The Controversial Role of Retinoic Acid in Fibrotic Diseases: Analysis of Involved Signaling Pathways


Abstract
:1. Introduction
2. Role of RA in Diseases
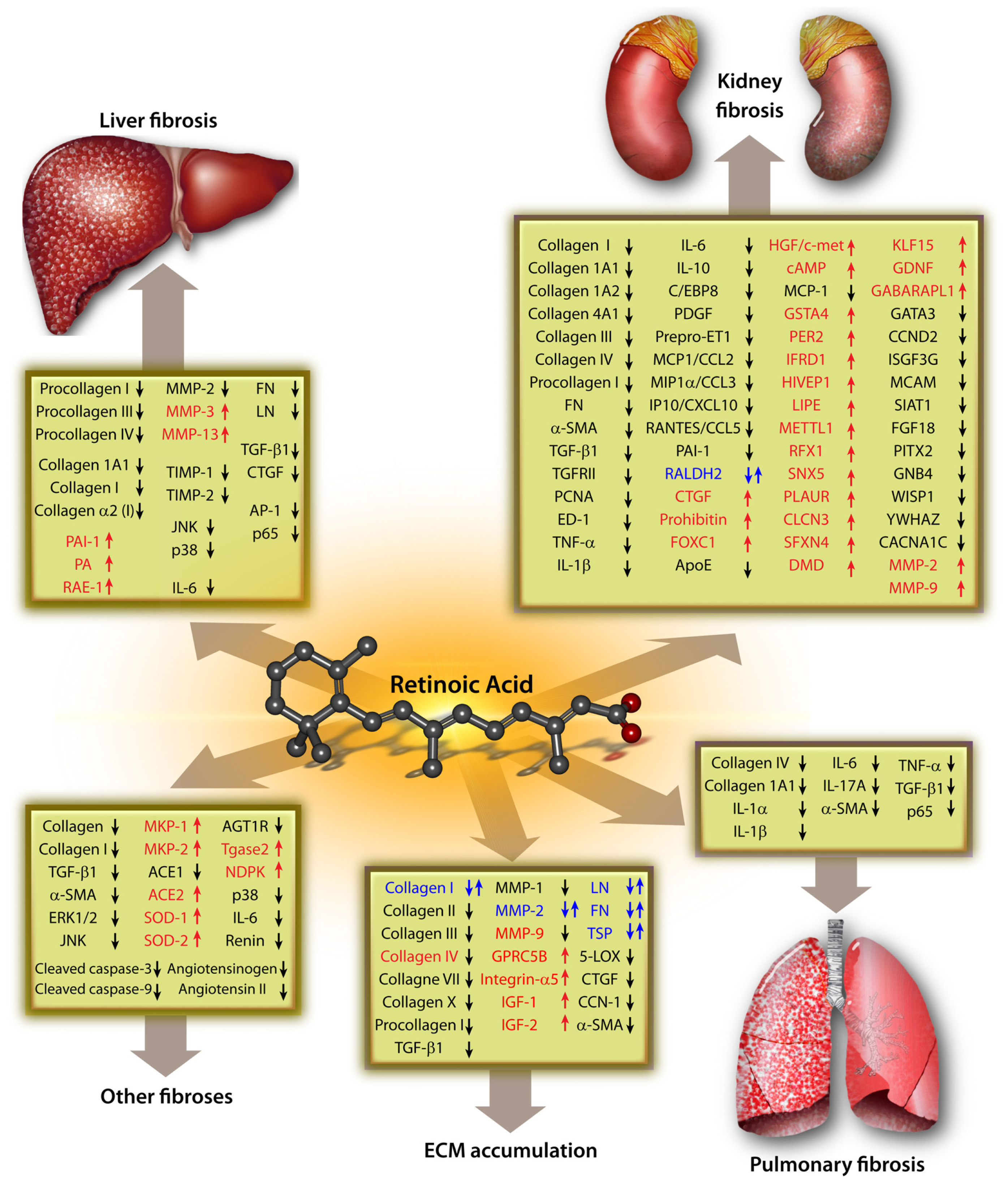
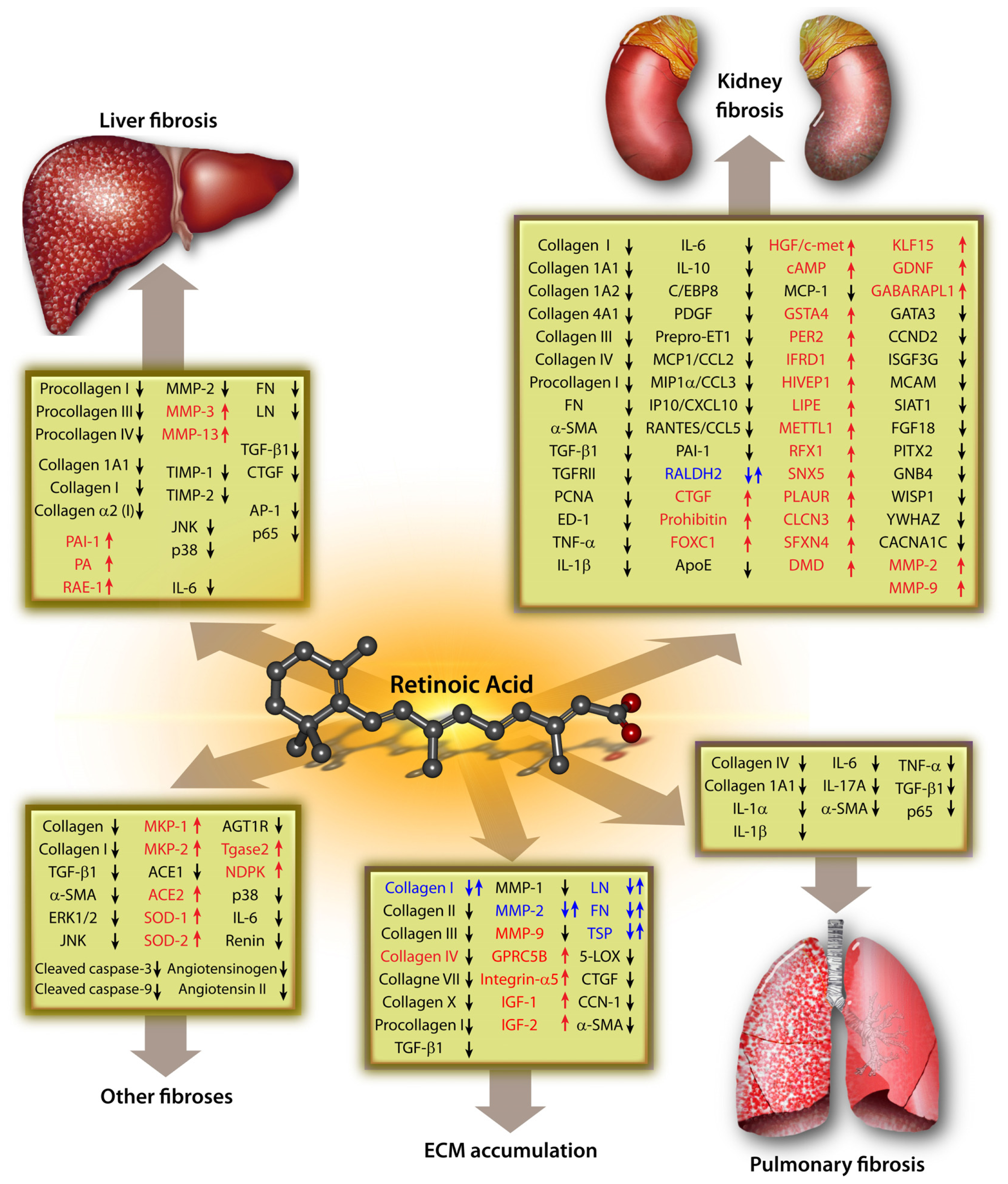
2.1. Role of RA in Liver Fibrosis
2.2. Role of RA in Pulmonary Fibrosis
2.3. Role of RA in Kidney Fibrosis
2.4. Role of RA in Other Fibrosis
3. Signaling Pathways of Role of RA in the ECM Metabolism
4. Conclusions and Perspectives
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Kong, X.; Horiguchi, N.; Mori, M.; Gao, B. Cytokines and STATs in liver fibrosis. Front. Physiol 2012, 3, 69. [Google Scholar]
- Meinecke, A.K.; Nagy, N.; D’Amico, L.G.; Kirmse, S.; Klose, R.; Schrodter, K.; Zimmermann, A.; Helfrich, I.; Rundqvist, H.; Theegarten, D.; et al. Aberrant mural cell recruitment to lymphatic vessels and impaired lymphatic drainage in a murine model of pulmonary fibrosis. Blood 2012, 119, 5931–5942. [Google Scholar]
- Wang, B.; Komers, R.; Carew, R.; Winbanks, C.E.; Xu, B.; Herman-Edelstein, M.; Koh, P.; Thomas, M.; Jandeleit-Dahm, K.; Gregorevic, P.; et al. Suppression of microRNA-29 expression by TGF-β1 promotes collagen expression and renal fibrosis. J. Am. Soc. Nephrol 2012, 23, 252–265. [Google Scholar]
- Varani, J.; Nickoloff, B.J.; Dixit, V.M.; Mitra, R.S.; Voorhees, J.J. All-trans retinoic acid stimulates growth of adult human keratinocytes cultured in growth factor-deficient medium, inhibits production of thrombospondin and fibronectin, and reduces adhesion. J. Investig. Dermatol 1989, 93, 449–454. [Google Scholar]
- Zhang, K.H.; Yu, Q.Z.; Mo, X.M. Fabrication and intermolecular interactions of silk fibroin/hydroxybutyl chitosan blended nanofibers. Int. J. Mol. Sci 2011, 12, 2187–2199. [Google Scholar]
- Zhang, Z.; Miao, L.; Wang, L. Inflammation amplification by versican: The first mediator. Int. J. Mol. Sci 2012, 13, 6873–6882. [Google Scholar]
- Zhang, Z.; Zhang, J.; Miao, L.; Liu, K.; Yang, S.; Pan, C.; Jiao, B. Interleukin-11 promotes the progress of gastric carcinoma via abnormally expressed versican. Int. J. Biol. Sci 2012, 8, 383–393. [Google Scholar]
- Marletaz, F.; Holland, L.Z.; Laudet, V.; Schubert, M. Retinoic acid signaling and the evolution of chordates. Int. J. Biol. Sci 2006, 2, 38–47. [Google Scholar]
- Zhou, T.B.; Qin, Y.H. The potential mechanism for the different expressions of gelatinases induced by all-trans retinoic acid in different cells. J. Recept. Signal. Transduct. Res 2012, 32, 129–133. [Google Scholar]
- Zhou, T.B.; Qin, Y.H.; Lei, F.Y.; Su, L.N.; Zhao, Y.J.; Huang, W.F. All-trans retinoic acid regulates the expression of apolipoprotein E in rats with glomerulosclerosis induced by Adriamycin. Exp. Mol. Pathol 2011, 90, 287–294. [Google Scholar]
- Aguilar, R.P.; Genta, S.; Oliveros, L.; Anzulovich, A.; Gimenez, M.S.; Sanchez, S.S. Vitamin A deficiency injures liver parenchyma and alters the expression of hepatic extracellular matrix. J. Appl. Toxicol 2009, 29, 214–222. [Google Scholar]
- Li, J.; Fan, R.; Zhao, S.; Liu, L.; Guo, S.; Wu, N.; Zhang, W.; Chen, P. Reactive oxygen species released from hypoxic hepatocytes regulates MMP-2 expression in hepatic stellate cells. Int. J. Mol. Sci 2011, 12, 2434–2447. [Google Scholar]
- Xia, J.R.; Liu, N.F.; Zhu, N.X. Specific siRNA targeting the receptor for advanced glycation end products inhibits experimental hepatic fibrosis in rats. Int. J. Mol. Sci 2008, 9, 638–661. [Google Scholar]
- Wang, L.; Potter, J.J.; Rennie-Tankersley, L.; Novitskiy, G.; Sipes, J.; Mezey, E. Effects of retinoic acid on the development of liver fibrosis produced by carbon tetrachloride in mice. Biochim. Biophys. Acta 2007, 1772, 66–71. [Google Scholar]
- Hisamori, S.; Tabata, C.; Kadokawa, Y.; Okoshi, K.; Tabata, R.; Mori, A.; Nagayama, S.; Watanabe, G.; Kubo, H.; Sakai, Y. All-trans-retinoic acid ameliorates carbon tetrachloride-induced liver fibrosis in mice through modulating cytokine production. Liver Int 2008, 28, 1217–1225. [Google Scholar]
- Yang, K.L.; Chang, W.T.; Chuang, C.C.; Hung, K.C.; Li, E.I. Antagonizing TGF-β induced liver fibrosis by a retinoic acid derivative through regulation of ROS and calcium influx. Biochem. Biophys. Res. Commun 2008, 365, 484–489. [Google Scholar]
- Wang, H.; Dan, Z.; Jiang, H. Effect of all-trans retinoic acid on liver fibrosis induced by common bile duct ligation in rats. J. Huazhong Univ. Sci. Technol. Med. Sci 2008, 28, 553–557. [Google Scholar]
- He, H.; Mennone, A.; Boyer, J.L.; Cai, S.Y. Combination of retinoic acid and ursodeoxycholic acid attenuates liver injury in bile duct-ligated rats and human hepatic cells. Hepatology 2011, 53, 548–557. [Google Scholar]
- Hellemans, K.; Grinko, I.; Rombouts, K.; Schuppan, D.; Geerts, A. All-trans and 9-cis retinoic acid alter rat hepatic stellate cell phenotype differentially. Gut 1999, 45, 134–142. [Google Scholar]
- Ye, Y.; Dan, Z. All-trans retinoic acid diminishes collagen production in a hepatic stellate cell line via suppression of active protein-1 and c-Jun N-terminal kinase signal. J. Huazhong Univ. Sci. Technol. Med. Sci 2010, 30, 726–733. [Google Scholar]
- Radaeva, S.; Wang, L.; Radaev, S.; Jeong, W.I.; Park, O.; Gao, B. Retinoic acid signaling sensitizes hepatic stellate cells to NK cell killing via upregulation of NK cell activating ligand RAE1. Am. J. Physiol. Gastrointest. Liver Physiol 2007, 293, 809–816. [Google Scholar]
- Okuno, M.; Moriwaki, H.; Imai, S.; Muto, Y.; Kawada, N.; Suzuki, Y.; Kojima, S. Retinoids exacerbate rat liver fibrosis by inducing the activation of latent TGF-β in liver stellate cells. Hepatology 1997, 26, 913–921. [Google Scholar]
- Okuno, M.; Kojima, S.; Akita, K.; Matsushima-Nishiwaki, R.; Adachi, S.; Sano, T.; Takano, Y.; Takai, K.; Obora, A.; Yasuda, I.; et al. Retinoids in liver fibrosis and cancer. Front. Biosci 2002, 7, 204–218. [Google Scholar]
- Inage, M.; Nakamura, H.; Saito, H.; Abe, S.; Hino, T.; Takabatake, N.; Terashita, K.; Ogura, M.; Kato, S.; Hosokawa, T.; et al. Vesnarinone represses the fibrotic changes in murine lung injury induced by bleomycin. Int. J. Biol. Sci 2009, 5, 304–310. [Google Scholar]
- Tabata, C.; Kadokawa, Y.; Tabata, R.; Takahashi, M.; Okoshi, K.; Sakai, Y.; Mishima, M.; Kubo, H. All-trans-retinoic acid prevents radiation- or bleomycin-induced pulmonary fibrosis. Am. J. Respir. Crit. Care Med 2006, 174, 1352–1360. [Google Scholar]
- Esteban-Pretel, G.; Marin, M.P.; Renau-Piqueras, J.; Barber, T.; Timoneda, J. Vitamin A deficiency alters rat lung alveolar basement membrane: Reversibility by retinoic acid. J. Nutr. Biochem 2010, 21, 227–236. [Google Scholar]
- Ozer, E.A.; Kumral, A.; Ozer, E.; Duman, N.; Yilmaz, O.; Ozkal, S.; Ozkan, H. Effect of retinoic acid on oxygen-induced lung injury in the newborn rat. Pediatr. Pulmonol 2005, 39, 35–40. [Google Scholar]
- Tabata, C.; Kubo, H.; Tabata, R.; Wada, M.; Sakuma, K.; Ichikawa, M.; Fujita, S.; Mio, T.; Mishima, M. All-trans retinoic acid modulates radiation-induced proliferation of lung fibroblasts via IL-6/IL-6R system. Am. J. Physiol. Lung Cell Mol. Physiol 2006, 290, 597–606. [Google Scholar]
- Dong, Z.; Tai, W.; Yang, Y.; Zhang, T.; Li, Y.; Chai, Y.; Zhong, H.; Zou, H.; Wang, D. The role of all-trans retinoic acid in bleomycin-induced pulmonary fibrosis in mice. Exp. Lung Res 2012, 38, 82–89. [Google Scholar]
- Lan, H.Y. Diverse roles of TGF-β/Smads in renal fibrosis and inflammation. Int. J. Biol. Sci 2011, 7, 1056–1067. [Google Scholar]
- Wagner, J.; Dechow, C.; Morath, C.; Lehrke, I.; Amann, K.; Waldherr, R.; Floege, J.; Ritz, E. Retinoic acid reduces glomerular injury in a rat model of glomerular damage. J. Am. Soc. Nephrol 2000, 11, 1479–1487. [Google Scholar]
- Morath, C.; Dechow, C.; Lehrke, I.; Haxsen, V.; Waldherr, R.; Floege, J.; Ritz, E.; Wagner, J. Effects of retinoids on the TGF-β system and extracellular matrix in experimental glomerulonephritis. J. Am. Soc. Nephrol 2001, 12, 2300–2309. [Google Scholar]
- Lehrke, I.; Schaier, M.; Schade, K.; Morath, C.; Waldherr, R.; Ritz, E.; Wagner, J. Retinoid receptor-specific agonists alleviate experimental glomerulonephritis. Am. J. Physiol. Ren. Physiol 2002, 282, 741–751. [Google Scholar]
- Oseto, S.; Moriyama, T.; Kawada, N.; Nagatoya, K.; Takeji, M.; Ando, A.; Yamamoto, T.; Imai, E.; Hori, M. Therapeutic effect of all-trans retinoic acid on rats with anti-GBM antibody glomerulonephritis. Kidney Int 2003, 64, 1241–1252. [Google Scholar]
- Schaier, M.; Liebler, S.; Schade, K.; Shimizu, F.; Kawachi, H.; Grone, H.J.; Chandraratna, R.; Ritz, E.; Wagner, J. Retinoic acid receptor α and retinoid X receptor specific agonists reduce renal injury in established chronic glomerulonephritis of the rat. J. Mol. Med 2004, 82, 116–125. [Google Scholar]
- Adams, J.; Kiss, E.; Arroyo, A.B.; Bonrouhi, M.; Sun, Q.; Li, Z.; Gretz, N.; Schnitger, A.; Zouboulis, C.C.; Wiesel, M.; et al. 13-cis retinoic acid inhibits development and progression of chronic allograft nephropathy. Am. J. Pathol 2005, 167, 285–298. [Google Scholar]
- He, J.C.; Lu, T.C.; Fleet, M.; Sunamoto, M.; Husain, M.; Fang, W.; Neves, S.; Chen, Y.; Shankland, S.; Iyengar, R.; et al. Retinoic acid inhibits HIV-1-induced podocyte proliferation through the cAMP pathway. J. Am. Soc. Nephrol 2007, 18, 93–102. [Google Scholar]
- Xu, Q.; Hendry, B.M.; Maden, M.; Lu, H.; Wong, Y.F.; Rankin, A.C.; Noor, M.; Kopp, J.B. Kidneys of Alb/TGF-β1 transgenic mice are deficient in retinoic acid and exogenous retinoic acid shows dose-dependent toxicity. Nephron Exp. Nephrol 2010, 114, 127–132. [Google Scholar]
- Wen, X.; Li, Y.; Hu, K.; Dai, C.; Liu, Y. Hepatocyte growth factor receptor signaling mediates the anti-fibrotic action of 9-cis-retinoic acid in glomerular mesangial cells. Am. J. Pathol 2005, 167, 947–957. [Google Scholar]
- Liu, X.; Lu, L.; Tao, B.B.; Zhou, A.L.; Zhu, Y.C. Amelioration of glomerulosclerosis with all-trans retinoic acid is linked to decreased plasminogen activator inhibitor-1 and α-smooth muscle actin. Acta Pharmacol. Sin 2011, 32, 70–78. [Google Scholar]
- Kishimoto, K.; Kinoshita, K.; Hino, S.; Yano, T.; Nagare, Y.; Shimazu, H.; Nozaki, Y.; Sugiyama, M.; Ikoma, S.; Funauchi, M. Therapeutic effect of retinoic acid on unilateral ureteral obstruction model. Nephron Exp. Nephrol 2011, 118, 69–78. [Google Scholar]
- Mallipattu, S.K.; Liu, R.; Zheng, F.; Narla, G.; Ma’ayan, A.; Dikman, S.; Jain, M.K.; Saleem, M.; D’Agati, V.; Klotman, P.; et al. Kruppel-Like factor 15 (KLF15) is a key regulator of podocyte differentiation. J. Biol. Chem 2012, 287, 19122–19135. [Google Scholar]
- Zhou, T.B.; Qin, Y.H.; Ou, C.; Lei, F.Y.; Su, L.N.; Huang, W.F.; Zhao, Y.J. All-trans retinoic acid can regulate the expressions of gelatinases and apolipoprotein E in glomerulosclerosis rats. Vasc. Pharmacol 2011, 55, 169–177. [Google Scholar]
- Zhou, T.B.; Qin, Y.H.; Li, Z.Y.; Xu, H.L.; Zhao, Y.J.; Lei, F.Y. All-trans retinoic Acid treatment is associated with prohibitin expression in renal interstitial fibrosis rats. Int. J. Mol. Sci 2012, 13, 2769–2782. [Google Scholar]
- Iyoda, M.; Hudkins, K.L.; Wietecha, T.A.; Banas, M.C.; Guo, S.; Liu, G.; Wang, L.; Kowalewska, J.; Alpers, C.E. All-trans-retinoic acid aggravates cryoglobulin-associated membranoproliferative glomerulonephritis in mice. Nephrol. Dial. Transpl 2007, 22, 3451–3461. [Google Scholar]
- Moulder, J.E.; Fish, B.L.; Regner, K.R.; Cohen, E.P.; Raife, T.J. Retinoic acid exacerbates experimental radiation nephropathy. Radiat. Res 2002, 157, 199–203. [Google Scholar]
- Alique, M.; Moreno-Manzano, V.; Sepulveda-Munoz, J.C.; Reyes-Martin, P.; Parra-Cid, T.; Calvino, M.; de Lucio-Cazana, F.J. All-trans retinoic acid and glycated albumin reciprocally influence their effects in human mesangial cells. Int. J. Vitam. Nutr. Res 2005, 75, 47–53. [Google Scholar]
- Patatanian, E.; Thompson, D.F. Retinoic acid syndrome: A review. J. Clin. Pharm. Ther 2008, 33, 331–338. [Google Scholar]
- Choudhary, R.; Palm-Leis, A.; Scott, R.R.; Guleria, R.S.; Rachut, E.; Baker, K.M.; Pan, J. All-trans retinoic acid prevents development of cardiac remodeling in aortic banded rats by inhibiting the renin-angiotensin system. Am. J. Physiol. Heart Circ. Physiol 2008, 294, 633–644. [Google Scholar]
- Klopcic, B.; Appelbee, A.; Raye, W.; Lloyd, F.; Jooste, J.C.; Forrest, C.H.; Lawrance, I.C. Indomethacin and retinoic acid modify mouse intestinal inflammation and fibrosis: A role for SPARC. Dig. Dis. Sci 2008, 53, 1553–1563. [Google Scholar]
- Okoshi, K.; Kubo, H.; Nagayama, S.; Tabata, C.; Kadokawa, Y.; Hisamori, S.; Yonenaga, Y.; Fujimoto, A.; Mori, A.; Onodera, H.; et al. All-trans-retinoic acid attenuates radiation-induced intestinal fibrosis in mice. J. Surg. Res 2008, 150, 53–59. [Google Scholar]
- Wang, G.H.; Tang, X.H.; Ma, Y.L.; Gao, Y.C. Effects of all-trans retinoic acid on the expression of TGF-β 1 and COL-I in rat model of peritoneal dialysis. Sichuan Da Xue Xue Bao Yi Xue Ban 2008, 39, 575–578. [Google Scholar]
- Treharne, K.J.; Giles, B.O.; Mehta, A. Transglutaminase 2 and nucleoside diphosphate kinase activity are correlated in epithelial membranes and are abnormal in cystic fibrosis. FEBS Lett 2009, 583, 2789–2792. [Google Scholar]
- Xiao, R.; Yoshida, N.; Higashi, Y.; Lu, Q.J.; Fukushige, T.; Kanzaki, T.; Kanekura, T. Retinoic acids exhibit anti-fibrotic activity through the inhibition of 5-lipoxygenase expression in scleroderma fibroblasts. J. Dermatol 2011, 38, 345–353. [Google Scholar]
- McCarroll, J.A.; Phillips, P.A.; Santucci, N.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Vitamin A inhibits pancreatic stellate cell activation: Implications for treatment of pancreatic fibrosis. Gut 2006, 55, 79–89. [Google Scholar]
- Nakajima, Y.; Morishima, M.; Nakazawa, M.; Momma, K. Inhibition of outflow cushion mesenchyme formation in retinoic acid-induced complete transposition of the great arteries. Cardiovasc. Res 1996, 31, 77–85. [Google Scholar]
- Touhami, M.; Bourge, J.F.; Legrand, C. Increased adhesion of the promyelocytic leukaemia cell line, NB4, to fibronectin and thrombospondin upon all-trans-retinoic acid treatment. Br. J. Haematol 1999, 104, 706–714. [Google Scholar]
- Malik, M.; Webb, J.; Catherino, W.H. Retinoic acid treatment of human leiomyoma cells transformed the cell phenotype to one strongly resembling myometrial cells. Clin. Endocrinol. (Oxf. ) 2008, 69, 462–470. [Google Scholar]
- Wu, L.N.; Ishikawa, Y.; Nie, D.; Genge, B.R.; Wuthier, R.E. Retinoic acid stimulates matrix calcification and initiates type I collagen synthesis in primary cultures of avian weight-bearing growth plate chondrocytes. J. Cell Biochem 1997, 65, 209–230. [Google Scholar]
- Chang, Y.C.; Kao, Y.H.; Hu, D.N.; Tsai, L.Y.; Wu, W.C. All-trans retinoic acid remodels extracellular matrix and suppresses laminin-enhanced contractility of cultured human retinal pigment epithelial cells. Exp. Eye Res 2009, 88, 900–909. [Google Scholar]
- Scita, G.; Darwiche, N.; Greenwald, E.; Rosenberg, M.; Politi, K.; de Luca, L.M. Retinoic acid down-regulation of fibronectin and retinoic acid receptor α proteins in NIH-3T3 cells. Blocks of this response by ras transformation. J. Biol. Chem 1996, 271, 6502–6508. [Google Scholar]
- Miller, M.G.; Kapron, C.M.; Metcalfe, C.D.; Lee, L.E. Down-regulation of fibronectin in rainbow trout gonadal cells exposed to retinoic acid. Aquat. Toxicol 2000, 48, 119–125. [Google Scholar]
- Quan, T.; Qin, Z.; Shao, Y.; Xu, Y.; Voorhees, J.J.; Fisher, G.J. Retinoids suppress cysteine-rich protein 61 (CCN1), a negative regulator of collagen homeostasis, in skin equivalent cultures and aged human skin in vivo. Exp. Dermatol 2011, 20, 572–576. [Google Scholar]
- Lenkowski, J.R.; McLaughlin, K.A. Acute atrazine exposure disrupts matrix metalloproteinases and retinoid signaling during organ morphogenesis in Xenopus laevis. J. Appl. Toxicol 2010, 30, 582–589. [Google Scholar]
- Oikarinen, H.; Oikarinen, A.I.; Tan, E.M.; Abergel, R.P.; Meeker, C.A.; Chu, M.L.; Prockop, D.J.; Uitto, J. Modulation of procollagen gene expression by retinoids. Inhibition of collagen production by retinoic acid accompanied by reduced type I procollagen messenger ribonucleic acid levels in human skin fibroblast cultures. J. Clin. Investig 1985, 75, 1545–1553. [Google Scholar]
- Pan, L.; Eckhoff, C.; Brinckerhoff, C.E. Suppression of collagenase gene expression by all-trans and 9-cis retinoic acid is ligand dependent and requires both RARs and RXRs. J. Cell Biochem 1995, 57, 575–589. [Google Scholar]
- Takahashi, N.; Takasu, S. A close relationship between type 1 diabetes and vitamin A-deficiency and matrix metalloproteinase and hyaluronidase activities in skin tissues. Exp. Dermatol 2011, 20, 899–904. [Google Scholar]
- Nakajima, M.; Lotan, D.; Baig, M.M.; Carralero, R.M.; Wood, W.R.; Hendrix, M.J.; Lotan, R. Inhibition by retinoic acid of type IV collagenolysis and invasion through reconstituted basement membrane by metastatic rat mammary adenocarcinoma cells. Cancer Res 1989, 49, 1698–1706. [Google Scholar]
- Lafage-Proust, M.H.; Wesolowski, G.; Ernst, M.; Rodan, G.A.; Rodan, S.B. Retinoic acid effects on an SV-40 large T antigen immortalized adult rat bone cell line. J. Cell Physiol 1999, 179, 267–275. [Google Scholar]
- Varani, J.; Larson, B.K.; Perone, P.; Inman, D.R.; Fligiel, S.E.; Voorhees, J.J. All-trans retinoic acid and extracellular Ca2+ differentially influence extracellular matrix production by human skin in organ culture. Am. J. Pathol 1993, 142, 1813–1822. [Google Scholar]
- Park, I.S.; Kim, W.S. Modulation of gelatinase activity correlates with the dedifferentiation profile of regenerating salamander limbs. Mol. Cells 1999, 9, 119–126. [Google Scholar]
- Imanishi, S.; Sugimoto, M.; Morita, M.; Kume, S.; Manabe, N. Changes in expression and localization of GPRC5B and RARα in the placenta and yolk sac during middle to late gestation in mice. J. Reprod. Dev 2007, 53, 1131–1136. [Google Scholar]
- Bohnsack, B.L.; Lai, L.; Dolle, P.; Hirschi, K.K. Signaling hierarchy downstream of retinoic acid that independently regulates vascular remodeling and endothelial cell proliferation. Genes Dev 2004, 18, 1345–1358. [Google Scholar]
- Shim, J.H.; Shin, D.W.; Lee, T.R.; Kang, H.H.; Jin, S.H.; Noh, M. The retinoic acid-induced up-regulation of insulin-like growth factor 1 and 2 is associated with prolidase-dependent collagen synthesis in UVA-irradiated human dermal equivalents. J. Dermatol. Sci 2012, 66, 51–59. [Google Scholar]
- Moro, B.J.; Gato, A.; Alonso, R.M.; Pastor, J.F.; Repressa, J.J.; Barbosa, E. Retinoic acid induces changes in the rhombencephalic neural crest cells migration and extracellular matrix composition in chick embryos. Teratology 1993, 48, 197–206. [Google Scholar]
- Varani, J.; Fisher, G.J.; Kang, S.; Voorhees, J.J. Molecular mechanisms of intrinsic skin aging and retinoid-induced repair and reversal. J. Investig. Dermatol. Symp. Proc 1998, 3, 57–60. [Google Scholar]
- Varani, J.; Mitra, R.S.; Gibbs, D.; Phan, S.H.; Dixit, V.M.; Mitra, R., Jr; Wang, T.; Siebert, K.J.; Nickoloff, B.J.; Voorhees, J.J. All-trans retinoic acid stimulates growth extracellular matrix production in growth-inhibited cultured human skin fibroblasts. J. Investig. Dermatol. 1990, 94, 717–723. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | IUPAC name | Molecular structure | Chemical formula | Molecular weight | Exp. Log Pow | ACD/Pred. Log Pow | Pharmaceutical classification |
|---|---|---|---|---|---|---|---|
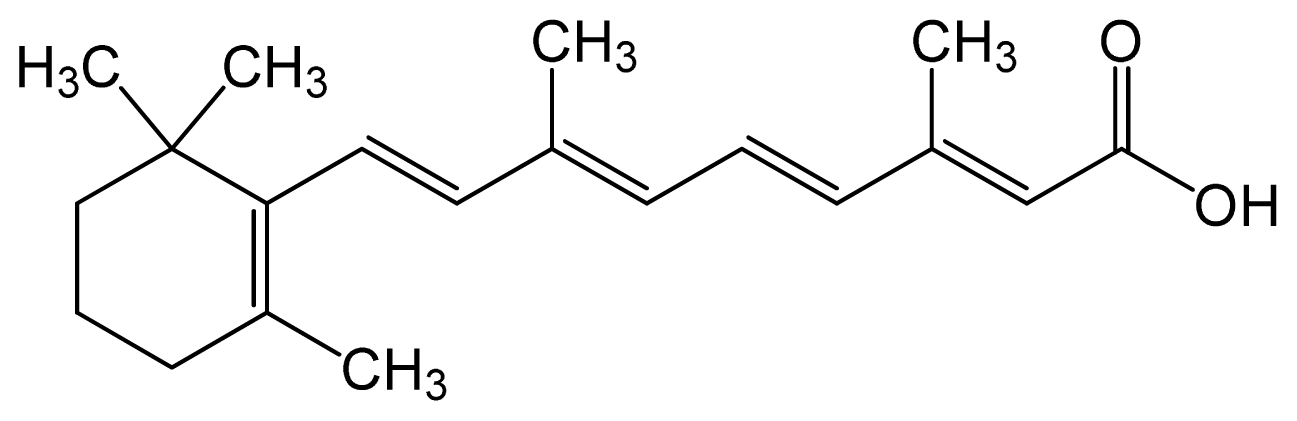
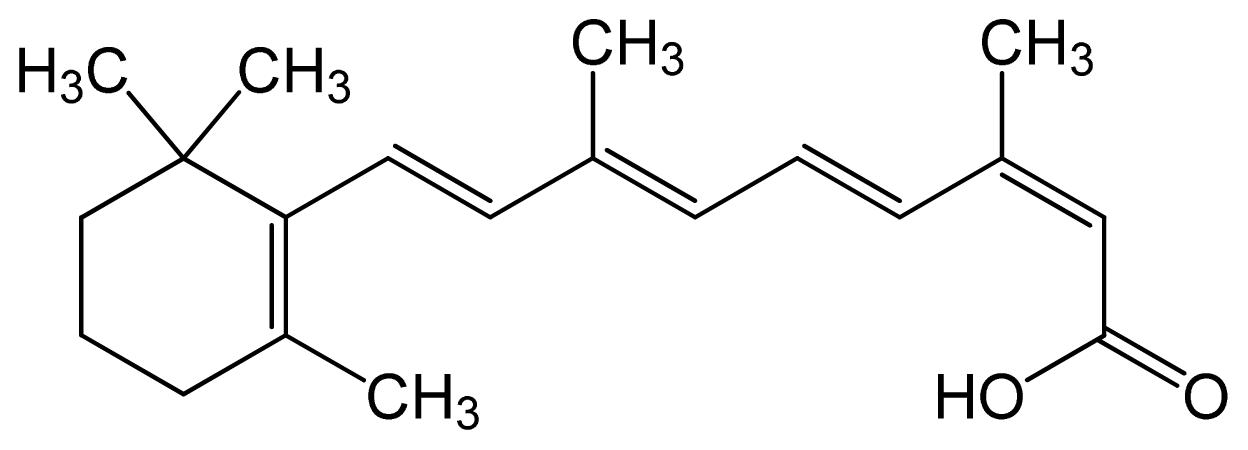
| Tretinoin (all-trans retinoic acid) | (2E,4E,6E,8E)-3,7-dim ethyl-9-(2,6,6-trimethy l-1-cyclohexenyl)nona- 2,4,6,8-tetraenoic acid | 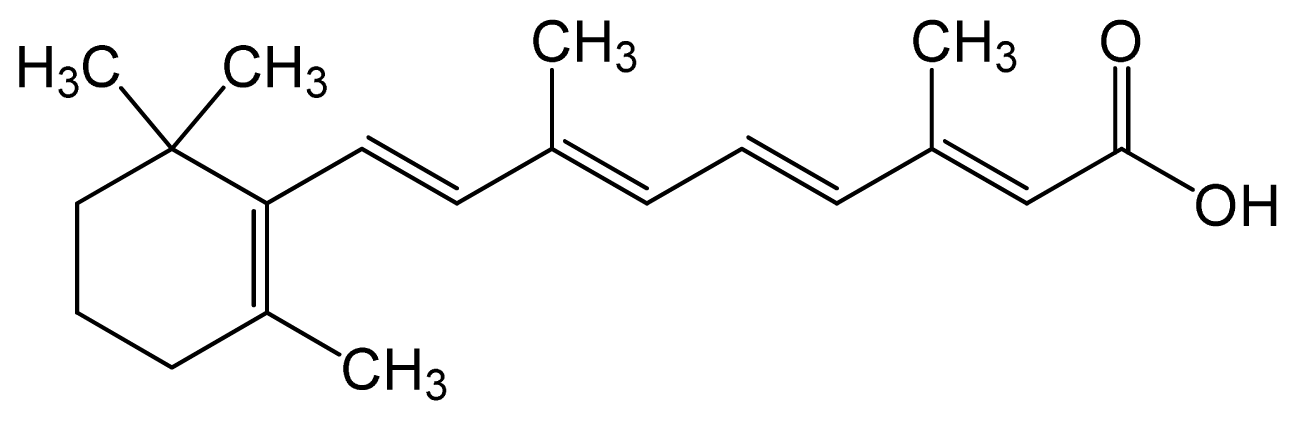 | C20H28O2 | 300.44 (g/mol) | – | 6.83 | Antineoplastic Agents [D27.505.954.248] Keratolytic Agents [D27.505.954.444.400] |
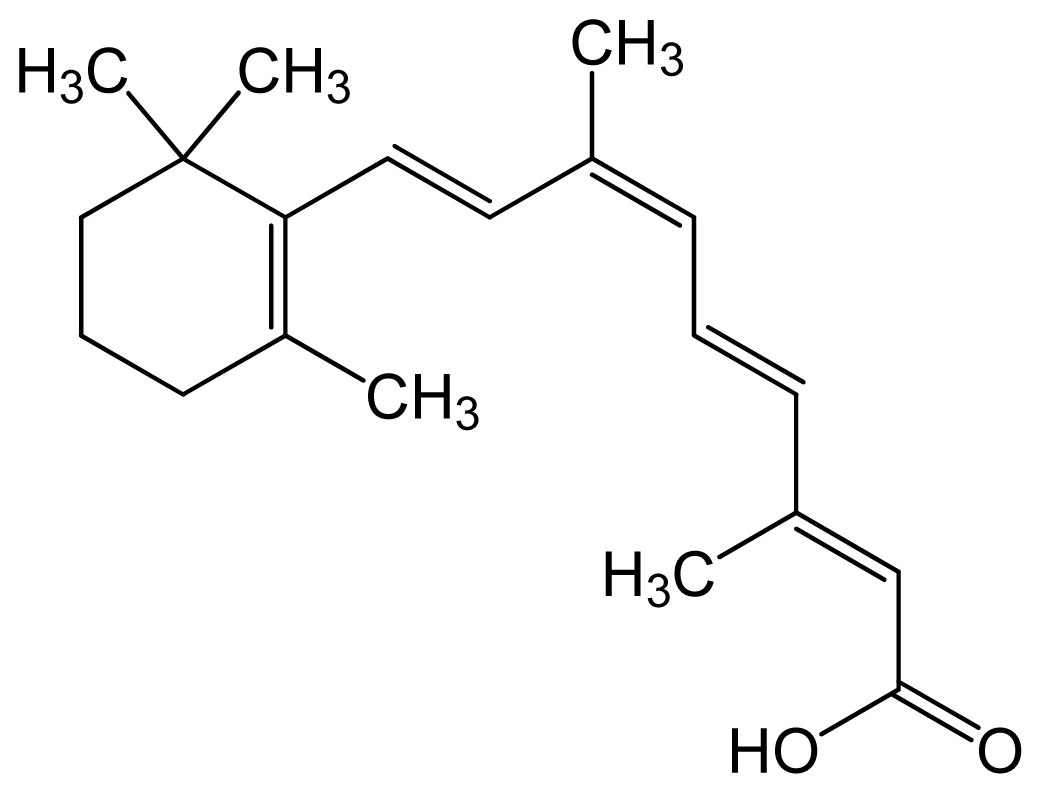
| Alitretinoin (9-cis retinoic acid) | (2E,4E,6Z,8E)-3,7-dim ethyl-9-(2,6,6-trimethy lcyclohexen-1-yl)nona- 2,4,6,8-tetraenoic acid | 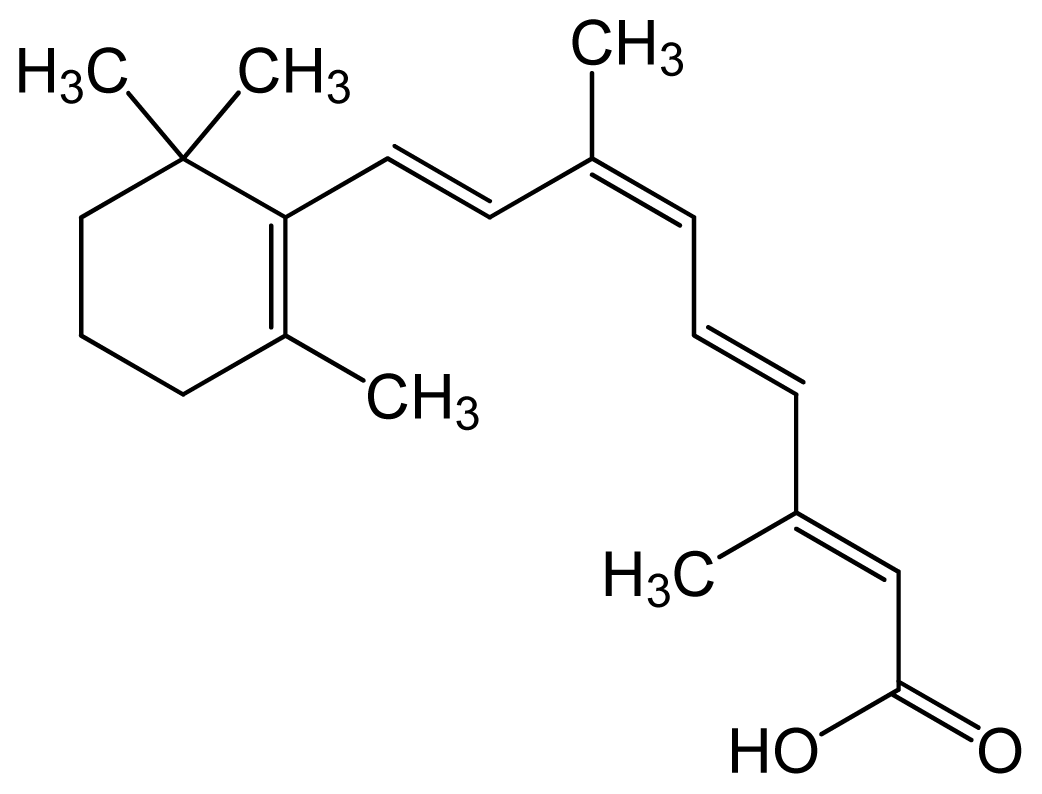 | 6.84 | 6.26 | Panretin® gel: topical treatment of cutaneous lesions in patients with AIDS-related Kaposi’s sarcoma | ||
| Isotretinoin (13-cis retinoic acid) | (2Z,4E,6E,8E)-3,7-dim ethyl-9-(2,6,6-trimethy lcyclohexen-1-yl)nona- 2,4,6,8-tetraenoic acid | 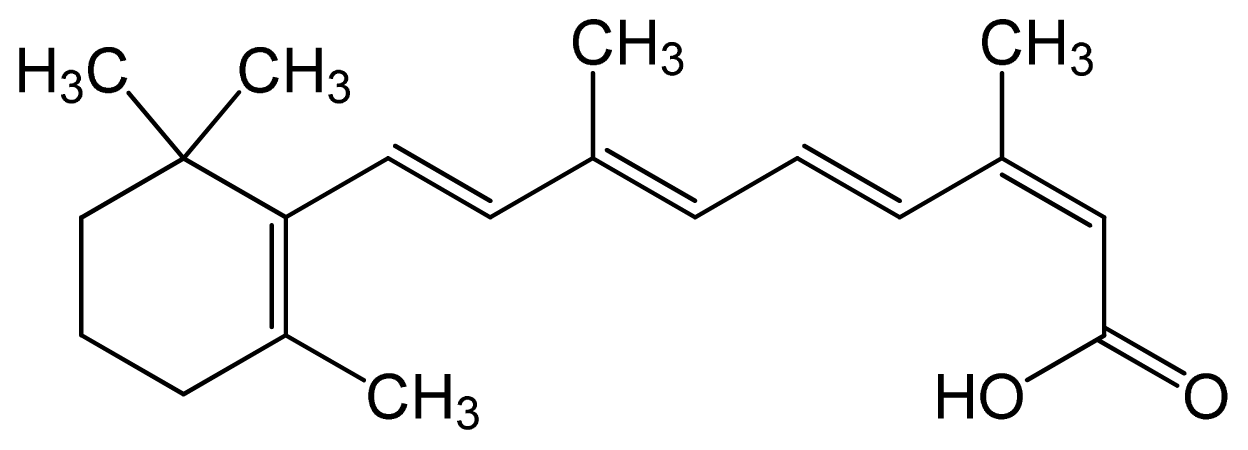 | – | 6.26 | Topical dermatologic agent for treatment of Acne Vulgaris and several other skin diseases |
| Author | Year | Type of animal/cells | Weight of animal/Age | Type of RA | Dose of RA | Effect |
|---|---|---|---|---|---|---|
| Hellemans et al. | 1999 | Hepatic stellate cells | – | ATRA or 9-cis-RA | 0.01, 0.1, and 1 μM | + |
| Ye et al. | 2010 | Hepatic stellate cells | – | ATRA | 0.01–10 μM | + |
| Wang et al. | 2007 | C57BL/6J mice | 20–25 g | ATRA | 1 mg/kg, three times per week | + |
| Yang et al. | 2008 | BALB/c mice | Eight-week-old | PL | 30, 150 or 300 μg/kg.day | + |
| Hepatic stellate cells | – | PL | 2 ng/mL | + | ||
| Wang et al. | 2008 | Wistar rats | 180–200 g | ATRA | 0.1, 1.5 and 7.5 mg/kg, four consecutive weeks | + |
| He et al. | 2011 | Sprague-Dawley rats | 200–230 g | ATRA | 5 mg/kg, 15 consecutive days | + |
| Hepatic stellate cells | – | ATRA | 5 μM | + | ||
| Hisamori et al. | 2008 | BALB/c mice | Eight-week-old | ATRA | 0.5 mg/mice, three times per week | + |
| Hepatic stellate cells | – | ATRA | 1 μM | + | ||
| Radaeva et al. | 2007 | Hepatic stellate cells | – | ATRA | NA | + |
| Okuno et al. | 1997 | Wistar rats | 100–120g body weight | 9-cis-RA | 40 mg/kg, 5 times per week | − |
| Hepatic stellate cells | – | 9-cis-RA | 0–10 μM | − |
| Author | Year | Type of animal/cells | Weight of animal/Age | Type of RA | Dose of RA | Effect |
|---|---|---|---|---|---|---|
| Tabata et al. | 2006 | C57BL/6 mice | Eight-week-old | ATRA | 0.5 mg/mice, repeated three times weekly | + |
| Lung fibroblasts | – | ATRA | 1 μM | + | ||
| Esteban-Pretel et al. | 2010 | Wistar rats | 60-day-old | ATRA | 100 μg/rat, 10 consecutive days | + |
| Ozer et al. | 2005 | Wistar rats | Postnatal day 3 | ATRA | 500 ug/kg, 10 consecutive days | + |
| Tabata et al. | 2005 | C57BL/6 mice | Eight-week-old | ATRA | 0.5 mg/mice, repeated three times weekly | + |
| Lung fibroblasts | – | ATRA | 1 μM | + | ||
| Dong et al. | 2012 | C57BL/6 mice | 18–22 g | ATRA | Repeated 3 times weekly, for 28 days | + |
| Author | Year | Type of animal/cells | Weight of animal/Age | Type of RA | Dose of RA | Effect |
|---|---|---|---|---|---|---|
| Wagner et al. | 2000 | Wistar rats | 180–200 g | ATRA | 10 mg/kg per day | + |
| Morath et al. | 2001 | Wistar rats | 180–200 g | ATRA | 10 mg/kg per day | + |
| Lehrke et al. | 2002 | Wistar rats | 180–200 g | Ro-257386 | 80 mg/kg per day | + |
| Oseto et al. | 2003 | Wistar rats | Twelve-week-old | ATRA | 30 mg/kg per day | + |
| Schaier et al. | 2004 | Wistar rats | 145–150 g | AGN 195183 or AGN 194204 | 4 mg/kg, 20 mg/kg per day AGN 195183; 0.4 mg/kg,, 2 mg/kg per day AGN 194204 | + |
| Adams et al. | 2005 | Rats | 200–220 g | 13-cis-RA | 2 mg/kg per day | + |
| Macrophages, Fibroblasts | – | 13-cis-RA | 10 μM | + | ||
| Wen et al. | 2005 | Glomerular mesangial cells | – | 9-cis-RA | 0.01–1 μM | + |
| He et al. | 2007 | Mice | – | ATRA | NA | + |
| Podocytes | – | ATRA or 9-cis-RA | 0.1–10 μM | + | ||
| Iyoda et al. | 2007 | Mice | – | ATRA | 20 mg/kg per day | − |
| Xu et al. | 2010 | C57BL/6J × CBA F1 mice | One-week-old | ATRA | 6–10.7 mg/kg per day; 12.7–18.8 mg/kg per day, 20.1–27.4 mg/kg per day | + |
| Liu et al. | 2011 | Sprague-Dawley rats | 250–330 g | ATRA | 5 mg/kg per day, 10mg/kg per day | + |
| Kishimoto et al. | 2011 | C57BL/B6 mice | 25–30 g (Eight-week-old) | ATRA | 20 mg/kg | + |
| Zhong et al. | 2012 | Mice | Four-week-old | Am580 | 0.3 mg/kg per day | + |
| Mallipattu et al. | 2012 | Podocytes | – | ATRA | 1 μM | + |
| Moulder et al. | 2002 | Rats | – | ATRA | 15 mg/kg per day | + |
| Zhou et al. | 2011, 2012 | Wistar rats | 180–200 g | ATRA | 15 mg/kg per day | + |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, T.-B.; Drummen, G.P.C.; Qin, Y.-H. The Controversial Role of Retinoic Acid in Fibrotic Diseases: Analysis of Involved Signaling Pathways. Int. J. Mol. Sci. 2013, 14, 226-243. https://doi.org/10.3390/ijms14010226
Zhou T-B, Drummen GPC, Qin Y-H. The Controversial Role of Retinoic Acid in Fibrotic Diseases: Analysis of Involved Signaling Pathways. International Journal of Molecular Sciences. 2013; 14(1):226-243. https://doi.org/10.3390/ijms14010226
Chicago/Turabian StyleZhou, Tian-Biao, Gregor P. C. Drummen, and Yuan-Han Qin. 2013. "The Controversial Role of Retinoic Acid in Fibrotic Diseases: Analysis of Involved Signaling Pathways" International Journal of Molecular Sciences 14, no. 1: 226-243. https://doi.org/10.3390/ijms14010226
APA StyleZhou, T.-B., Drummen, G. P. C., & Qin, Y.-H. (2013). The Controversial Role of Retinoic Acid in Fibrotic Diseases: Analysis of Involved Signaling Pathways. International Journal of Molecular Sciences, 14(1), 226-243. https://doi.org/10.3390/ijms14010226