The Role of Photolabile Dermal Nitric Oxide Derivates in Ultraviolet Radiation (UVR)-Induced Cell Death

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. UVR-Induced Cell Damage
3. Physiology of Nitric Oxide
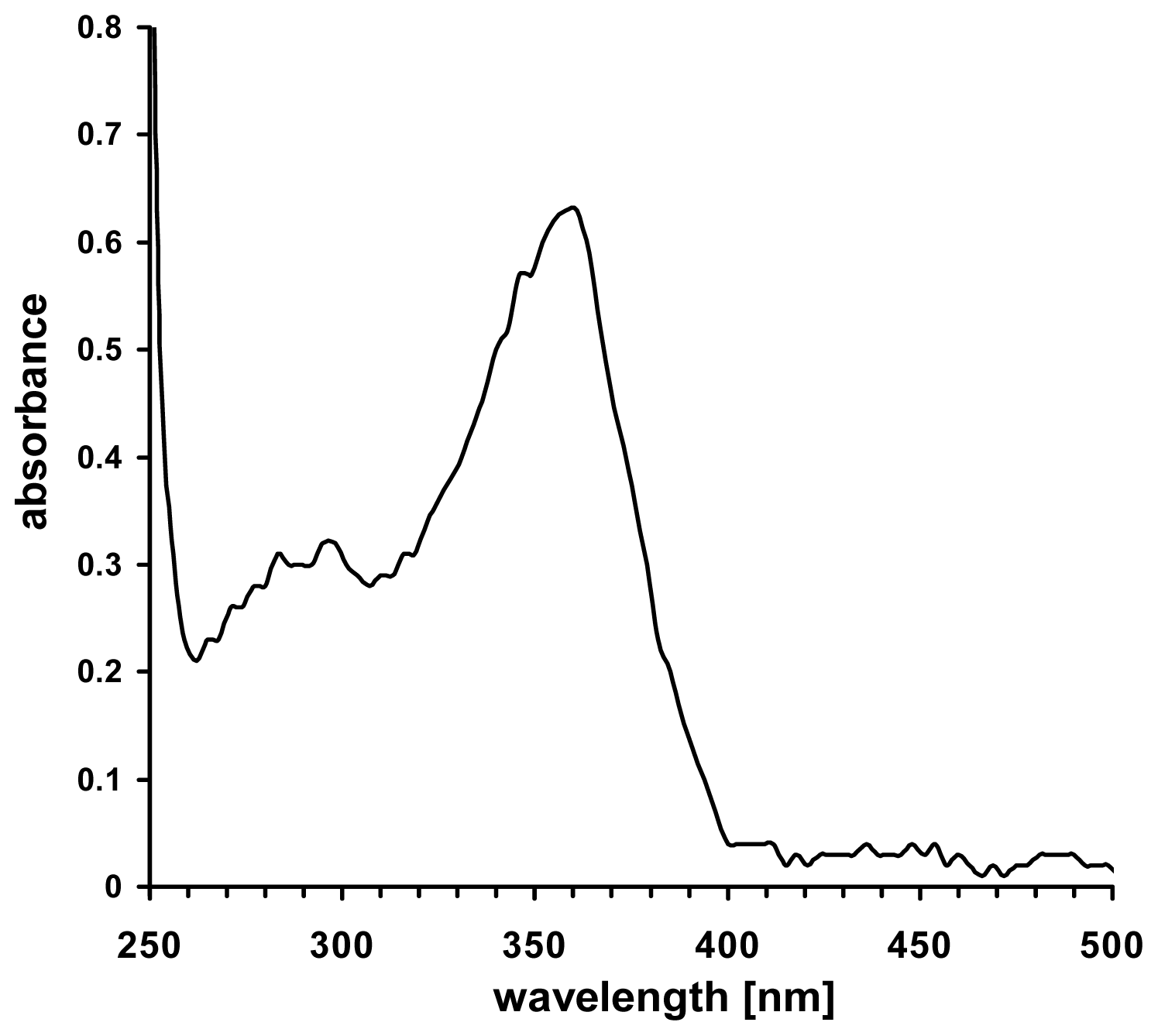
4. Nitrogen Oxide Derivates as Source of Nitric Oxide
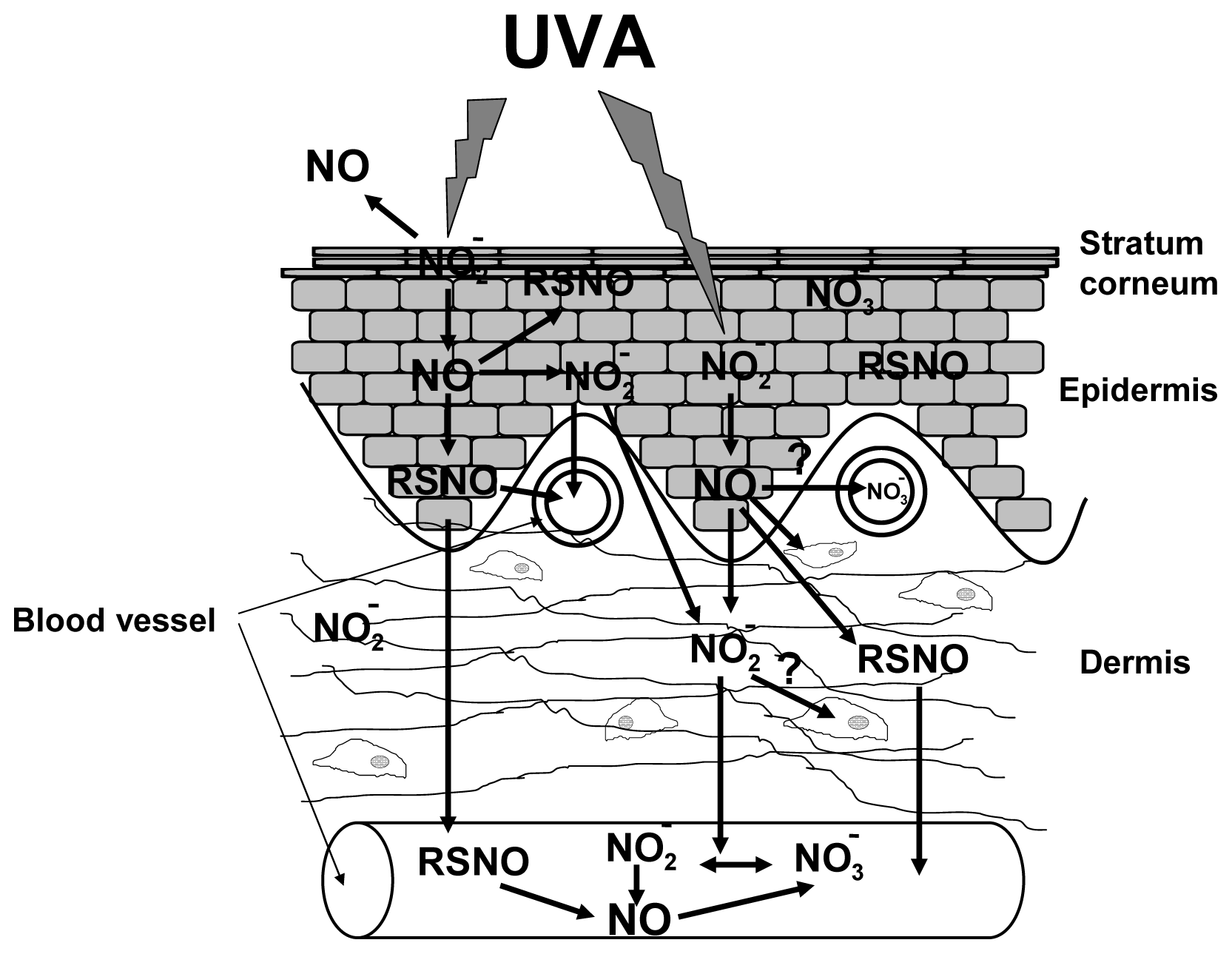
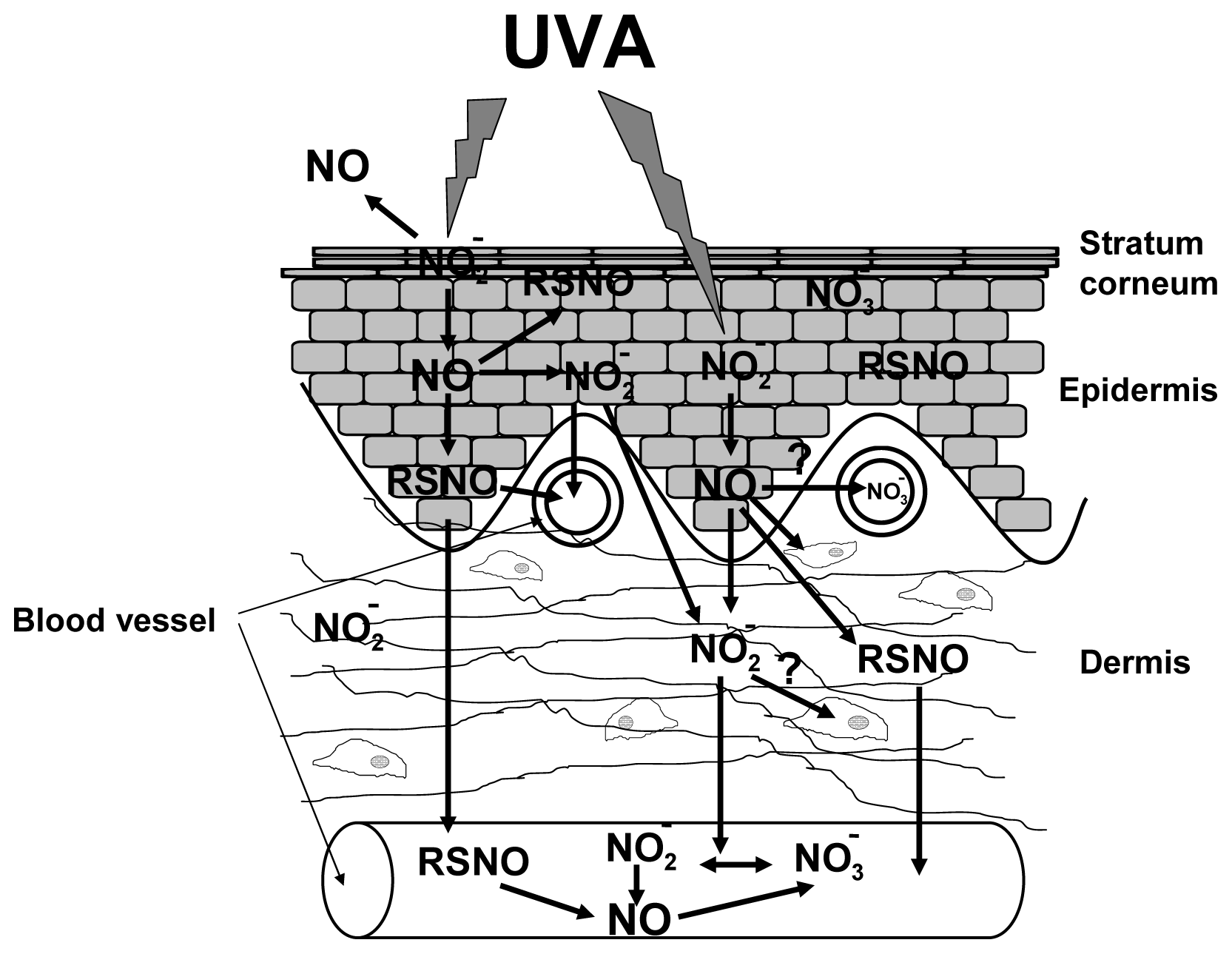
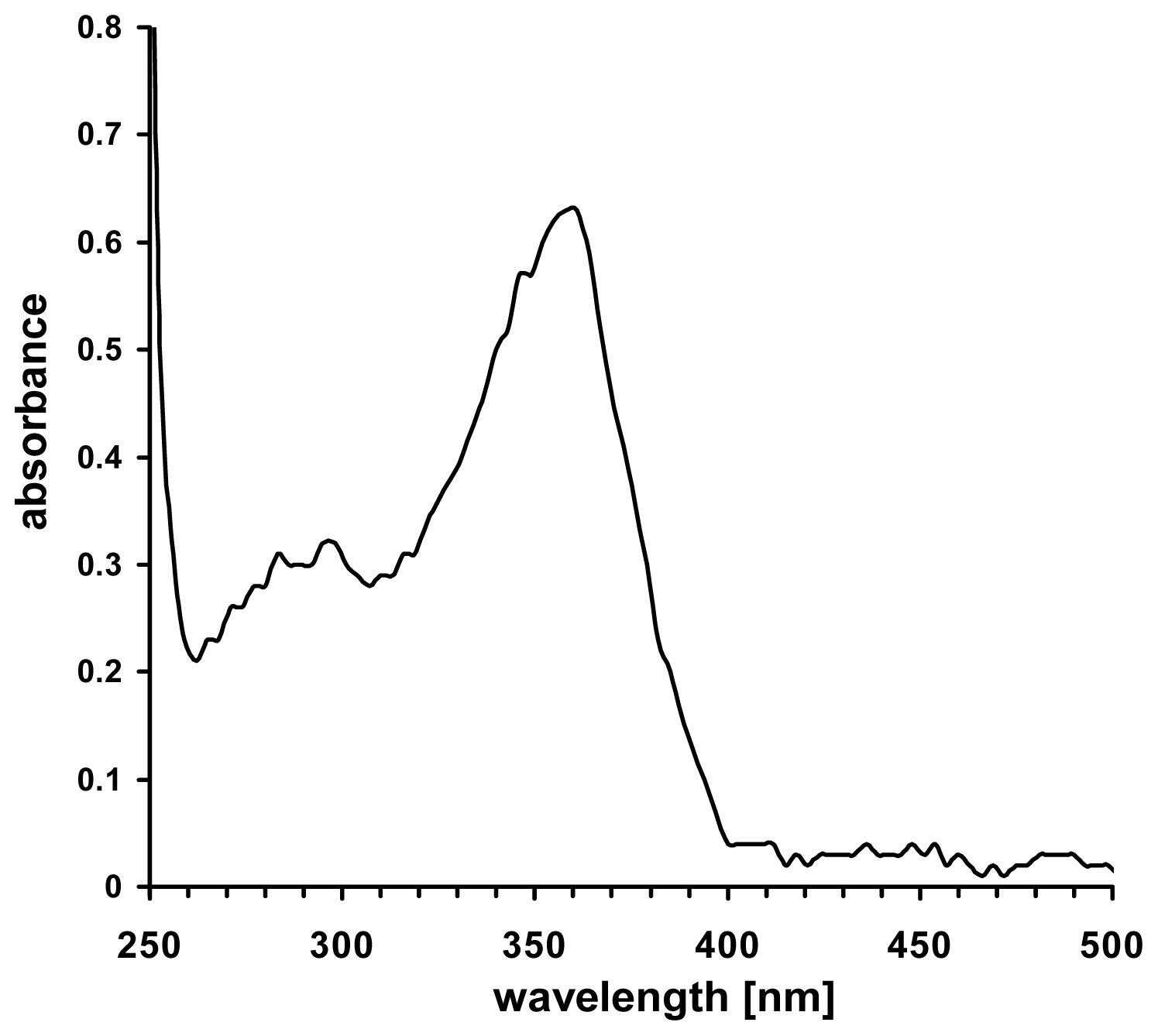
5. UVA Induces Non-Enzymatic NO-Formation in Human Skin
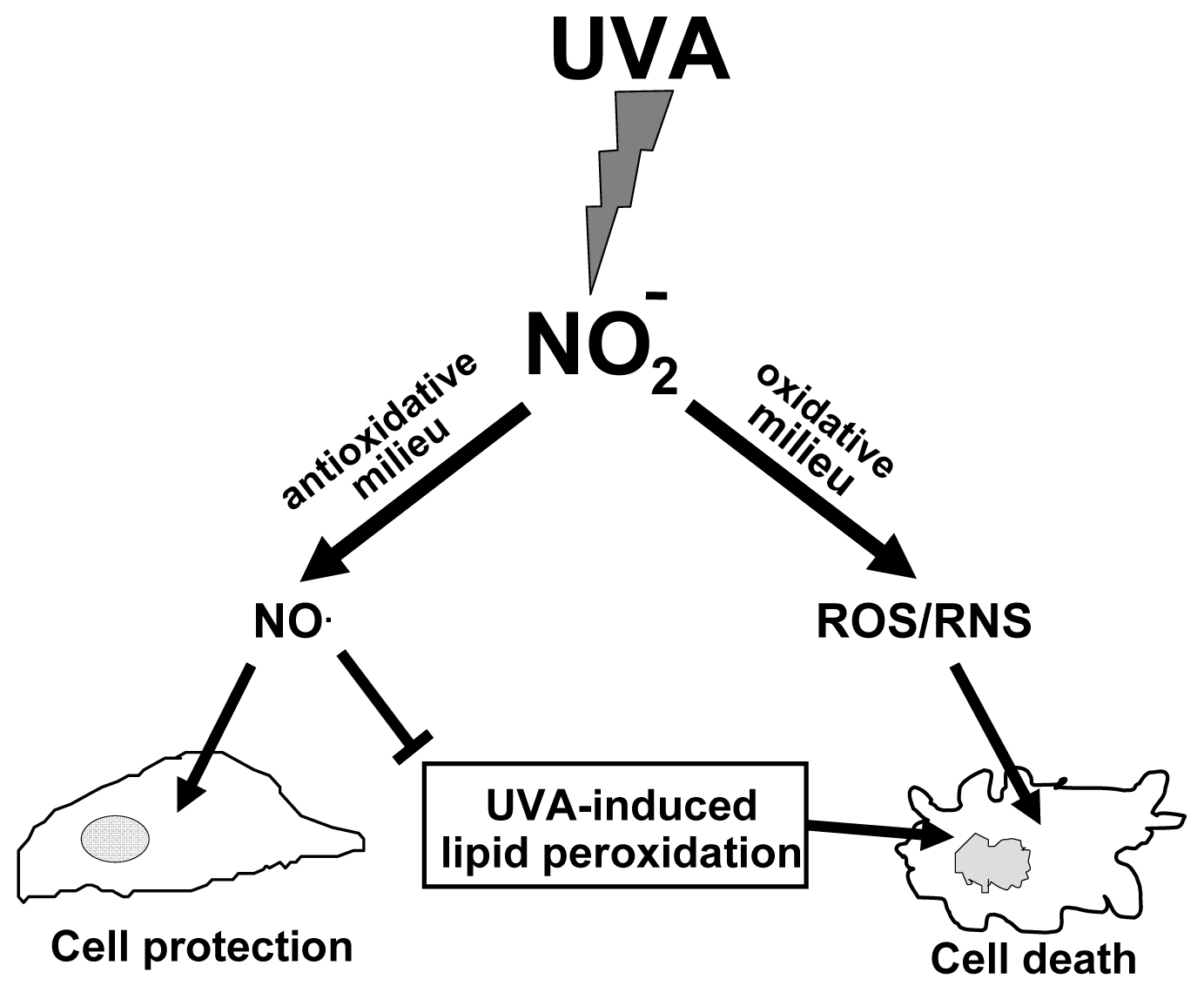
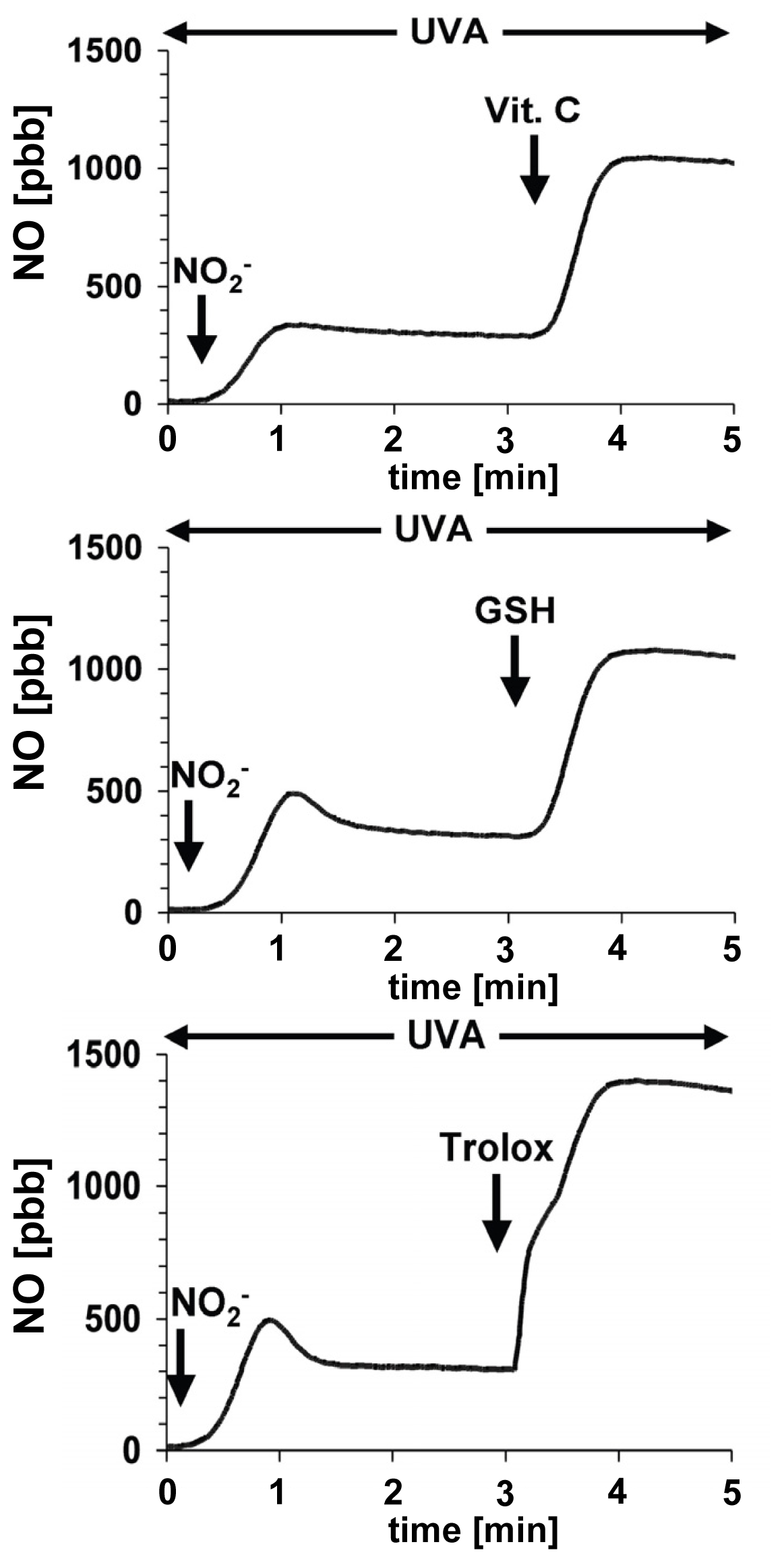
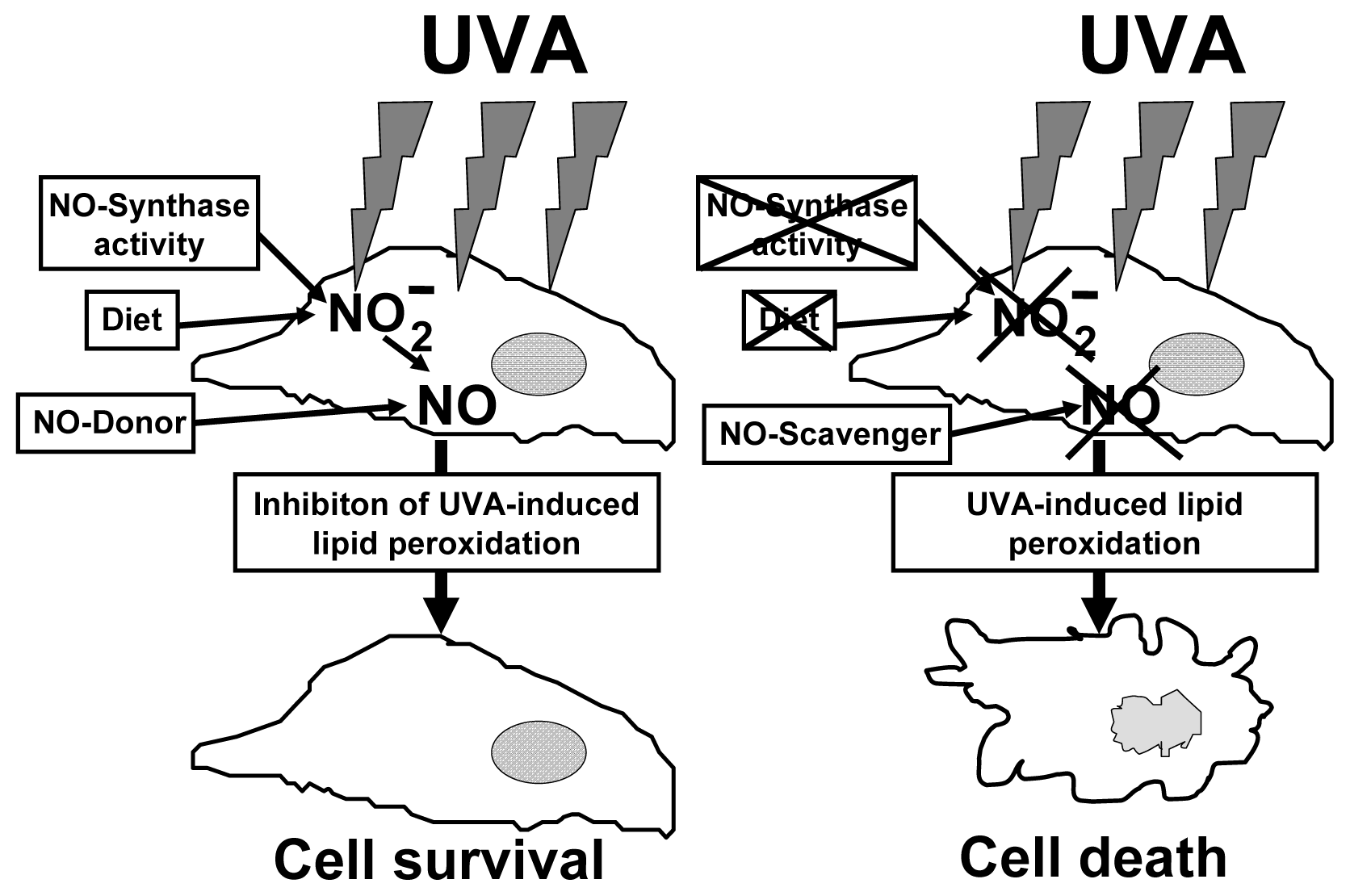
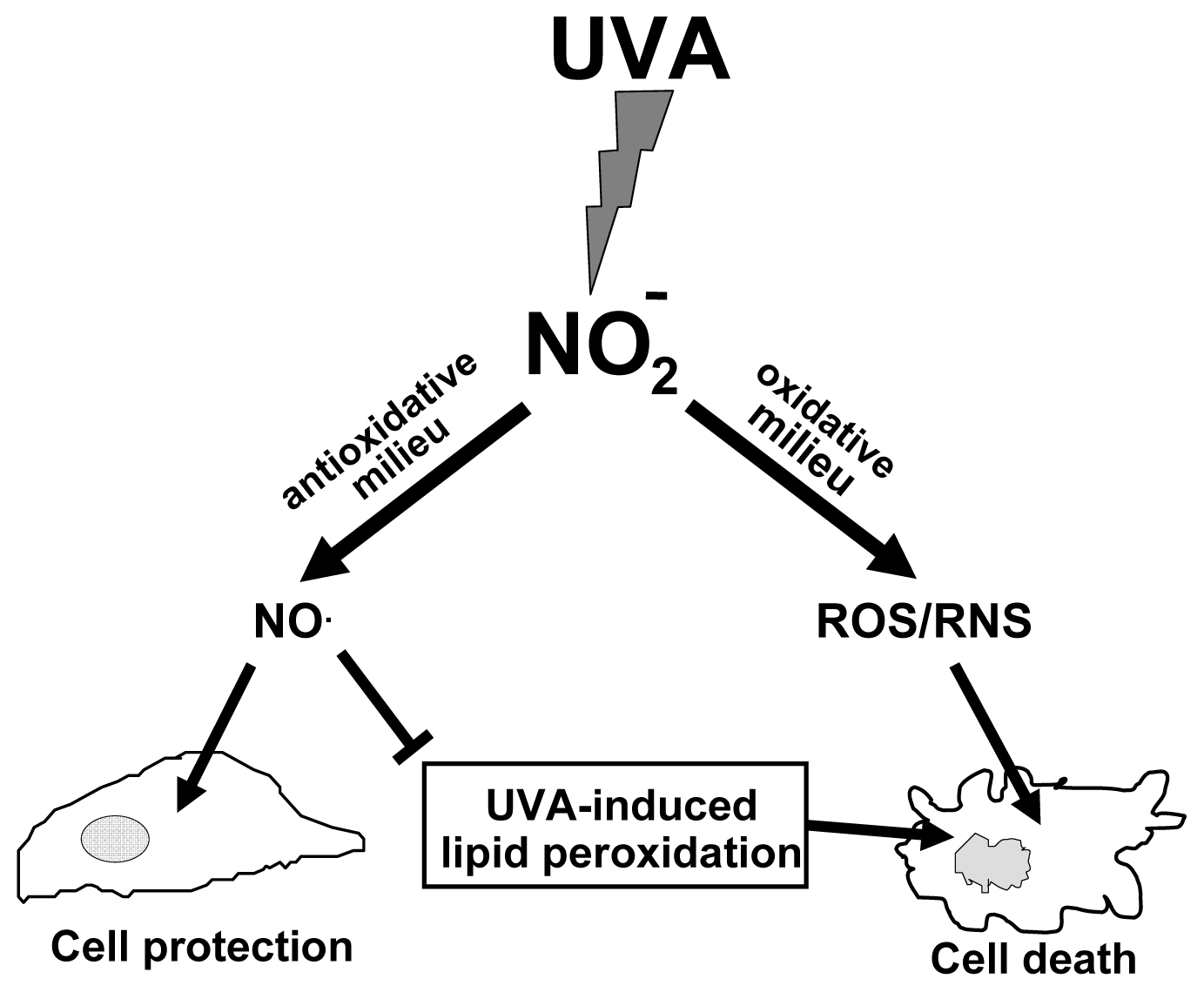
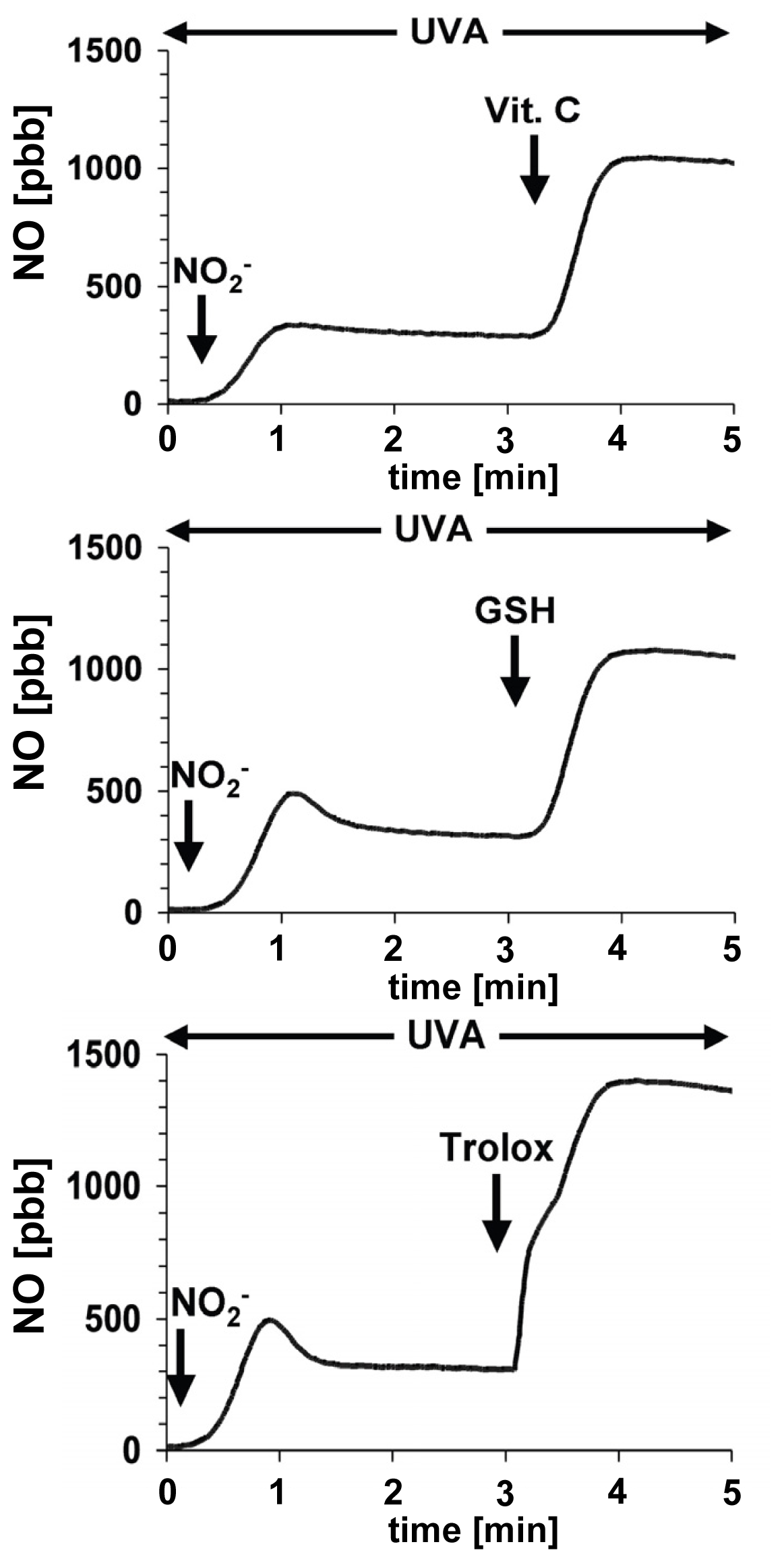
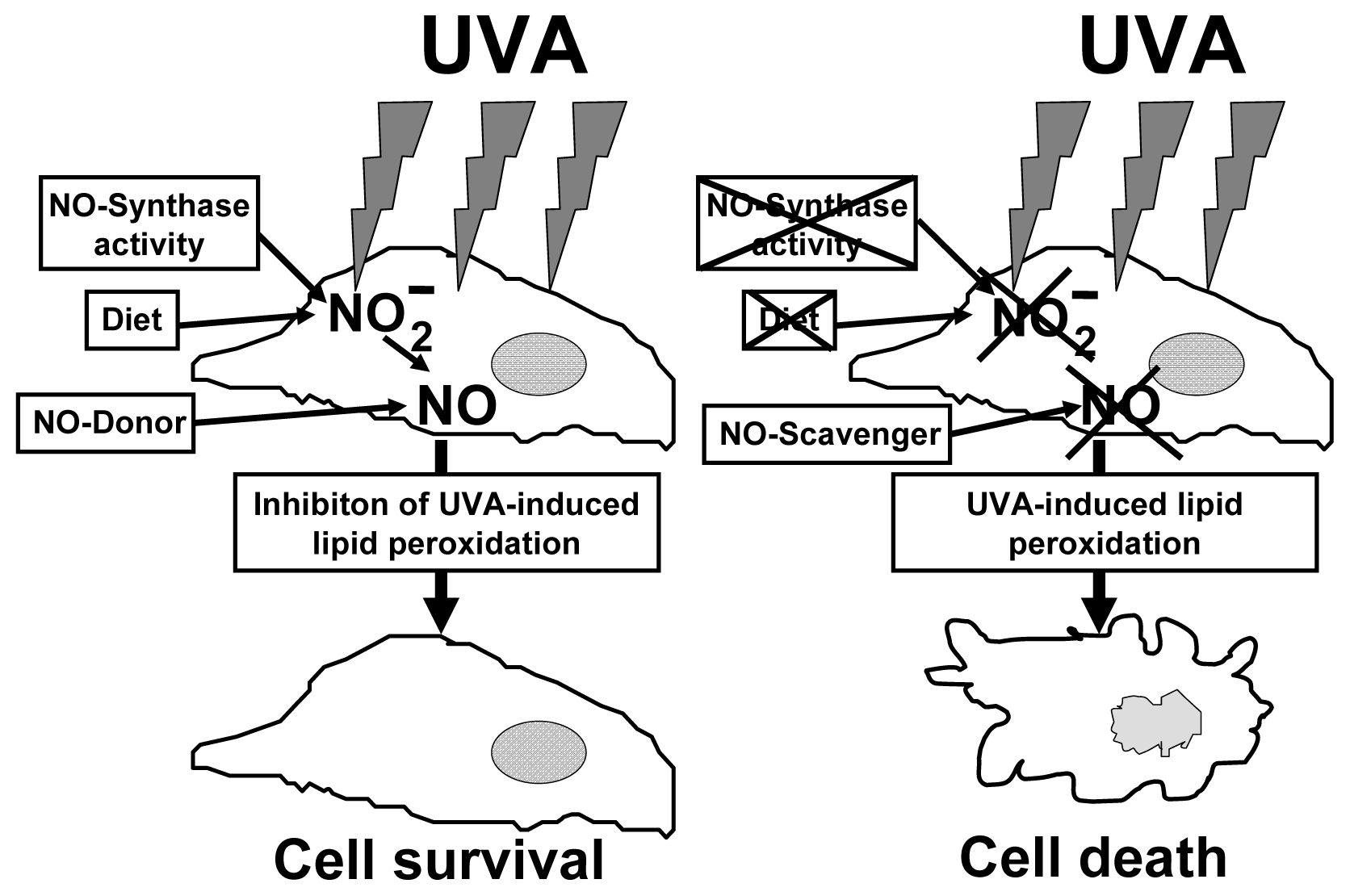
6. The Protective Role of Intracellular Nitrite during UVA-Challenge
Acknowledgments
References
- Rigel, D.S. Cutaneous ultraviolet exposure and its relationship to the development of skin cancer. J. Am. Acad. Dermatol 2008, 58, S129–S132. [Google Scholar]
- Helfrich, Y.R.; Sachs, D.L.; Voorhees, J.J. Overview of skin aging and photoaging. Dermatol. Nurs. 2008, 20, 177–183, quiz 184. [Google Scholar]
- Moan, J.; Porojnicu, A.C.; Dahlback, A.; Setlow, R.B. Addressing the health benefits and risks, involving vitamin D or skin cancer, of increased sun exposure. Proc. Natl. Acad. Sci. USA 2008, 105, 668–673. [Google Scholar]
- Van der Rhee, H.J.; de Vries, E.; Coebergh, J.W.W. Does sunlight prevent cancer? A systematic review. Eur. J. Cancer 2006, 42, 2222–2232. [Google Scholar]
- Grant, W.B.; Boucher, B.J. Requirements for vitamin D across the life span. Biol. Res. Nurs 2011, 13, 120–133. [Google Scholar]
- Brennan, P.J.; Greenberg, G.; Miall, W.E.; Thompson, S.G. Seasonal variation in arterial blood pressure. Br. Med. J. (Clin. Res. Ed.) 1982, 285, 919–923. [Google Scholar]
- Feelisch, M.; Kolb-Bachofen, V.; Liu, D.; Lundberg, J.O.; Revelo, L.P.; Suschek, C.V.; Weller, R.B. Is sunlight good for our heart? Eur. Heart J 2010, 31, 1041–1045. [Google Scholar]
- Oplander, C.; Volkmar, C.M.; Paunel-Gorgulu, A.; van Faassen, E.E.; Heiss, C.; Kelm, M.; Halmer, D.; Murtz, M.; Pallua, N.; Suschek, C.V. Whole body UVA irradiation lowers systemic blood pressure by release of nitric oxide from intracutaneous photolabile nitric oxide derivates. Circ. Res 2009, 105, 1031–1040. [Google Scholar]
- Pustisek, N.; Situm, M. UV-radiation, apoptosis and skin. Coll. Antropol 2012, 35, 339–341. [Google Scholar]
- Yang, Y.; Wang, H.; Wang, S.; Xu, M.; Liu, M.; Liao, M.; Frank, J.A.; Adhikari, S.; Bower, K.A.; Shi, X.; et al. GSK3beta signaling is involved in ultraviolet B-induced activation of autophagy in epidermal cells. Int. J. Oncol 2012, 41, 1782–1788. [Google Scholar]
- Lamore, S.D.; Wondrak, G.T. Autophagic-lysosomal dysregulation downstream of cathepsin B inactivation in human skin fibroblasts exposed to UVA. Photochem. Photobiol. Sci 2012, 11, 163–172. [Google Scholar]
- Matsunaga, T.; Hieda, K.; Nikaido, O. Wavelength dependent formation of thymine dimers and (6-4) photoproducts in DNA by monochromatic ultraviolet light ranging from 150 to 365 nm. Photochem. Photobiol 1991, 54, 403–410. [Google Scholar]
- Ziegler, A.; Leffell, D.J.; Kunala, S.; Sharma, H.W.; Gailani, M.; Simon, J.A.; Halperin, A.J.; Baden, H.P.; Shapiro, P.E.; Bale, A.E.; et al. Mutation hotspots due to sunlight in the p53 gene of nonmelanoma skin cancers. Proc. Natl. Acad. Sci. USA 1993, 90, 4216–4220. [Google Scholar]
- Dumaz, N.; van Kranen, H.J.; de Vries, A.; Berg, R.J.; Wester, P.W.; van Kreijl, C.F.; Sarasin, A.; Daya-Grosjean, L.; de Gruijl, F.R. The role of UV-B light in skin carcinogenesis through the analysis of p53 mutations in squamous cell carcinomas of hairless mice. Carcinogenesis 1997, 18, 897–904. [Google Scholar]
- Marrot, L.; Meunier, J.R. Skin DNA photodamage and its biological consequences. J. Am. Acad. Dermatol 2008, 58, S139–S148. [Google Scholar]
- Nakanishi, M.; Niida, H.; Murakami, H.; Shimada, M. DNA damage responses in skin biology—implications in tumor prevention and aging acceleration. J. Dermatol. Sci 2009, 56, 76–81. [Google Scholar]
- Devary, Y.; Rosette, C.; DiDonato, J.A.; Karin, M. NF-kappa B activation by ultraviolet light not dependent on a nuclear signal. Science 1993, 261, 1442–1445. [Google Scholar]
- Rosette, C.; Karin, M. Ultraviolet light and osmotic stress: Activation of the JNK cascade through multiple growth factor and cytokine receptors. Science 1996, 274, 1194–1197. [Google Scholar]
- Fritsche, E.; Schafer, C.; Calles, C.; Bernsmann, T.; Bernshausen, T.; Wurm, M.; Hubenthal, U.; Cline, J.E.; Hajimiragha, H.; Schroeder, P.; et al. Lightening up the UV response by identification of the arylhydrocarbon receptor as a cytoplasmatic target for ultraviolet B radiation. Proc. Natl. Acad. Sci. USA 2007, 104, 8851–8856. [Google Scholar]
- Ji, C.; Yang, B.; Yang, Z.; Tu, Y.; Yang, Y.L.; He, L.; Bi, Z.G. Ultra-violet B (UVB)-induced skin cell death occurs through a cyclophilin D intrinsic signaling pathway. Biochem. Biophys. Res. Commun 2012, 425, 825–829. [Google Scholar]
- George, K.S.; Elyassaki, W.; Wu, Q.; Wu, S. The role of cholesterol in UV light B-induced apoptosis. Photochem. Photobiol 2012, 88, 1191–1197. [Google Scholar]
- Wu, Q.; Wu, S. Lipid rafts association and anti-apoptotic function of prohibitin in ultraviolet B light-irradiated HaCaT keratinocytes. Exp. Dermatol 2012, 21, 640–642. [Google Scholar]
- Zhou, B.R.; Xu, Y.; Luo, D. Effect of UVB irradiation on microRNA expression in mouse epidermis. Oncol. Lett 2012, 3, 560–564. [Google Scholar]
- Pattison, D.I.; Rahmanto, A.S.; Davies, M.J. Photo-oxidation of proteins. Photochem. Photobiol. Sci 2012, 11, 38–53. [Google Scholar]
- Paz, M.L.; Gonzalez Maglio, D.H.; Weill, F.S.; Bustamante, J.; Leoni, J. Mitochondrial dysfunction and cellular stress progression after ultraviolet B irradiation in human keratinocytes. Photodermatol. Photoimmunol. Photomed 2008, 24, 115–122. [Google Scholar]
- Punnonen, K.; Puntala, A.; Jansen, C.T.; Ahotupa, M. UVB irradiation induces lipid peroxidation and reduces antioxidant enzyme activities in human keratinocytes in vitro. Acta Derm. Venereol 1991, 71, 239–242. [Google Scholar]
- Stadtman, E.R.; Berlett, B.S. Reactive oxygen-mediated protein oxidation in aging and disease. Drug Metab. Rev 1998, 30, 225–243. [Google Scholar]
- Polefka, T.G.; Meyer, T.A.; Agin, P.P.; Bianchini, R.J. Effects of solar radiation on the skin. J. Cosmet. Dermatol 2012, 11, 134–143. [Google Scholar]
- Uitto, J.; Bernstein, E.F. Molecular mechanisms of cutaneous aging: Connective tissue alterations in the dermis. J. Investig. Dermatol. Symp. Proc 1998, 3, 41–44. [Google Scholar]
- Krutmann, J. Ultraviolet A radiation-induced biological effects in human skin: Relevance for photoaging and photodermatosis. J. Dermatol. Sci 2000, 23, S22–S26. [Google Scholar]
- Wondrak, G.T.; Jacobson, M.K.; Jacobson, E.L. Endogenous UVA-photosensitizers: Mediators of skin photodamage and novel targets for skin photoprotection. Photochem. Photobiol. Sci 2006, 5, 215–237. [Google Scholar]
- Cadet, J.; Mouret, S.P.; Ravanat, J.-L.; Douki, T. Photoinduced Damage to Cellular DNA: Direct and Photosensitized Reactions. Photochem. Photobiol 2012, 88, 1048–1065. [Google Scholar]
- McMillan, T.J.; Leatherman, E.; Ridley, A.; Shorrocks, J.; Tobi, S.E.; Whiteside, J.R. Cellular effects of long wavelength UV light (UVA) in mammalian cells. J. Pharm. Pharmacol 2008, 60, 969–976. [Google Scholar]
- Morliere, P.; Moysan, A.; Tirache, I. Action spectrum for UV-induced lipid peroxidation in cultured human skin fibroblasts. Free Radic. Biol. Med 1995, 19, 365–371. [Google Scholar]
- Aroun, A.; Zhong, J.L.; Tyrrell, R.M.; Pourzand, C. Iron, oxidative stress and the example of solar ultraviolet A radiation. Photochem. Photobiol. Sci 2012, 11, 118–134. [Google Scholar] [Green Version]
- Suschek, C.V.; Paunel, A.; Kolb-Bachofen, V. Nonenzymatic nitric oxide formation during UVA irradiation of human skin: Experimental setups and ways to measure. Methods Enzymol 2005, 396, 568–578. [Google Scholar]
- Suschek, C.V.; Schroeder, P.; Aust, O.; Sies, H.; Mahotka, C.; Horstjann, M.; Ganser, H.; Murtz, M.; Hering, P.; Schnorr, O.; et al. The presence of nitrite during UVA irradiation protects from apoptosis. FASEB J 2003, 17, 2342–2344. [Google Scholar]
- Suschek, C.V.; Briviba, K.; Bruch-Gerharz, D.; Sies, H.; Kroncke, K.D.; Kolb-Bachofen, V. Even after UVA-exposure will nitric oxide protect cells from reactive oxygen intermediate-mediated apoptosis and necrosis. Cell Death Differ 2001, 8, 515–527. [Google Scholar]
- Suschek, C.V.; Krischel, V.; Bruch-Gerharz, D.; Berendji, D.; Krutmann, J.; Kroncke, K.D.; Kolb-Bachofen, V. Nitric oxide fully protects against UVA-induced apoptosis in tight correlation with Bcl-2 up-regulation. J. Biol. Chem 1999, 274, 6130–6137. [Google Scholar]
- Suschek, C.V.; Oplander, C.; van Faassen, E.E. Non-enzymatic NO production in human skin: Effect of UVA on cutaneous NO stores. Nitric Oxide 2010, 22, 120–135. [Google Scholar]
- Hogg, N.; Kalyanaraman, B. Nitric oxide and lipid peroxidation. Biochim. Biophys. Acta 1999, 1411, 378–384. [Google Scholar]
- Kroncke, K.D.; Fehsel, K.; Suschek, C.; Kolb-Bachofen, V. Inducible nitric oxide synthase-derived nitric oxide in gene regulation, cell death and cell survival. Int. Immunopharmacol 2001, 1, 1407–1420. [Google Scholar]
- Kroncke, K.D.; Klotz, L.O.; Suschek, C.V.; Sies, H. Comparing nitrosative versus oxidative stress toward zinc finger-dependent transcription. Unique role for NO. Nitric Oxide 2002, 277, 13294–13301. [Google Scholar]
- Kroncke, K.D.; Suschek, C.V.; Kolb-Bachofen, V. Implications of inducible nitric oxide synthase expression and enzyme activity. Antioxid. Redox Signal 2000, 2, 585–605. [Google Scholar]
- Snyder, S.H. Nitric oxide: First in a new class of neurotransmitters. Science 1992, 257, 494–496. [Google Scholar]
- Forstermann, U.; Closs, E.I.; Pollock, J.S.; Nakane, M.; Schwarz, P.; Gath, I.; Kleinert, H. Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 1994, 23, 1121–1131. [Google Scholar]
- Peunova, N.; Enikolopov, G. Amplification of calcium-induced gene transcription by nitric oxide in neuronal cells. Nature 1993, 364, 450–453. [Google Scholar]
- Weiss, G.; Goossen, B.; Doppler, W.; Fuchs, D.; Pantopoulos, K.; Werner-Felmayer, G.; Wachter, H.; Hentze, M.W. Translational regulation via iron-responsive elements by the nitric oxide/NO-synthase pathway. EMBO J 1993, 12, 3651–3657. [Google Scholar]
- Brune, B.; Dimmeler, S.; Molina y Vedia, L.; Lapetina, E.G. Nitric oxide: A signal for ADP-ribosylation of proteins. Life Sci 1994, 54, 61–70. [Google Scholar]
- Nathan, C.F.; Hibbs, J.B., Jr. Role of nitric oxide synthesis in macrophage antimicrobial activity. Curr. Opin. Immunol. 1991, 3, 65–70. [Google Scholar]
- Wink, D.A.; Kasprzak, K.S.; Maragos, C.M.; Elespuru, R.K.; Misra, M.; Dunams, T.M.; Cebula, T.A.; Koch, W.H.; Andrews, A.W.; Allen, J.S.; et al. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science 1991, 254, 1001–1003. [Google Scholar]
- Frank, S.; Kampfer, H.; Wetzler, C.; Pfeilschifter, J. Nitric oxide drives skin repair: Novel functions of an established mediator. Kidney Int. 2002, 61, 882–888. [Google Scholar]
- Krischel, V.; Bruch-Gerharz, D.; Suschek, C.; Kroncke, K.D.; Ruzicka, T.; Kolb-Bachofen, V. Biphasic effect of exogenous nitric oxide on proliferation and differentiation in skin derived keratinocytes but not fibroblasts. J. Invest. Dermatol 1998, 111, 286–291. [Google Scholar]
- Bruch-Gerharz, D.; Ruzicka, T.; Kolb-Bachofen, V. Nitric oxide in human skin: Current status and future prospects. J. Invest. Dermatol 1998, 110, 1–7. [Google Scholar]
- Ehrt, S.; Schnappinger, D.; Bekiranov, S.; Drenkow, J.; Shi, S.; Gingeras, T.R.; Gaasterland, T.; Schoolnik, G.; Nathan, C. Reprogramming of the macrophage transcriptome in response to interferon-gamma and Mycobacterium tuberculosis: Signaling roles of nitric oxide synthase-2 and phagocyte oxidase. J. Exp. Med 2001, 194, 1123–1140. [Google Scholar]
- Hemish, J.; Nakaya, N.; Mittal, V.; Enikolopov, G. Nitric oxide activates diverse signaling pathways to regulate gene expression. J. Biol. Chem 2003, 278, 42321–42329. [Google Scholar]
- Niziolek, M.; Korytowski, W.; Girotti, A.W. Nitric oxide-induced resistance to lethal photooxidative damage in a breast tumor cell line. Free Radic. Biol. Med 2006, 40, 1323–1331. [Google Scholar]
- Moncada, S.; Higgs, A. The L-arginine-nitric oxide pathway. N. Engl. J. Med 1993, 329, 2002–2012. [Google Scholar]
- Warren, J.B. Nitric oxide and human skin blood flow responses to acetylcholine and ultraviolet light. FASEB J 1994, 8, 247–251. [Google Scholar]
- Weller, R.; Dykhuizen, R.; Leifert, C.; Ormerod, A. Nitric oxide release accounts for the reduced incidence of cutaneous infections in psoriasis. J. Am. Acad. Dermatol 1997, 36, 281–282. [Google Scholar]
- Cals-Grierson, M.M.; Ormerod, A.D. Nitric oxide function in the skin. Nitric Oxide 2004, 10, 179–193. [Google Scholar]
- Suschek, C.V.; Bruch-Gerharz, D.; Kleinert, H.; Forstermann, U.; Kolb-Bachofen, V. Ultraviolet A1 radiation induces nitric oxide synthase-2 expression in human skin endothelial cells in the absence of proinflammatory cytokines. J. Invest. Dermatol 2001, 117, 1200–1205. [Google Scholar]
- Paunel, A.N.; Dejam, A.; Thelen, S.; Kirsch, M.; Horstjann, M.; Gharini, P.; Murtz, M.; Kelm, M.; de Groot, H.; Kolb-Bachofen, V.; et al. Enzyme-independent nitric oxide formation during UVA challenge of human skin: Characterization, molecular sources, and mechanisms. Free Radic. Biol. Med 2005, 38, 606–615. [Google Scholar]
- Ehrreich, S.J.; Furchgott, R.F. Relaxation of mammalian smooth muscles by visible and ultraviolet radiation. Nature 1968, 218, 682–684. [Google Scholar]
- Fischer, M.; Warneck, P. Photodecomposition of nitrite and undissociated nitrous acid in aqueous solution. J. Phys. Chem 1996, 100, 18749–18756. [Google Scholar]
- Zhelyaskov, V.R.; Gee, K.R.; Godwin, D.W. Control of NO concentration in solutions of nitrosothiol compounds by light. Photochem. Photobiol 1998, 67, 282–288. [Google Scholar]
- Dejam, A.; Kleinbongard, P.; Rassaf, T.; Hamada, S.; Gharini, P.; Rodriguez, J.; Feelisch, M.; Kelm, M. Thiols enhance NO formation from nitrate photolysis. Free Radic. Biol. Med 2003, 35, 1551–1559. [Google Scholar]
- Mowbray, M.; McLintock, S.; Weerakoon, R.; Lomatschinsky, N.; Jones, S.; Rossi, A.G.; Weller, R.B. Enzyme-independent NO stores in human skin: quantification and influence of UV radiation. J. Invest. Dermatol 2009, 129, 834–842. [Google Scholar]
- Oplander, C.; Cortese, M.M.; Korth, H.-G.; Kirsch, M.; Mahotka, C.; Wetzel, W.; Pallua, N.; Suschek, C.V. The impact of nitrite and antioxidants on ultraviolet-A-induced cell death of human skin fibroblasts. Free Radic. Biol. Med 2007, 43, 818–829. [Google Scholar]
- Kirsch, M.; Korth, H.G.; Sustmann, R.; de Groot, H. The pathobiochemistry of nitrogen dioxide. Biol. Chem 2002, 383, 389–399. [Google Scholar]
- Borisenko, G.G.; Martin, I.; Zhao, Q.; Amoscato, A.A.; Tyurina, Y.Y.; Kagan, V.E. Glutathione propagates oxidative stress triggered by myeloperoxidase in HL-60 cells. Evidence for glutathionyl radical-induced peroxidation of phospholipids and cytotoxicity. J. Biol. Chem 2004, 279, 23453–23462. [Google Scholar]
- Volkmar, C.M.; Vukadinovic-Walter, B.; Oplander, C.; Bozkurt, A.; Korth, H.G.; Kirsch, M.; Mahotka, C.; Pallua, N.; Suschek, C.V. UVA-induced phenoxyl radical formation: A new cytotoxic principle in photodynamic therapy. Free Radic. Biol. Med 2010, 49, 1129–1137. [Google Scholar]
- Alessi, M.; Paul, T.; Scaiano, J.C.; Ingold, K.U. The contrasting kinetics of peroxidation of vitamin E-containing phospholipid unilamellar vesicles and human low-density lipoprotein. J. Am. Chem. Soc 2002, 124, 6957–6965. [Google Scholar]
- Oplander, C.; Wetzel, W.; Cortese, M.M.; Pallua, N.; Suschek, C.V. Evidence for a physiological role of intracellularly occurring photolabile nitrogen oxides in human skin fibroblasts. Free Radic. Biol. Med 2008, 44, 1752–1761. [Google Scholar]
- Sarkar, S.; Korolchuk, V.I.; Renna, M.; Imarisio, S.; Fleming, A.; Williams, A.; Garcia-Arencibia, M.; Rose, C.; Luo, S.; Underwood, B.R.; et al. Complex inhibitory effects of nitric oxide on autophagy. Mol. Cell 2011, 43, 19–32. [Google Scholar]
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Opländer, C.; Suschek, C.V. The Role of Photolabile Dermal Nitric Oxide Derivates in Ultraviolet Radiation (UVR)-Induced Cell Death. Int. J. Mol. Sci. 2013, 14, 191-204. https://doi.org/10.3390/ijms14010191
Opländer C, Suschek CV. The Role of Photolabile Dermal Nitric Oxide Derivates in Ultraviolet Radiation (UVR)-Induced Cell Death. International Journal of Molecular Sciences. 2013; 14(1):191-204. https://doi.org/10.3390/ijms14010191
Chicago/Turabian StyleOpländer, Christian, and Christoph V. Suschek. 2013. "The Role of Photolabile Dermal Nitric Oxide Derivates in Ultraviolet Radiation (UVR)-Induced Cell Death" International Journal of Molecular Sciences 14, no. 1: 191-204. https://doi.org/10.3390/ijms14010191
APA StyleOpländer, C., & Suschek, C. V. (2013). The Role of Photolabile Dermal Nitric Oxide Derivates in Ultraviolet Radiation (UVR)-Induced Cell Death. International Journal of Molecular Sciences, 14(1), 191-204. https://doi.org/10.3390/ijms14010191