Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
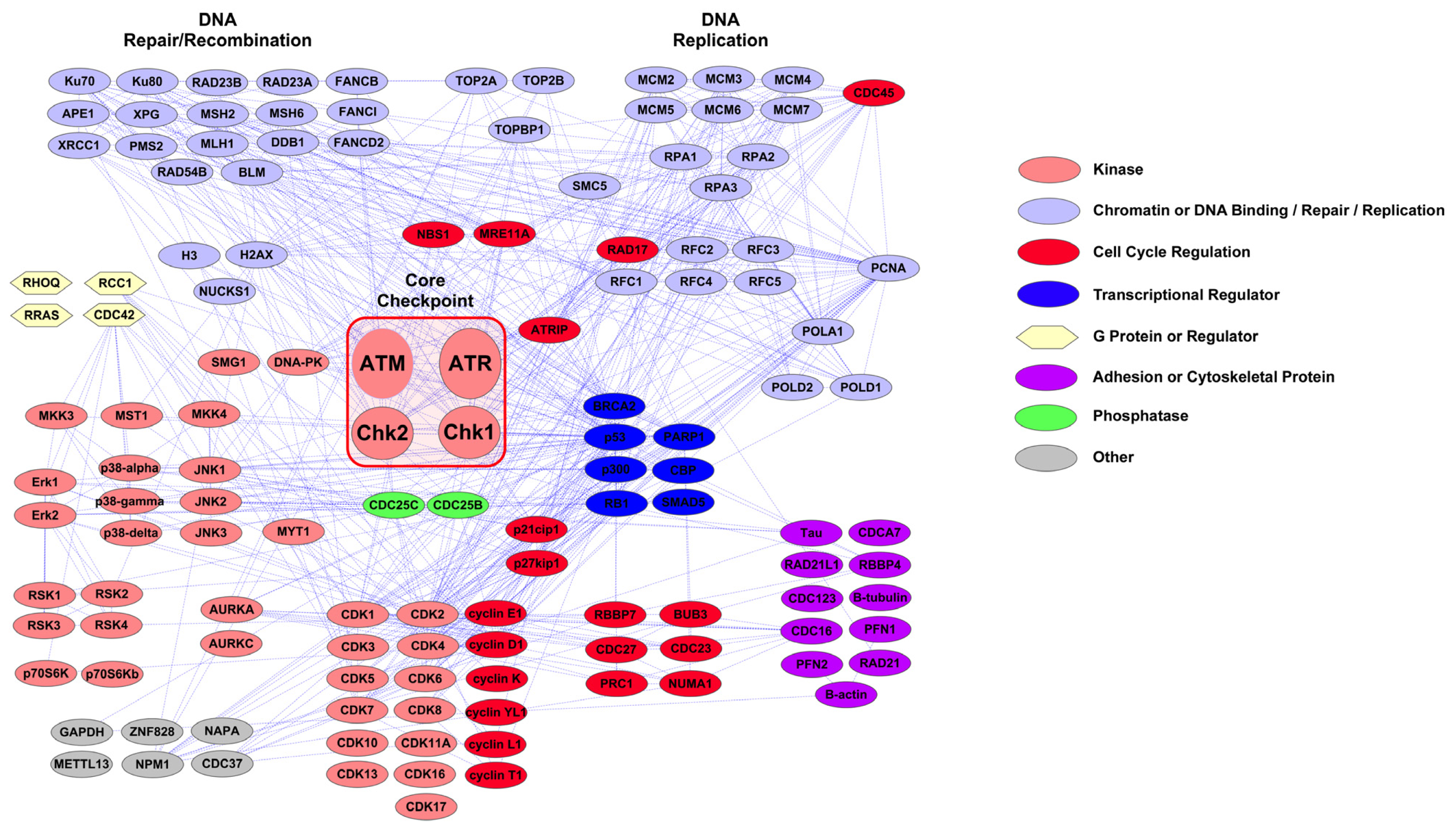
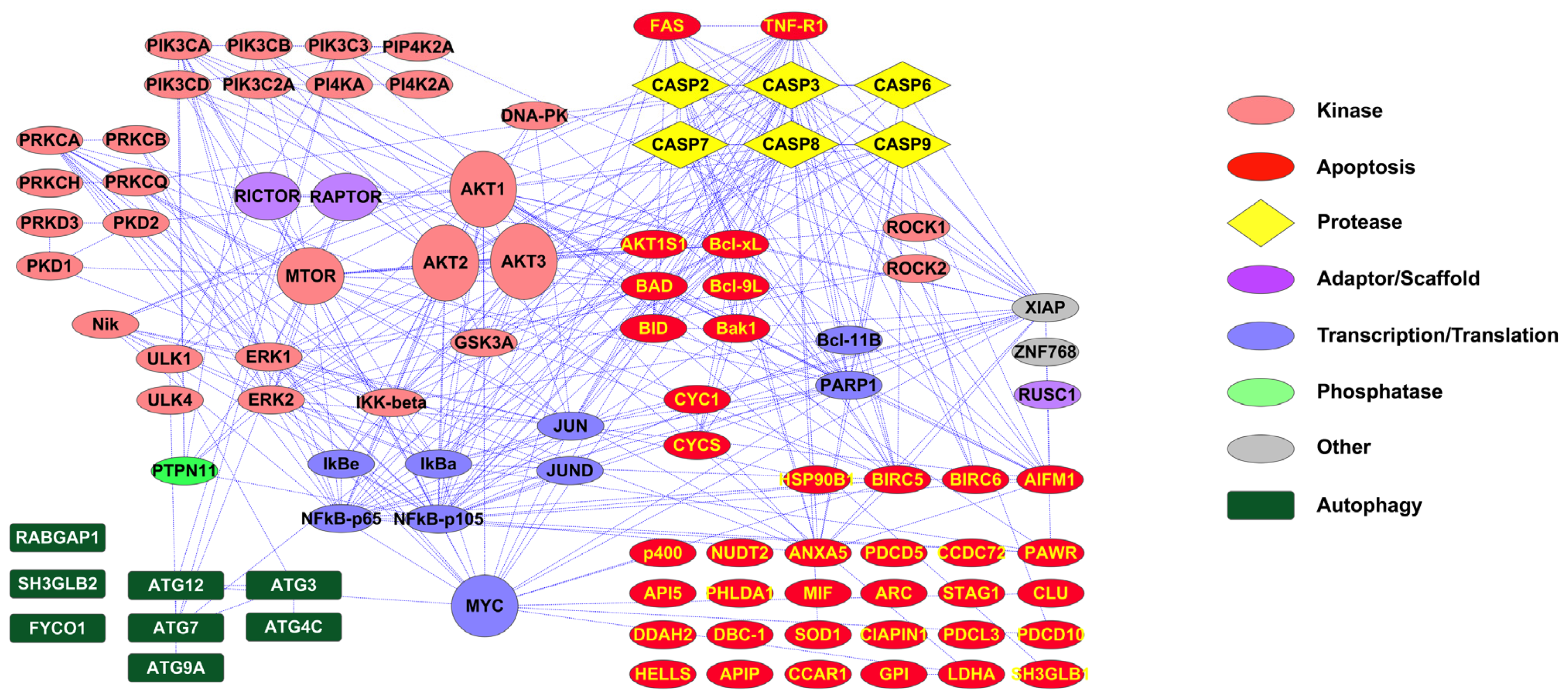
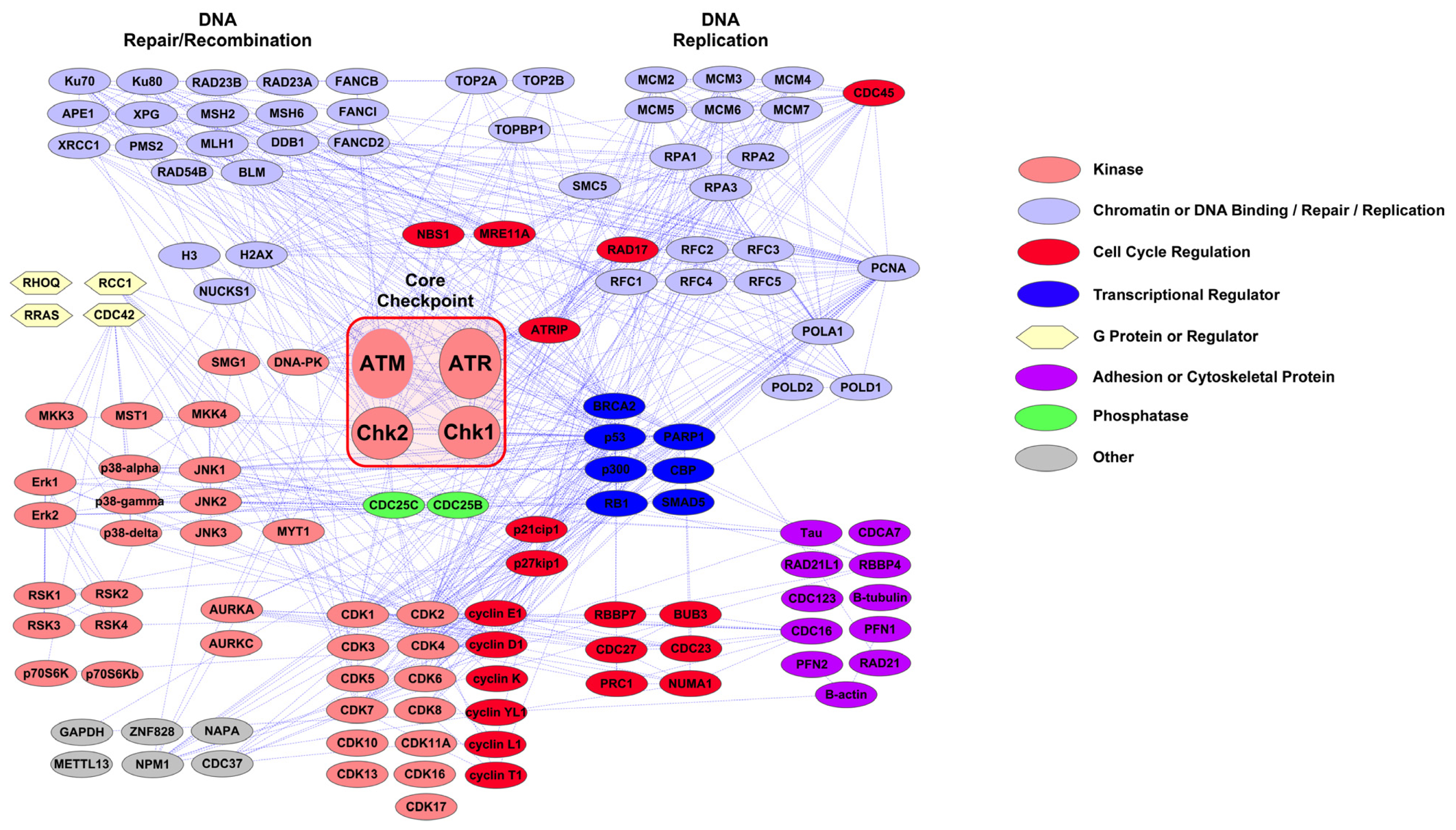
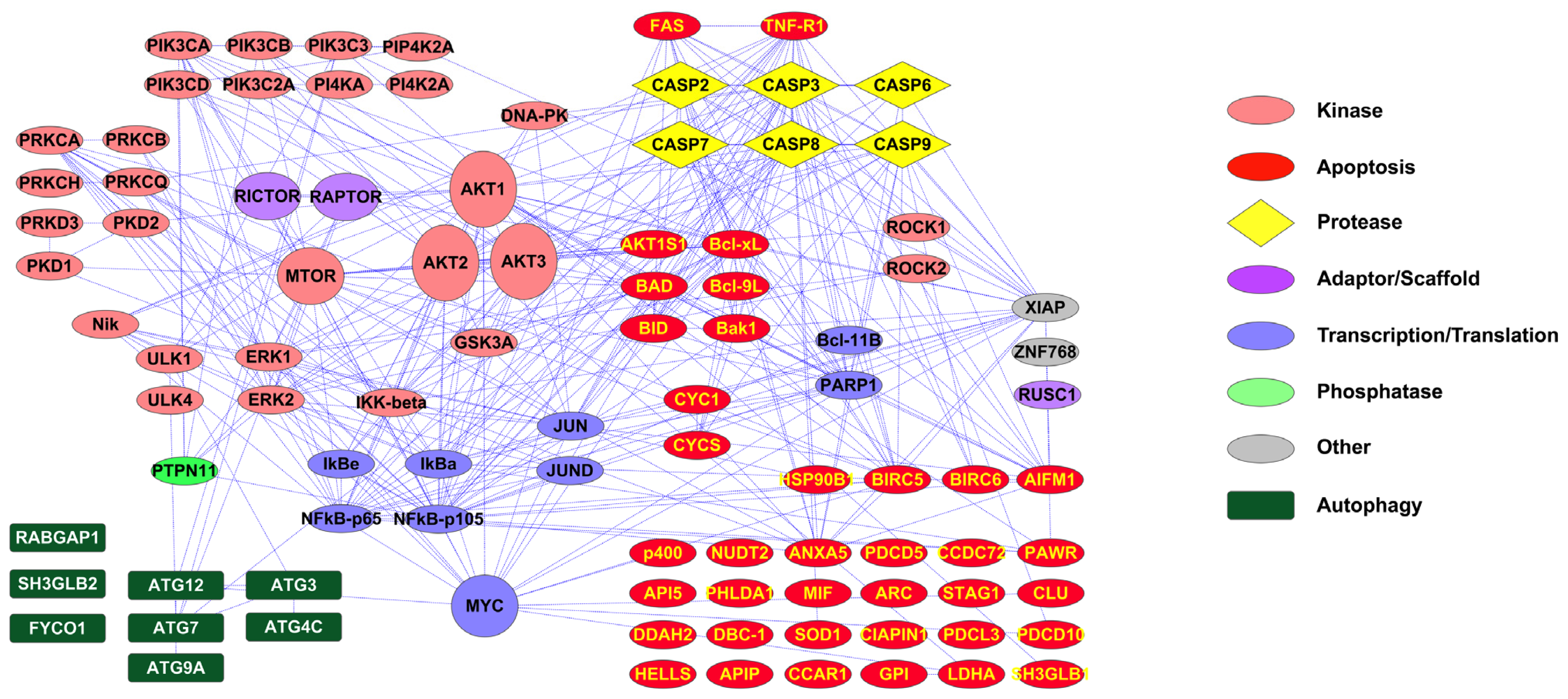
2.1. PTMScan Direct: DNA Damage/Cell Cycle and Apoptosis/Autophagy Reagents
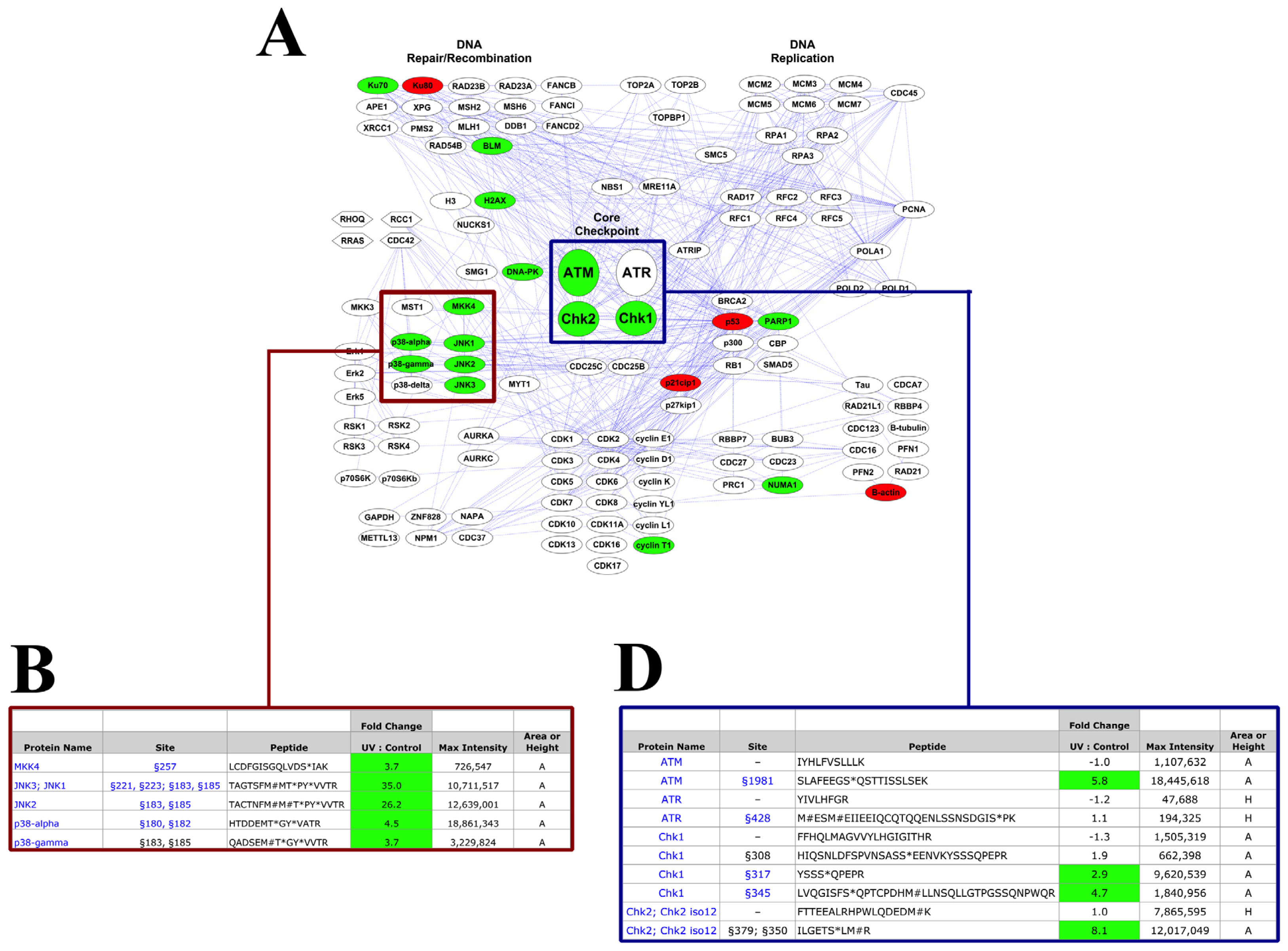
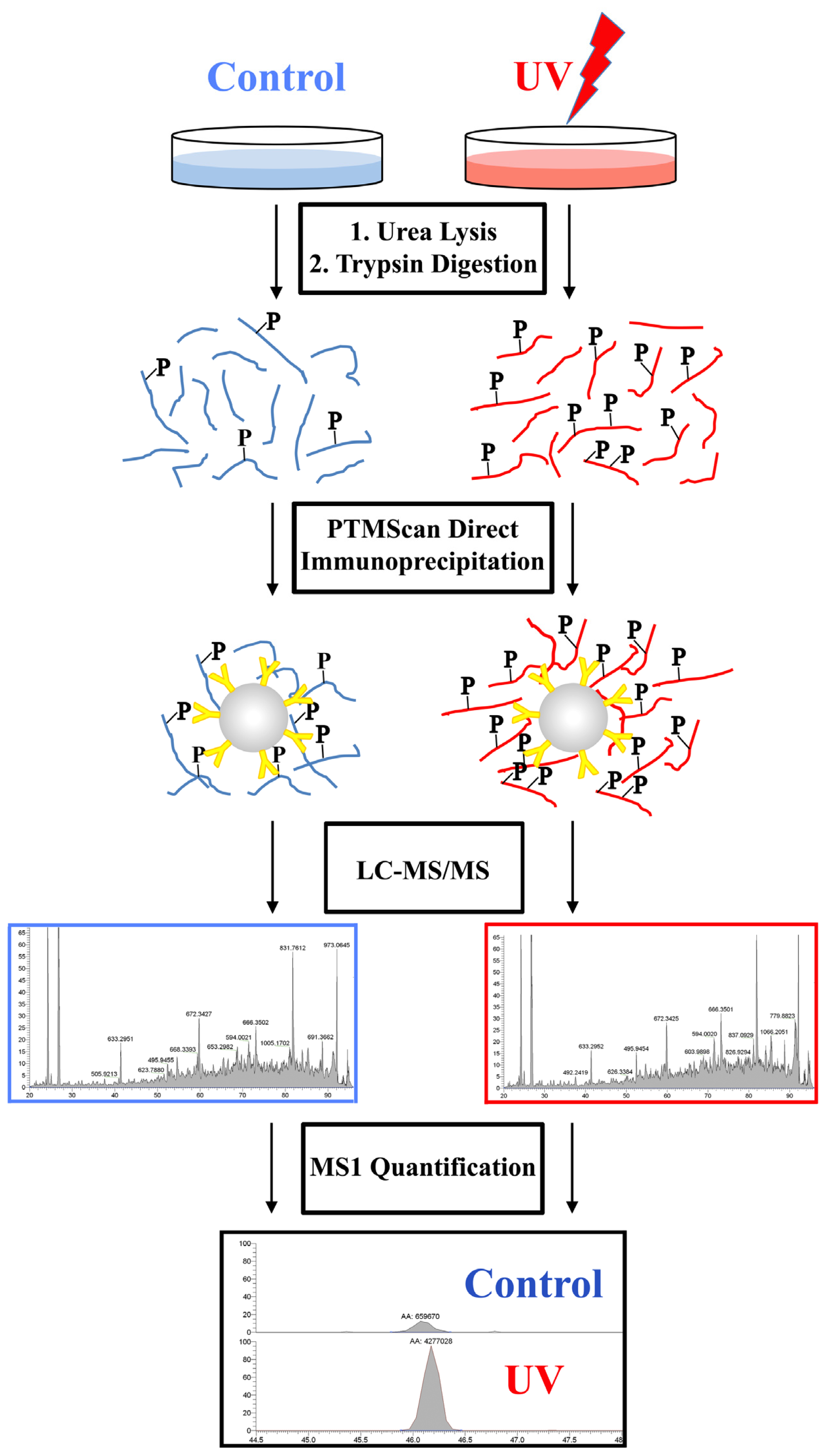
2.2. PTMScan Direct Profiling of UV Damage in HeLa Cells
3. Discussion
3.1. Novel PTMScan Direct Reagents
3.2. DNA Damage Response Signaling
3.3. Induction of Apoptosis
4. Experimental Section
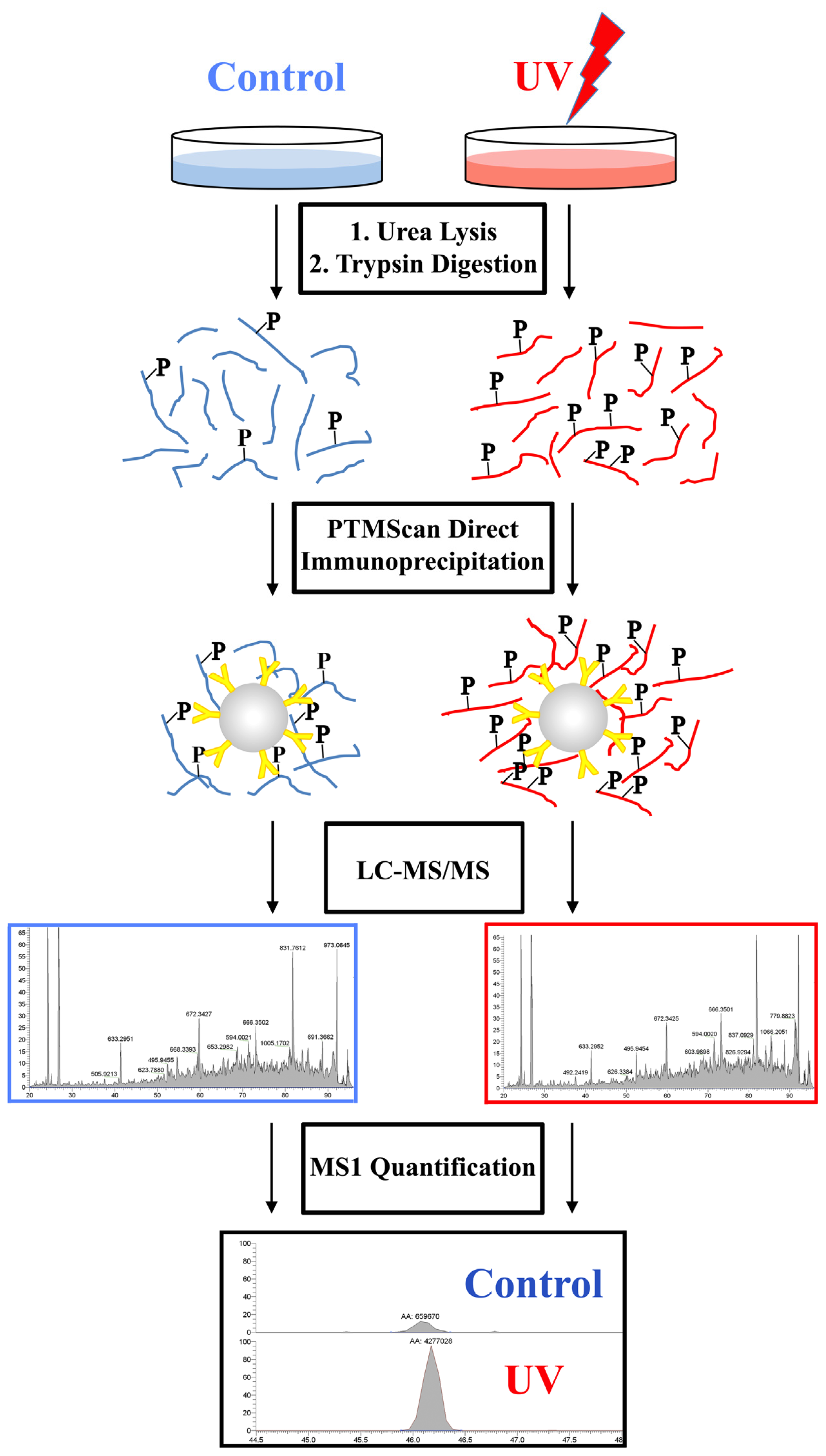
4.1. Overview
4.2. Cell Lines
4.3. LC-MS/MS Analysis
4.4. Data Analysis
4.5. Western Blotting
5. Conclusions
Abbreviations
| CST | Cell Signaling Technology |
| DDR | DNA damage response |
| IAP | Immunoaffinity purification |
| LC-MS/MS | Liquid chromatography tandem mass spectrometry |
| PI3K | Phosphatidylinositol 3-kinase |
| PTM | Post-translational modification |
- Conflict of InterestThe authors declare no conflict of interest.
References
- Abraham, R.T. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001, 15, 2177–2196. [Google Scholar]
- Hartwell, L.H.; Weinert, T.A. Checkpoints: Controls that ensure the order of cell cycle events. Science 1989, 246, 629–634. [Google Scholar]
- Zhou, B.B.; Elledge, S.J. The DNA damage response: putting checkpoints in perspective. Nature 2000, 408, 433–439. [Google Scholar]
- Batista, L.F.; Kaina, B.; Meneghini, R.; Menck, C.F. How DNA lesions are turned into powerful killing structures: insights from UV-induced apoptosis. Mutat. Res 2009, 681, 197–208. [Google Scholar]
- Izumaru, S.; Arima, N.; Toyozumi, Y.; Kato, S.; Morimatsu, M.; Nakashima, T. Down-regulation of p21Waf-1 protein facilitates IR- and UV-induced apoptosis in human squamous carcinoma cells. Int. J. Oncol 2004, 24, 1245–1255. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced cell death by apoptosis. Trends Mol. Med 2006, 12, 440–450. [Google Scholar]
- Roos, W.P.; Kaina, B. DNA damage-induced apoptosis: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2012, in press. [Google Scholar]
- Sheikh, M.S.; Antinore, M.J.; Huang, Y.; Fornace, A.J., Jr. Ultraviolet-irradiation-induced apoptosis is mediated via ligand independent activation of tumor necrosis factor receptor 1. Oncogene 1998, 17, 2555–2563. [Google Scholar]
- Abraham, R.T. PI 3-kinase related kinases: “big” players in stress-induced signaling pathways. DNA Repair 2004, 3, 883–887. [Google Scholar]
- Banin, S.; Moyal, L.; Shieh, S.; Taya, Y.; Anderson, C.W.; Chessa, L.; Smorodinsky, N.I.; Prives, C.; Reiss, Y.; Shiloh, Y.; et al. Enhanced phosphorylation of p53 by ATM in response to DNA damage. Science 1998, 281, 1674–1677. [Google Scholar]
- Canman, C.E.; Lim, D.S.; Cimprich, K.A.; Taya, Y.; Tamai, K.; Sakaguchi, K.; Appella, E.; Kastan, M.B.; Siliciano, J.D. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998, 281, 1677–1679. [Google Scholar]
- Cliby, W.A.; Roberts, C.J.; Cimprich, K.A.; Stringer, C.M.; Lamb, J.R.; Schreiber, S.L.; Friend, S.H. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J 1998, 17, 159–169. [Google Scholar]
- Guo, Z.; Kumagai, A.; Wang, S.X.; Dunphy, W.G. Requirement for Atr in phosphorylation of Chk1 and cell cycle regulation in response to DNA replication blocks and UV-damaged DNA in Xenopus egg extracts. Genes Dev 2000, 14, 2745–2756. [Google Scholar]
- Heffernan, T.P.; Simpson, D.A.; Frank, A.R.; Heinloth, A.N.; Paules, R.S.; Cordeiro-Stone, M.; Kaufmann, W.K. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol. Cell. Biol 2002, 22, 8552–8561. [Google Scholar]
- Kastan, M.B.; Lim, D.S. The many substrates and functions of ATM. Nat. Rev. Mol. Cell. Biol 2000, 1, 179–186. [Google Scholar]
- Khanna, K.K.; Keating, K.E.; Kozlov, S.; Scott, S.; Gatei, M.; Hobson, K.; Taya, Y.; Gabrielli, B.; Chan, D.; Lees-Miller, S.P.; et al. ATM associates with and phosphorylates p53: Mapping the region of interaction. Nat. Genet 1998, 20, 398–400. [Google Scholar]
- Khosravi, R.; Maya, R.; Gottlieb, T.; Oren, M.; Shiloh, Y.; Shkedy, D. Rapid ATM-dependent phosphorylation of MDM2 precedes p53 accumulation in response to DNA damage. Proc. Natl. Acad. Sci. USA 1999, 96, 14973–14977. [Google Scholar]
- Kim, S.T.; Lim, D.S.; Canman, C.E.; Kastan, M.B. Substrate specificities and identification of putative substrates of ATM kinase family members. J. Biol. Chem 1999, 274, 37538–37543. [Google Scholar]
- Lavin, M.F.; Khanna, K.K. ATM: The protein encoded by the gene mutated in the radiosensitive syndrome ataxia-telangiectasia. Int. J. Radiat. Biol 1999, 75, 1201–1214. [Google Scholar]
- Lavin, M.F.; Shiloh, Y. The genetic defect in ataxia-telangiectasia. Annu. Rev. Immunol 1997, 15, 177–202. [Google Scholar]
- Liu, Q.; Guntuku, S.; Cui, X.S.; Matsuoka, S.; Cortez, D.; Tamai, K.; Luo, G.; Carattini-Rivera, S.; DeMayo, F.; Bradley, A.; et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000, 14, 1448–1459. [Google Scholar]
- Matsuoka, S.; Rotman, G.; Ogawa, A.; Shiloh, Y.; Tamai, K.; Elledge, S.J. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc. Natl. Acad. Sci. USA 2000, 97, 10389–10394. [Google Scholar]
- Oakley, G.G.; Loberg, L.I.; Yao, J.; Risinger, M.A.; Yunker, R.L.; Zernik-Kobak, M.; Khanna, K.K.; Lavin, M.F.; Carty, M.P.; Dixon, K. UV-induced hyperphosphorylation of replication protein a depends on DNA replication and expression of ATM protein. Mol. Biol. Cell 2001, 12, 1199–1213. [Google Scholar]
- Sarkaria, J.N.; Busby, E.C.; Tibbetts, R.S.; Roos, P.; Taya, Y.; Karnitz, L.M.; Abraham, R.T. Inhibition of ATM and ATR kinase activities by the radiosensitizing agent, caffeine. Cancer Res 1999, 59, 4375–4382. [Google Scholar]
- Tibbetts, R.S.; Brumbaugh, K.M.; Williams, J.M.; Sarkaria, J.N.; Cliby, W.A.; Shieh, S.Y.; Taya, Y.; Prives, C.; Abraham, R.T. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999, 13, 152–157. [Google Scholar]
- Wright, J.A.; Keegan, K.S.; Herendeen, D.R.; Bentley, N.J.; Carr, A.M.; Hoekstra, M.F.; Concannon, P. Protein kinase mutants of human ATR increase sensitivity to UV and ionizing radiation and abrogate cell cycle checkpoint control. Proc. Natl. Acad. Sci. USA 1998, 95, 7445–7450. [Google Scholar]
- Zhao, H.; Piwnica-Worms, H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol. Cell. Biol 2001, 21, 4129–4139. [Google Scholar]
- Chehab, N.H.; Malikzay, A.; Appel, M.; Halazonetis, T.D. Chk2/hCds1 functions as a DNA damage checkpoint in G(1) by stabilizing p53. Genes Dev 2000, 14, 278–288. [Google Scholar]
- Reinhardt, H.C.; Yaffe, M.B. Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell. Biol 2009, 21, 245–255. [Google Scholar]
- Shieh, S.Y.; Ahn, J.; Tamai, K.; Taya, Y.; Prives, C. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 2000, 14, 289–300. [Google Scholar]
- Tominaga, K.; Morisaki, H.; Kaneko, Y.; Fujimoto, A.; Tanaka, T.; Ohtsubo, M.; Hirai, M.; Okayama, H.; Ikeda, K.; Nakanishi, M. Role of human Cds1 (Chk2) kinase in DNA damage checkpoint and its regulation by p53. J. Biol. Chem 1999, 274, 31463–31467. [Google Scholar]
- Mailand, N.; Falck, J.; Lukas, C.; Syljuasen, R.G.; Welcker, M.; Bartek, J.; Lukas, J. Rapid destruction of human Cdc25A in response to DNA damage. Science 2000, 288, 1425–1429. [Google Scholar]
- Sanchez, Y.; Wong, C.; Thoma, R.S.; Richman, R.; Wu, Z.; Piwnica-Worms, H.; Elledge, S.J. Conservation of the Chk1 checkpoint pathway in mammals: linkage of DNA damage to Cdk regulation through Cdc25. Science 1997, 277, 1497–1501. [Google Scholar]
- Ciccia, A.; Elledge, S.J. The DNA damage response: making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar]
- Finn, K.; Lowndes, N.F.; Grenon, M. Eukaryotic DNA damage checkpoint activation in response to double-strand breaks. Cell. Mol. Life Sci 2011, 69, 1447–1473. [Google Scholar]
- Nam, E.A.; Cortez, D. ATR signalling: More than meeting at the fork. Biochem. J 2011, 436, 527–536. [Google Scholar]
- Matsuoka, S.; Ballif, B.A.; Smogorzewska, A.; McDonald, E.R., III; Hurov, K.E.; Luo, J.; Bakalarski, C.E.; Zhao, Z.; Solimini, N.; Lerenthal, Y.; et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007, 316, 1160–1166. [Google Scholar]
- Pines, A.; Kelstrup, C.D.; Vrouwe, M.G.; Puigvert, J.C.; Typas, D.; Misovic, B.; de Groot, A.; von Stechow, L.; van de Water, B.; Danen, E.H.; et al. Global phosphoproteome profiling reveals unanticipated networks responsive to cisplatin treatment of embryonic stem cells. Mol. Cell. Biol 2011, 31, 4964–4977. [Google Scholar]
- Stokes, M.P.; Comb, M.J. A wide-ranging cellular response to UV damage of DNA. Cell. Cycle 2008, 7, 2097–2099. [Google Scholar]
- Stokes, M.P.; Rush, J.; Macneill, J.; Ren, J.M.; Sprott, K.; Nardone, J.; Yang, V.; Beausoleil, S.A.; Gygi, S.P.; Livingstone, M.; et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 19855–19860. [Google Scholar]
- Bonnette, P.C.; Robinson, B.S.; Silva, J.C.; Stokes, M.P.; Brosius, A.D.; Baumann, A.; Buckbinder, L. Phosphoproteomic characterization of PYK2 signaling pathways involved in osteogenesis. J. Proteomics 2010, 73, 1306–1320. [Google Scholar]
- Brave, S.R.; Ratcliffe, K.; Wilson, Z.; James, N.H.; Ashton, S.; Wainwright, A.; Kendrew, J.; Dudley, P.; Broadbent, N.; Sproat, G.; et al. Assessing the activity of cediranib, a VEGFR-2/3 tyrosine kinase inhibitor, against VEGFR-1 and members of the structurally related PDGFR family. Mol. Cancer Ther 2011, 10, 861–873. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Lee, K.A.; Hammerle, L.P.; Andrews, P.S.; Stokes, M.P.; Mustelin, T.; Silva, J.C.; Black, R.A.; Doedens, J.R. Ubiquitin ligase substrate identification through quantitative proteomics at both the protein and peptide levels. J. Biol. Chem 2011, 286, 41530–41538. [Google Scholar]
- Moritz, A.; Li, Y.; Guo, A.; Villen, J.; Wang, Y.; MacNeill, J.; Kornhauser, J.; Sprott, K.; Zhou, J.; Possemato, A.; et al. Akt-RSK-S6 kinase signaling networks activated by oncogenic receptor tyrosine kinases. Sci. Signal. 2010, 3, ra64. [Google Scholar]
- Peng, J.; Schwartz, D.; Elias, J.E.; Thoreen, C.C.; Cheng, D.; Marsischky, G.; Roelofs, J.; Finley, D.; Gygi, S.P. A proteomics approach to understanding protein ubiquitination. Nat. Biotechnol 2003, 21, 921–926. [Google Scholar]
- Rush, J.; Moritz, A.; Lee, K.A.; Guo, A.; Goss, V.L.; Spek, E.J.; Zhang, H.; Zha, X.M.; Polakiewicz, R.D.; Comb, M.J. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat. Biotechnol 2005, 23, 94–101. [Google Scholar]
- Xu, G.; Paige, J.S.; Jaffrey, S.R. Global analysis of lysine ubiquitination by ubiquitin remnant immunoaffinity profiling. Nat. Biotechnol 2010, 28, 868–873. [Google Scholar]
- Zhang, H.; Zha, X.; Tan, Y.; Hornbeck, P.V.; Mastrangelo, A.J.; Alessi, D.R.; Polakiewicz, R.D.; Comb, M.J. Phosphoprotein analysis using antibodies broadly reactive against phosphorylated motifs. J. Biol. Chem 2002, 277, 39379–39387. [Google Scholar]
- Andersen, J.N.; Sathyanarayanan, S.; Di Bacco, A.; Chi, A.; Zhang, T.; Chen, A.H.; Dolinski, B.; Kraus, M.; Roberts, B.; Arthur, W.; et al. Pathway-based identification of biomarkers for targeted therapeutics: Personalized oncology with PI3K pathway inhibitors. Sci. Transl. Med. 2010, 2, ra55. [Google Scholar]
- Anderson, N.L.; Anderson, N.G.; Haines, L.R.; Hardie, D.B.; Olafson, R.W.; Pearson, T.W. Mass spectrometric quantitation of peptides and proteins using Stable Isotope Standards and Capture by Anti-Peptide Antibodies (SISCAPA). J. Proteome Res 2004, 3, 235–244. [Google Scholar]
- Whiteaker, J.R.; Zhao, L.; Anderson, L.; Paulovich, A.G. An automated and multiplexed method for high throughput peptide immunoaffinity enrichment and multiple reaction monitoring mass spectrometry-based quantification of protein biomarkers. Mol. Cell. Proteomics 2010, 9, 184–196. [Google Scholar]
- Cantin, G.T.; Yi, W.; Lu, B.; Park, S.K.; Xu, T.; Lee, J.D.; Yates, J.R., III. Combining protein-based IMAC, peptide-based IMAC, and MudPIT for efficient phosphoproteomic analysis. J. Proteome Res. 2008, 7, 1346–1351. [Google Scholar]
- Cao, P.; Stults, J.T. Mapping the phosphorylation sites of proteins using on-line immobilized metal affinity chromatography/capillary electrophoresis/electrospray ionization multiple stage tandem mass spectrometry. Rapid Comm. Mass Spectrom 2000, 14, 1600–1606. [Google Scholar]
- Choudhary, C.; Mann, M. Decoding signalling networks by mass spectrometry-based proteomics. Nat. Rev. Mol. Cell. Biol 2010, 11, 427–439. [Google Scholar]
- Eyrich, B.; Sickmann, A.; Zahedi, R.P. Catch me if you can: mass spectrometry-based phosphoproteomics and quantification strategies. Proteomics 2011, 11, 554–570. [Google Scholar]
- Feng, S.; Ye, M.; Zhou, H.; Jiang, X.; Zou, H.; Gong, B. Immobilized zirconium ion affinity chromatography for specific enrichment of phosphopeptides in phosphoproteome analysis. Mol. Cell. Proteomics 2007, 6, 1656–1665. [Google Scholar]
- Larsen, M.R.; Thingholm, T.E.; Jensen, O.N.; Roepstorff, P.; Jorgensen, T.J. Highly selective enrichment of phosphorylated peptides from peptide mixtures using titanium dioxide microcolumns. Mol. Cell. Proteomics 2005, 4, 873–886. [Google Scholar]
- Mann, M.; Ong, S.E.; Gronborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol 2002, 20, 261–268. [Google Scholar]
- Monetti, M.; Nagaraj, N.; Sharma, K.; Mann, M. Large-scale phosphosite quantification in tissues by a spike-in SILAC method. Nat. Methods 2011, 8, 655–658. [Google Scholar]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell 2006, 127, 635–648. [Google Scholar]
- Rigbolt, K.T.; Prokhorova, T.A.; Akimov, V.; Henningsen, J.; Johansen, P.T.; Kratchmarova, I.; Kassem, M.; Mann, M.; Olsen, J.V.; Blagoev, B. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci. Signal. 2011, 4, rs3. [Google Scholar]
- Stensballe, A.; Andersen, S.; Jensen, O.N. Characterization of phosphoproteins from electrophoretic gels by nanoscale Fe(III) affinity chromatography with off-line mass spectrometry analysis. Proteomics 2001, 1, 207–222. [Google Scholar]
- Tan, F.; Zhang, Y.; Mi, W.; Wang, J.; Wei, J.; Cai, Y.; Qian, X. Enrichment of phosphopeptides by Fe3+-immobilized magnetic nanoparticles for phosphoproteome analysis of the plasma membrane of mouse liver. J. Proteome Res 2008, 7, 1078–1087. [Google Scholar]
- Villen, J.; Beausoleil, S.A.; Gerber, S.A.; Gygi, S.P. Large-scale phosphorylation analysis of mouse liver. Proc. Natl. Acad. Sci. USA 2007, 104, 1488–1493. [Google Scholar]
- Zhou, H.; Tian, R.; Ye, M.; Xu, S.; Feng, S.; Pan, C.; Jiang, X.; Li, X.; Zou, H. Highly specific enrichment of phosphopeptides by zirconium dioxide nanoparticles for phosphoproteome analysis. Electrophoresis 2007, 28, 2201–2215. [Google Scholar]
- Stokes, M.P.; Farnsworth, C.L.; Moritz, A.; Silva, J.C.; Jia, X.; Lee, K.A.; Guo, A.; Polakiewicz, R.D.; Comb, M.J. PTMScan direct: identification and quantification of peptides from critical signaling proteins by immunoaffinity enrichment coupled with LC-MS/MS. Mol. Cell. Proteomics 2012, 11, 187–201. [Google Scholar]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res 2003, 13, 2498–2504. [Google Scholar]
- Von Mering, C.; Huynen, M.; Jaeggi, D.; Schmidt, S.; Bork, P.; Snel, B. STRING: A database of predicted functional associations between proteins. Nucleic Acids Res 2003, 31, 258–261. [Google Scholar]
- Lundgren, D.H.; Martinez, H.; Wright, M.E.; Han, D.K. Protein identification using Sorcerer 2 and SEQUEST. Curr. Protoc. Bioinforma. 2009. [Google Scholar] [CrossRef]
- Pandey, P.; Raingeaud, J.; Kaneki, M.; Weichselbaum, R.; Davis, R.J.; Kufe, D.; Kharbanda, S. Activation of p38 mitogen-activated protein kinase by c-Abl-dependent and -independent mechanisms. J. Biol. Chem 1996, 271, 23775–23779. [Google Scholar]
- Hibi, M.; Lin, A.; Smeal, T.; Minden, A.; Karin, M. Identification of an oncoprotein- and UV-responsive protein kinase that binds and potentiates the c-Jun activation domain. Genes Dev 1993, 7, 2135–2148. [Google Scholar]
- Derijard, B.; Hibi, M.; Wu, I.H.; Barrett, T.; Su, B.; Deng, T.; Karin, M.; Davis, R.J. JNK1: A protein kinase stimulated by UV light and Ha-Ras that binds and phosphorylates the c-Jun activation domain. Cell 1994, 76, 1025–1037. [Google Scholar]
- Raingeaud, J.; Gupta, S.; Rogers, J.S.; Dickens, M.; Han, J.; Ulevitch, R.J.; Davis, R.J. Pro-inflammatory cytokines and environmental stress cause p38 mitogen-activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem 1995, 270, 7420–7426. [Google Scholar]
- Han, J.; Lee, J.D.; Bibbs, L.; Ulevitch, R.J. A MAP kinase targeted by endotoxin and hyperosmolarity in mammalian cells. Science 1994, 265, 808–811. [Google Scholar]
- Yuan, J.; Adamski, R.; Chen, J. Focus on histone variant H2AX: to be or not to be. FEBS Lett 2010, 584, 3717–3724. [Google Scholar]
- Rogakou, E.P.; Pilch, D.R.; Orr, A.H.; Ivanova, V.S.; Bonner, W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem 1998, 273, 5858–5868. [Google Scholar]
- Flores-Rozas, H.; Kelman, Z.; Dean, F.B.; Pan, Z.Q.; Harper, J.W.; Elledge, S.J.; O’Donnell, M.; Hurwitz, J. Cdk-interacting protein 1 directly binds with proliferating cell nuclear antigen and inhibits DNA replication catalyzed by the DNA polymerase delta holoenzyme. Proc. Natl. Acad. Sci. USA 1994, 91, 8655–8659. [Google Scholar]
- Pestell, R.G.; Albanese, C.; Reutens, A.T.; Segall, J.E.; Lee, R.J.; Arnold, A. The cyclins and cyclin-dependent kinase inhibitors in hormonal regulation of proliferation and differentiation. Endocr. Rev 1999, 20, 501–534. [Google Scholar]
- Cmielova, J.; Rezacova, M. p21Cip1/Waf1 protein and its function based on a subcellular localization. J. Cell. Biochem 2011, 112, 3502–3506. [Google Scholar]
- Harper, J.W.; Adami, G.R.; Wei, N.; Keyomarsi, K.; Elledge, S.J. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993, 75, 805–816. [Google Scholar]
- Bakkenist, C.J.; Kastan, M.B. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature 2003, 421, 499–506. [Google Scholar]
- Nam, E.A.; Zhao, R.; Glick, G.G.; Bansbach, C.E.; Friedman, D.B.; Cortez, D. Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase. J. Biol. Chem 2011, 286, 28707–28714. [Google Scholar]
- Liu, S.; Shiotani, B.; Lahiri, M.; Marechal, A.; Tse, A.; Leung, C.C.; Glover, J.N.; Yang, X.H.; Zou, L. ATR autophosphorylation as a molecular switch for checkpoint activation. Mol. Cell 2011, 43, 192–202. [Google Scholar]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Cadenas, E.; Spencer, J.P. Inhibition of cellular proliferation by the genistein metabolite 5,7,3′,4′-tetrahydroxyisoflavone is mediated by DNA damage and activation of the ATR signalling pathway. Arch. Biochem. Biophys 2007, 468, 159–166. [Google Scholar]
- Helt, C.E.; Cliby, W.A.; Keng, P.C.; Bambara, R.A.; O’Reilly, M.A. Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage. J. Biol. Chem 2005, 280, 1186–1192. [Google Scholar]
- Douglas, P.; Cui, X.; Block, W.D.; Yu, Y.; Gupta, S.; Ding, Q.; Ye, R.; Morrice, N.; Lees-Miller, S.P.; Meek, K. The DNA-dependent protein kinase catalytic subunit is phosphorylated in vivo on threonine 3950, a highly conserved amino acid in the protein kinase domain. Mol. Cell. Biol 2007, 27, 1581–1591. [Google Scholar]
- Beausoleil, S.A.; Jedrychowski, M.; Schwartz, D.; Elias, J.E.; Villen, J.; Li, J.; Cohn, M.A.; Cantley, L.C.; Gygi, S.P. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc. Natl. Acad. Sci. USA 2004, 101, 12130–12135. [Google Scholar]
- Hartley, K.O.; Gell, D.; Smith, G.C.; Zhang, H.; Divecha, N.; Connelly, M.A.; Admon, A.; Lees-Miller, S.P.; Anderson, C.W.; Jackson, S.P. DNA-dependent protein kinase catalytic subunit: A relative of phosphatidylinositol 3-kinase and the ataxia telangiectasia gene product. Cell 1995, 82, 849–856. [Google Scholar]
- Chan, D.W.; Chen, B.P.; Prithivirajsingh, S.; Kurimasa, A.; Story, M.D.; Qin, J.; Chen, D.J. Autophosphorylation of the DNA-dependent protein kinase catalytic subunit is required for rejoining of DNA double-strand breaks. Genes Dev 2002, 16, 2333–2338. [Google Scholar]
- Mukherjee, B.; Kessinger, C.; Kobayashi, J.; Chen, B.P.; Chen, D.J.; Chatterjee, A.; Burma, S. DNA-PK phosphorylates histone H2AX during apoptotic DNA fragmentation in mammalian cells. DNA Repair 2006, 5, 575–590. [Google Scholar]
- Solier, S.; Sordet, O.; Kohn, K.W.; Pommier, Y. Death receptor-induced activation of the Chk2- and histone H2AX-associated DNA damage response pathways. Mol. Cell. Biol 2009, 29, 68–82. [Google Scholar]
- Lu, C.; Zhu, F.; Cho, Y.Y.; Tang, F.; Zykova, T.; Ma, W.Y.; Bode, A.M.; Dong, Z. Cell apoptosis: requirement of H2AX in DNA ladder formation, but not for the activation of caspase-3. Mol. Cell 2006, 23, 121–132. [Google Scholar]
- Lu, C.; Shi, Y.; Wang, Z.; Song, Z.; Zhu, M.; Cai, Q.; Chen, T. Serum starvation induces H2AX phosphorylation to regulate apoptosis via p38 MAPK pathway. FEBS Lett 2008, 582, 2703–2708. [Google Scholar]
- Li, A.; Yu, Y.; Lee, S.C.; Ishibashi, T.; Lees-Miller, S.P.; Ausio, J. Phosphorylation of histone H2A.X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. J. Biol. Chem 2010, 285, 17778–17788. [Google Scholar]
- Liu, M.; Pelling, J.C. UV-B/A irradiation of mouse keratinocytes results in p53-mediated WAF1/CIP1 expression. Oncogene 1995, 10, 1955–1960. [Google Scholar]
- Savio, M.; Coppa, T.; Cazzalini, O.; Perucca, P.; Necchi, D.; Nardo, T.; Stivala, L.A.; Prosperi, E. Degradation of p21CDKN1A after DNA damage is independent of type of lesion, and is not required for DNA repair. DNA Repair 2009, 8, 778–785. [Google Scholar]
- Zhao, H.; Traganos, F.; Darzynkiewicz, Z. Kinetics of the UV-induced DNA damage response in relation to cell cycle phase. Correlation with DNA replication. Cytometry A 2010, 77, 285–293. [Google Scholar]
- Bendjennat, M.; Boulaire, J.; Jascur, T.; Brickner, H.; Barbier, V.; Sarasin, A.; Fotedar, A.; Fotedar, R. UV irradiation triggers ubiquitin-dependent degradation of p21(WAF1) to promote DNA repair. Cell 2003, 114, 599–610. [Google Scholar]
- Soria, G.; Gottifredi, V. PCNA-coupled p21 degradation after DNA damage: The exception that confirms the rule? DNA Repair 2010, 9, 358–364. [Google Scholar]
- Derijard, B.; Raingeaud, J.; Barrett, T.; Wu, I.H.; Han, J.; Ulevitch, R.J.; Davis, R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science 1995, 267, 682–685. [Google Scholar]
- Olsen, J.V.; Vermeulen, M.; Santamaria, A.; Kumar, C.; Miller, M.L.; Jensen, L.J.; Gnad, F.; Cox, J.; Jensen, T.S.; Nigg, E.A.; et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci. Signal. 2010, 3, ra3. [Google Scholar]
- Dephoure, N.; Zhou, C.; Villen, J.; Beausoleil, S.A.; Bakalarski, C.E.; Elledge, S.J.; Gygi, S.P. A quantitative atlas of mitotic phosphorylation. Proc. Natl. Acad. Sci. USA 2008, 105, 10762–10767. [Google Scholar]
- Kallunki, T.; Deng, T.; Hibi, M.; Karin, M. c-Jun can recruit JNK to phosphorylate dimerization partners via specific docking interactions. Cell 1996, 87, 929–939. [Google Scholar]
- Devary, Y.; Gottlieb, R.A.; Lau, L.F.; Karin, M. Rapid and preferential activation of the c-jun gene during the mammalian UV response. Mol. Cell. Biol 1991, 11, 2804–2811. [Google Scholar]
- Adler, V.; Fuchs, S.Y.; Kim, J.; Kraft, A.; King, M.P.; Pelling, J.; Ronai, Z. jun-NH2-terminal kinase activation mediated by UV-induced DNA lesions in melanoma and fibroblast cells. Cell Growth Differ 1995, 6, 1437–1446. [Google Scholar]
- O’Dea, E.L.; Kearns, J.D.; Hoffmann, A. UV as an amplifier rather than inducer of NF-kappaB activity. Mol. Cell 2008, 30, 632–641. [Google Scholar]
- Huang, T.T.; Feinberg, S.L.; Suryanarayanan, S.; Miyamoto, S. The zinc finger domain of NEMO is selectively required for NF-kappa B activation by UV radiation and topoisomerase inhibitors. Mol. Cell. Biol 2002, 22, 5813–5825. [Google Scholar]
- Fan, C.; Yang, J.; Engelhardt, J.F. Temporal pattern of NFkappaB activation influences apoptotic cell fate in a stimuli-dependent fashion. J. Cell. Sci 2002, 115, 4843–4853. [Google Scholar]
- Tsuchiya, Y.; Asano, T.; Nakayama, K.; Kato, T., Jr; Karin, M.; Kamata, H. Nuclear IKKbeta is an adaptor protein for IkappaBalpha ubiquitination and degradation in UV-induced NF-kappaB activation. Mol. Cell. 2010, 39, 570–582. [Google Scholar]
- Kanarek, N.; Ben-Neriah, Y. Regulation of NF-kappaB by ubiquitination and degradation of the IkappaBs. Immunol. Rev 2012, 246, 77–94. [Google Scholar]
- Wu, Z.H.; Shi, Y.; Tibbetts, R.S.; Miyamoto, S. Molecular linkage between the kinase ATM and NF-kappaB signaling in response to genotoxic stimuli. Science 2006, 311, 1141–1146. [Google Scholar]
- Olsen, J.V.; de Godoy, L.M.; Li, G.; Macek, B.; Mortensen, P.; Pesch, R.; Makarov, A.; Lange, O.; Horning, S.; Mann, M. Parts per million mass accuracy on an Orbitrap mass spectrometer via lock mass injection into a C-trap. Mol. Cell. Proteomics 2005, 4, 2010–2021. [Google Scholar]



© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Stokes, M.P.; Silva, J.C.; Jia, X.; Lee, K.A.; Polakiewicz, R.D.; Comb, M.J. Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct. Int. J. Mol. Sci. 2013, 14, 286-307. https://doi.org/10.3390/ijms14010286
Stokes MP, Silva JC, Jia X, Lee KA, Polakiewicz RD, Comb MJ. Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct. International Journal of Molecular Sciences. 2013; 14(1):286-307. https://doi.org/10.3390/ijms14010286
Chicago/Turabian StyleStokes, Matthew P., Jeffrey C. Silva, Xiaoying Jia, Kimberly A. Lee, Roberto D. Polakiewicz, and Michael J. Comb. 2013. "Quantitative Profiling of DNA Damage and Apoptotic Pathways in UV Damaged Cells Using PTMScan Direct" International Journal of Molecular Sciences 14, no. 1: 286-307. https://doi.org/10.3390/ijms14010286