [Google Scholar]
- Hopkins, C.R.; Trowbridge, I.S. Internalization and processing of transferrin and the transferrin receptor in human carcinoma A431 cells. J. Cell Biol 1983, 97, 508–521. [Google Scholar]
- Roepstorff, K.; Grandal, M.V.; Henriksen, L.; Knudsen, S.L.; Lerdrup, M.; Grovdal, L.; Willumsen, B.M.; van Deurs, B. Differential effects of EGFR ligands on endocytic sorting of the receptor. Traffic 2009, 10, 1115–1127. [Google Scholar]
- Kempiak, S.J.; Yip, S.-C.; Backer, J.M.; Segall, J.E. Local signaling by the EGF receptor. J. Cell Biol 2003, 162, 781–787. [Google Scholar]
- Reynolds, A.R.; Tischer, C.; Verveer, P.J.; Rocks, O.; Bastiaens, P.I.H. EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat. Cell Biol 2003, 5, 447–453. [Google Scholar]
- Verveer, P.J.; Wouters, F.S.; Reynolds, A.R.; Bastiaens, P.I.H. Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 2000, 290, 1567–1570. [Google Scholar]
- Stefonek, T.J.; Masters, K.S. Immobilized gradients of epidermal growth factor promote accelerated and directed keratinocyte migration. Wound Repair Regen 2007, 15, 847–855. [Google Scholar]
- Singh, A.B.; Sugimoto, K.; Harris, R.C. Juxtacrine activation of epidermal growth factor (EGF) receptor by membrane-anchored heparin-binding EGF-like growth factor protects cells from anoikis while maintainaing an epithelial phenotype. J. Biol. Chem 2007, 282, 32890–32901. [Google Scholar]
- Alvarado, D.; Klein, D.E.; Lemmon, M.A. Structural basis for negative cooperativity in growth factor binding to an EGF receptor. Cell 2010, 142, 568–579. [Google Scholar]
- Liu, P.; Cleveland, T.E.T.; Bouyain, S.; Byrne, P.O.; Longo, P.A.; Leahy, D.J. A single ligand is sufficient to activate EGFR dimers. Proc. Natl. Acad. Sci. USA 2012, 109, 10861–10866. [Google Scholar]
- Omerovic, J.; Hammond, D.E.; Prior, I.A.; Clague, M.J. Global snapshot of the influence of endocytosis upon EGF receptor signaling output. J. Proteome Res 2012, 11, 5157–5166. [Google Scholar]
- Luwor, R.B.; Chin, X.; McGeachie, A.B.; Robinson, P.J.; Zhu, H.J. Dynamin II function is required for EGF-mediated Stat3 activation but not Erk1/2 phosphorylation. Growth Factors 2012, 30, 220–229. [Google Scholar]
- Haugh, J.M.; Huang, A.C.; Wiley, H.S.; Wells, A.; Lauffenburger, D.A. Internalized epidermal growth factor receptors participate in the activation of p21(ras) in fibroblasts. J. Biol. Chem 1999, 274, 34350–343560. [Google Scholar]
- Haugh, J.M.; Schooler, K.; Wells, A.; Wiley, H.S.; Lauffenburger, D.A. Effect of epidermal growth factor receptor internalization on regulation of the phospholipase C-gamma1 signaling pathway. J. Biol. Chem 1999, 274, 8958–8965. [Google Scholar]
- Kyriakis, J.M.; Avruch, J. Mammalian MAPK signal transduction pathways activated by stress and inflammation: A 10-year update. Physiol. Rev 2012, 92, 689–737. [Google Scholar]
- Ceresa, B.P.; Kao, A.W.; Santeler, S.R.; Pessin, J.E. Regulation of insulin receptor signaling pathways by clathrin mediated endocytosis. Mol. Cell. Biol 1998, 18, 3862–3870. [Google Scholar]
- Galperin, E.; Sorkin, A. Endosomal targeting of MEK2 requires RAF, MEK kinase activity and clathrin-dependent endocytosis. Traffic 2008, 9, 1776–1790. [Google Scholar]
- Printen, J.A.; Sprague, G.F., Jr. Protein-protein interactions in the yeast pheromone response pathway: Ste5p interacts with all members of the MAP kinase cascade. Genetics 1994, 138, 609–619. [Google Scholar]
- Galperin, E.; Abdelmoti, L.; Sorkin, A. Shoc2 is targeted to late endosomes and required for Erk1/2 activation in EGF-stimulated cells. PLoS One 2012, 7, e36469. [Google Scholar]
- Dai, P.; Xiong, W.C.; Mei, L. Erbin inhibits RAF activation by disrupting the sur-8-Ras-Raf complex. J. Biol. Chem 2006, 281, 927–933. [Google Scholar]
- Brankatschk, B.; Wichert, S.P.; Johnson, S.D.; Schaad, O.; Rossner, M.J.; Gruenberg, J. Regulation of the EGF transcriptional response by endocytic sorting. Sci. Signal. 2012, 5. [Google Scholar] [CrossRef]
- Wu, P.; Wee, P.; Jiang, J.; Chen, X.; Wang, Z. Differential regulation of transcription factors by location-specific EGF receptor signaling via a spatio-temporal interplay of ERK activation. PLoS One 2012, 7, e41354. [Google Scholar]
- Singh, A.B.; Tsukada, T.; Zent, R.; Harris, R.C. Membrane-associated HB-EGF modulates HGF-induced cellular responses in MDCK cells. J. Cell Sci 2004, 117, 1365–1379. [Google Scholar]
- Hyatt, D.C.; Ceresa, B.P. Cellular localization of the activated EGFR determines its effect on cell growth in MDA-MB-468 cells. Exp. Cell Res 2008, 314, 3415–3425. [Google Scholar]
- Filmus, J.; Pollak, M.N.; Cailleau, R.; Buick, R.N. MDA-468, a human breast cancer cell line with a high number of epidermal growth factor (EGF) receptors, has an amplified EGF receptor gene and is growth inhibited by EGF. Biochem. Biophys. Res. Commun 1985, 128, 898–905. [Google Scholar]
- Rush, J.S.; Quinalty, L.M.; Engelman, L.; Sherry, D.M.; Ceresa, B.P. Endosomal accumulation of the activated epidermal growth factor receptor (EGFR) induces apoptosis. J. Biol. Chem 2012, 287, 712–722. [Google Scholar]
- Overmeyer, J.H.; Kaul, A.; Johnson, E.E.; Maltese, W.A. Active ras triggers death in glioblastoma cells through hyperstimulation of macropinocytosis. Mol. Cancer Res 2008, 6, 965–977. [Google Scholar]
- Robinson, M.W.; Overmeyer, J.H.; Young, A.M.; Erhardt, P.W.; Maltese, W.A. Synthesis and evaluation of indole-based chalcones as inducers of methuosis, a novel type of nonapoptotic cell death. J. Med. Chem 2012, 55, 1940–1956. [Google Scholar]
- Overmeyer, J.H.; Young, A.M.; Bhanot, H.; Maltese, W.A. A chalcone-related small molecule that induces methuosis, a novel form of non-apoptotic cell death, in glioblastoma cells. Mol. Cancer 2011, 10, 1–17. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ceresa, B.P. Spatial Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis. Int. J. Mol. Sci. 2013, 14, 72-87. https://doi.org/10.3390/ijms14010072
Ceresa BP. Spatial Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis. International Journal of Molecular Sciences. 2013; 14(1):72-87. https://doi.org/10.3390/ijms14010072
Chicago/Turabian StyleCeresa, Brian P. 2013. "Spatial Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis" International Journal of Molecular Sciences 14, no. 1: 72-87. https://doi.org/10.3390/ijms14010072
APA StyleCeresa, B. P. (2013). Spatial Regulation of Epidermal Growth Factor Receptor Signaling by Endocytosis. International Journal of Molecular Sciences, 14(1), 72-87. https://doi.org/10.3390/ijms14010072