Potential Applications of Carbohydrases Immobilization in the Food Industry


 ,
,
Abstract
:1. Introduction
2. Enzyme Immobilization Techniques
3. Immobilized Enzymes in Syrup Production
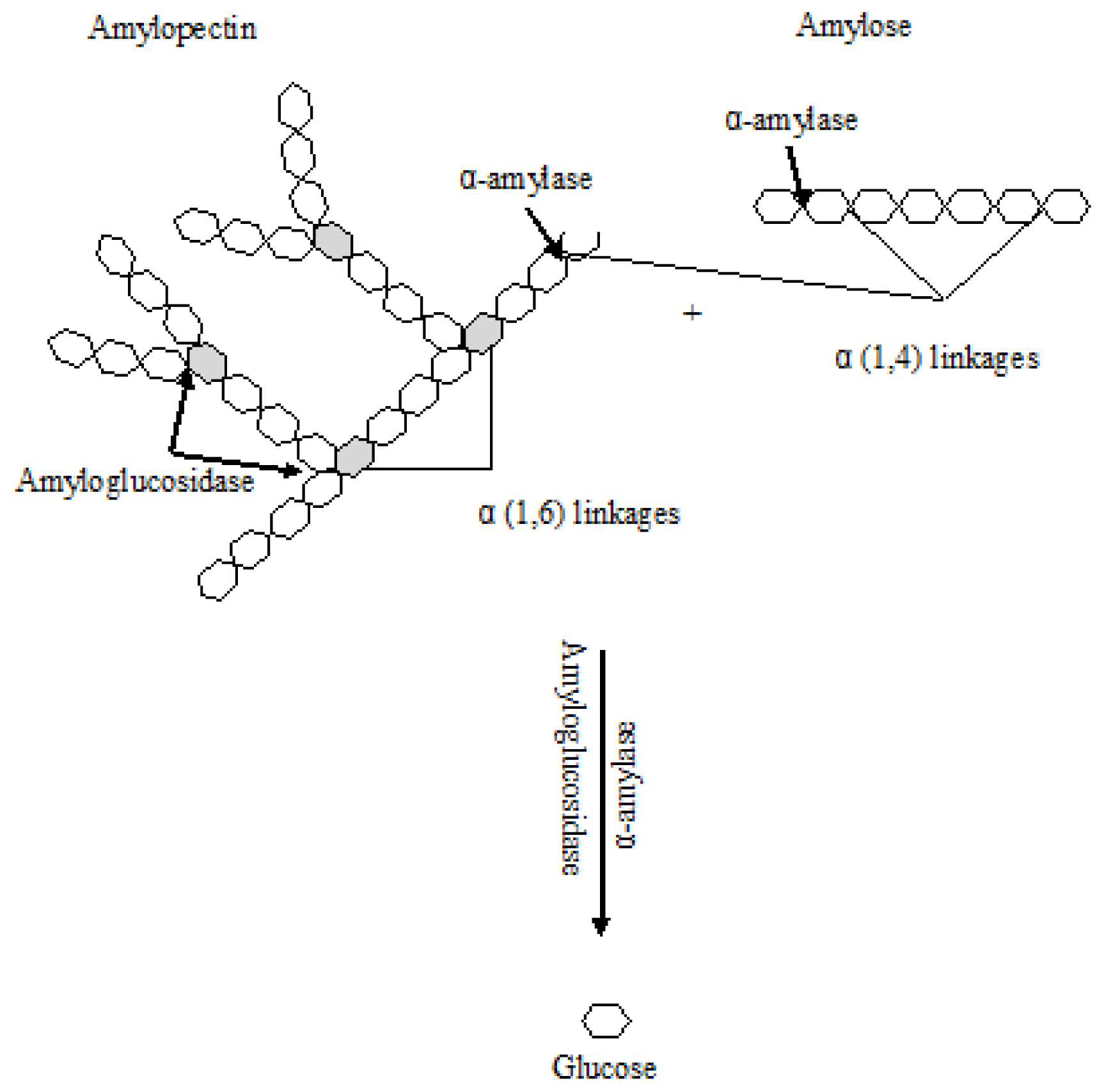
3.1. Amylolytic Enzymes
3.2. Invertases
3.3. Inulinases
4. Immobilized Carbohydrases in the Beverage Industry
5. Immobilized Carbohydrases in the Production of Prebiotics
5.1. Galactooligosaccharides
5.2. Fructooligosaccharides
6. Other Immobilized Carbohydrases
6.1. Dairy Products
6.2. Isomaltulose Production
7. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Simpson, B.K.; Rui, X.; Klomklao, S. Enzymes in Food Processing. In Food Biochemistry and Food Processing, 2nd ed; Simpson, B.K., Ed.; Wiley-Blackwell: Oxford, UK, 2012; pp. 181–206. [Google Scholar]
- Jana, M.; Maity, C.; Samanta, S.; Pati, B.R.; Islam, S.S.; Mohapatra, P.K.D.; Mondal, K.C. Salt-independent thermophilic α-amylase from Bacillus megaterium VUMB109: An efficacy testing for preparation of maltooligosaccharides. Ind. Crop. Prod 2013, 41, 386–391. [Google Scholar]
- Akgöl, S.; Kaçar, Y.; Denizli, A.; Arıca, M.Y. Hydrolysis of sucrose by invertase immobilized onto novel magnetic polyvinylalcohol microspheres. Food Chem 2001, 74, 281–288. [Google Scholar]
- Nakamura, T.; Ogata, Y.; Shitara, A.; Nakamura, A.; Ohta, K. Continuous production of fructose syrups from inulin by immobilized inulinase from Aspergillus niger mutant 817. J. Ferm. Bioeng 1995, 80, 164–169. [Google Scholar]
- Vasiljevic, T.; Jelen, P. Production of β-galactosidase for lactose hydrolysis in milk and dairy products using thermophilic lactic acid bacteria. Innov. Food Sci. Emerg 2001, 2, 75–85. [Google Scholar]
- Vera, C.; Guerrero, C.; Conejeros, R.; Illanes, A. Synthesis of galacto-oligosaccharides by β-galactosidase from Aspergillus oryzae using partially dissolved and supersaturated solution of lactose. Enzyme Microb. Technol 2012, 50, 188–194. [Google Scholar]
- Krisch, J.; Bencsik, O.; Papp, T.; Vágvölgyi, C.; Takó, M. Characterization of a β-glucosidase with transgalactosylation capacity from the zygomycete Rhizomucor miehei. Bioresour. Technol 2012, 114, 555–560. [Google Scholar]
- Kawaguti, H.Y.; Sato, H.H. Palatinose production by free and Ca-alginate gel immobilized cells of Erwinia sp. Biochem. Eng. J 2007, 36, 202–208. [Google Scholar]
- Tufvesson, P.; Lima-Ramos, J.; Nordblad, M.; Woodley, J.M. Guidelines and cost analysis for catalyst production in biocatalytic processes. Org. Process Res. Dev 2010, 15, 266–274. [Google Scholar]
- Dodge, T. Production of Industrial Enzymes. In Enzymes in Food Technology, 2nd ed; Amauri, A.B., Ed.; Wiley-Blackwell: Hoboken, NJ, USA, 2009; pp. 44–58. [Google Scholar]
- Wen, F.; McLachlan, M.; Zhao, H. Directed Evolution: Novel and Improved Enzymes. In Wiley Encyclopedia of Chemical Biology; John Wiley & Sons, Inc: Hoboken, NJ, USA, 2008; pp. 1–10. [Google Scholar]
- Martins, D.A.B.; Prado, H.F.A.; Leite, R.S.R.; Ferreira, H.; Moretti, M.M.S.; Silva, R.; Gomes, E. Agroindustrial wastes as substrates for microbial enzymes production and source of sugar for bioethanol production. In Integrated Waste Management; Kumar, S., Ed.; In Tech: Rijeka, Croatia, 2011; Volume 2, pp. 319–361. [Google Scholar]
- Hanefeld, U.; Gardossi, L.; Magner, E. Understanding enzyme immobilisation. Chem. Soc. Rev 2009, 38, 453–468. [Google Scholar]
- Torres-Salas, P.; del Monte-Martinez, A.; Cutiño-Avila, B.; Rodriguez-Colinas, B.; Alcalde, M.; Ballesteros, A.O.; Plou, F.J. Immobilized biocatalysts: Novel approaches and tools for binding enzymes to supports. Adv. Mater 2011, 23, 5275–5282. [Google Scholar]
- Kahraman, M.V.; Bayramoğlu, G.; Kayaman-Apohan, N.; Güngör, A. α-Amylase immobilization on functionalized glass beads by covalent attachment. Food Chem 2007, 104, 1385–1392. [Google Scholar]
- Silva, R.N.; Asquieri, E.R.; Fernandes, K.F. Immobilization of Aspergillus niger glucoamylase onto a polyaniline polymer. Process Biochem 2005, 40, 1155–1159. [Google Scholar]
- Zhang, L.; Zhu, X.; Zheng, S.; Sun, H. Photochemical preparation of magnetic chitosan beads for immobilization of pullulanase. Biochem. Eng. J 2009, 46, 83–87. [Google Scholar]
- Guiraud, J.; Demeulle, S.; Galzy, P. Inulin hydrolysis by the Debaryomyces phaffii inulinase immobilized on DEAE cellulose. Biotechnol. Lett 1981, 3, 683–688. [Google Scholar]
- Tanriseven, A.; Doğan, Ş. Immobilization of invertase within calcium alginate gel capsules. Process Biochem. 2001, 36, 1081–1083. [Google Scholar]
- Neri, D.F.M.; Balcão, V.M.; Cunha, M.G.C.; Carvalho, L.B., Jr; Teixeira, J.A. Immobilization of β-galactosidase from Kluyveromyces lactis onto a polysiloxane–polyvinyl alcohol magnetic (mPOS–PVA) composite for lactose hydrolysis. Catal. Commun. 2008, 9, 2334–2339. [Google Scholar]
- González-Pombo, P.; Fariña, L.; Carrau, F.; Batista-Viera, F.; Brena, B.M. A novel extracellular β-glucosidase from Issatchenkia terricola: Isolation, immobilization and application for aroma enhancement of white Muscat wine. Process Biochem 2011, 46, 385–389. [Google Scholar]
- Li, T.; Wang, N.; Li, S.; Zhao, Q.; Guo, M.; Zhang, C. Optimization of covalent immobilization of pectinase on sodium alginate support. Biotechnol. Lett 2007, 29, 1413–1416. [Google Scholar]
- Tanriseven, A.; Aslan, Y. Immobilization of Pectinex Ultra SP-L to produce fructooligosaccharides. Enzyme Microb. Technol 2005, 36, 550–554. [Google Scholar]
- Mateo, C.; Palomo, J.M.; Fernandez-Lorente, G.; Guisan, J.M.; Fernandez-Lafuente, R. Improvement of enzyme activity, stability and selectivity via immobilization techniques. Enzyme Microb. Technol 2007, 40, 1451–1463. [Google Scholar]
- Garcia-Galan, C.; Berenguer-Murcia, Á.; Fernandez-Lafuente, R.; Rodrigues, R.C. Potential of different enzyme immobilization strategies to improve enzyme performance. Adv. Synth. Catal. 2011, 353, 2885–2904. [Google Scholar]
- Rodrigues, R.C.; Ortiz, C.; Berenguer-Murcia, A.; Torres, R.; Fernandez-Lafuente, R. Modifying enzyme activity and selectivity by immobilization. Chem. Soc. Rev. 2013. [Google Scholar] [CrossRef]
- Iyer, P.V.; Ananthanarayan, L. Enzyme stability and stabilization—Aqueous and non-aqueous environment. Process Biochem 2008, 43, 1019–1032. [Google Scholar]
- Bes, M.T.; Carlos-Moreno, C.; Guisan, J.M.; Fernandez-Lafuente, R.; Gomez-Moreno, C. Selective oxidation: Stabilisation by multipoint attachment of ferredoxin NADP+ reductase, an interesting cofactor recycling enzyme. J. Mol. Catal. A Chem 1995, 98, 161–169. [Google Scholar]
- Sheldon, R.A. Enzyme immobilization: The quest for optimum performance. Adv. Synth. Catal 2007, 349, 1289–1307. [Google Scholar]
- Singh, A.K.; Flounders, A.W.; Volponi, J.V.; Ashley, C.S.; Wally, K.; Schoeniger, J.S. Development of sensors for direct detection of organophosphates Part I immobilization characterization and stabilization of acetylcholinesterase and organophosphate hydrolase on silica supports. Biosens. Bioelectron 1999, 14, 703–713. [Google Scholar]
- Takahashi, H.; Li, B.; Sasaki, T.; Miyazaki, C.; Kajino, T.; Inagaki, S. Catalytic activity in organic solvents and stability of immobilized enzymes depend on the pore size and surface characteristics of mesoporous silica. Chem. Mater 2000, 12, 3301–3305. [Google Scholar]
- Hsu, A.-F.; Foglia, T.A.; Shen, S. Immobilization of Pseudomonas cepacia lipase in a phyllosilicate sol–gel matrix: Effectiveness as a biocatalyst. Biotechnol. Appl. Biochem 2000, 31, 179–183. [Google Scholar]
- Betancor, L.; Fuentes, M.; Dellamora-Ortiz, G.; López-Gallego, F.; Hidalgo, A.; Alonso-Morales, N.; Mateo, C.; Guisán, J.M.; Fernández-Lafuente, R. Dextran aldehyde coating of glucose oxidase immobilized on magnetic nanoparticles prevents its inactivation by gas bubbles. J. Mol. Catal. B Enzym 2005, 32, 97–101. [Google Scholar]
- Brady, D.; Jordaan, J. Advances in enzyme immobilisation. Biotechnol. Lett 2009, 31, 1639–1650. [Google Scholar]
- Fernandez-Lafuente, R. Stabilization of multimeric enzymes: Strategies to prevent subunit dissociation. Enzyme Microb. Technol 2009, 45, 405–418. [Google Scholar]
- Dib, I.; Nidetzky, B. The stabilizing effects of immobilization in d-amino acid oxidase from Trigonopsis variabilis. BMC Biotechnol 2008, 8, 1–11. [Google Scholar]
- Bolivar, J.M.; Mateo, C.; Grazu, V.; Carrascosa, A.V.; Pessela, B.C.; Guisan, J.M. Heterofunctional supports for the one-step purification, immobilization and stabilization of large multimeric enzymes: Amino-glyoxyl versus amino-epoxy supports. Process Biochem 2010, 45, 1692–1698. [Google Scholar]
- Da Silva, V.; Contesini, F.; de Oliveira Carvalho, P. Enantioselective behavior of lipases from Aspergillus niger immobilized in different supports. J. Ind. Microbiol. Biotechnol 2009, 36, 949–954. [Google Scholar]
- Gómez de Segura, A.; Alcalde, M.; Plou, F.J.; Remaud-simeon, M.; Monsan, P.; Ballesteros, A. Encapsulation in lentikats of dextransucrase from leuconostoc mesenteroides nrrl b-1299, and its effect on product selectivity. Biocatal. Biotransform 2003, 21, 325–331. [Google Scholar]
- Guidini, C.Z.; Fischer, J.; Resende, M.M.; Cardoso, V.L.; Ribeiro, E.J. β-Galactosidase of Aspergillus oryzae immobilized in an ion exchange resin combining the ionic-binding and crosslinking methods: Kinetics and stability during the hydrolysis of lactose. J. Mol. Catal. B Enzym 2011, 71, 139–145. [Google Scholar]
- Ray, R.R.; Jana, S.C.; Nanda, G. Biochemical approaches of increasing thermostability of β-amylase from Bacillus megaterium B6. FEBS Lett 1994, 356, 30–32. [Google Scholar]
- Mateo, C.; Fernández-Lorente, G.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Multifunctional epoxy supports: A new tool to improve the covalent immobilization of proteins. The promotion of physical adsorptions of proteins on the supports before their covalent linkage. Biomacromolecules 2000, 1, 739–745. [Google Scholar]
- Mateo, C.; Pessela, B.C.; Grazu, V.; López-Gallego, F.; Torres, R.; Fuentes, M.; Hidalgo, A.; Palomo, J.M.; Betancor, L.; Fernández-Lorente, G.; et al. Immobilization and stabilization of proteins by multipoint covalent attachment on novel amino-epoxy-sepabeads®. 2006, 22, 153–162. [Google Scholar]
- Basso, A.; Spizzo, P.; Ferrario, V.; Knapic, L.; Savko, N.; Braiuca, P.; Ebert, C.; Ricca, E.; Calabrò, V.; Gardossi, L. Endo- and exo-inulinases: Enzyme-substrate interaction and rational immobilization. Biotechnol. Prog 2010, 26, 397–405. [Google Scholar]
- Bolivar, J.M.; Nidetzky, B. Positively charged mini-protein zbasic2 as a highly efficient silica binding module: Opportunities for enzyme immobilization on unmodified silica supports. Langmuir 2012, 28, 10040–10049. [Google Scholar]
- Gopinath, S.; Sugunan, S. Leaching studies over immobilized a-amylase. importance of the nature of enzyme attachment. React. Kinet. Catal. Lett 2004, 83, 79–83. [Google Scholar]
- Ivanov, A.E.; Schneider, M.P. Methods for the immobilization of lipases and their use for ester synthesis. J. Mol. Catal. B Enzym 1997, 3, 303–309. [Google Scholar]
- Brena, B.M.; Batista-Viera, F. Immobilization of Enzymes. In Methods in Biotechnology. Immobilization of Enzymes and Cells; Guisan, M.J., Ed.; Humana Press Inc: Totowa, NJ, USA, 2006; pp. 15–30. [Google Scholar]
- Cao, L.; Schmid, R.D. Carrier-bound Immobilized Enzymes: Principles, Application and Design; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2006. [Google Scholar]
- Torres, R.; Mateo, C.; Fernández-Lorente, G.; Ortiz, C.; Fuentes, M.; Palomo, J.M.; Guisan, J.M.; Fernández-Lafuente, R. A novel heterofunctional epoxy-amino sepabeads for a new enzyme immobilization protocol: Immobilization-stabilization of β-galactosidase from Aspergillus oryzae. Biotechnol. Prog 2003, 19, 1056–1060. [Google Scholar]
- Hannibal-Friedrich, O.; Chun, M.; Sernetz, M. Immobilization of beta-galactosidase, albumin, and gamma-globulin on epoxy-activated acrylic beads. Biotechnol. Bioeng 1980, 22, 157–175. [Google Scholar]
- Katchalski-Katzir, E.; Kraemer, D.M. Eupergit® C, a carrier for immobilization of enzymes of industrial potential. J. Mol. Catal. B Enzym 2000, 10, 157–176. [Google Scholar]
- Mateo, C.; Grazú, V.; Pessela, B.C.; Montes, T.; Palomo, J.M.; Torres, R.; López-Gallego, F.; Fernández-Lafuente, R.; Guisán, J.M. Advances in the design of new epoxy supports for enzyme immobilization–stabilization. Biochem. Soc. Trans 2007, 35, 1593–1601. [Google Scholar]
- Mateo, C.; Abian, O.; Fernández-Lorente, G.; Pedroche, J.; Fernández-Lafuente, R.; Guisan, J.M.; Tam, A.; Daminati, M. Epoxy Sepabeads: A novel epoxy support for stabilization of industrial enzymes via very intense multipoint covalent attachment. Biotechnol. Prog 2002, 18, 629–634. [Google Scholar]
- Grazú, V.; Abian, O.; Mateo, C.; Batista-Viera, F.; Fernández-Lafuente, R.; Guisán, J.M. Novel bifunctional epoxy/thiol-reactive support to immobilize thiol containing proteins by the epoxy chemistry. Biomacromolecules 2003, 4, 1495–1501. [Google Scholar]
- Abian, O.; Fernández-Lafuente, R.; García, J.L.; González García, R.; Grazú, V.; Guisán, J.M.; Hermoso, J.A. Stabilization of penicillin G acylase from Escherichia coli: Site-directed mutagenesis of the protein surface to increase multipoint covalent attachment. Appl. Environ. Microb 2004, 70, 1249–1251. [Google Scholar]
- Bolivar, J.M.; López-Gallego, F.; Godoy, C.; Rodrigues, D.S.; Rodrigues, R.C.; Batalla, P.; Rocha-Martín, J.; Mateo, C.; Giordano, R.L.C.; Guisán, J.M. The presence of thiolated compounds allows the immobilization of enzymes on glyoxyl agarose at mild pH values: New strategies of stabilization by multipoint covalent attachment. Enzyme Microb. Technol 2009, 45, 477–483. [Google Scholar]
- Godoy, C.A.; Rivas, B.; Grazú, V.; Montes, T.; Guisán, J.M.; López-Gallego, F. Glyoxyl-disulfide agarose: A tailor-made support for site-directed rigidification of proteins. Biomacromolecules 2011, 12, 1800–1809. [Google Scholar]
- Hernandez, K.; Fernandez-Lafuente, R. Control of protein immobilization: Coupling immobilization and site-directed mutagenesis to improve biocatalyst or biosensor performance. Enzyme Microb. Technol 2011, 48, 107–122. [Google Scholar]
- Bernardino, S.; Estrela, N.; Ochoa-Mendes, V.; Fernandes, P.; Fonseca, L. Optimization in the immobilization of penicillin G acylase by entrapment in xerogel particles with magnetic properties. J. Sol-Gel Sci. Technol 2011, 58, 545–556. [Google Scholar]
- Pierre, A.C. The sol-gel encapsulation of enzymes. Biocatal. Biotransform 2004, 22, 145–170. [Google Scholar]
- Shah, S.; Sharma, A.; Gupta, M.N. Preparation of cross-linked enzyme aggregates by using bovine serum albumin as a proteic feeder. Anal. Biochem 2006, 351, 207–213. [Google Scholar]
- Dong, T.; Zhao, L.; Huang, Y.; Tan, X. Preparation of cross-linked aggregates of aminoacylase from Aspergillus melleus by using bovine serum albumin as an inert additive. Bioresour. Technol 2010, 101, 6569–6571. [Google Scholar]
- Cabana, H.; Jones, J.P.; Agathos, S.N. Preparation and characterization of cross-linked laccase aggregates and their application to the elimination of endocrine disrupting chemicals. J. Biotechnol 2007, 132, 23–31. [Google Scholar]
- López-Gallego, F.; Betancor, L.; Hidalgo, A.; Alonso, N.; Fernández-Lafuente, R.; Guisán, J.M. Co-aggregation of enzymes and polyethyleneimine: A simple method to prepare stable and immobilized derivatives of glutaryl acylase. Biomacromolecules 2005, 6, 1839–1842. [Google Scholar]
- Wilson, L.; Illanes, A.; Abián, O.; Pessela, B.C.C.; Fernández-Lafuente, R.; Guisán, J.M. Co-aggregation of penicillin G acylase and polyionic polymers: An easy methodology to prepare enzyme biocatalysts stable in organic media. Biomacromolecules 2004, 5, 852–857. [Google Scholar]
- Wilson, L.; Illanes, A.; Abián, O.; Fernández-Lafuente, R.; Guisan, J.M. Encapsulation of very soft cross-linked enzyme aggregates (CLEAs) into very rigid LentiKats. FAL Agric. Res 2002, 241, 121–125. [Google Scholar]
- Wilson, L.; Illanes, A.; Pessela, B.C.; Abian, O.; Fernández-Lafuente, R.; Guisán, J.M. Encapsulation of crosslinked penicillin G acylase aggregates in lentikats: Evaluation of a novel biocatalyst in organic media. Biotechnol. Bioeng 2004, 86, 558–562. [Google Scholar]
- Kim, J.; Lee, J.; Na, H.B.; Kim, B.C.; Youn, J.K.; Kwak, J.H.; Moon, K.; Lee, E.; Kim, J.; Park, J.; et al. A Magnetically separable, highly stable enzyme system based on nanocomposites of enzymes and magnetic nanoparticles shipped in hierarchically ordered, mesocellular, mesoporous silica. Small 2005, 1, 1203–1207. [Google Scholar]
- Hobbs, L. Sweeteners from Starch: Production, Properties and Uses. In Starch, 3rd ed; James, B., Roy, W., Eds.; Academic Press: San Diego, CA, USA, 2009; pp. 797–832. [Google Scholar]
- Roy, I.; Gupta, M.N. Hydrolysis of starch by a mixture of glucoamylase and pullulanase entrapped individually in calcium alginate beads. Enzyme Microb. Technol 2004, 34, 26–32. [Google Scholar]
- Al-Mayah, A.M.R. Simulation of enzyme catalysis in calcium alginate beads. Enzyme Res. 2012. [Google Scholar] [CrossRef]
- Ivanova, V.; Dobreva, E.; Legoy, M.D. Characteristics of immobilized thermostable amylases from two Bacillus licheniformis strains. Acta Biotechnol 1998, 18, 339–351. [Google Scholar]
- Shewale, S.D.; Pandit, A.B. Hydrolysis of soluble starch using Bacillus licheniformis α-amylase immobilized on superporous CELBEADS. Carbohydr. Res 2007, 342, 997–1008. [Google Scholar]
- Singh, V.; Kumar, P. Carboxymethyl tamarind gum–silica nanohybrids for effective immobilization of amylase. J. Mol. Catal. B Enzym 2011, 70, 67–73. [Google Scholar]
- Tüzmen, N.; Kalburcu, T.; Denizli, A. A-amylase immobilization onto dye attached magnetic beads: Optimization and characterization. J. Mol. Catal. B Enzym 2012, 78, 16–23. [Google Scholar]
- Talekar, S.; Chavare, S. Optimization of immobilization of α-amylase in alginate gel and its comparative biochemical studies with free α-amylase. Rec. Res. Sci. Technol 2012, 4, 1–5. [Google Scholar]
- Carpio, C.; Escobar, F.; Batista-Viera, F.; Ruales, J. Bone-bound glucoamylase as a biocatalyst in bench-scale production of glucose syrups from liquefied cassava starch. Food Bioprocess Tech 2011, 4, 566–577. [Google Scholar]
- Zhao, G.; Wang, J.; Li, Y.; Huang, H.; Chen, X. Reversible immobilization of glucoamylase onto metal–ligand functionalized magnetic FeSBA-15. Biochem. Engin. J 2012, 68, 159–166. [Google Scholar]
- Singh, R.S.; Saini, G.K.; Kennedy, J.F. Covalent immobilization and thermodynamic characterization of pullulanase for the hydrolysis of pullulan in batch system. Carbohydr. Polym 2010, 81, 252–259. [Google Scholar]
- Singh, R.S.; Saini, G.K.; Kennedy, J.F. Maltotriose syrup preparation from pullulan using pullulanase. Carbohydr. Polym 2010, 80, 401–407. [Google Scholar]
- Talekar, S.; Ghodake, V.; Ghotage, T.; Rathod, P.; Deshmukh, P.; Nadar, S.; Mulla, M.; Ladole, M. Novel magnetic cross-linked enzyme aggregates (magnetic CLEAs) of alpha amylase. Bioresour. Technol 2012, 123, 542–547. [Google Scholar]
- Emregul, E.; Sungur, S.; Akbulut, U. Polyacrylamide–gelatine carrier system used for invertase immobilization. Food Chem 2006, 97, 591–597. [Google Scholar]
- Kotwal, S.M.; Shankar, V. Immobilized invertase. Biotechnol. Adv 2009, 27, 311–322. [Google Scholar]
- Cadena, P.G.; Wiggers, F.N.; Silva, R.A.; Lima Filho, J.L.; Pimentel, M.C.B. Kinetics and bioreactor studies of immobilized invertase on polyurethane rigid adhesive foam. Bioresour. Technol 2011, 102, 513–518. [Google Scholar]
- Smaali, I.; Soussi, A.; Bouallagui, H.; Chaira, N.; Hamdi, M.; Marzouki, M.N. Production of high-fructose syrup from date by-products in a packed bed bioreactor using a novel thermostable invertase from Aspergillus awamori. Biocatal. Biotransform 2011, 29, 253–261. [Google Scholar]
- Albertini, A.V.P.; Cadena, P.G.; Silva, J.L.; Nascimento, G.A.; Reis, A.L.S.; Freire, V.N.; Santos, R.P.; Martins, J.L.; Cavada, B.S.; Neto, P.J.R.; et al. Performance of invertase immobilized on glass–ceramic supports in batch bioreactor. Chem. Eng. J 2012, 187, 341–350. [Google Scholar]
- Albertini, A.V.P.; Reis, A.L.S.; Teles, F.R.R.; Souza, J.C.; Filho, J.L.R.; Freire, V.N.; Santos, R.P.; Martins, J.L.; Cavada, B.S.; Martins, D.B.G.; et al. The new flow system approach in packed bed reactor applicable for immobilized enzyme. J. Mol. Catal. B Enzyme 2012, 79, 1–7. [Google Scholar]
- Mirzarakhmetova, D.; Dekhkonov, D.; Rakhimov, M.; Abdurazakova, S.; Akhmedova, Z. The properties of invertase, covalently immobilized at activated carbon. Appl. Biochem. Microbiol 2009, 45, 258–261. [Google Scholar]
- Vujčić, Z.; Milovanović, A.; Božić, N.; Dojnov, B.; Vujčić, M.; Andjelković, U.; Lončar, N. Immobilization of cell wall invertase modified with glutaraldehyde for continuous production of invert sugar. J. Agric. Food Chem 2010, 58, 11896–11900. [Google Scholar]
- Mohd Zain, N.A.; Mohd Suardi, S.; Idris, A. Hydrolysis of liquid pineapple waste by invertase immobilized in PVA–alginate matrix. Biochem. Eng. J 2010, 50, 83–89. [Google Scholar]
- Talekar, S.; Shah, V.; Patil, S.; Nimbalkar, M. Porous cross linked enzyme aggregates (p-CLEAs) of Saccharomyces cerevisiae invertase. Catal. Sci. Technol 2012, 2, 1575–1579. [Google Scholar]
- Cho, Y.J.; Sinha, J.; Park, J.P.; Yun, J.W. Production of inulooligosaccharides from inulin by a dual endoinulinase system. Enzyme Microb. Technol 2001, 29, 428–433. [Google Scholar]
- Vandamme, E.J.; Derycke, D.G. Microbial Inulinases: Fermentation Process, Properties, and Applications. In Advances in Applied Microbiology; Allen, I.L., Ed.; Academic Press: New York, NY, USA, 1983; Volume 29, pp. 139–176. [Google Scholar]
- Ricca, E.; Calabrò, V.; Curcio, S.; Iorio, G. The state of the art in the production of fructose from inulin enzymatic hydrolysis. Crit. Rev. Biotechnol 2007, 27, 129–145. [Google Scholar]
- Santa, G.; Bernardino, S.; Magalhães, S.; Mendes, V.; Marques, M.; Fonseca, L.; Fernandes, P. From inulin to fructose syrups using sol–gel immobilized inulinase. Appl. Biochem. Biotechnol 2011, 165, 1–12. [Google Scholar]
- Singh, R.; Dhaliwal, R.; Puri, M. Production of high fructose syrup from Asparagus inulin using immobilized exoinulinase from Kluyveromyces marxianus YS-1. J. Ind. Microbiol. Biotechnol 2007, 34, 649–655. [Google Scholar]
- Singh, R.; Dhaliwal, R.; Puri, M. Development of a stable continuous flow immobilized enzyme reactor for the hydrolysis of inulin. J. Ind. Microbiol. Biotechnol 2008, 35, 777–782. [Google Scholar]
- Ricca, E.; Calabrò, V.; Curcio, S.; Basso, A.; Gardossi, L.; Iorio, G. Fructose production by inulinase covalently immobilized on sepabeads in batch and fluidized bed bioreactor. Int. J. Mol. Sci 2010, 11, 1180–1189. [Google Scholar]
- De Paula, F.C.; Cazetta, M.L.; Monti, R.; Contiero, J. Sucrose hydrolysis by gelatin-immobilized inulinase from Kluyveromyces marxianus var. bulgaricus. Food Chem 2008, 111, 691–695. [Google Scholar]
- Ettalibi, M.; Baratti, J.C. Sucrose hydrolysis by thermostable immobilized inulinases from Aspergillus ficuum. Enzyme Microb. Technol 2001, 28, 596–601. [Google Scholar]
- Barranco-Florido, E.; Garcıía-Garibay, M.; Gómez-Ruiz, L.; Azaola, A. Immobilization system of Kluyveromyces marxianus cells in barium alginate for inulin hydrolysis. Process Biochem 2001, 37, 513–519. [Google Scholar]
- Makino, Y.; Lima, P.S.C.; Filho, F.M.; Rodrigues, M.I. Adsorption of the inulinase from Kluyveromyces marxianus NRRL Y-7571 on Streamline® DEAE resin. Braz. J. Chem. Eng 2005, 22, 539–545. [Google Scholar]
- Nguyen, Q.D.; Rezessy-Szabó, J.M.; Czukor, B.; Hoschke, Á. Continuous production of oligofructose syrup from Jerusalem artichoke juice by immobilized endo-inulinase. Process Biochem. 2011, 46, 298–303. [Google Scholar]
- Yun, J.W.; Park, J.P.; Song, C.H.; Lee, C.Y.; Kim, J.H.; Song, S.K. Continuous production of inulo-oligosaccharides from chicory juice by immobilized endoinulinase. Bioprocess Eng 2000, 22, 189–194. [Google Scholar]
- Spagna, G.; Pifferi, P.G.; Tramontini, M. Immobilization and stabilization of pectinlyase on synthetic polymers for application in the beverage industry. J. Mol. Catal. A Chem 1995, 101, 99–105. [Google Scholar]
- Wilson, B.; Strauss, C.R.; Williams, P.J. The distribution of free and glycosidically-bound monoterpenes among skin, juice, and pulp fractions of some white grape varieties. Am. J. Enol. Vitic 1986, 37, 107–111. [Google Scholar]
- Voirin, S.G.; Baumes, R.L.; Bitteur, S.M.; Gunata, Z.Y.; Bayonove, C.L. Novel monoterpene disaccharide glycosides of Vitis vinifera grapes. J. Agric. Food Chem 1990, 38, 1373–1378. [Google Scholar]
- Gueguen, Y.; Chemardin, P.; Janbon, G.; Arnaud, A.; Galzy, P. A very efficient beta-glucosidase catalyst for the hydrolysis of flavor precursors of wines and fruit juices. J. Agric. Food Chem 1996, 44, 2336–2340. [Google Scholar]
- Spagna, G.; Barbagallo, R.N.; Greco, E.; Manenti, I.; Pifferi, P.G. A mixture of purified glycosidases from Aspergillus niger for oenological application immobilised by inclusion in chitosan gels. Enzyme Microb. Technol 2002, 30, 80–89. [Google Scholar]
- Su, E.; Xia, T.; Gao, L.; Dai, Q.; Zhang, Z. Immobilization of β-glucosidase and its aroma-increasing effect on tea beverage. Food Bioprod. Process 2010, 88, 83–89. [Google Scholar]
- Figueira, J.A.; Dias, F.F.G.; Sato, H.H.; Fernandes, P. Screening of supports for the immobilization β-glucosidase. Enzyme Res 2011, 2011, 8. [Google Scholar]
- Whitaker, J.R. New and future uses of enzymes in food processing. Food Biotechnol 1990, 4, 669–697. [Google Scholar]
- Alaña, A.; Gabilondo, A.; Hernando, F.; Moragues, M.D.; Dominguez, J.B.; Llama, M.J.; Serra, J.L. Pectin lyase production by a penicillium italicum strain. Appl. Environ. Microbiol 1989, 55, 1612–1616. [Google Scholar]
- Lanzarini, G.; Pifferi, P.G. Enzymes in the Fruit Juice Industry. In Biotechnology Applications in Beverage Production; Cantarelli, C., Ed.; Elsevier Applied Science: New York, NY, USA, 1989. [Google Scholar]
- Alkorta, I.; Garbisu, C.; María Llama, J.; Serra, J.L. Immobilization of pectin lyase from Penicillium italicum by covalent binding to nylon. Enzyme Microb. Technol. 1996, 18, 141–146. [Google Scholar]
- Torres, D.P.M.; Gonçalves, M.P.F.; Teixeira, J.A.; Rodrigues, L.R. Galacto-Oligosaccharides: Production, properties, applications, and significance as prebiotics. Compr. Rev. Food Sci. Food Saf 2010, 9, 438–454. [Google Scholar]
- Panesar, P.S.; Kumari, S.; Panesar, R. Potential applications of immobilized β-galactosidase in food processing industries. Enzyme Res 2010, 2010, 1–16. [Google Scholar]
- Boon, M.A.; Janssen, A.E.M.; van’t Riet, K. Effect of temperature and enzyme origin on the enzymatic synthesis of oligosaccharides. Enzyme Microb. Technol 2000, 26, 271–281. [Google Scholar]
- Boon, M.A.; Janssen, A.E.M.; van der Padt, A. Modelling and parameter estimation of the enzymatic synthesis of oligosaccharides by β-galactosidase from Bacillus circulans. Biotechnol. Bioeng 1999, 64, 558–567. [Google Scholar]
- Neri, D.F.M.; Balcão, V.M.; Costa, R.S.; Rocha, I.C.A.P.; Ferreira, E.M.F.C.; Torres, D.P.M.; Rodrigues, L.R.M.; Carvalho, L.B., Jr; Teixeira, J.A. Galacto-oligosaccharides production during lactose hydrolysis by free Aspergillus oryzae β-galactosidase and immobilized on magnetic polysiloxane-polyvinyl alcohol. Food Chem. 2009, 115, 92–99. [Google Scholar]
- Vera, C.; Guerrero, C.; Illanes, A.; Conejeros, R. A pseudo steady-state model for galacto-oligosaccharides synthesis with β-galactosidase from Aspergillus oryzae. Biotechnol. Bioeng 2011, 108, 2270–2279. [Google Scholar]
- Gosling, A.; Stevens, G.W.; Barber, A.R.; Kentish, S.E.; Gras, S.L. Effect of the substrate concentration and water activity on the yield and rate of the transfer reaction of β-galactosidase from Bacillus circulans. J. Agric. Food Chem 2011, 59, 3366–3372. [Google Scholar]
- Gaur, R.; Pant, H.; Jain, R.; Khare, S.K. Galacto-oligosaccharide synthesis by immobilized Aspergillus oryzae β-galactosidase. Food Chem 2006, 97, 426–430. [Google Scholar]
- Shin, H.-J.; Park, J.-M.; Yang, J.-W. Continuous production of galacto-oligosaccharides from lactose by Bullera singularis β-galactosidase immobilized in chitosan beads. Process Biochem 1998, 33, 787–792. [Google Scholar]
- Neri, D.F.M.; Balcão, V.M.; Dourado, F.O.Q.; Oliveira, J.M.B.; Carvalho, L.B., Jr; Teixeira, J.A. Immobilized β-galactosidase onto magnetic particles coated with polyaniline: Support characterization and galactooligosaccharides production. J. Mol. Catal. B Enzyme 2011, 70, 74–80. [Google Scholar]
- Liu, H.; Liu, J.; Tan, B.; Zhou, F.; Qin, Y.; Yang, R. Covalent immobilization of Kluyveromyces fragilis β-galactosidase on magnetic nanosized epoxy support for synthesis of galacto-oligosaccharide. Bioprocess Biosyst. Eng. 2012, 35, 1287–1295. [Google Scholar]
- Albayrak, N.; Yang, S.-T. Immobilization of β-galactosidase on fibrous matrix by polyethyleneimine for production of galacto-oligosaccharides from lactose. Biotechnol. Prog 2002, 18, 240–251. [Google Scholar]
- Güleç, H.; Gürdaş, S.; Albayrak, N.; Mutlu, M. Immobilization of Aspergillus oryzae β-galactosidase on low-pressure plasma-modified cellulose acetate membrane using polyethyleneimine for production of galactooligosaccharide. Biotechnol. Bioprocess Eng 2010, 15, 1006–1015. [Google Scholar]
- Nakkharat, P.; Haltrich, D. β-Galactosidase from Talaromyces thermophilus immobilized on to Eupergit C for production of galacto-oligosaccharides during lactose hydrolysis in batch and packed-bed reactor. World J. Microbiol. Biotechnol 2007, 23, 759–764. [Google Scholar]
- Zheng, P.; Yu, H.; Sun, Z.; Ni, Y.; Zhang, W.; Fan, Y.; Xu, Y. Production of galacto-oligosaccharides by immobilized recombinant β-galactosidase from Aspergillus candidus. Biotechnol. J 2006, 1, 1464–1470. [Google Scholar]
- Albayrak, N.; Yang, S.-T. Production of galacto-oligosaccharides from lactose by Aspergillus oryzae β-galactosidase immobilized on cotton cloth. Biotechnol. Bioeng 2002, 77, 8–19. [Google Scholar]
- Mozaffar, Z.; Nakanishi, K.; Matsuno, R. Mechanism for reversible inactivation of immobilized β-galactosidase from Bacillus circulans during continuous production of galactooligosaccharides. Appl. Microbiol. Biotechnol 1986, 25, 229–231. [Google Scholar]
- Nakkharat, P.; Kulbe, K.D.; Yamabhai, M.; Haltrich, D. Formation of galacto-oligosaccharides during lactose hydrolysis by a novel β-galactosidase from the moderately thermophilic fungus Talaromyces thermophilus. Biotechnol. J 2006, 1, 633–638. [Google Scholar]
- Maiorano, A.; Piccoli, R.; da Silva, E.; de Andrade Rodrigues, M. Microbial production of fructosyltransferases for synthesis of pre-biotics. Biotechnol. Lett 2008, 30, 1867–1877. [Google Scholar]
- Alvarado-Huallanco, M.B.; Maugeri Filho, F. Kinetic studies and modelling of the production of fructooligosaccharides by fructosyltransferase from Rhodotorula sp. Catal. Sci. Technol 2011, 1, 1043–1050. [Google Scholar]
- Sánchez, O.; Rodriguez, A.; Silva, E.; Caicedo, L. Sucrose biotransformation to fructooligosaccharides by Aspergillus sp. N74 free cells. Food Bioprocess Tech 2010, 3, 662–673. [Google Scholar]
- Vaňková, K.; Onderková, Z.; Antošová, M.; Polakovič, M. Design and economics of industrial production of fructooligosaccharides. Chem. Pap 2008, 62, 375–381. [Google Scholar]
- Ning, Y.; Wang, J.; Chen, J.; Yang, N.; Jin, Z.; Xu, X. Production of neo-fructooligosaccharides using free-whole-cell biotransformation by Xanthophyllomyces dendrorhous. Bioresour. Technol 2010, 101, 7472–7478. [Google Scholar]
- Cruz, R.; Cruz, V.D.; Belini, M.Z.; Belote, J.G.; Vieira, C.R. Production of fructooligosaccharides by the mycelia of Aspergillus japonicus immobilized in calcium alginate. Bioresour. Technol 1998, 65, 139–143. [Google Scholar]
- Jung, K.H.; Bang, S.H.; Oh, T.K.; Park, H.J. Industrial production of fructooligosaccharides by immobilized cells of Aureobasidium pullulans in a packed bed reactor. Biotechnol. Lett 2011, 33, 1621–1624. [Google Scholar]
- Shin, H.T.; Park, K.M.; Kang, K.H.; Oh, D.J.; Lee, S.W.; Baig, S.Y.; Lee, J.H. Novel method for cell immobilization and its application for production of oligosaccharides from sucrose. Lett. Appl. Microbiol 2004, 38, 176–179. [Google Scholar]
- Smaali, I.; Jazzar, S.; Soussi, A.; Muzard, M.; Aubry, N.; Marzouki, M. Enzymatic synthesis of fructooligosaccharides from date by-products using an immobilized crude enzyme preparation of β-d-fructofuranosidase from Aspergillus awamori NBRC 4033. Biotechnol. Bioprocess Eng 2012, 17, 385–392. [Google Scholar]
- Kovaleva, T.; Holyavka, M.; Bogdanova, S. Inulinase immobilization on macroporous anion-exchange resins by different methods. Bull. Exp. Biol. Med 2009, 148, 39–41. [Google Scholar]
- Ghazi, I.; Segura, A.G.D.; Fernández-Arrojo, L.; Alcalde, M.; Yates, M.; Rojas-Cervantes, M.L.; Plou, F.J.; Ballesteros, A. Immobilisation of fructosyltransferase from Aspergillus aculeatus on epoxy-activated Sepabeads EC for the synthesis of fructo-oligosaccharides. J. Mol. Catal. B Enzyme 2005, 35, 19–27. [Google Scholar]
- Alvarado-Huallanco, M.B.; Maugeri-Filho, F. Kinetics and modeling of fructooligosaccharide synthesis by immobilized fructosyltransferase from Rhodotorula sp. J. Chem. Technol. Biotechnol 2010, 85, 1654–1662. [Google Scholar]
- Husain, Q. β Galactosidases and their potential applications: A review. Crit. Rev. Biotechnol 2010, 30, 41–62. [Google Scholar]
- Guidini, C.Z.; Fischer, J.; Santana, L.N.S.; Cardoso, V.L.; Ribeiro, E.J. Immobilization of Aspergillus oryzae β-galactosidase in ion exchange resins by combined ionic-binding method and cross-linking. Biochem. Eng. J 2010, 52, 137–143. [Google Scholar]
- Pessela, B.C.C.; Fernandez-Lafuente, R.; Fuentes, M.; Vian, A.; Garca, J.L.; Carrascosa, A.V.; Mateo, C.; Guisan, J.M. Reversible immobilization of a thermophilic beta-galactosidase via ionic adsorption on PEI-coated Sepabeads. Enzyme Microb. Technol 2003, 32, 369–374. [Google Scholar]
- Pessela, B.C.C.; Mateo, C.; Fuentes, M.; Vian, A.; Garcıía, J.L.; Carrascosa, A.V.; Guisán, J.M.; Fernández-Lafuente, R. The immobilization of a thermophilic β-galactosidase on Sepabeads supports decreases product inhibition: Complete hydrolysis of lactose in dairy products. Enzyme Microb. Technol 2003, 33, 199–205. [Google Scholar]
- Mateo, C.; Monti, R.; Pessela, B.C.C.; Fuentes, M.; Torres, R.; Manuel Guisán, J.; Fernández-Lafuente, R. Immobilization of lactase from Kluyveromyces lactis greatly reduces the inhibition promoted by glucose. Full hydrolysis of lactose in milk. Biotechnol. Prog 2004, 20, 1259–1262. [Google Scholar]
- Tanriseven, A.; Doğan, Ş. A novel method for the immobilization of β-galactosidase. Process Biochem. 2002, 38, 27–30. [Google Scholar]
- Jochems, P.; Satyawali, Y.; van Roy, S.; Doyen, W.; Diels, L.; Dejonghe, W. Characterization and optimization of β-galactosidase immobilization process on a mixed-matrix membrane. Enzyme Microb. Technol 2011, 49, 580–588. [Google Scholar]
- Takazoe, G. Palatinose—An Isomeric Alternative to Sucrose. In Progress in Sweeteners; Grenby, T.H., Ed.; Elsevier: London, UK, 1989; pp. 143–167. [Google Scholar]
- Sasaki, N.; Topitsoglou, V.; Takazoe, I.; Frostell, G. Cariogenicity of isomaltulose (palatinose), sucrose and mixture of these sugars in rats infected with Streptococcus mutans E-49. Swed. Dent. J 1985, 9, 9149–9155. [Google Scholar]
- Achten, J.; Jentjens, R.L.; Brouns, F.; Jeukendrup, A.E. Exogenous oxidation of isomaltulose is lower than that of sucrose during exercise in men. J. Nutr 2007, 137, 1143–1148. [Google Scholar]
- Contesini, F.J.; Ibarguren, C.; Grosso, C.R.F.; Carvalho, P.O.; Sato, H.H. Immobilization of glucosyltransferase from Erwinia sp. using two different techniques. J. Biotechnol 2012, 158, 137–143. [Google Scholar]


{kind=link}
| Carbohydrase | Microbial source | Application | Reference |
|---|---|---|---|
| Amylases | Bacillus megaterium VUMB109 | Maltooligosaccharides production | [2] |
| Invertases | Sacharomyces cereviseae | Hydrolysis of sucrose | [3] |
| Inulinases | Aspergillus niger | Fructose syrup production | [4] |
| β-galactosidase | Lactobacillus delbrueckii subsp. bulgaricus ATCC 11842 | Lactose hydrolysis in milk | [5] |
| Aspergillus oryzae | Synthesis of galactooligosaccharides | [6] | |
| β-glucosidase | Rhizomucor miehei NRRL 528 | Enhancement of the amounts of free phenolic antioxidants in sour cherry pomace | [7] |
| Glucosyltransferase | Erwinia sp. | Isomaltulose production | [8] |
| Enzyme | Support | Immobilization method | Improvement compared to the free form | Reference |
|---|---|---|---|---|
| α-amylase | Functionalized glass beads | Covalent binding | Better thermostability and reuse after six runs | [15] |
| Glucoamylase | Polyaniline polymer | Covalent binding | Better thermostability and higher stability in the alkaline range | [16] |
| Pullulanase | Magnetic chitosan beads | Covalent binding | Higher relative activity, stabilization of enzyme over a broader pH range | [17] |
| Inulinase | DEAE-Cellulose | Adsorption | Better thermostability | [18] |
| Invertase | Calcium alginate gel capsules | Entrapment | Better stability at high pH and temperatures | [19] |
| β-Galactosidase | Polysiloxane–polyvinyl alcohol magnetic (mPOS–PVA) composite | Covalent binding | Higher operational and thermal stability | [20] |
| β-glucosidase | Eupergit C | Covalent binding | Improvement of the stability | [21] |
| Pectinase | Sodium alginate support using glutaraldehyde | Covalent binding | Higher thermostability and reusability | [22] |
| Fructosyltransferase | Eupergit C | Covalent binding | Reuse for 20 batch reactions, better stability at high pH and temperatures | [23] |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Contesini, F.J.; De Alencar Figueira, J.; Kawaguti, H.Y.; De Barros Fernandes, P.C.; De Oliveira Carvalho, P.; Da Graça Nascimento, M.; Sato, H.H. Potential Applications of Carbohydrases Immobilization in the Food Industry. Int. J. Mol. Sci. 2013, 14, 1335-1369. https://doi.org/10.3390/ijms14011335
Contesini FJ, De Alencar Figueira J, Kawaguti HY, De Barros Fernandes PC, De Oliveira Carvalho P, Da Graça Nascimento M, Sato HH. Potential Applications of Carbohydrases Immobilization in the Food Industry. International Journal of Molecular Sciences. 2013; 14(1):1335-1369. https://doi.org/10.3390/ijms14011335
Chicago/Turabian StyleContesini, Fabiano Jares, Joelise De Alencar Figueira, Haroldo Yukio Kawaguti, Pedro Carlos De Barros Fernandes, Patrícia De Oliveira Carvalho, Maria Da Graça Nascimento, and Hélia Harumi Sato. 2013. "Potential Applications of Carbohydrases Immobilization in the Food Industry" International Journal of Molecular Sciences 14, no. 1: 1335-1369. https://doi.org/10.3390/ijms14011335