pH Dependence of the Fluorescence Lifetime of FAD in Solution and in Cells

Abstract
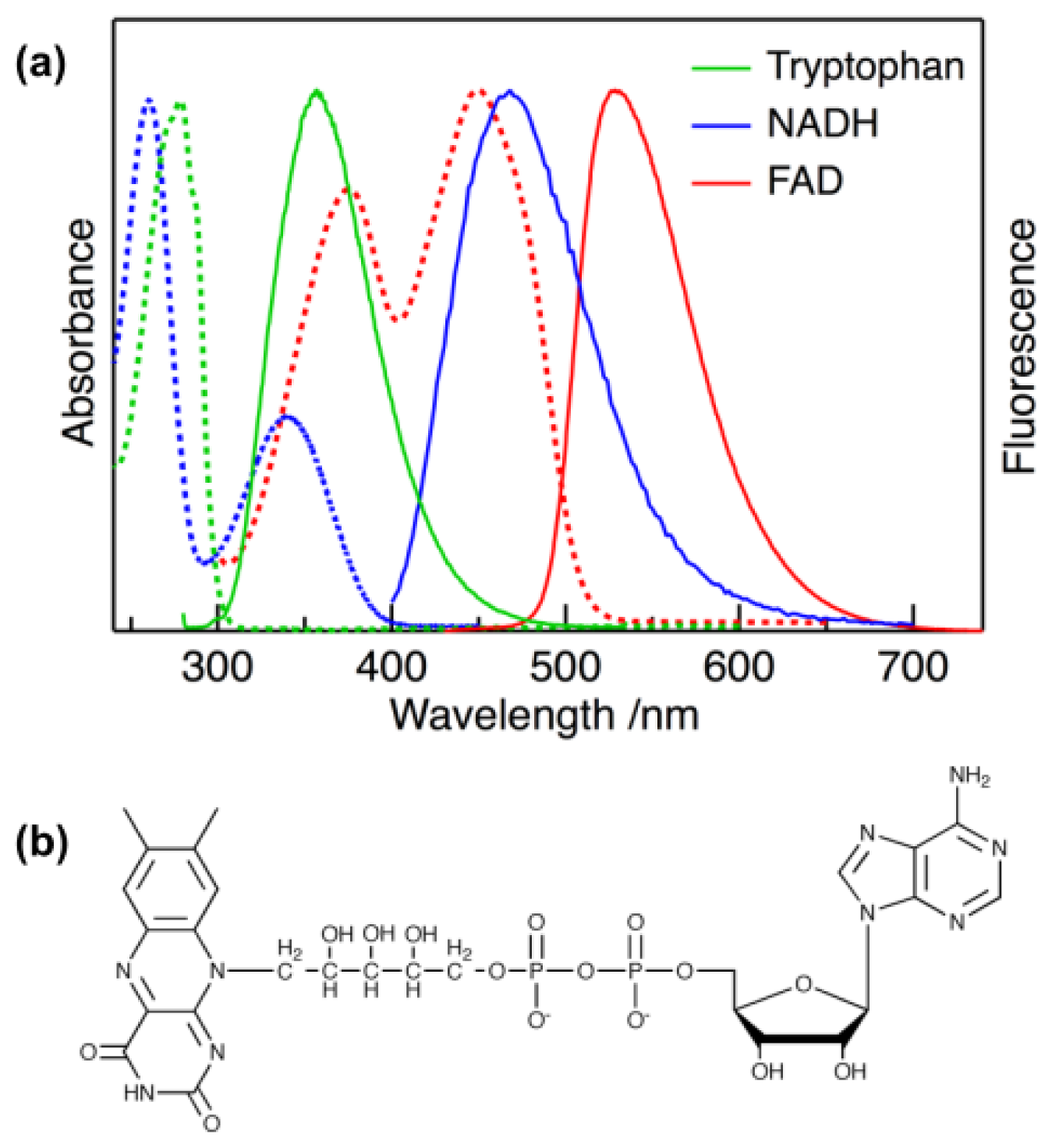
:1. Introduction
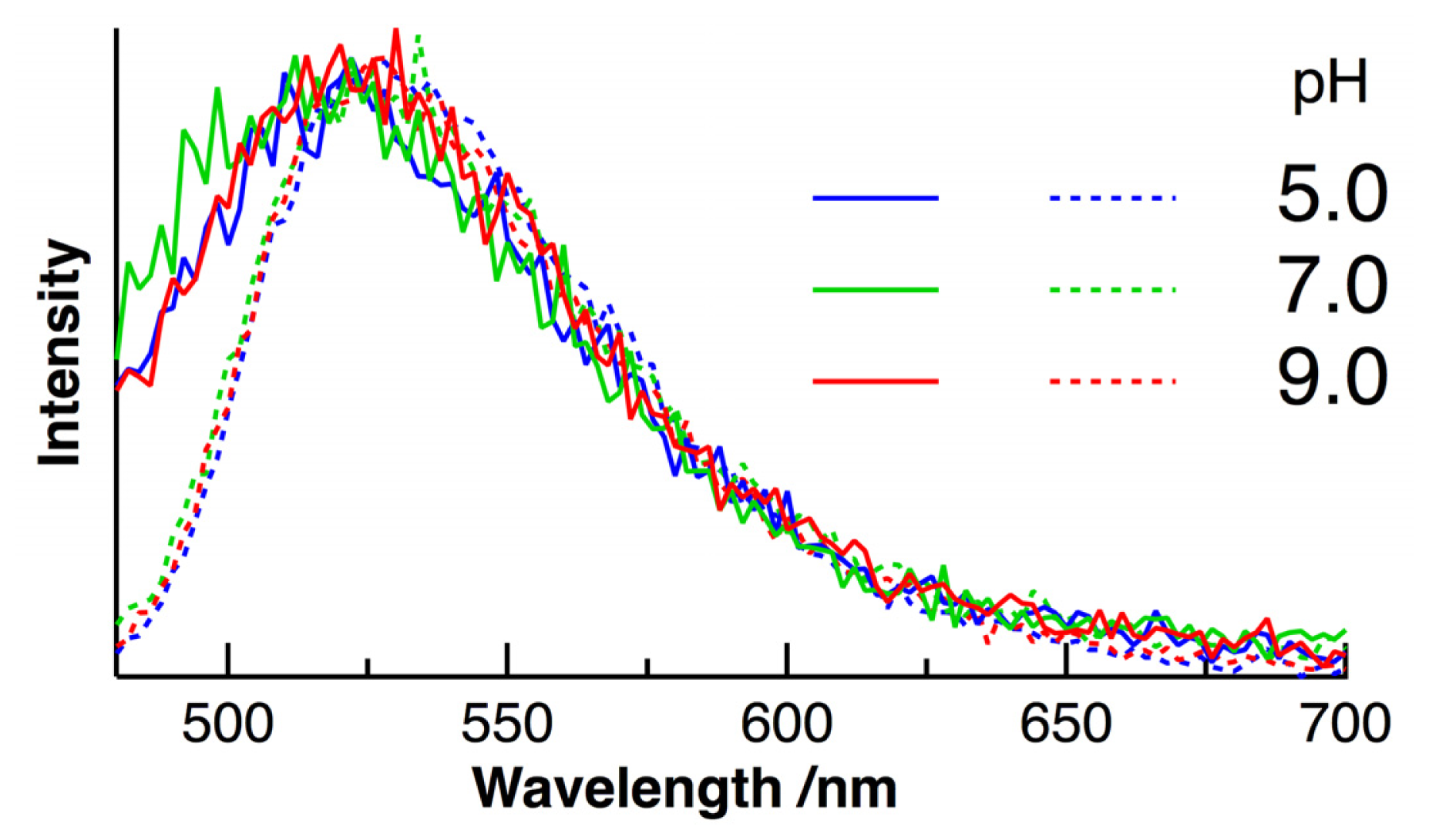
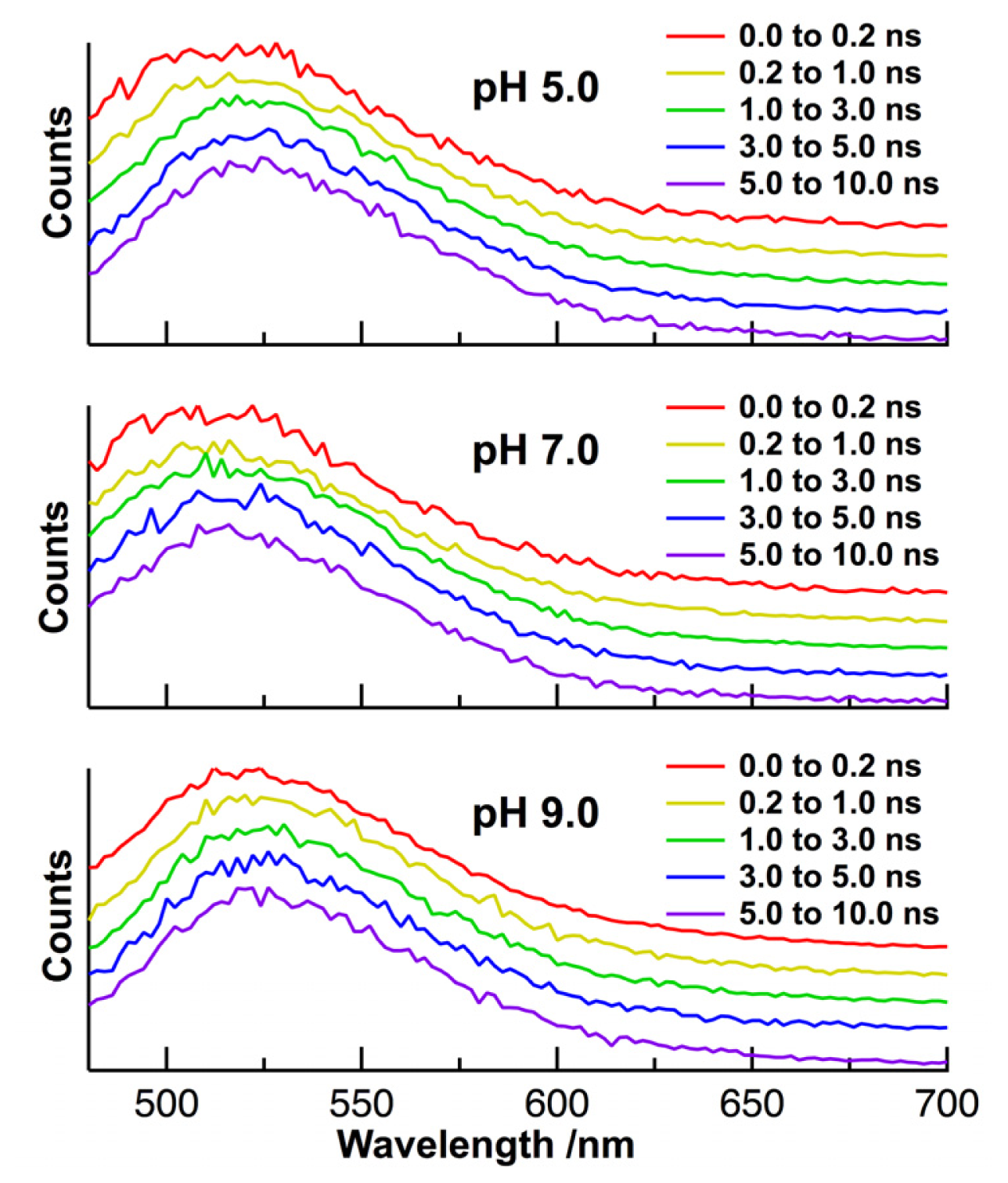
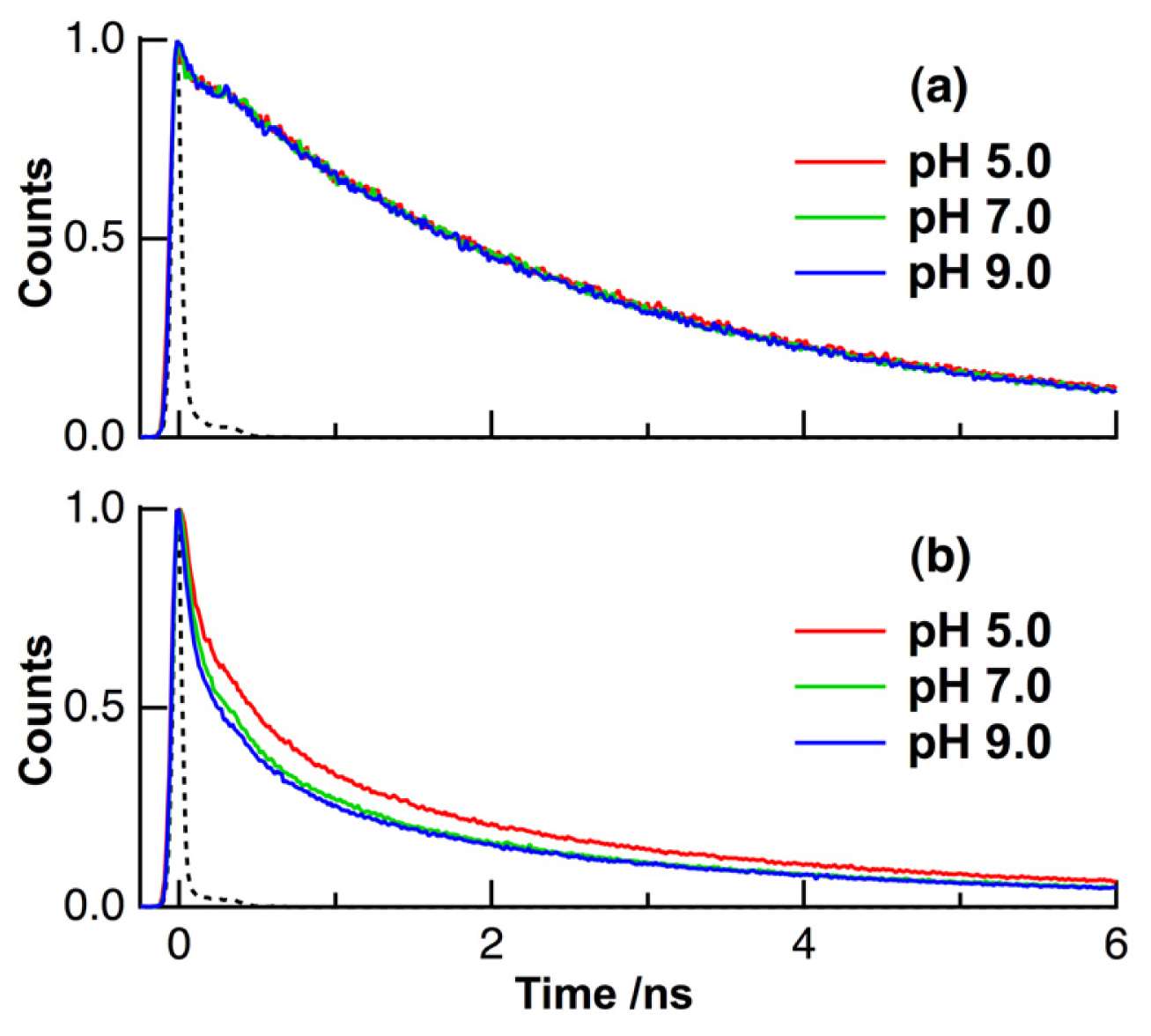
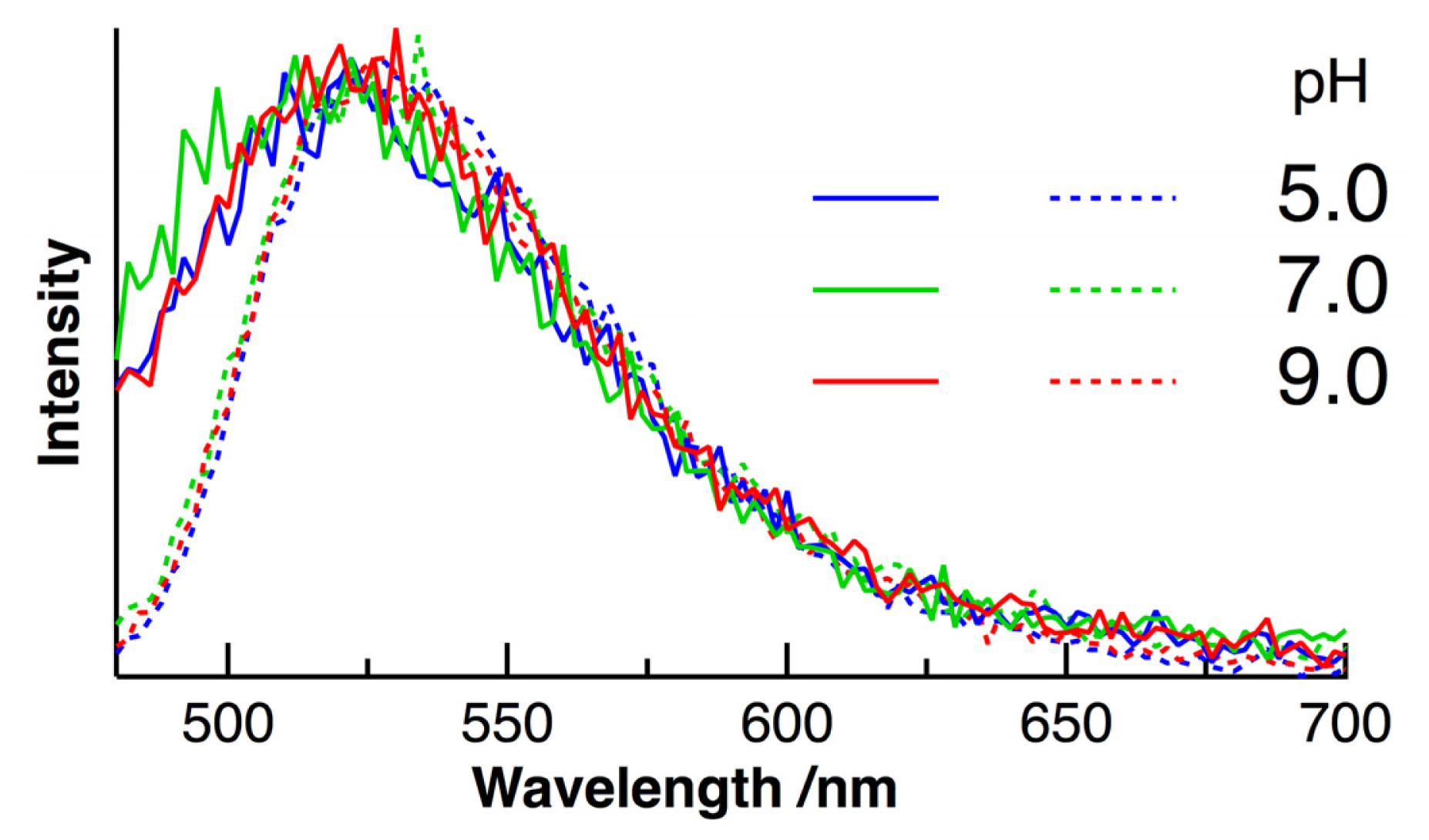
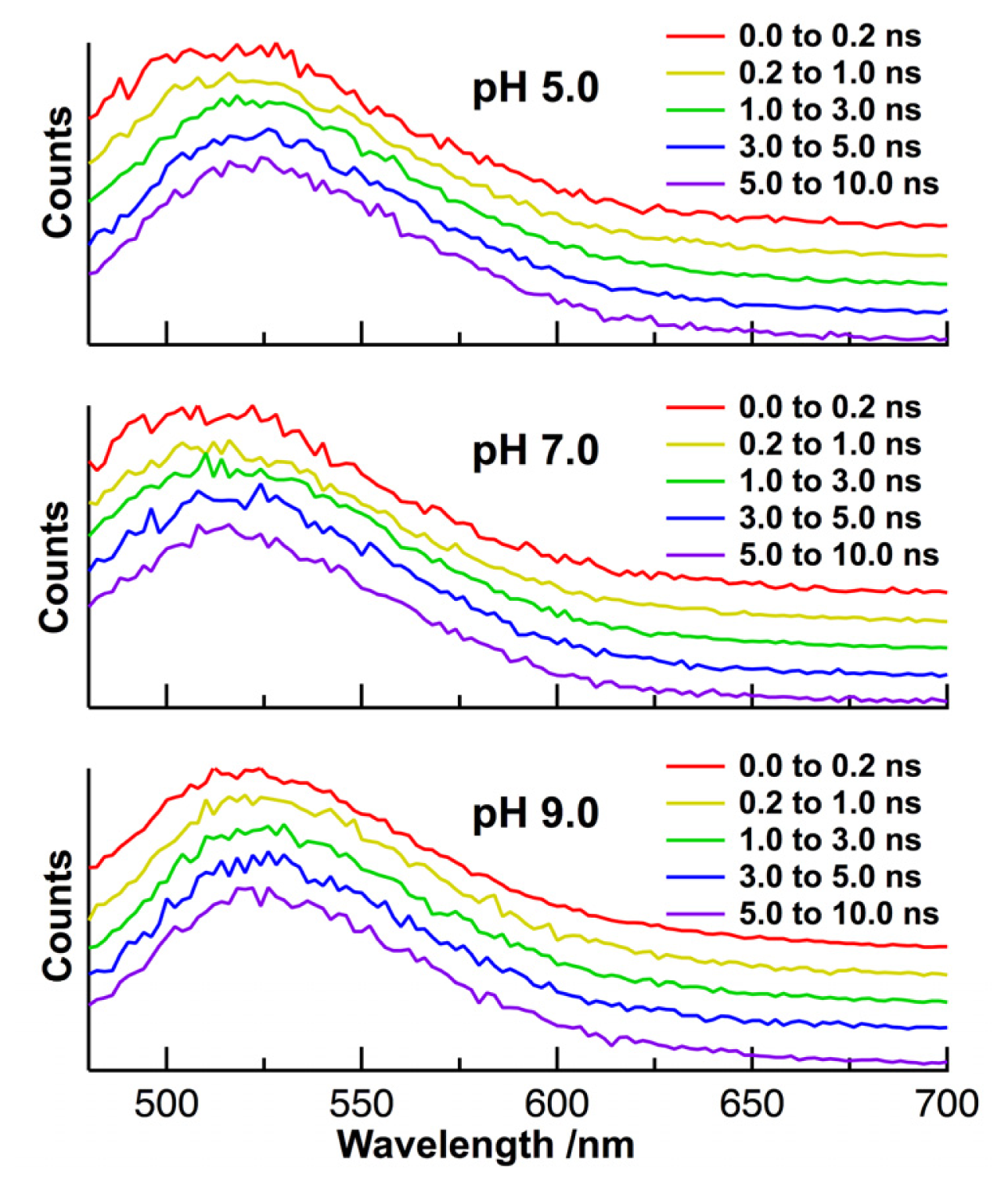
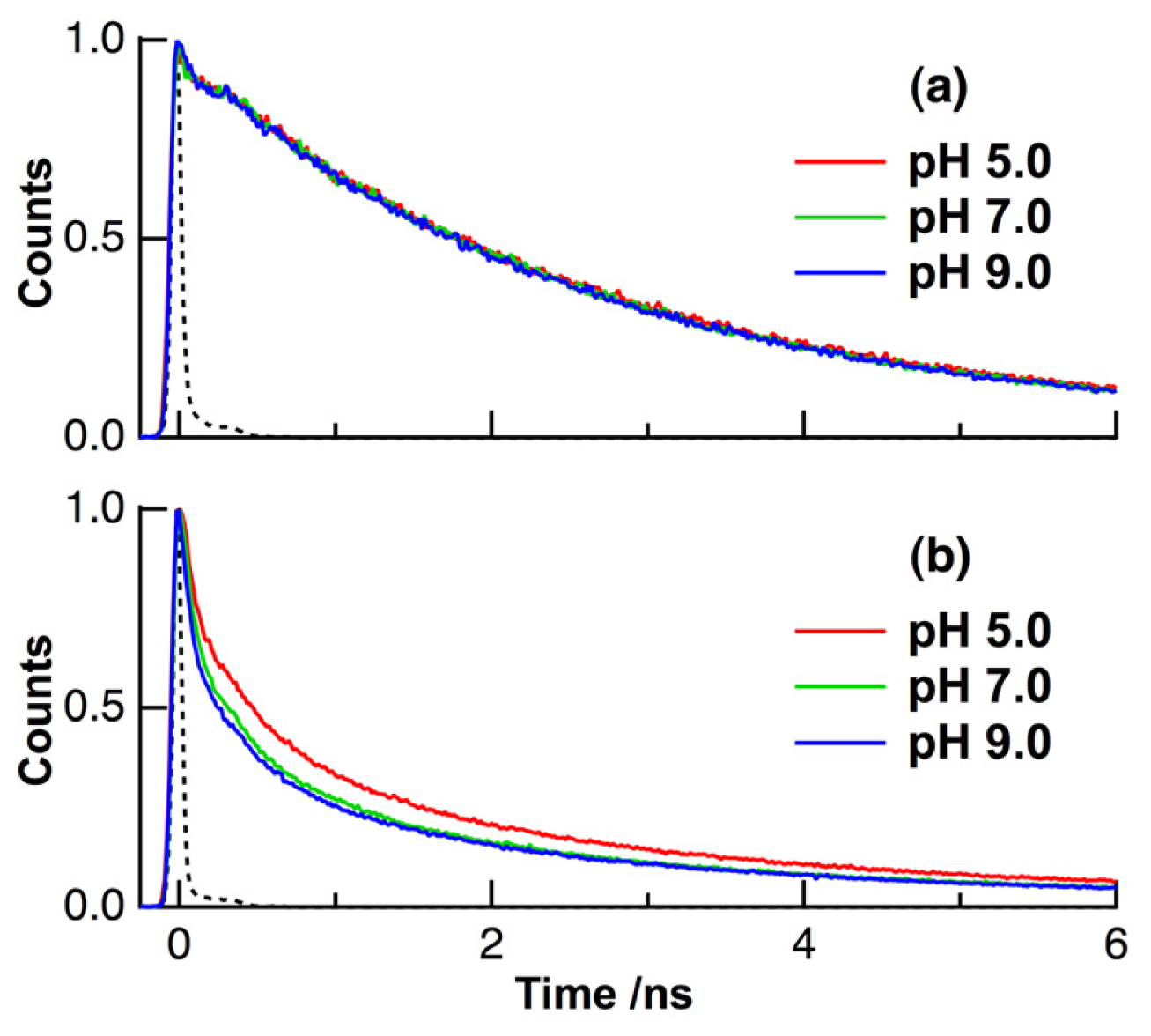
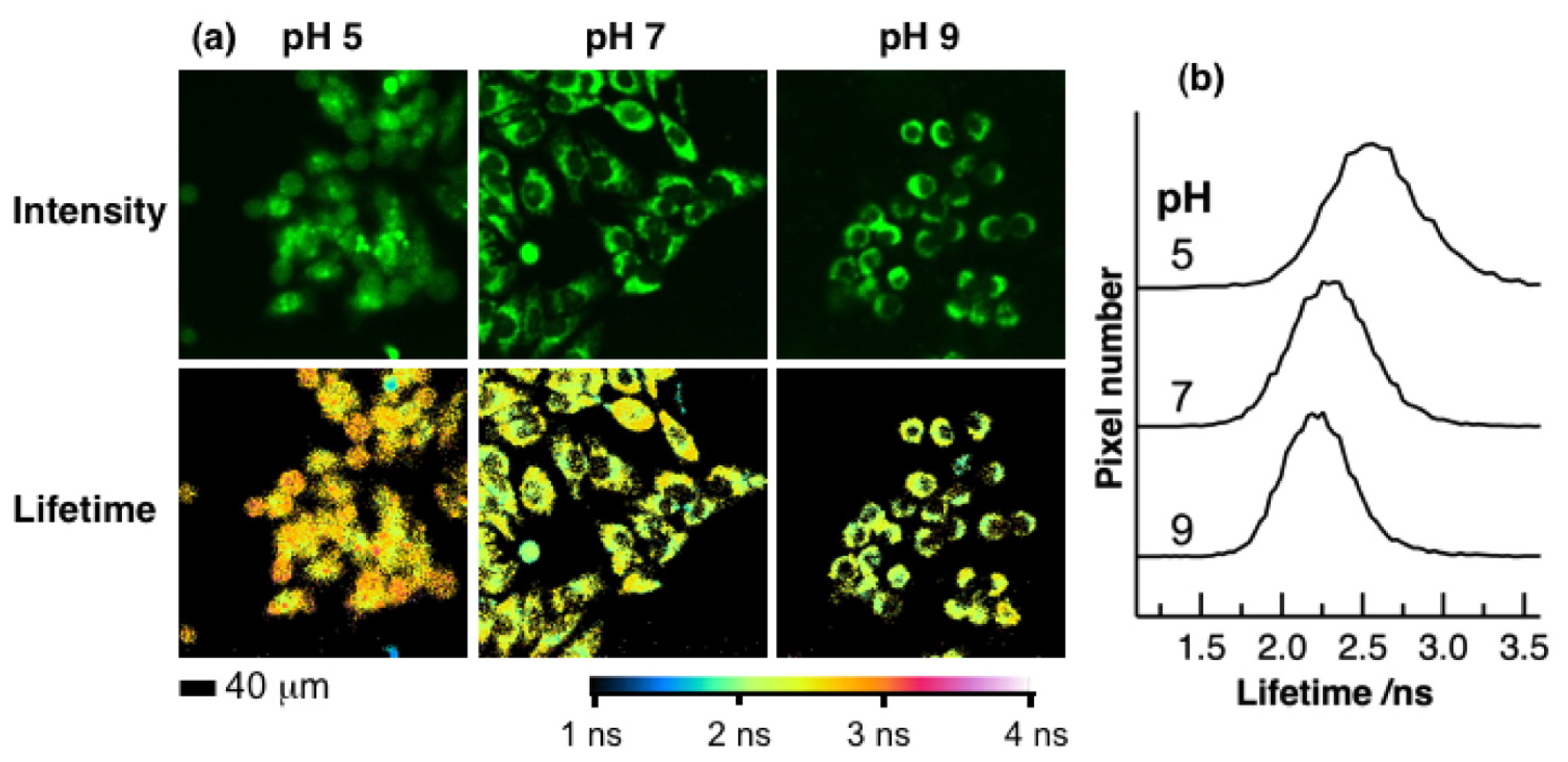
2. Results and Discussion
3. Experimental Section
3.1. Sample Preparation
3.2. Time-Resolved Fluorescence Measurements
4. Conclusions
Acknowledgments
References
- Huang, S.; Heikal, A.A.; Webb, W.W. Two-photon fluorescence spectroscopy and microscopy of NAD(P)H and flavoprotein. Biophys. J 2002, 82, 2811–2825. [Google Scholar]
- Niesner, R.; Peker, B.; Schlüsche, P.; Gericke, K.-H. Noniterative biexponential fluorescence lifetime imaging in the investigation of cellular metabolism by means of NAD(P)H autofluorescence. ChemPhysChem 2004, 5, 1141–1149. [Google Scholar]
- Skala, M.C.; Riching, K.M.; Gendron-Fitzpatrick, A.; Eickhoff, J.; Eliceiri, K.W.; White, J.G.; Ramanujam, N. In vivo multiphoton microscopy of NADH and FAD redox states, fluorescence lifetimes, and cellular morphology in precancerous epithelia. Proc. Natl. Acad. Sci. USA 2007, 104, 19494–19499. [Google Scholar]
- Schweitzer, D.; Schenke, S.; Hammer, M.; Schweitzer, F.; Jentsch, S.; Birckner, E.; Becker, W.; Bergmann, A. Towards metabolic mapping of the human retina. Microsc. Res. Tech 2007, 70, 410–419. [Google Scholar]
- Yu, Q.; Heikal, A.A. Two-photon autofluorescence dynamics imaging reveals sensitivity of intracellular NADH concentration and conformation to cell physiology at the single-cell level. J. Photochem. Photobiol. B 2009, 95, 46–57. [Google Scholar]
- Ghukasyan, V.V.; Kao, F.J. Monitoring cellular metabolism with fluorescence lifetime of reduced nicotinamide adenine dinucleotide. J. Phys. Chem. C 2009, 113, 11532–11540. [Google Scholar]
- Chorvat, D., Jr; Chorvatova, A. Multi-wavelength fluorescence lifetime spectroscopy: A new approach to the study of endogenous fluorescence in living cells and tissues. Laser Phys. Lett. 2009, 6, 175–193. [Google Scholar]
- Berezin, M.Y.; Achilefu, S. Fluorescence lifetime measurements and biological imaging. Chem. Rev 2010, 110, 2641–2684. [Google Scholar]
- Wang, H.-P.; Nakabayashi, T.; Tsujimoto, K.; Miyauchi, S.; Kamo, N.; Ohta, N. Fluorescence lifetime image of a single halobacterium. Chem. Phys. Lett 2007, 442, 441–444. [Google Scholar]
- Nakabayashi, T.; Wang, H.-P.; Kinjo, M.; Ohta, N. Application of fluorescence lifetime imaging of enhanced green fluorescent protein to intracellular pH measurements. Photochem. Photobiol. Sci 2008, 7, 668–670. [Google Scholar]
- Ogikubo, S.; Nakabayashi, T.; Adachi, T.; Islam, M.S.; Yoshizawa, T.; Kinjo, M.; Ohta, N. Intracellular pH sensing using autofluorescence lifetime microscopy. J. Phys. Chem. B 2011, 115, 10385–10390. [Google Scholar]
- Nakabayashi, T.; Nagao, I.; Kinjo, M.; Aoki, Y.; Tanaka, M.; Ohta, N. Stress-induced environmental changes in a single cell as revealed by fluorescence lifetime imaging. Photochem. Photobiol. Sci 2008, 7, 671–674. [Google Scholar]
- Ito, T.; Oshita, S.; Nakabayashi, T.; Sun, F.; Kinjo, M.; Ohta, N. Fluorescence lifetime images of green fluorescent protein in HeLa cells during TNF-alpha induced apoptosis. Photochem. Photobiol. Sci 2009, 8, 763–767. [Google Scholar]
- Awasthi, K.; Nakabayashi, T.; Ohta, N. Application of nanosecond pulsed electric fields into HeLa cells expressing enhanced green fluorescent protein and fluorescence lifetime microscopy. J. Phys. Chem. B 2012, 116, 11159–11165. [Google Scholar]
- Wallrabe, H.; Periasamy, A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol 2005, 16, 19–27. [Google Scholar]
- Becker, W.; Bergmann, A.; Biskup, C. Multispectral fluorescence lifetime imaging by TCSPC. Microsc. Res. Tech 2007, 70, 403–409. [Google Scholar]
- Levitt, J.A.; Matthews, D.R.; Ameer-Beg, S.M.; Suhling, K. Fluorescence lifetime and polarization-resolved imaging in cell biology. Curr. Opin. Biotechnol 2009, 20, 28–36. [Google Scholar]
- Borst, J.W.; Visser, A.J.W.G. Fluorescence lifetime imaging microscopy in life sciences. Meas. Sci. Technol 2010, 21, 102002. [Google Scholar]
- Hanson, K.M.; Behne, M.J.; Barry, N.P.; Mauro, T.M.; Gratton, E.; Clegg, R.M. Two-photon fluorescence lifetime imaging of the skin stratum corneum pH gradient. Biophys. J 2002, 83, 1682–1690. [Google Scholar]
- Niesner, R.; Peker, B.; Schlüsche, P.; Gericke, K.-H.; Hoffmann, C.; Hahne, D.; Müller-Goymann, C. 3D-resolved investigation of the pH gradient in artificial skin constructs by means of fluorescence lifetime imaging. Pharmaceu. Res 2005, 22, 1079–1087. [Google Scholar]
- Lin, H.-J.; Herman, P.; Lakowicz, J.R. Fluorescence lifetime-resolved pH imaging of living cells. Cytometry 2003, 52A, 77–89. [Google Scholar]
- Ghisla, S.; Massey, V. Mechanisms of flavoprotein-catalyzed reactions. Eur. J. Biochem 1989, 181, 1–17. [Google Scholar]
- Mattevi, A. To be or not to be an oxidase: Challenging the oxygen reactivity of flavoenzymes. Trends Biochem. Sci 2006, 31, 276–283. [Google Scholar]
- Mataga, N.; Chosrowjan, H.; Shibata, Y.; Tanaka, F.; Nishima, Y.; Shiga, K. Dynamics and mechanisms of ultrafast fluorescence quenching reactions of flavin chromophores in protein nanospace. J. Phys. Chem. B 2000, 104, 10667–10677. [Google Scholar]
- Chosrowjan, H.; Taniguchi, S.; Mataga, N.; Nakanishi, T.; Haruyama, Y.; Sato, S.; Kitamura, M.; Tanaka, F. Effects of the disappearance of one charge on ultrafast fluorescence dynamics of the FMN binding protein. J. Phys. Chem. B 2010, 114, 6175–6182. [Google Scholar]
- Hall, C.L.; Kamin, H. The purification and some properties of electron transfer flavoprotein and general fatty acyl coenzyme A dehydrogenase from pig liver mitochondria. J. Biol. Chem 1975, 250, 3476–3486. [Google Scholar]
- Kunz, W.S.; Kunz, W. Contribution of different enzymes to flavoprotein fluorescence of isolated rat liver mitochondria. Biochim. Biophys. Acta 1985, 841, 237–246. [Google Scholar]
- Kunz, W.S. Spectral properties of fluorescent flavoproteins of isolated rat liver mitochondria. FEBS Lett 1986, 195, 92–96. [Google Scholar]
- Romashko, D.N.; Marban, E.; O’Rourke, B. Subcellular metabolic transients and mitochondrial redox waves in heart cells. Proc. Natl. Acad. Sci. USA 1998, 95, 1618–1623. [Google Scholar]
- Chorvat, D., Jr; Chorvatova, A. Spectrally resolved time-correlated single photon counting: A novel approach for characterization of endogenous fluorescence in isolated cardiac myocytes. Eur. Biophys. J. 2006, 36, 73–83. [Google Scholar]
- Barrio, J.R.; Tolman, G.L.; Leonard, N.J.; Spencer, R.D.; Weber, G. Flavin 1, N6-ethenoadenine dinucleotide: Dynamic and static quenching of fluorescence. Proc. Natl. Acad. Sci. USA 1973, 70, 941–943. [Google Scholar]
- Stanley, R.J.; MacFarlane, A.W., IV. Ultrafast excited state dynamics of oxidized flavins: Direct observations of quenching by purines. J. Phys. Chem. A 2000, 104, 6899–6906. [Google Scholar]
- Van den Berg, P.A.W.; Feenstra, K.A.; Mark, A.E.; Berendsen, H.J.C.; Visser, A.J.W.G. Dynamic conformations of flavin adenine dinucleotide: Simulated molecular dynamics of the flavin cofactor related to the time-resolved fluorescence characteristics. J. Phys. Chem. B 2002, 106, 8858–8869. [Google Scholar]
- Kao, Y.-T.; Saxena, C.; He, T.-F.; Guo, L.; Wang, L.; Sancar, A.; Zhong, D. Ultrafast dynamics of flavins in five redox states. J. Am. Chem. Soc 2008, 130, 13132–13139. [Google Scholar]
- Li, G.; Glusac, K.D. Light-triggered proton and electron transfer in flavin cofactors. J. Phys. Chem. A 2008, 112, 4573–4583. [Google Scholar]
- Nakabayashi, T.; Islam, M.S.; Ohta, N. Fluorescence decay dynamics of flavin adenine dinucleotide in a mixture of alcohol and water in the femtosecond and nanosecond time range. J. Phys. Chem. B 2010, 114, 15254–15260. [Google Scholar]
- Islam, S.D.M.; Susdorf, T.; Penzkofer, A.; Hegemann, P. Fluorescence quenching of flavin adenine dinucleotide in aqueous solution by pH dependent isomerisation and photo-induced electron transfer. Chem. Phys 2003, 295, 137–149. [Google Scholar]
- Sengupta, A.; Khade, R.V.; Hazra, P. pH dependent dynamic behavior of flavin mononucleotide (FMN) and flavin adenine dinucleotide (FAD) in femtosecond to nanosecond time scale. J. Photochem. Photobiol. A 2011, 221, 105–112. [Google Scholar]
- Thomas, J.A.; Buchsbaum, R.N.; Zimniak, A.; Racker, E. Intracellular pH measurements in ehrlich ascites tumor cells utilizing spectroscopic probes generated in situ. Biochemistry 1979, 18, 2210–2218. [Google Scholar]
- Nedergaard, M.; Desai, S.; Pulsinelli, W. Dicarboxy-dichlorofluorescein: A new fluorescent probe for measuring acidic intracellular pH. Anal. Biochem 1990, 187, 109–114. [Google Scholar]
- Sanders, R.; Draaijer, A.; Gerritsen, H.C.; Houpt, P.M.; Levine, Y.K. Quantitative pH imaging in cells using confocal fluorescence lifetime imaging microscopy. Anal. Biochem 1995, 227, 302–308. [Google Scholar]
- Gardecki, J.A.; Maroncelli, M. Comparison of the single-wavelength and spectral-reconstruction methods for determining the solvation-response function. J. Phys. Chem. A 1999, 103, 1187–1197. [Google Scholar]
- Bastiaens, P.I.H.; van Hoek, A.; Wolkers, W.F.; Brochon, J.-C.; Visser, A.J.W.G. Comparison of the dynamical structures of lipoamide dehydrogenase and glutathione reductase by time-resolved polarized flavin fluorescence. Biochemistry 1992, 31, 7050–7060. [Google Scholar]
- Yamazaki, I.; Tamai, N.; Kume, H.; Tsuchiya, H.; Oba, K. Microchannel-plate photomultiplier applicability to the time-correlated photon-counting method. Rev. Sci. Instrum 1985, 56, 1187–1194. [Google Scholar]
- Matsuyama, S.; Llopis, J.; Deveraux, Q.L.; Tsien, R.Y.; Reed, J.C. Changes in intramitochondrial and cytosolic pH: Early events that modulate caspase activation during apoptosis. Nat. Cell Biol. 2000, 2, 318–325. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pH | τ1 (ps) | τ2 (ps) | τ3 (ns) | τ4 (ns) | τave (ns) | |
|---|---|---|---|---|---|---|
| solution | 5, 7, 9 | 7 (0.66) c | 220 (0.03) | 2.09 (0.17) | 3.97 (0.14) | 2.70 d |
| HeLa cells | 5 | 80 (0.52) | 700 (0.27) | 3.17 (0.18) | 10.2 (0.03) | 1.11 |
| 7 | 80 (0.60) | 700 (0.25) | 3.17 (0.13) | 10.2 (0.02) | 0.84 | |
| 9 | 80 (0.63) | 700 (0.23) | 3.17 (0.12) | 9.42 (0.02) | 0.78 |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Islam, M.S.; Honma, M.; Nakabayashi, T.; Kinjo, M.; Ohta, N. pH Dependence of the Fluorescence Lifetime of FAD in Solution and in Cells. Int. J. Mol. Sci. 2013, 14, 1952-1963. https://doi.org/10.3390/ijms14011952
Islam MS, Honma M, Nakabayashi T, Kinjo M, Ohta N. pH Dependence of the Fluorescence Lifetime of FAD in Solution and in Cells. International Journal of Molecular Sciences. 2013; 14(1):1952-1963. https://doi.org/10.3390/ijms14011952
Chicago/Turabian StyleIslam, Md. Serajul, Masato Honma, Takakazu Nakabayashi, Masataka Kinjo, and Nobuhiro Ohta. 2013. "pH Dependence of the Fluorescence Lifetime of FAD in Solution and in Cells" International Journal of Molecular Sciences 14, no. 1: 1952-1963. https://doi.org/10.3390/ijms14011952