Gamma-Aminobutyric Acid Production Using Immobilized Glutamate Decarboxylase Followed by Downstream Processing with Cation Exchange Chromatography

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
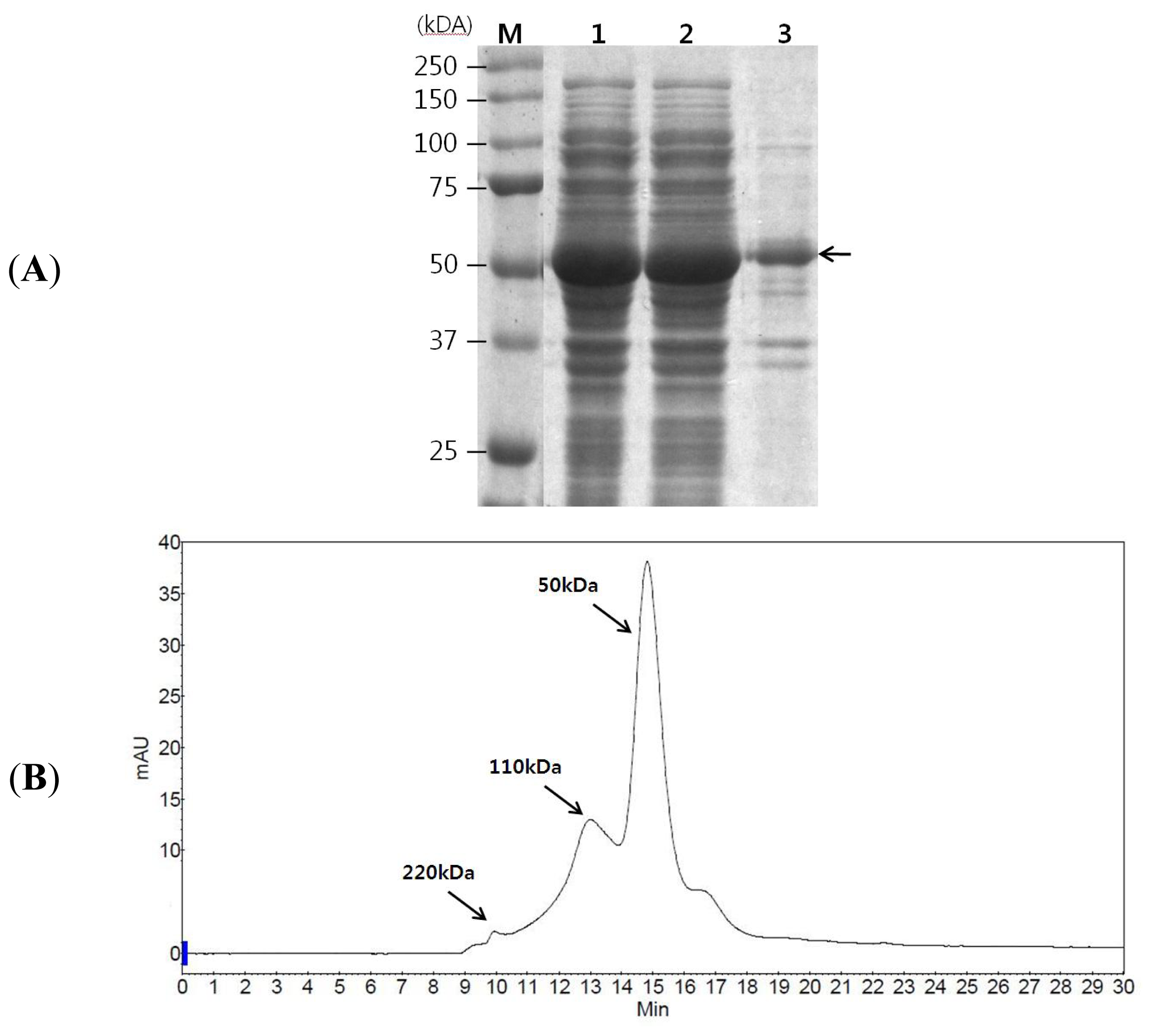
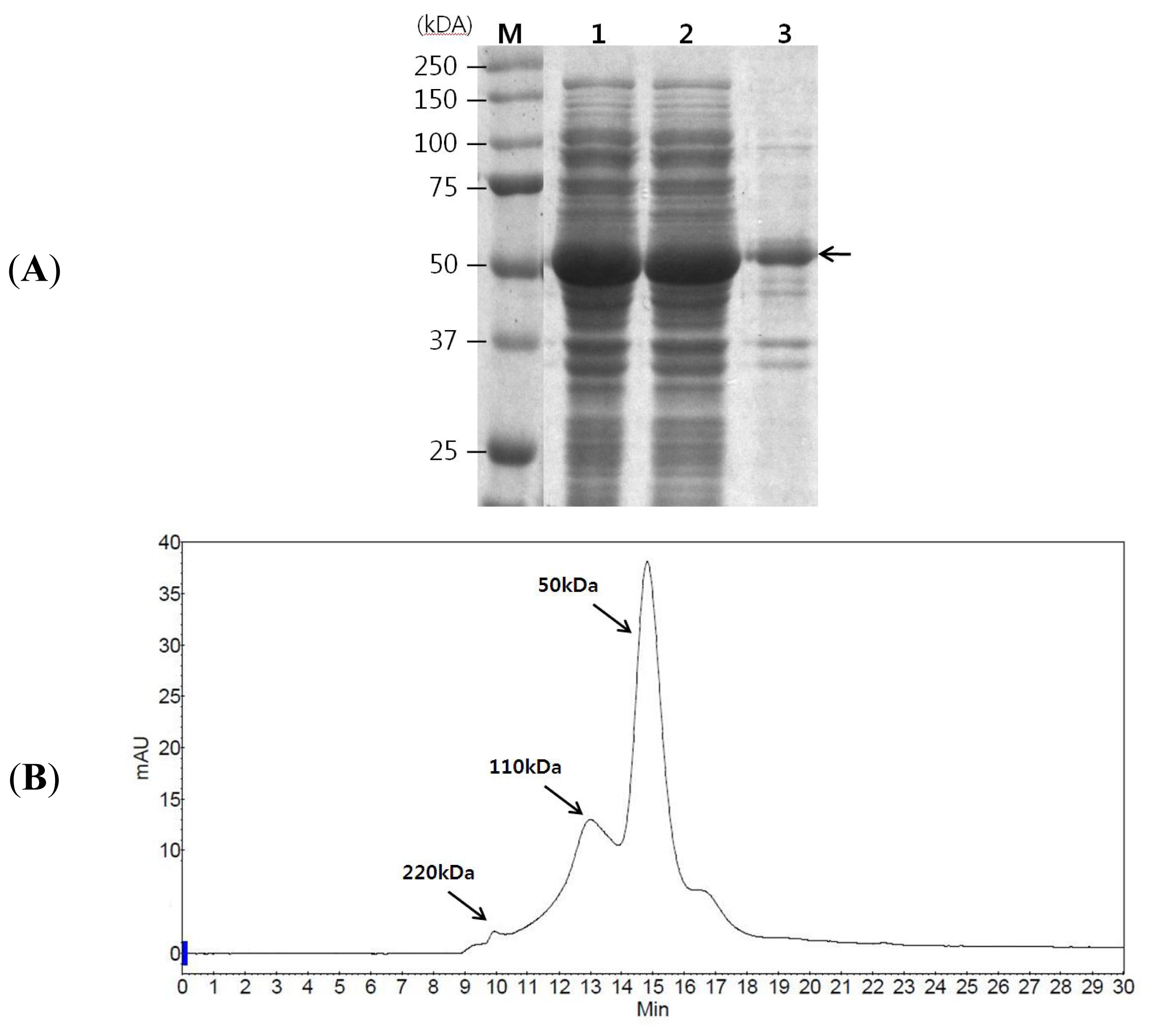
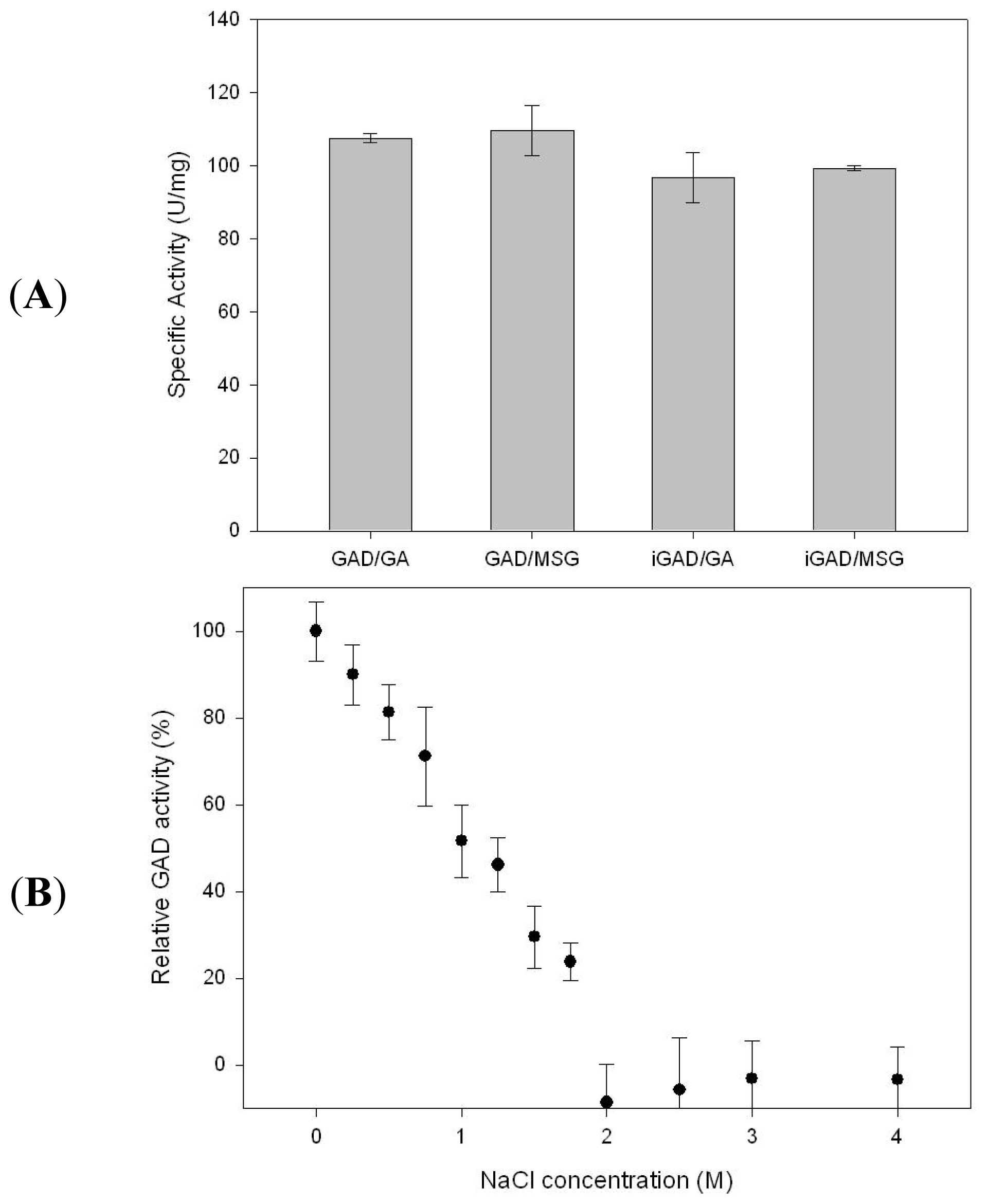
2.1. GAD Expression and Its Characterization
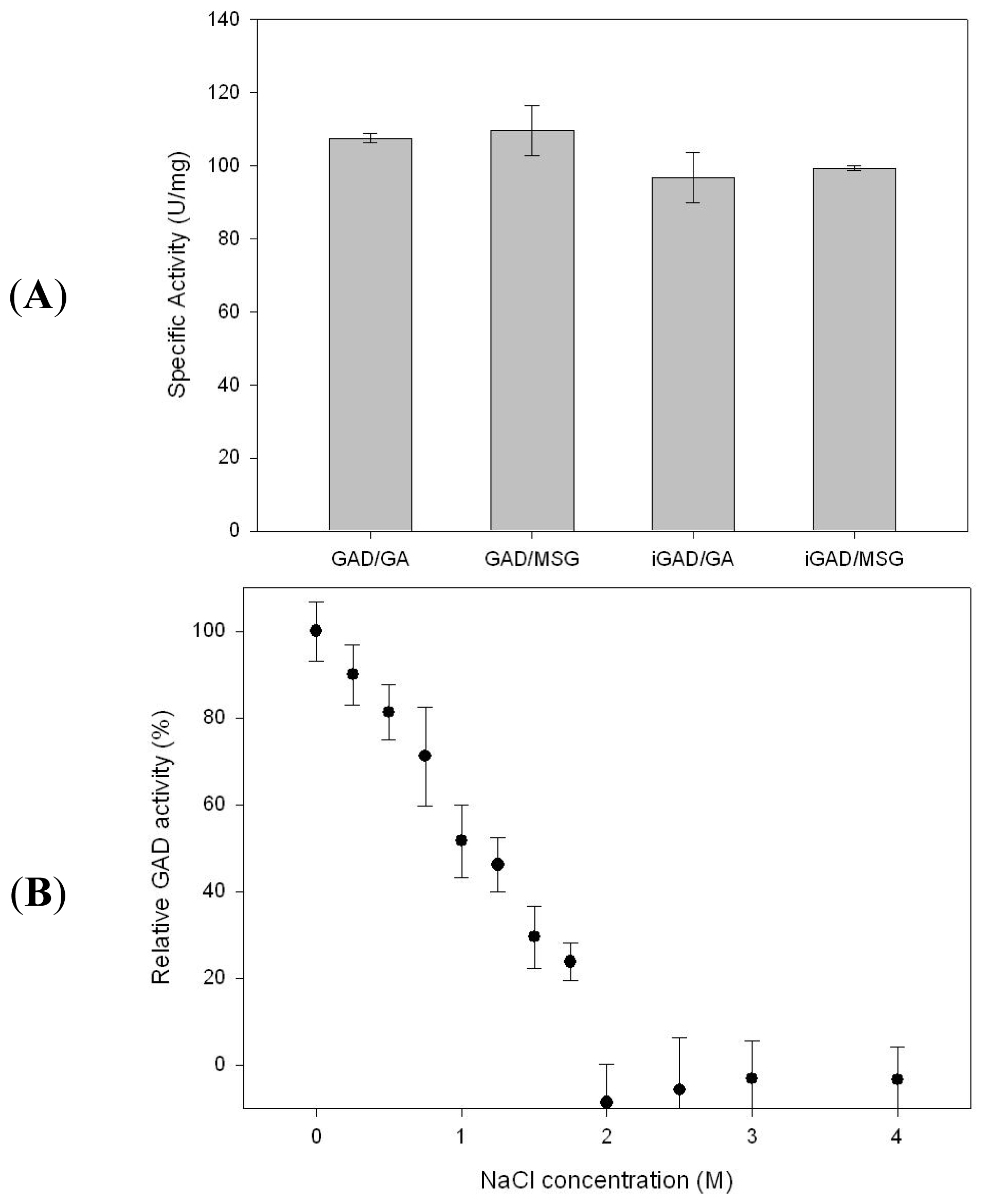
2.2. GAD Immobilization and Substrate Selection
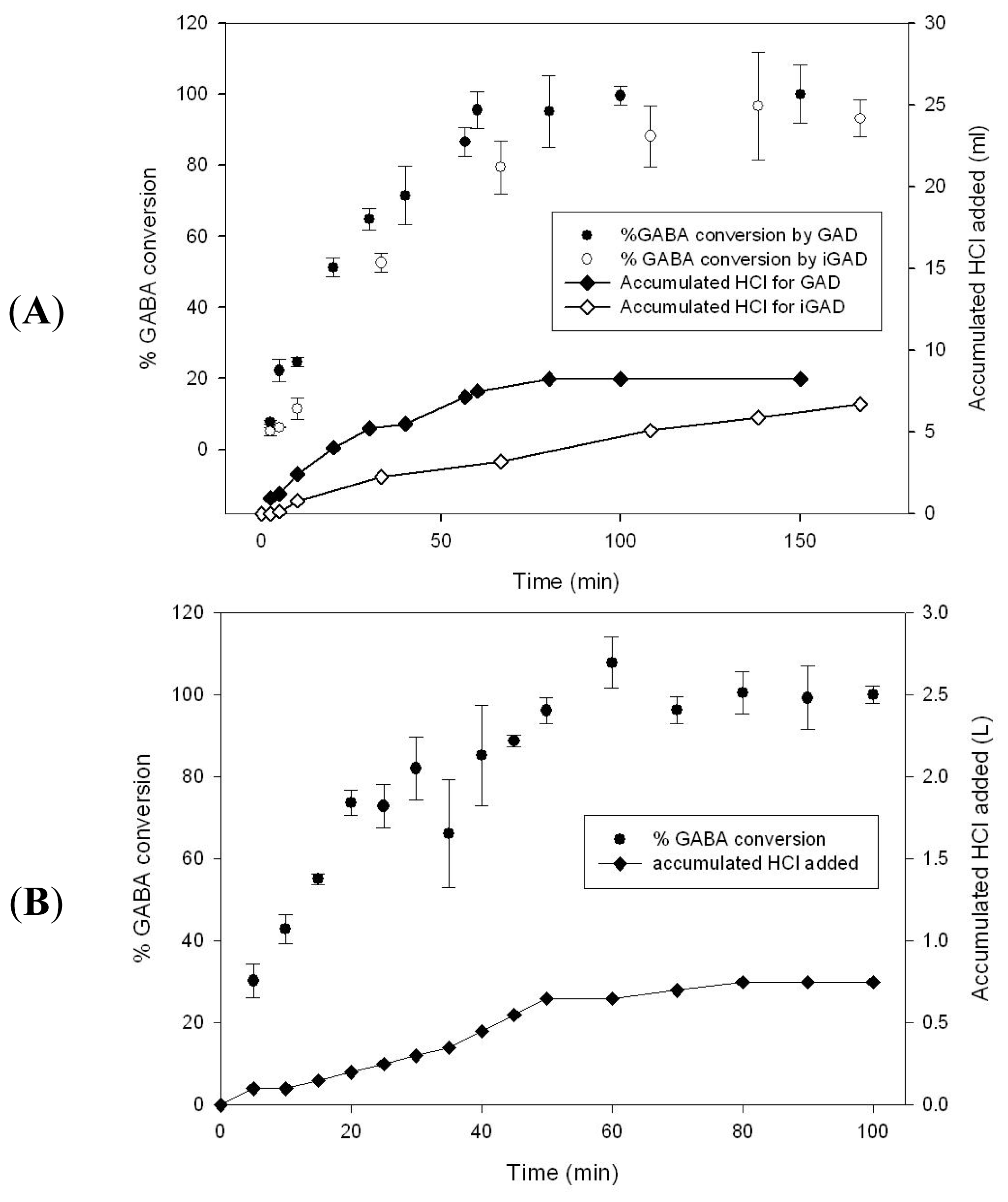
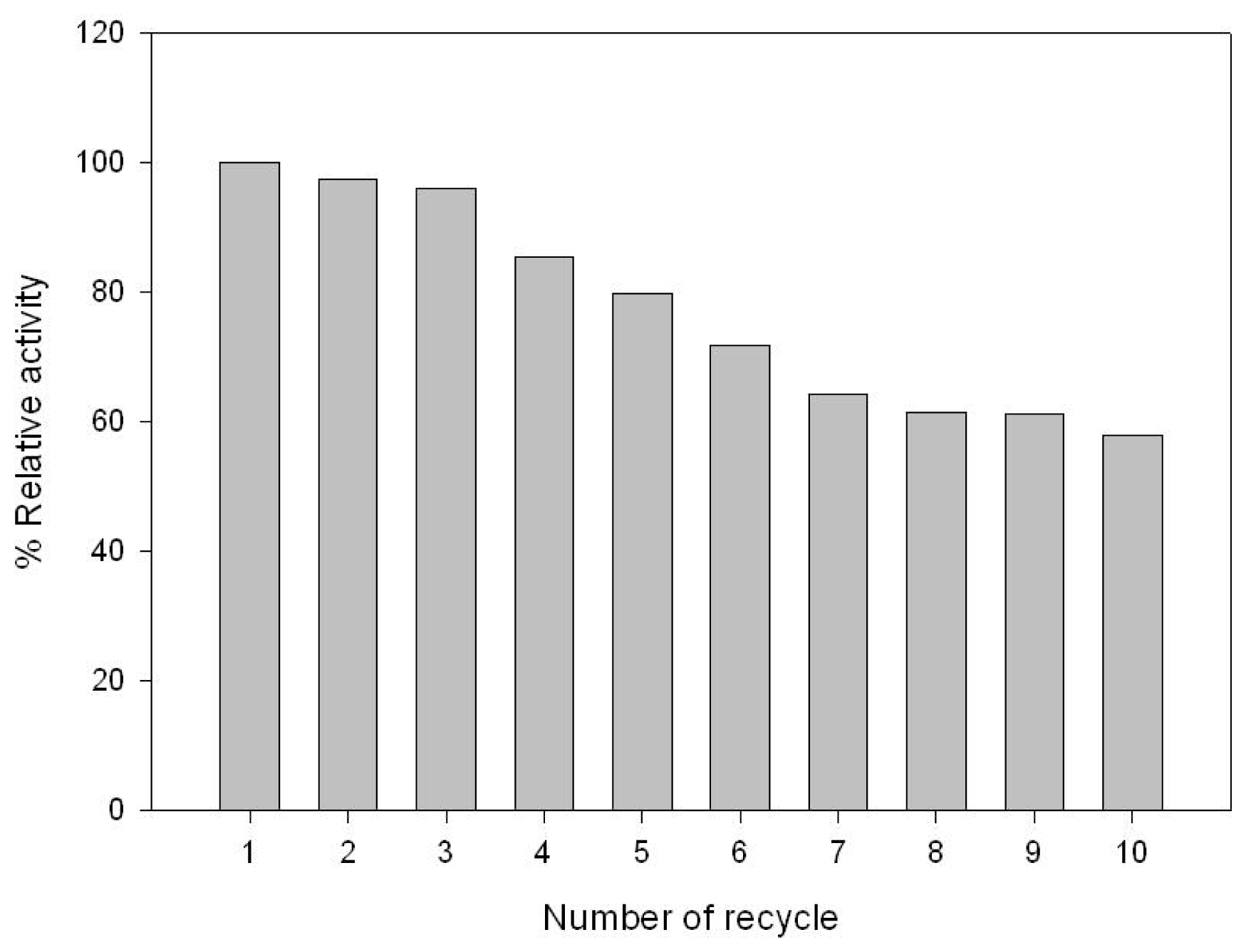
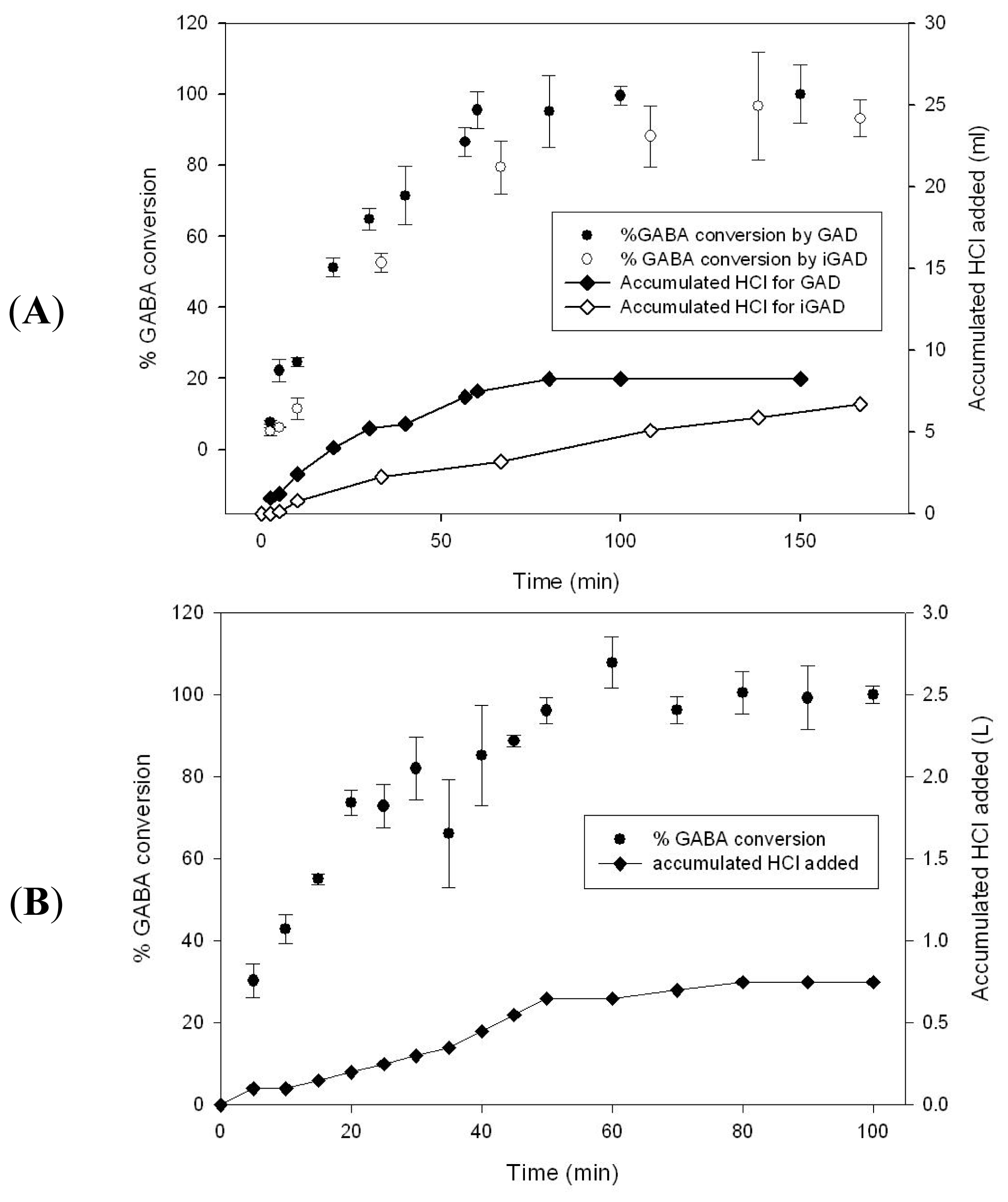
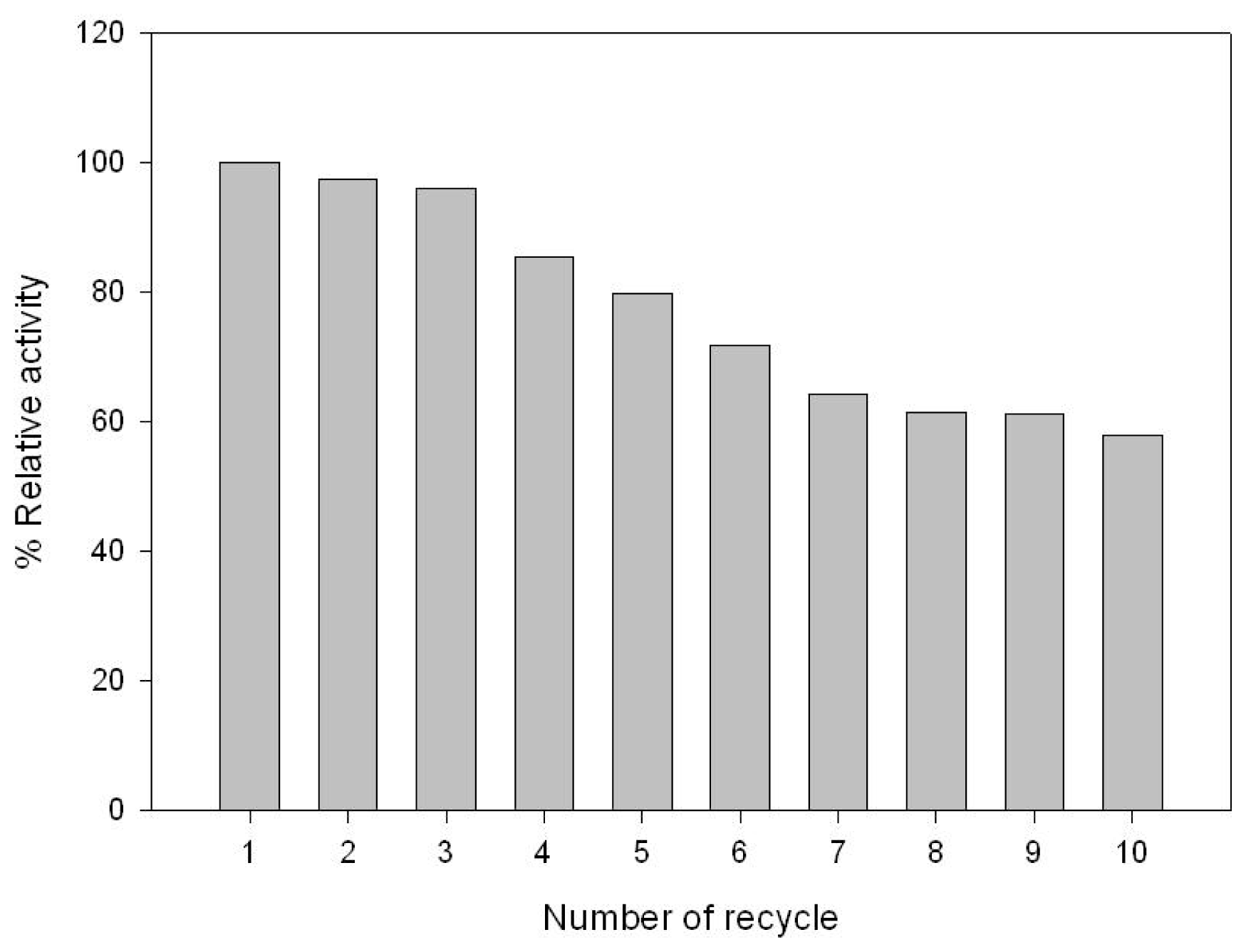
2.3. GABA Conversion and GAD Recycling
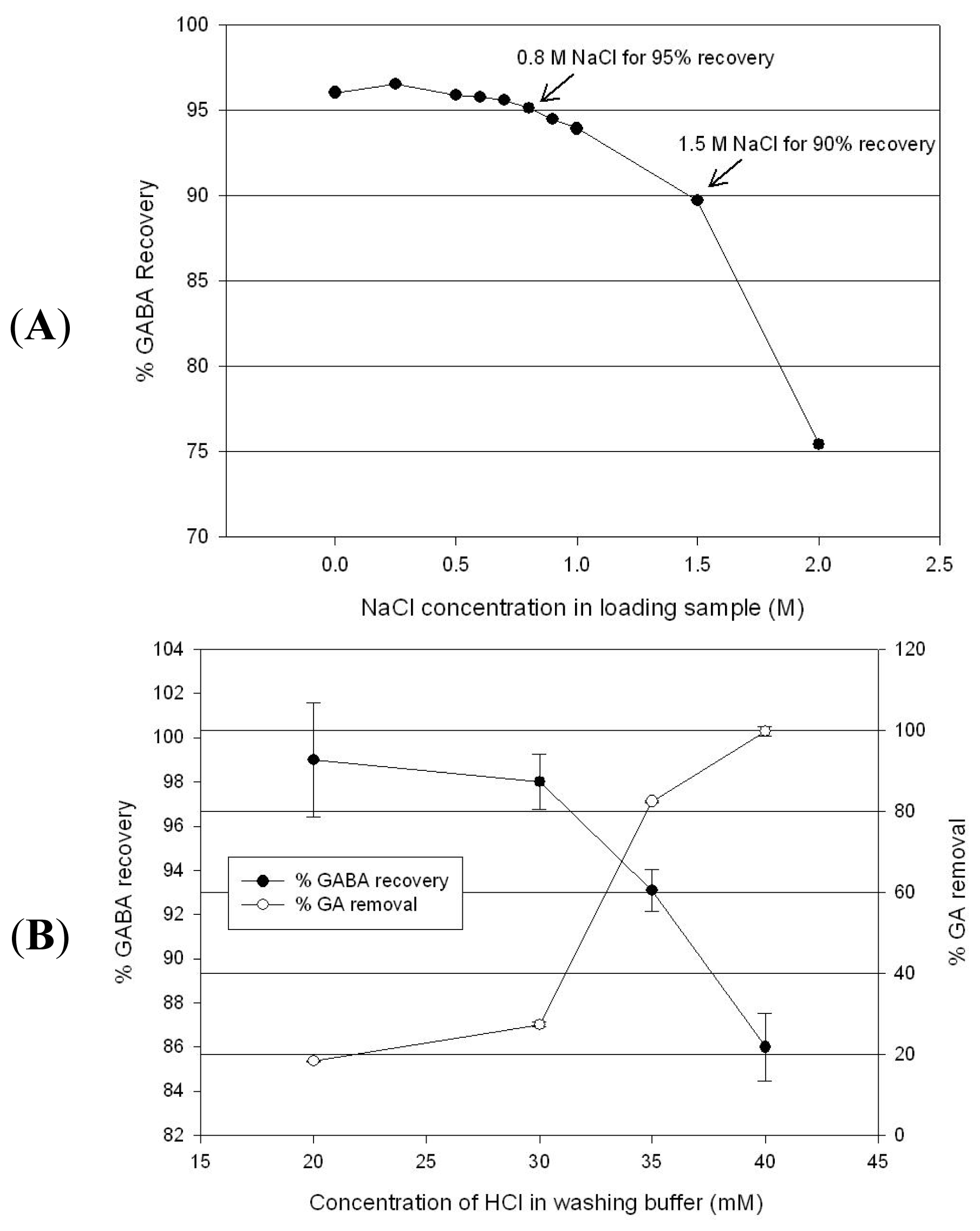
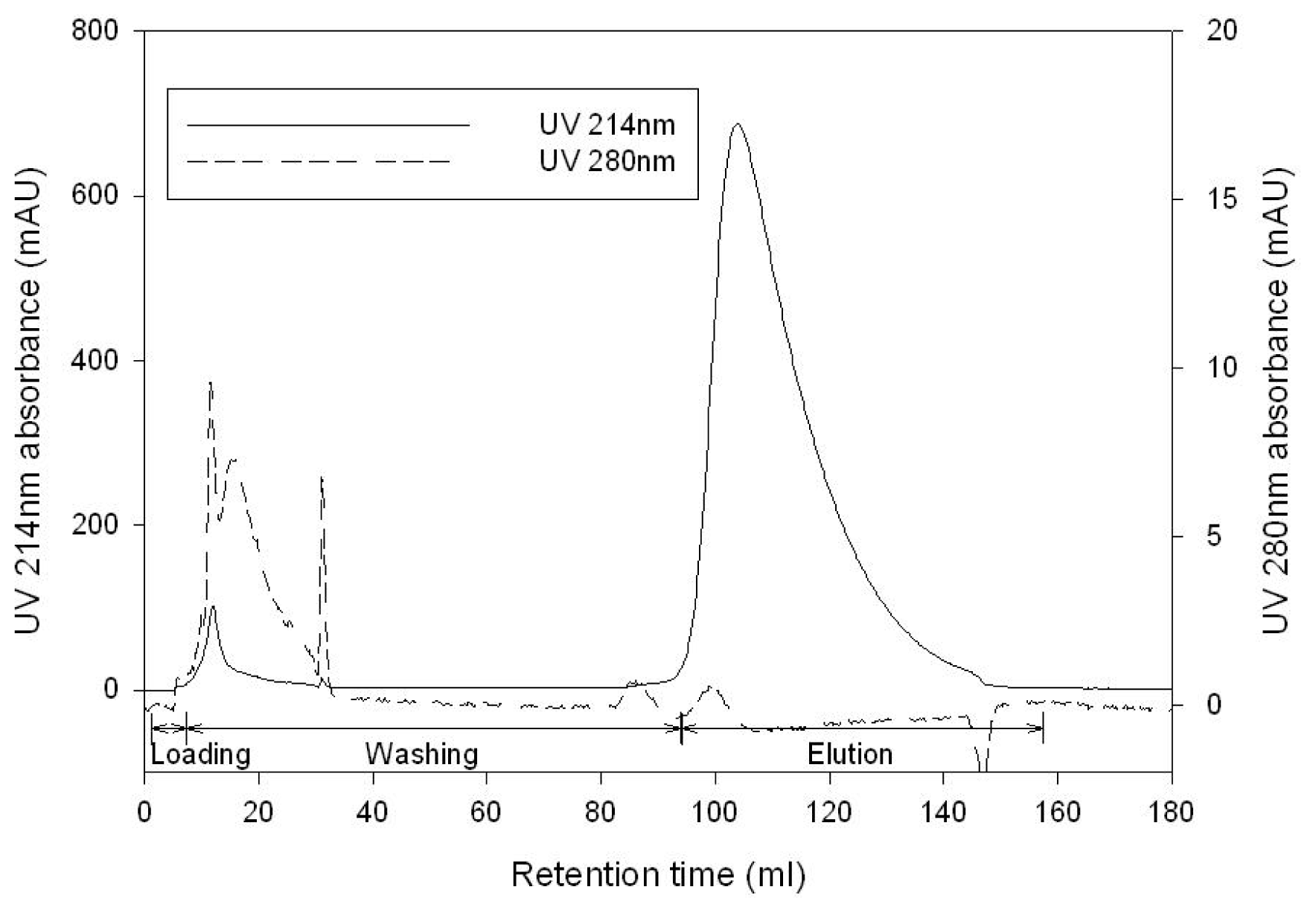
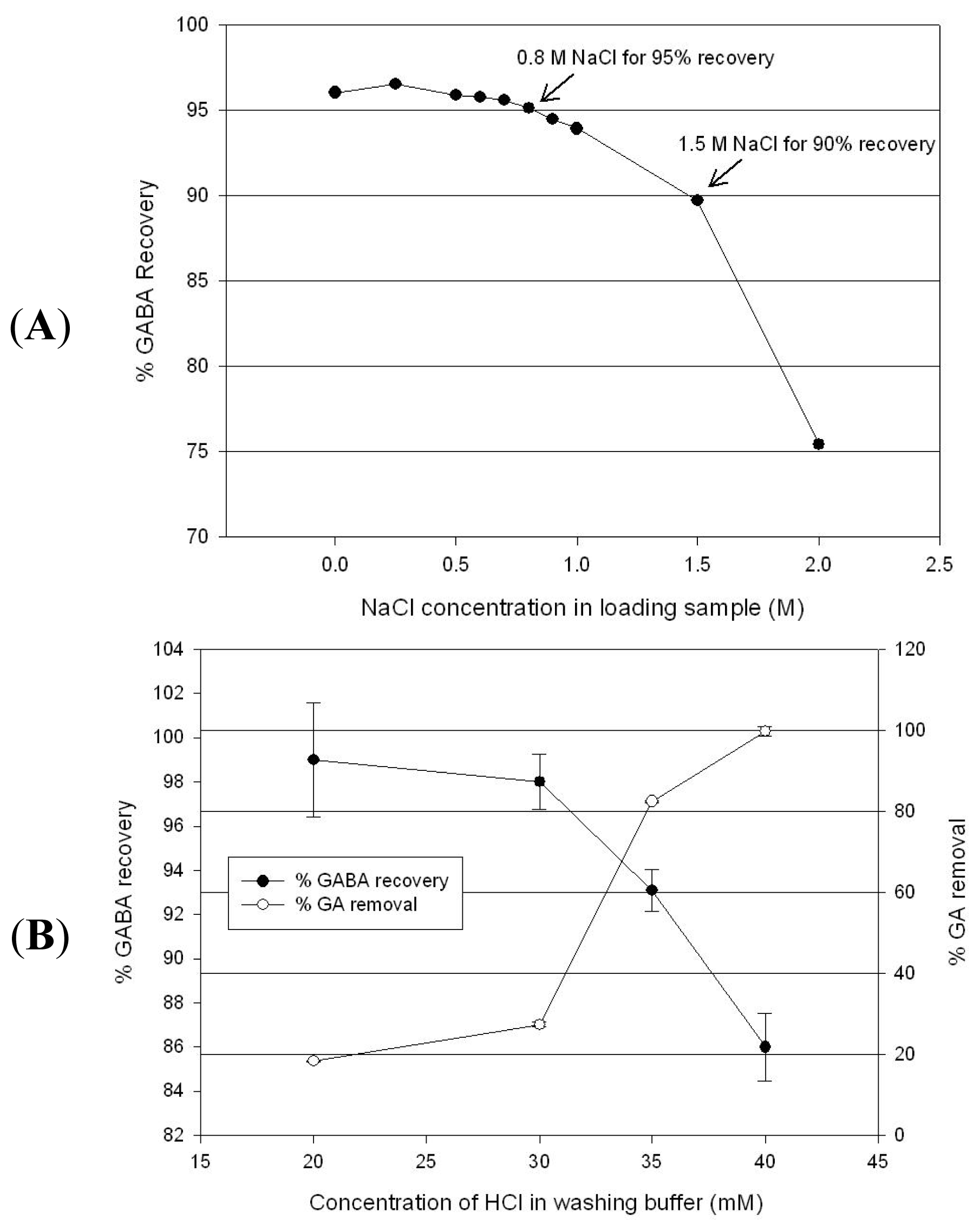
2.4. GABA Purification by Cation Exchange Chromatography
3. Experimental Section
3.1. Bacterial Strains and Plasmid Construction
3.2. GAD Expression and Purification
3.3. Immobilization
3.4. GABA Conversion and GAD Recycling
3.5. Cation Exchange Chromatography
3.6. Analysis of GABA, Glutamate and GAD Concentrations
3.7. Characterization of GAD
4. Conclusions
Acknowledgments
References
- Bhargava, K.P.; Bhattacharya, S.S.; Srimal, R.C. Central cardiovascular actions of GABA. Br. J. Pharmacol 1964, 23, 383–390. [Google Scholar]
- Erlander, M.; Tobin, A. The structural and functional heterogeneity of glutamic acid decarboxylase: A review. Neurochem. Res 1991, 16, 215–226. [Google Scholar]
- Fenalti, G.; Law, R.H.; Buckle, A.M.; Langendorf, C.; Tuck, K.; Rosado, C.J.; Faux, N.G.; Mahmood, K.; Hampe, C.S.; Banga, J.P.; et al. GABA production by glutamic acid decarboxylase is regulated by a dynamic catalytic loop. Nat. Struct. Mol. Biol 2007, 14, 280–286. [Google Scholar]
- Shelp, B.J.; Bown, A.W.; McLean, M.D. Metabolism and functions of gamma-aminobutyric acid. Trends Plant Sci 1999, 4, 446–452. [Google Scholar]
- Lammens, T.M.; de Biase, D.; Franssen, M.C.R.; Scott, E.L.; Sanders, J.P.M. The application of glutamic acid α-decarboxylase for the valorization of glutamic acid. Green Chem 2009, 11, 1562. [Google Scholar]
- Yamano, N.; Kawasaki, N.; Takeda, S.; Nakayama, A. Production of 2-pyrrolidone from biobased glutamate by using. Escherichia coli. J. Polym. Environ. 2012. Available online: http://link.springer.com/article/10.1007/s10924-012-0466-x# accessed on accessed on 15 September 2012. [CrossRef]
- MetaCyc. Available online: http://biocyc.org/META/NEW-IMAGE?type=REACTION&object=GLUTDECARBOX-RXN&redirect=T accessed on 15 September 2012.
- Hiraga, K.; Ueno, Y.; Oda, K. Glutamate decarboxylase from Lactobacillus brevis: Activation by ammonium sulfate. Biosci. Biotechnol. Biochem 2008, 72, 1299–1306. [Google Scholar]
- Komatsuzaki, N.; Nakamura, T.; Kimura, T.; Shima, J. Characterization of glutamate decarboxylase from a high γ-aminobutyric acid (GABA)-producer, Lactobacillus paracasei. Biosci. Biotechnol. Biochem 2008, 72, 278–285. [Google Scholar]
- Komatsuzaki, N.; Shima, J.; Kawamoto, S.; Momose, H.; Kimura, T. Production of γ-aminobutyric acid (GABA) by Lactobacillus paracasei isolated from traditional fermented foods. Food Microbiol 2005, 22, 497–504. [Google Scholar]
- Li, H.; Cao, Y. Lactic acid bacterial cell factories for gamma-aminobutyric acid. Amino Acids 2010, 39, 1107–1116. [Google Scholar]
- Li, H.; Cao, Y.; Cao, Y.; Xu, H. A high γ-aminobutyric acid-producing Lactobacillus brevis isolated from Chinese traditional paocai. Ann. Microbiol 2008, 58, 649–653. [Google Scholar]
- Li, H.; Qiu, T.; Chen, Y.; Cao, Y. Separation of gamma-aminobutyric acid from fermented broth. J. Ind. Microbiol. Biotechnol 2011, 38, 1955–1959. [Google Scholar]
- Ueno, Y.; Hayakawa, K.; Takahashi, S.; Oda, K. Purification and characterisation of glutamate decarboxylase from Lactobacillus brevis IFO 12005. Biosci. Biotechnol. Biochem 1997, 61, 1168–1171. [Google Scholar]
- Jiang, D.; Ji, H.; Ye, Y.; Hou, J. Studies on screening of higher γ-aminobutyric acid-producing Monascus and optimization of fermentative parameters. Eur. Food Res. Technol 2011, 232, 541–547. [Google Scholar]
- Lee, C.L.; Pan, T.M. Development of Monascus fermentation technology for high hypolipidemic effect. Appl. Microbiol. Biotechnol 2012, 94, 1449–1459. [Google Scholar]
- Su, Y.C.; Wang, J.J.; Lin, T.T.; Pan, T.M. Production of the secondary metabolites gamma-aminobutyric acid and monacolin K by Monascus. J. Ind. Microbiol. Biotechnol 2003, 30, 41–46. [Google Scholar]
- Wang, J.J.; Lee, C.L.; Pan, T.M. Improvement of monacolin K, gamma-aminobutyric acid and citrinin production ratio as a function of environmental conditions of Monascus purpureus NTU 601. J. Ind. Microbiol. Biotechnol 2003, 30, 669–676. [Google Scholar]
- Le Vo, T.D.; Kim, T.W.; Hong, S.H. Effects of glutamate decarboxylase and gamma-aminobutyric acid (GABA) transporter on the bioconversion of GABA in engineered Escherichia coli. Bioprocess Biosyst. Eng 2012, 35, 645–650. [Google Scholar]
- Plokhov, A.Y.; Gusyatiner, M.M.; Yampolskaya, T.A.; Kaluzhsky, V.E.; Sukhareva, B.S.; Schulga, A.A. Preparation of γ-aminobutyric acid using E. coli cells with high activity of glutamate decarboxylase. Appl. Biochem. Biotechnol 2000, 88, 257–265. [Google Scholar]
- Park, H.; Ahn, J.; Lee, J.; Lee, H.; Kim, C.; Jung, J.K.; Lee, E.G. Expression, immobilization and enzymatic properties of glutamate decarboxylase fused to a cellulose-binding domain. Int. J. Mol. Sci 2012, 13, 358–368. [Google Scholar]
- Lee, E.G.; Baek, J.-E.; Lee, S.-H.; Kim, T.-W.; Choi, J.H.; Rho, M.-C.; Ahn, J.-O.; Lee, H.-W.; Jung, J.-K. Efficient proteolytic cleavage by insertion of oligopeptide linkers and its application to production of recombinant human interleukin-6 in Escherichia coli. Enzyme Microb. Technol 2009, 44, 254–262. [Google Scholar]
- Capitani, G.; de Biase, D.; Aurizi, C.; Gut, H.; Bossa, F.; Grutter, M.G. Crystal structure and functional analysis of Escherichia coli glutamate decarboxylase. EMBO J 2003, 22, 4027–4037. [Google Scholar]
- Fonda, M.L. L-Glutamate decarboxylase from bacteria. Meth. Enzymol 1985, 113, 11–16. [Google Scholar]
- Nakanishi, K.; Sakiyama, T.; Kumada, Y.; Imamura, K.; Imanaka, H. Recent advances in controlled immobilization of proteins onto the surface of the solid substrate and its possible application to proteomics. Curr. Proteomics 2008, 5, 161–175. [Google Scholar]
- Wang, Q.; Xin, Y.; Zhang, F.; Feng, Z.; Fu, J.; Luo, L.; Yin, Z. Enhanced γ-aminobutyric acid-forming activity of recombinant glutamate decarboxylase (gadA) from Escherichia coli. World J. Microbiol. Biotechnol 2011, 27, 693–700. [Google Scholar]

© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Lee, S.; Ahn, J.; Kim, Y.-G.; Jung, J.-K.; Lee, H.; Lee, E.G. Gamma-Aminobutyric Acid Production Using Immobilized Glutamate Decarboxylase Followed by Downstream Processing with Cation Exchange Chromatography. Int. J. Mol. Sci. 2013, 14, 1728-1739. https://doi.org/10.3390/ijms14011728
Lee S, Ahn J, Kim Y-G, Jung J-K, Lee H, Lee EG. Gamma-Aminobutyric Acid Production Using Immobilized Glutamate Decarboxylase Followed by Downstream Processing with Cation Exchange Chromatography. International Journal of Molecular Sciences. 2013; 14(1):1728-1739. https://doi.org/10.3390/ijms14011728
Chicago/Turabian StyleLee, Seungwoon, Jungoh Ahn, Yeon-Gu Kim, Joon-Ki Jung, Hongweon Lee, and Eun Gyo Lee. 2013. "Gamma-Aminobutyric Acid Production Using Immobilized Glutamate Decarboxylase Followed by Downstream Processing with Cation Exchange Chromatography" International Journal of Molecular Sciences 14, no. 1: 1728-1739. https://doi.org/10.3390/ijms14011728
APA StyleLee, S., Ahn, J., Kim, Y.-G., Jung, J.-K., Lee, H., & Lee, E. G. (2013). Gamma-Aminobutyric Acid Production Using Immobilized Glutamate Decarboxylase Followed by Downstream Processing with Cation Exchange Chromatography. International Journal of Molecular Sciences, 14(1), 1728-1739. https://doi.org/10.3390/ijms14011728