Catalytic Mechanism of Short Ethoxy Chain Nonylphenol Dehydrogenase Belonging to a Polyethylene Glycol Dehydrogenase Group in the GMC Oxidoreductase Family

Abstract
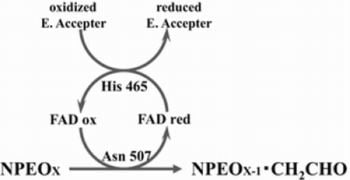
:
1. Introduction
2. Results
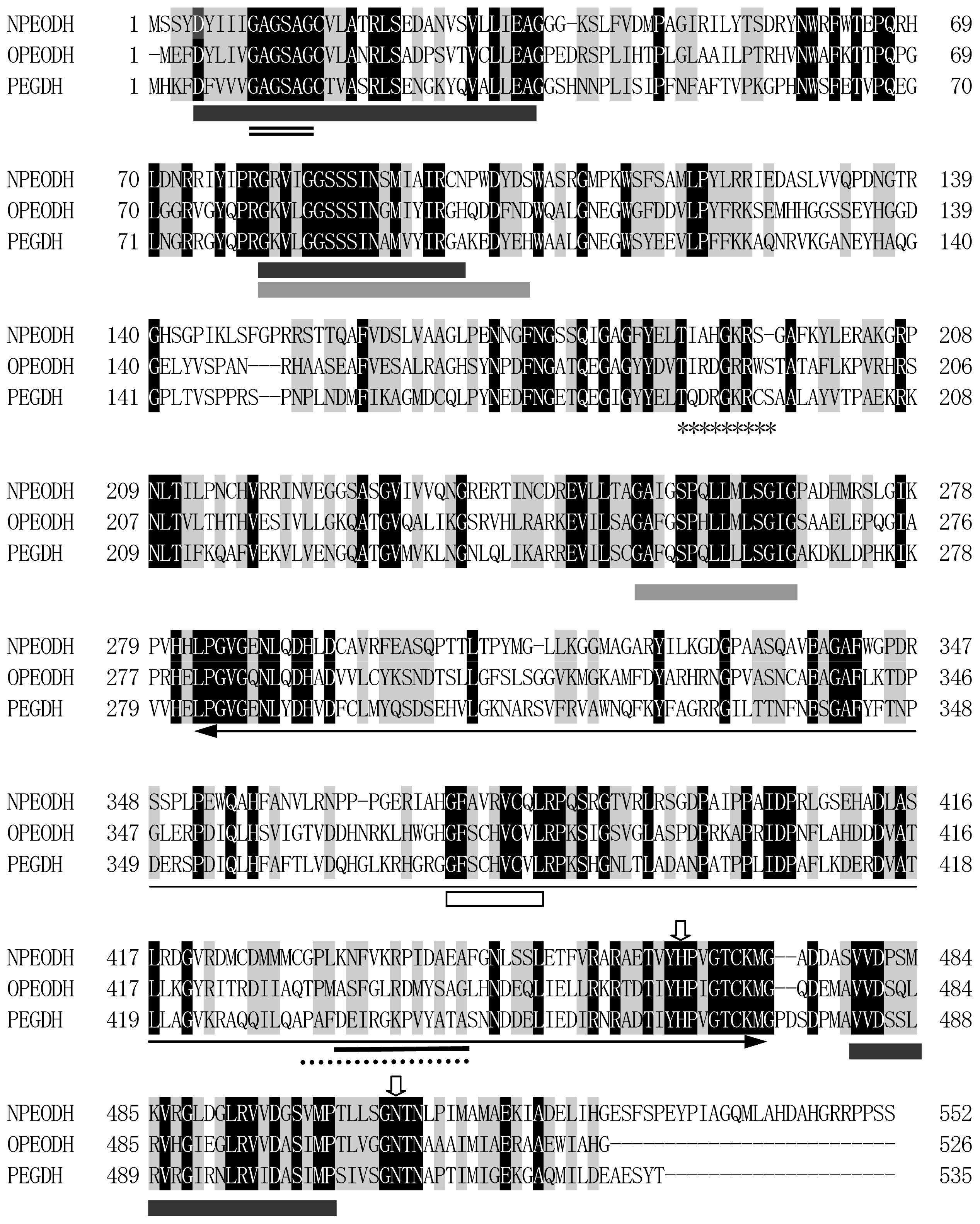
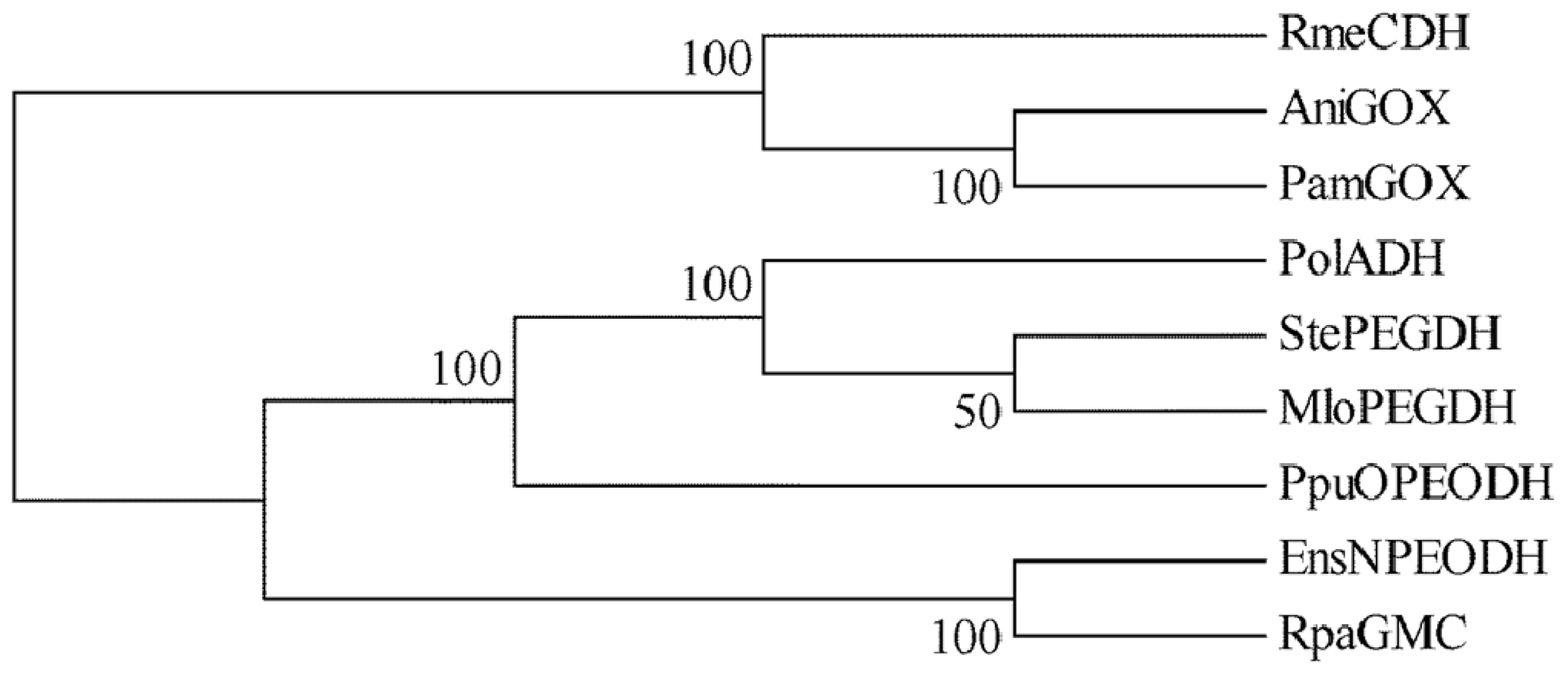
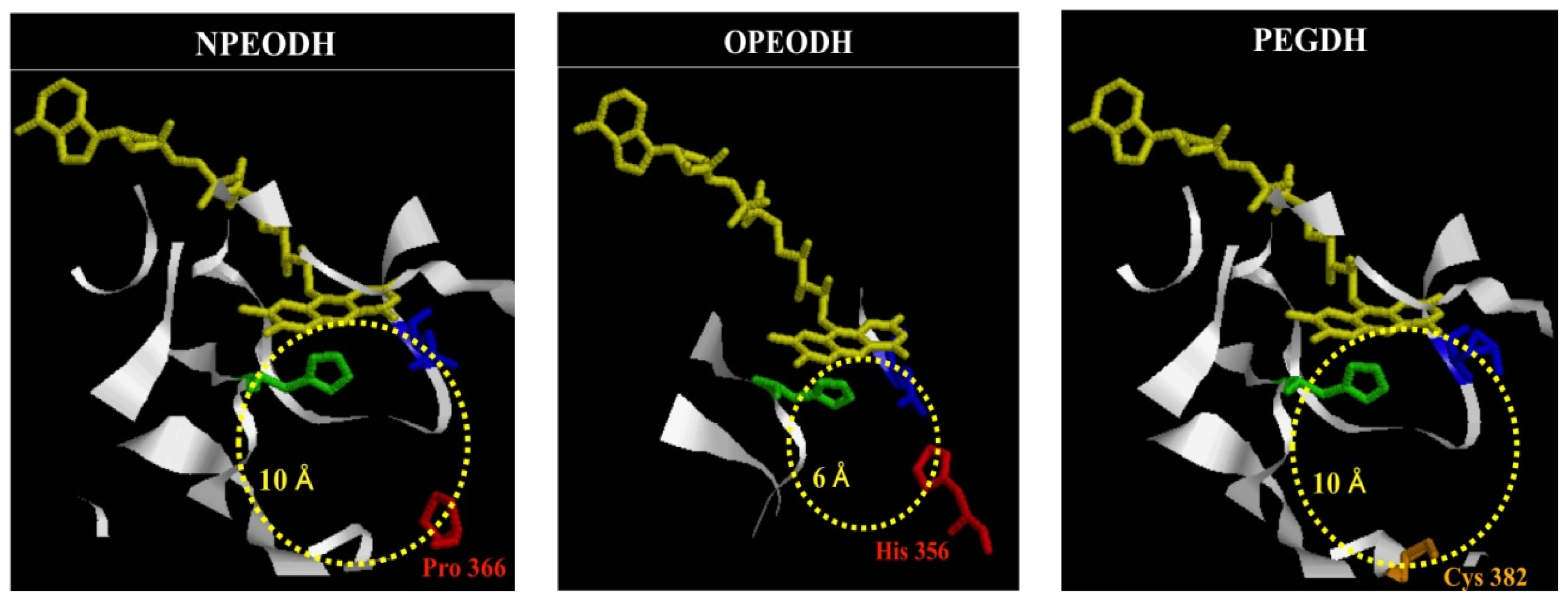
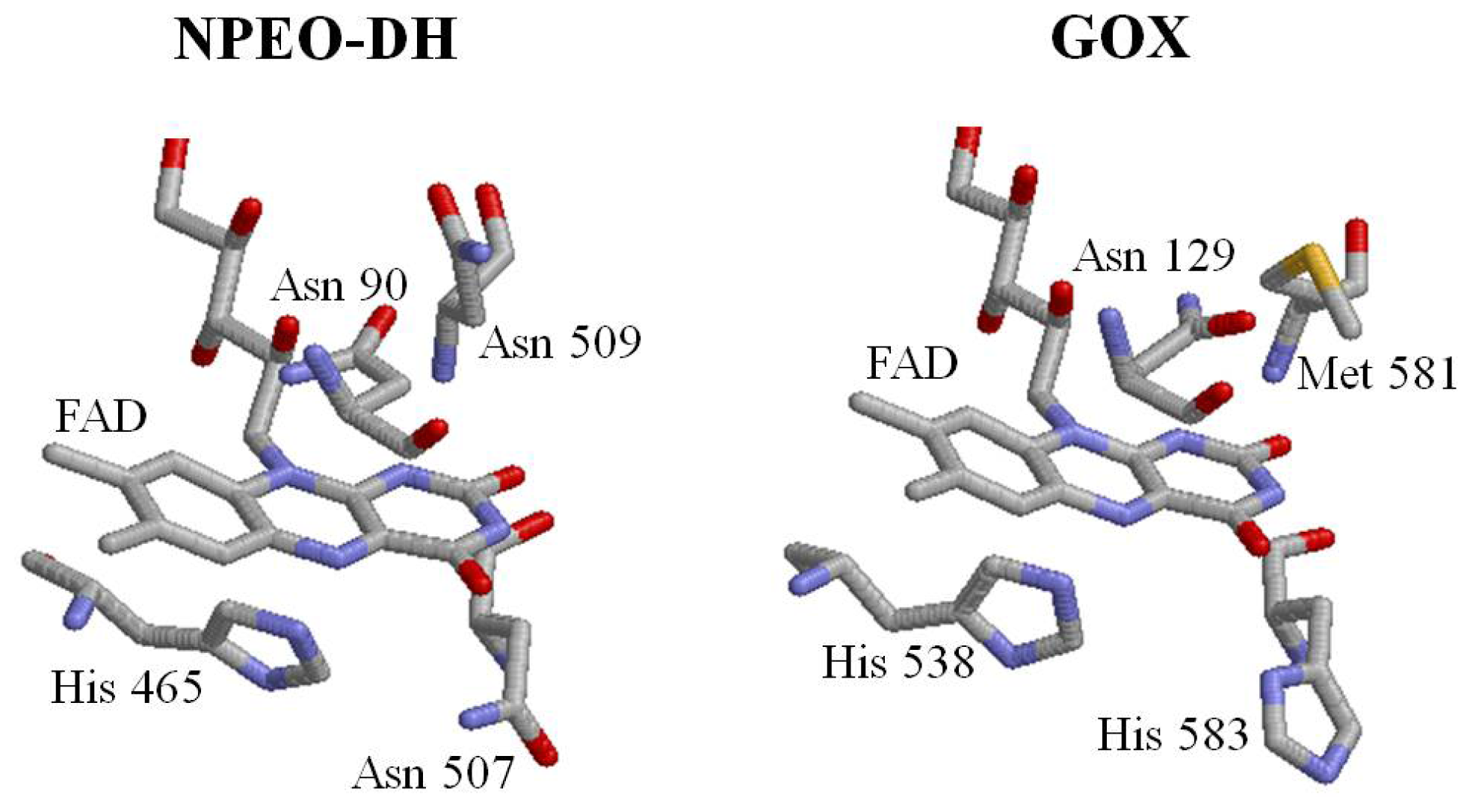
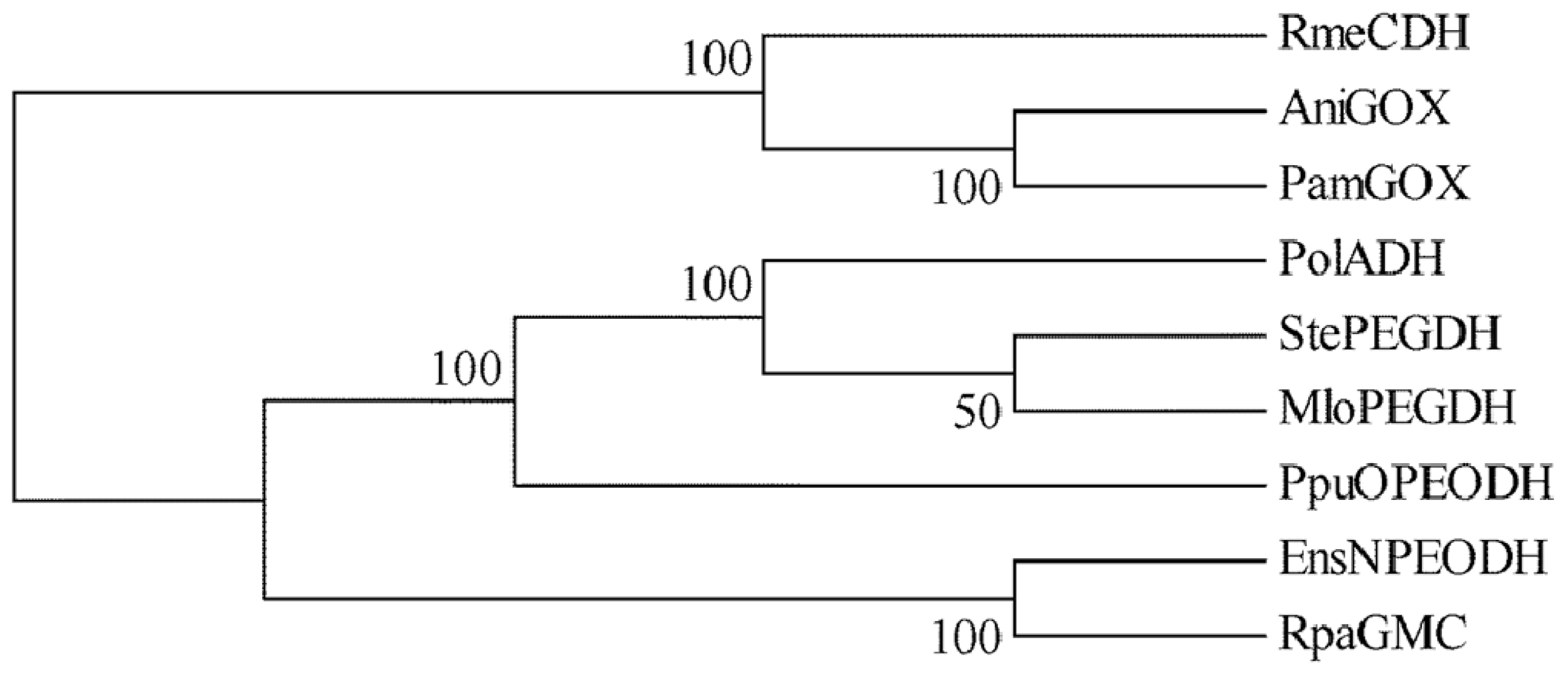
2.1. Comparison of Sequence Motifs and 3D Structure Models for NPEO-DH, OPEO-DH and PEG-DH
2.2. Mutation of NPEO-DH and Expression of Wild Type and Mutant NPEO-DHs
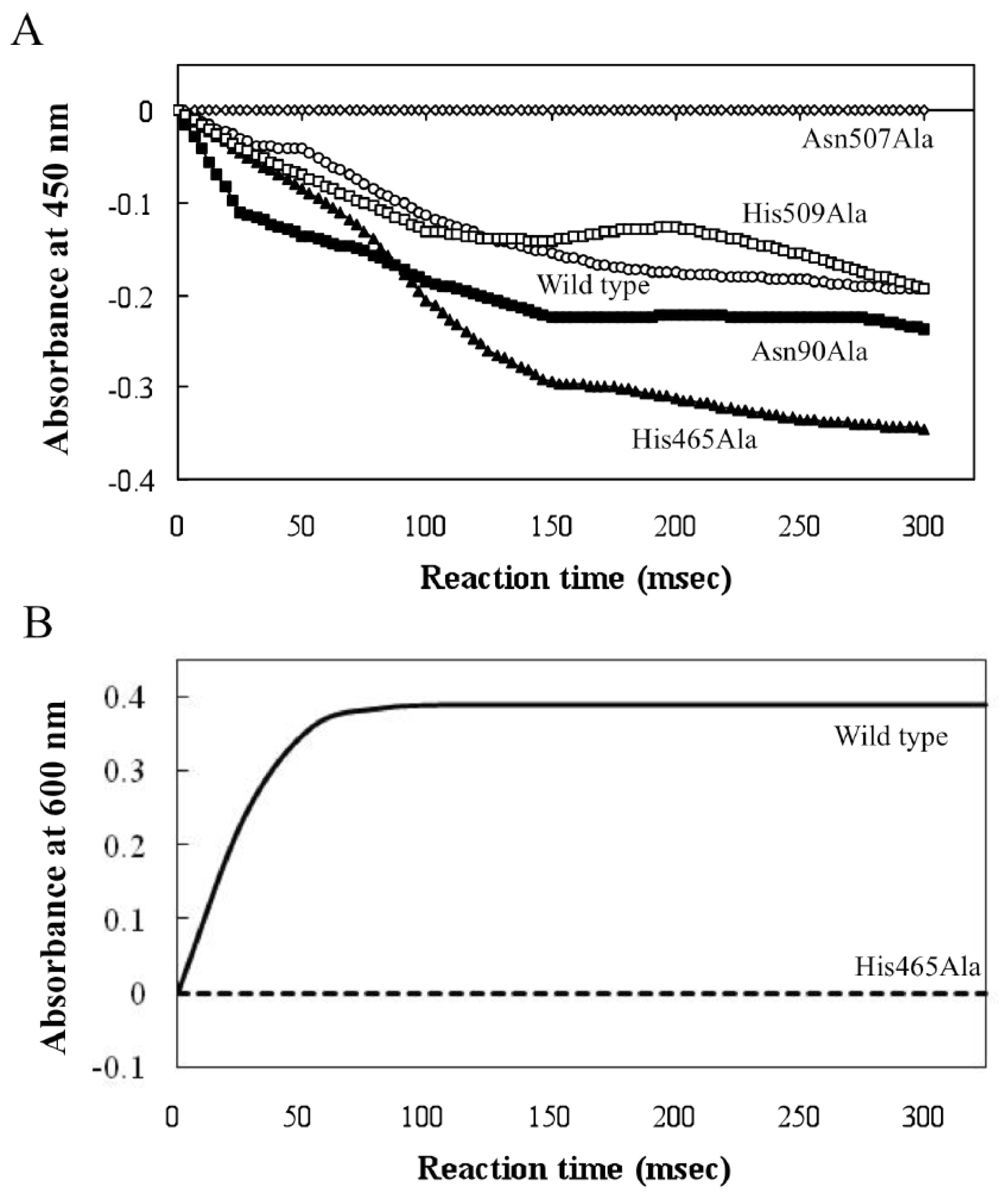
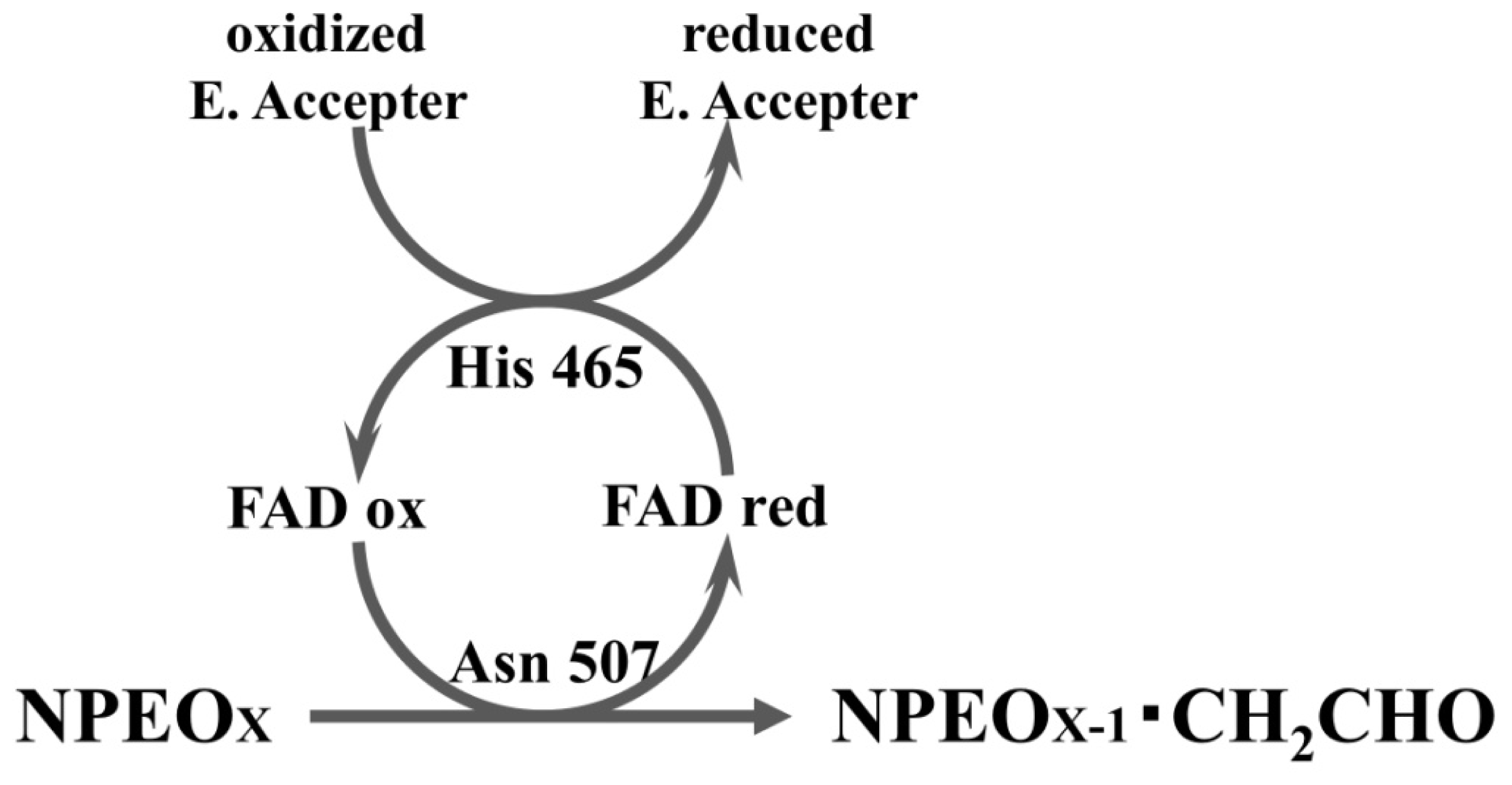
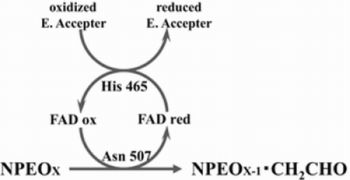
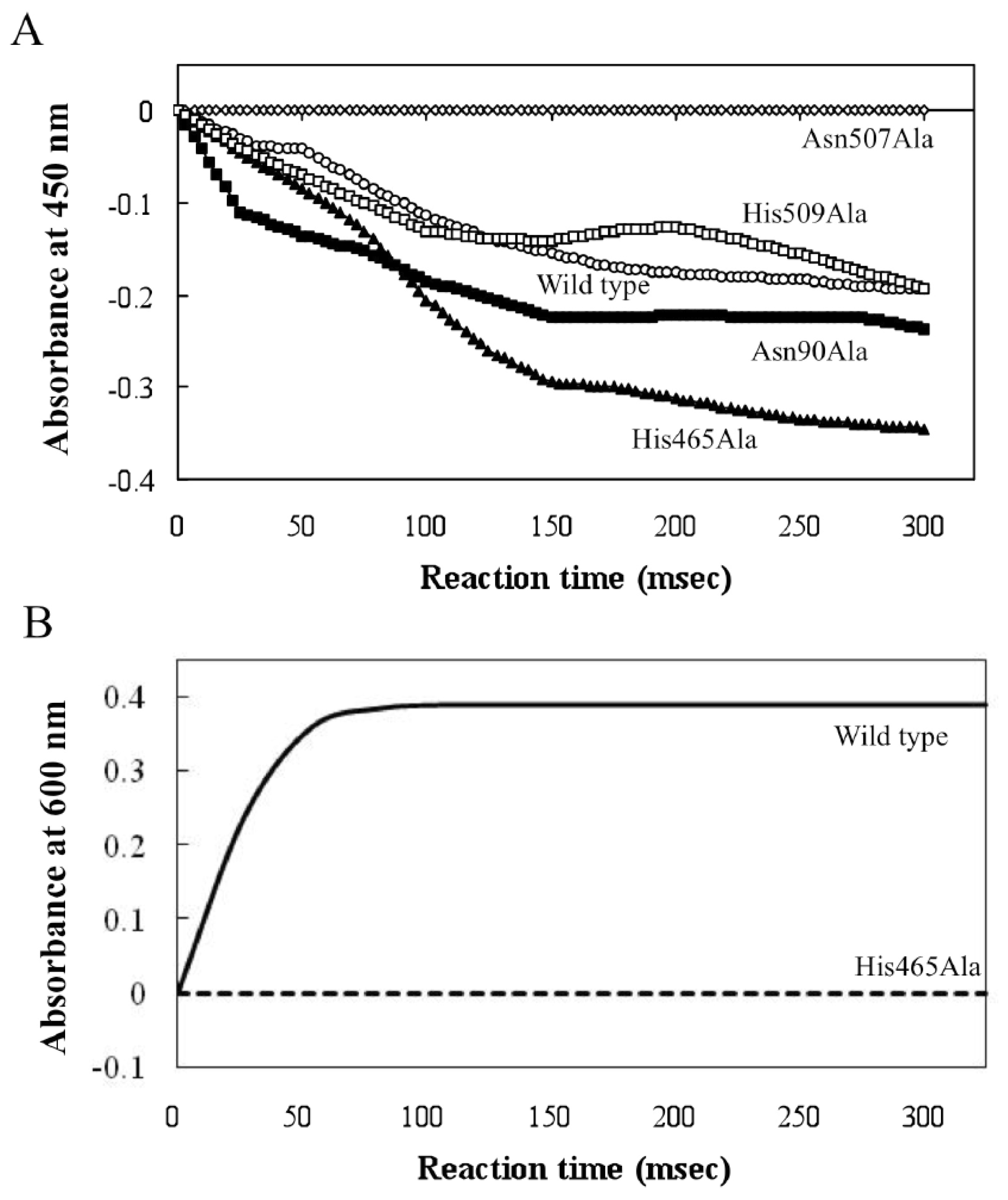
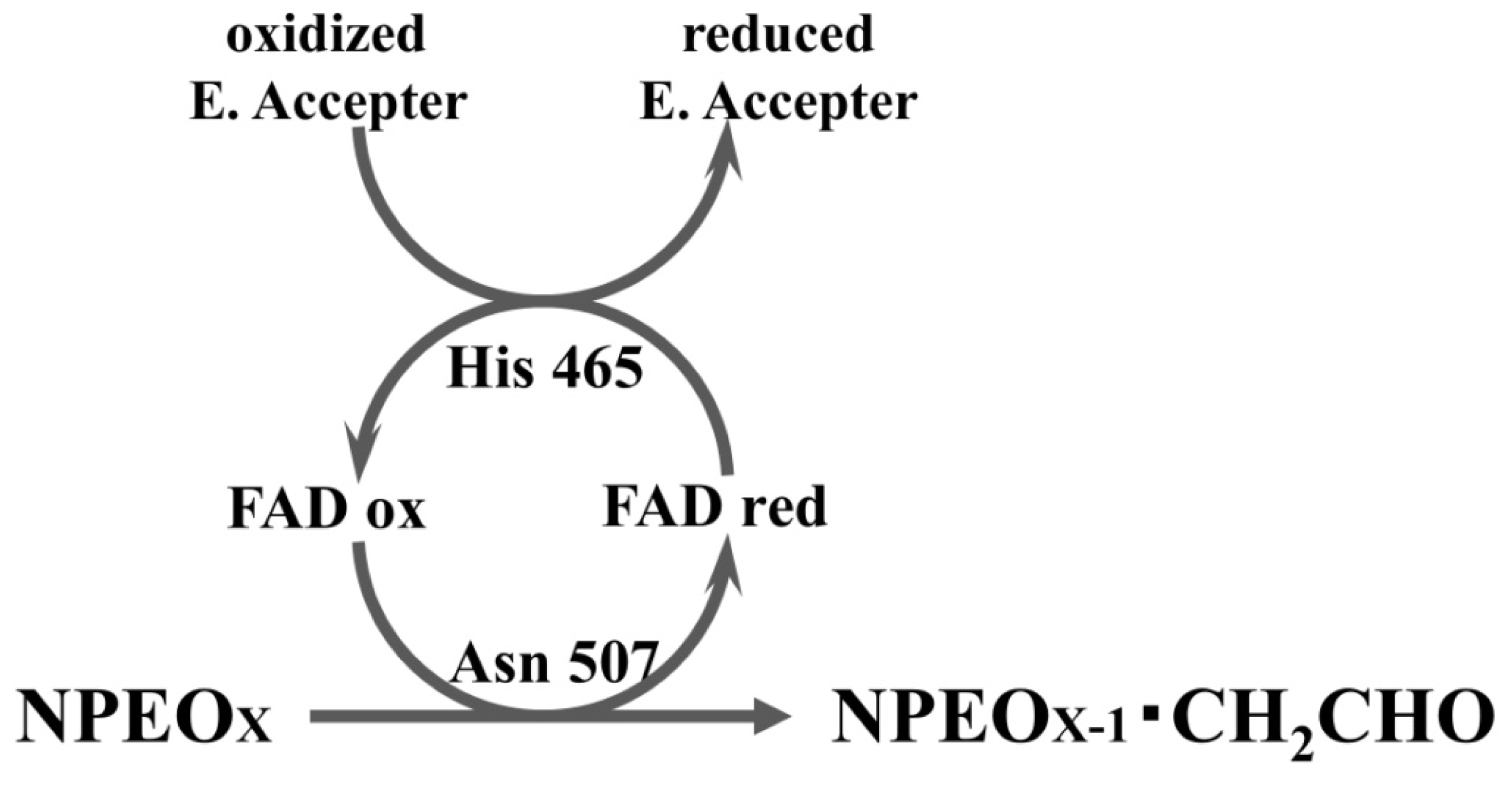
2.3. Roles of Asn507 and His465 in the Catalysis
3. Experimental Section
3.1. Materials, Bacterial Strains and Bacterial Cultivation
3.2. 3D Structure Modeling
3.3. Mutagenesis
3.4. Expression and Purification of Recombinant Proteins
3.5. Enzyme Assay
4. Discussion
5. Conclusions
Supplementary Information
ijms-14-01218-s001.pdfAcknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Cavener, D.R. GMC oxidoreductases. J. Mol. Biol 1992, 223, 811–814. [Google Scholar]
- Kiess, M.; Hecht, H.J.; Kalisz, H.M. Glucose oxidase from Penicillium amagasakiense. Eur. J. Biochem 1998, 252, 90–99. [Google Scholar]
- Mattevi, A. The PHBH fold: Not only flavoenzymes. Biophys. Chem 1998, 70, 217–222. [Google Scholar]
- Dreveny, L.; Gruber, K.; Glieder, A.; Thompson, A.; Kratky, C. The hydroxynitrile lyase from almond: A lyase that looks like an oxidoreductase. Structure 2001, 9, 803–815. [Google Scholar]
- Zámocky, M.; Hallberg, M.; Ludwig, R.; Divne, C.; Haltrich, D. Ancestal gene fusion in cellobiose dehydrogenases reflects a specific evolution of GMC oxidoreductases in fungi. Gene 2004, 338, 1–14. [Google Scholar]
- Massey, V.; Curti, B.; Muller, F.; Mayhew, S.G. On the reaction of borohydride with d- and l-amino acid oxidases. J. Biol. Chem 1968, 243, 1329–1330. [Google Scholar]
- Muller, F.; Massey, V.; Heizmann, C.; Hemmerich, P.; Lhoste, J.M.; Gould, D.C. The reduction of flavins by borohydride: 3,4-dihydroflavin. Eur. J. Biochem 1969, 9, 392–401. [Google Scholar]
- Fitzpatrick, P.F. Substrate dehydrogenation by flavoproteins. Acc. Chem. Res 2001, 34, 299–307. [Google Scholar]
- Gibson, Q.H.; Swoboda, B.E.P.; Massey, V. Kinetics and mechanism of action of glucose oxidase. J. Biol. Chem 1964, 239, 3927–3934. [Google Scholar]
- Wohlfahrt, G.; Witt, S.; Hendle, J.; Schomburg, D.; Kalisz, H.M.; Hecht, H.J. 1.8 and 1.9 Å resolution structures of the Penicillium amagasakiense and Aspergillus niger glucose oxidases as a basis for modeling substrate complexes. Acta Crystallogr. 1999, D55, 969–977. [Google Scholar]
- Witt, S.; Wohlfahrt, G.; Schomburg, D.; Hecht, H.-J.; Kalisz, H.M. Conserved arginine-516 of Penicillium amagasakiense glucose oxidase is essential for the efficient binding of β-d-glucose. Biochem. J 2000, 47, 553–559. [Google Scholar]
- Su, Q.; Klinman, J.P. Nature of oxygen activation in glucose oxidase from Aspergillus niger: The importance of electrostatic stabilization in superoxide formation. Biochemistry 1999, 38, 8572–8581. [Google Scholar]
- Roth, J.P.; Klinman, J.P. Catalysis of electron transfer during activation of O2 by the flavoproein glucose oxidase. Proc. Natl. Acad. Sci. USA 2003, 100, 62–67. [Google Scholar]
- Ghanem, M.; Gadda, G. Effects of reversing the protein positive charge in the proximity of the flavin N(1) locus of choline oxidase. Biochemistry 2006, 45, 3437–3447. [Google Scholar]
- Fan, F.; Gadda, G. On the catalytic mechanism of choline oxidase. J. Am. Chem. Soc 2005, 127, 2067–2074. [Google Scholar]
- Ghanem, M.; Gadda, G. On the catalytic role of the conserved active site residue His466 of choline oxidase. Biochemistry 2005, 44, 893–904. [Google Scholar]
- Ohta, T.; Kawabata, T.; Nishikawa, K.; Tani, A.; Kimbara, K.; Kawai, F. Analysis of amino acid residues involved in catalysis of polyethylene glycol dehydrogenase from Sphingopyxis terrae, using three-dimensional molecular modeling-based kinetic characterization of mutants. Appl. Environ. Microbiol 2006, 72, 4388–4396. [Google Scholar]
- Tasaki, Y.; Yoshikawa, H.; Tamura, H. Isolation and characterization of an alcohol dehydrogenase gene from the octylphenol polyethoxylate degrader Pseudomonas utida S-5. Biosci. Biotechnol. Biochem 2006, 70, 1855–1863. [Google Scholar]
- Liu, X.; Tani, A.; Kimbara, K.; Kawai, F. Xenoestrogenic short ethoxy chain nonylphenol is oxidized by a flavoprotein alcohol dehydrogenase from Ensifer sp. strain AS08. Appl. Microbiol. Biotechnol 2007, 73, 1414–1422. [Google Scholar]
- Sugimoto, M.; Tanabe, M.; Hataya, M.; Enokibara, S.; Duine, J.A.; Kawai, F. The first step in polyethylene glycol degradation by sphingomonads proceeds via a flavoprotein alcohol dehydrogenase containing flavin adenine dinucleotide. J. Bacteriol 2001, 183, 6694–6698. [Google Scholar]
- Kaneko, T.; Nakamura, Y.; Sato, S.; Asamizu, E.; Kato, T.; Sasamoto, S.; Watanabe, A.; Idesama, K.; Ishikawa, A.; Kawashima, K.; et al. Complete genome structure of the nitrogen-fixing symbiotic bacteriumMeshorhizobium loti. DNA Res 2000, 7, 331–338. [Google Scholar]
- Tamura, K.; Peterson, D.; Peterson, N.; Stecher, G.; Nei, M.; Kumar, S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol. Biol. Evol 2011, 28, 2731–2739. [Google Scholar]
- A sense of scale. Available online: http://www.falstad.com/scale/ accessed on 9 January 2013.
- Bowie, J.U.; Lüthy, R.; Eisenberg, D. A method to identify protein sequences that fold into a known three-dimensional structure. Science 1991, 253, 164–170. [Google Scholar]
- Wierenga, R.K.; Terpstra, P.; Hol, W.G.J. Prediction of the occurrence of the ADP-binding βαβ-fold in proteins, using an amino acid sequence finger-print. J. Mol. Biol 1986, 187, 101–107. [Google Scholar]
- Sali, A.; Blundell, T.L. Comparative protein modeling by satisfaction of spatial restraints. J. Mol. Biol 1993, 234, 779–815. [Google Scholar]
- Altchul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Matsudaira, P. Sequence form picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Biol. Chem 1987, 262, 10035–10038. [Google Scholar]
- Antonsen, K.P.; Hoffman, A.S. Poly (Ethylene Glycol) Chemistry; Harris, J.M., Ed.; Plenum Press: New York, NY, USA, 1992; p. 15. [Google Scholar]
- Kawai, F.; Kimura, T.; Tani, Y.; Yamada, H.; Kurachi, M. Purification and characterization of polyethylene glycol dehydrogenase involved in the bacterial metabolism of polyethylene glycol. Appl. Environ. Microbiol 1980, 40, 701–705. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NPEODH | OPEODH | PEGDH | |
|---|---|---|---|
| First Sequence | 365–370 β-strand PPPGER  (hydrophilic) | 355–360 β-strand LHSVIG  (hydrophobic) | 368–373 loop GLKRHG  (hydrophilic) |
| Secnod Sequence | 375–380 β-strand FAVRVC  (hydrophobic) | 375–380 loop FSCHVC 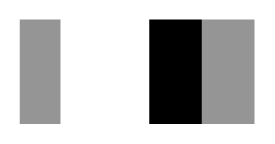 (hydrophobic) | 377–382 loop FSCHVC 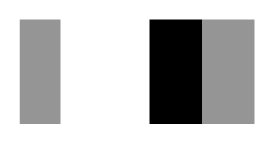 (hydrophobic) |
| Mutant | PEG1,000 | NPEOav10 | ||||||
|---|---|---|---|---|---|---|---|---|
| Vmax (units/mg) | Km mM | kcat s−1 | kcat/Km s−1 mM−1 | Vmax (units/mg) | Km mM | kcat s−1 | kcat/Km s−1 mM−1 | |
| Wild type | 3.9 | 3.3 | 12 | 3.6 | 3.6 | 2.1 | 11 | 5.2 |
| Asn90Ala | 2.8 | 4.5 | 8.4 | 1.9 | 1.8 | 4.1 | 5.5 | 1.3 |
| His465Ala | 0.21 | 2.5 | 0.63 | 0.24 | 0.43 | 2.4 | 1.3 | 0.54 |
| Asn507Ala | n.a. a | - | - | - | - | - | - | - |
| Asn90Ala | 1.6 | 2.8 | 9.1 | 3.3 | 1.2 | 1.7 | 3.6 | 2.1 |
| His465Ala–Asn507Ala | n.a. | - | - | - | - | - | - | - |
| Strain or Plasmid | Genotype and Description | Source or Reference |
|---|---|---|
| Strains | ||
| E. coli BL21(DE3)(pLysS) | Takara Bio | |
| E. coli DH5 α | Takara Bio | |
| Plasmids and primers | ||
| pCold I DNA | 4407 bp; Amp r; His.Tag; TEE; cspA Promotor | Takara Bio |
| pCold-npeA | 1659 bp complete ORF of NPEO-DH ligated with pCold vector | [19] |
| pCold-npeA-N90A | N90A-F1 5′-CATCAATCGCTTCGATGATCGCGAT-3′ N90A-R1 5′-ATCATCGAAGCGATTGATGACGACC-3′ | This study |
| pCold-npeA-H465A | H465A-F1 5′-CGGTATATGCTCCCGTTGGGACCTG-3′ H465A-R1 5′-ATCATCGAAGCGATTGATGACGACC-3′ | This study |
| pCold-npeA-N507A | N507A-F1 5′-TAAGCGGCGCTACAAACCTGCCCAT-3′ N507A-R1 5′-AGGTTTGTAGCGCCGCTTAGAAGCG-3′ | This study |
| Cold-npeA-N509A | N509A-F1 5′-GCAATACAGCTCTGCCCATTATGGC-3′ N509A-R1 5′-ATGGGCAGAGCTGTATTGCCGCTTA-3′ | This study |
| Cold-npeA-H465A-N509A | npeA double mutant in H465A (CAT→GCT) and N507A (AAT→GCT) | This study |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, X.; Ohta, T.; Kawabata, T.; Kawai, F. Catalytic Mechanism of Short Ethoxy Chain Nonylphenol Dehydrogenase Belonging to a Polyethylene Glycol Dehydrogenase Group in the GMC Oxidoreductase Family. Int. J. Mol. Sci. 2013, 14, 1218-1231. https://doi.org/10.3390/ijms14011218
Liu X, Ohta T, Kawabata T, Kawai F. Catalytic Mechanism of Short Ethoxy Chain Nonylphenol Dehydrogenase Belonging to a Polyethylene Glycol Dehydrogenase Group in the GMC Oxidoreductase Family. International Journal of Molecular Sciences. 2013; 14(1):1218-1231. https://doi.org/10.3390/ijms14011218
Chicago/Turabian StyleLiu, Xin, Takeshi Ohta, Takeshi Kawabata, and Fusako Kawai. 2013. "Catalytic Mechanism of Short Ethoxy Chain Nonylphenol Dehydrogenase Belonging to a Polyethylene Glycol Dehydrogenase Group in the GMC Oxidoreductase Family" International Journal of Molecular Sciences 14, no. 1: 1218-1231. https://doi.org/10.3390/ijms14011218