Structural Analysis of Cytochrome P450 105N1 Involved in the Biosynthesis of the Zincophore, Coelibactin


Abstract
:1. Introduction
2. Results and Discussion
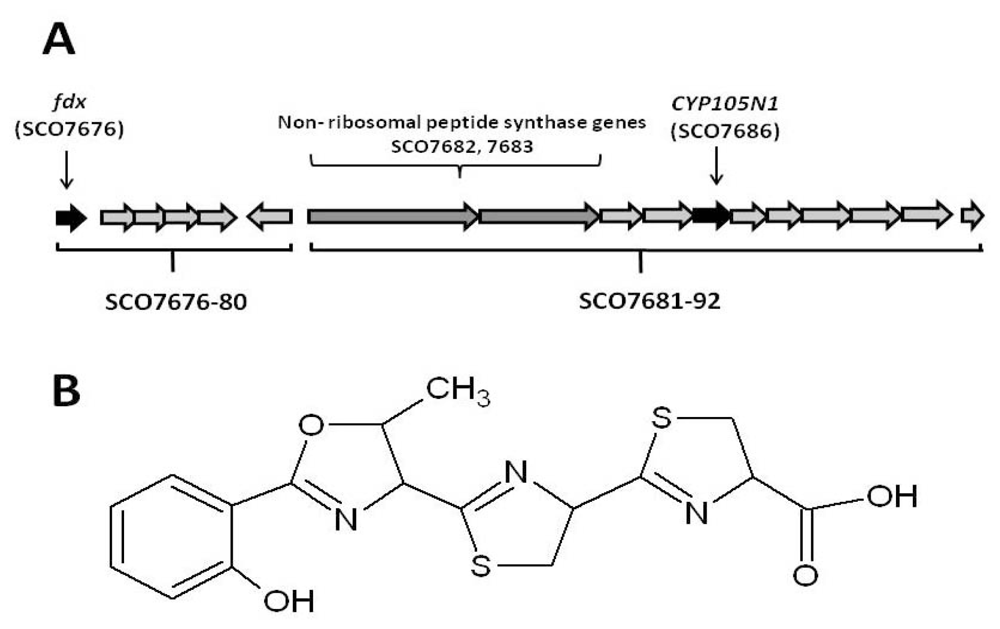
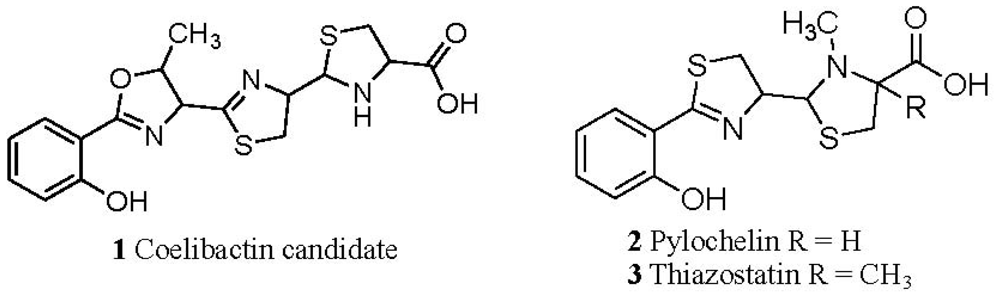
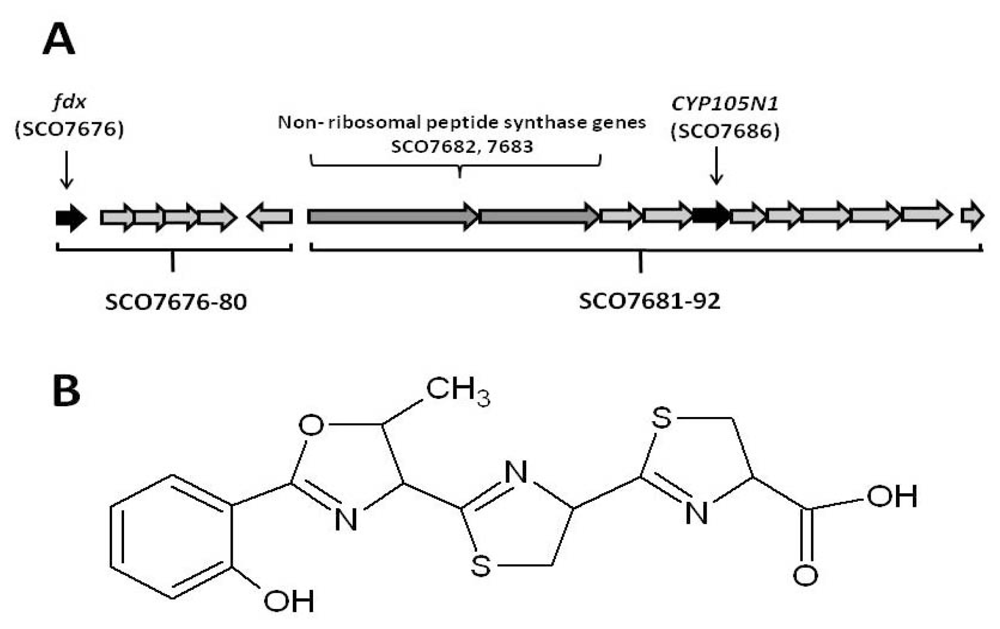
2.1. The Role of the Coelibactin Operon in Zinc Chelation and Bioinformatics Analysis
2.2. Bioinformatic Analysis of CYP105N1
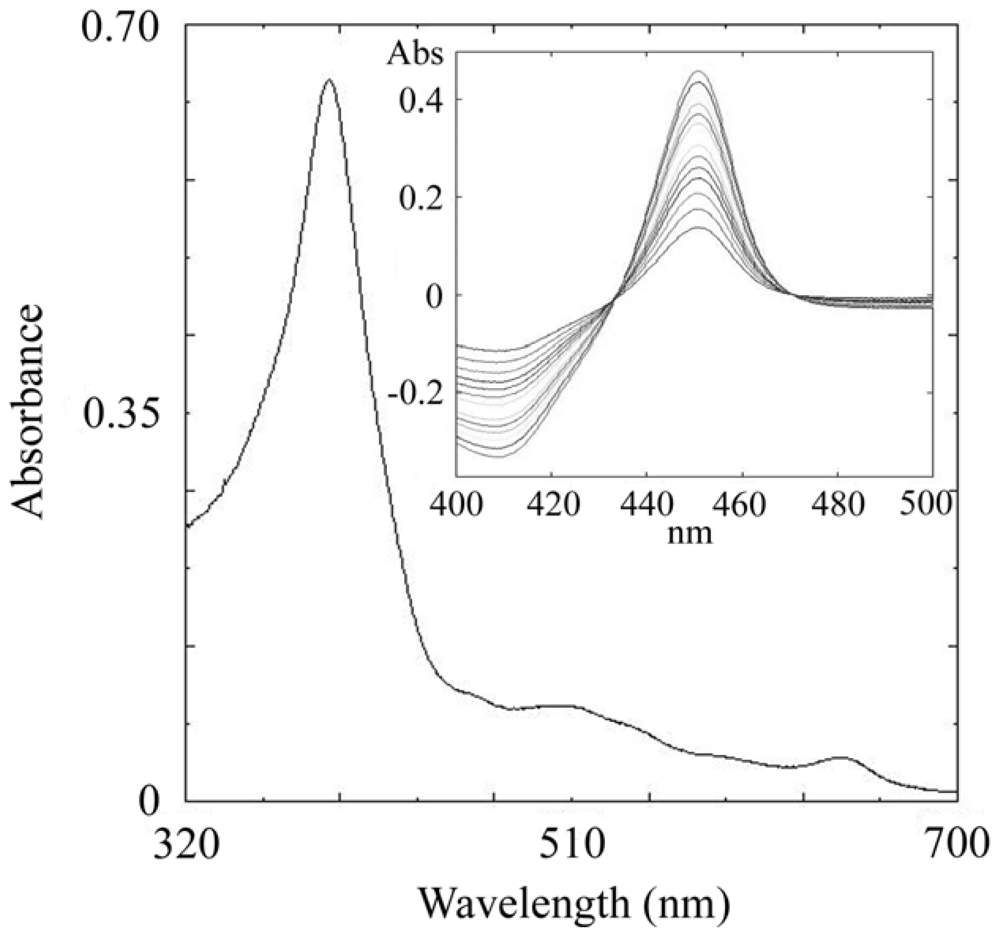
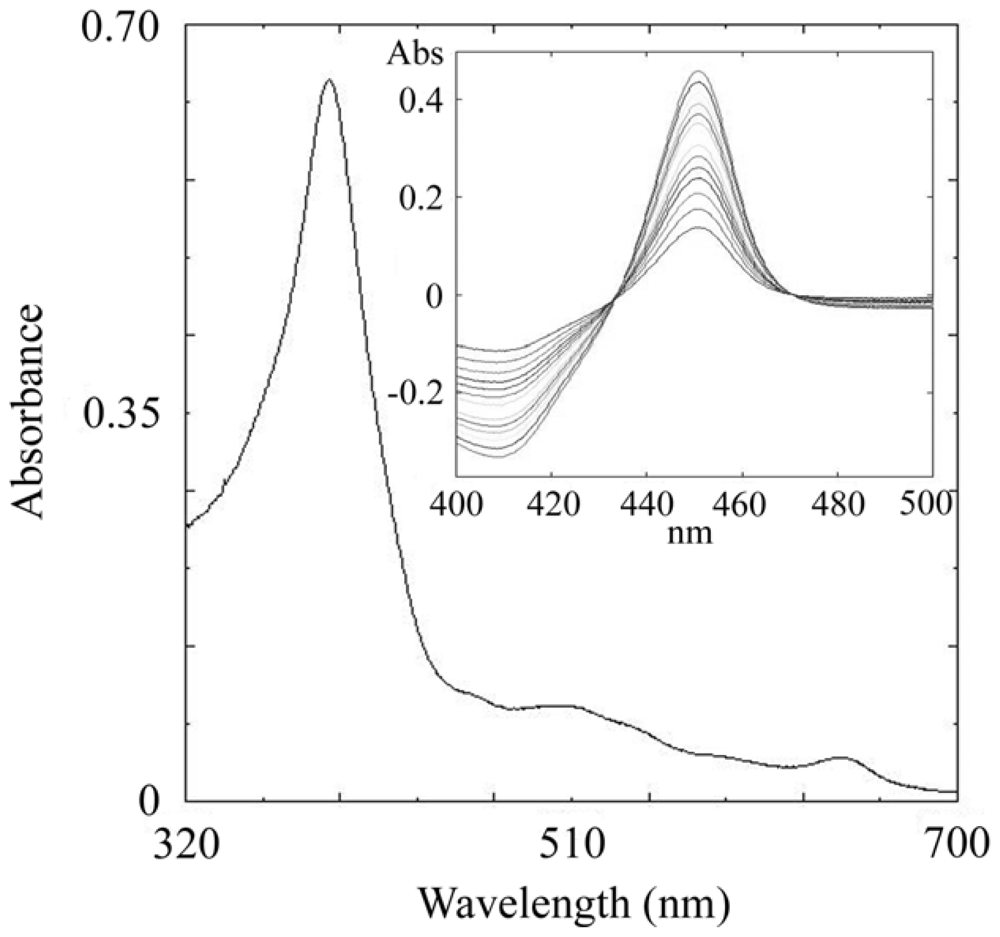
2.3. CYP105N1 Expression, Purification and Spectral Analysis
2.4. Features of the Crystal Structure of CYP105N1
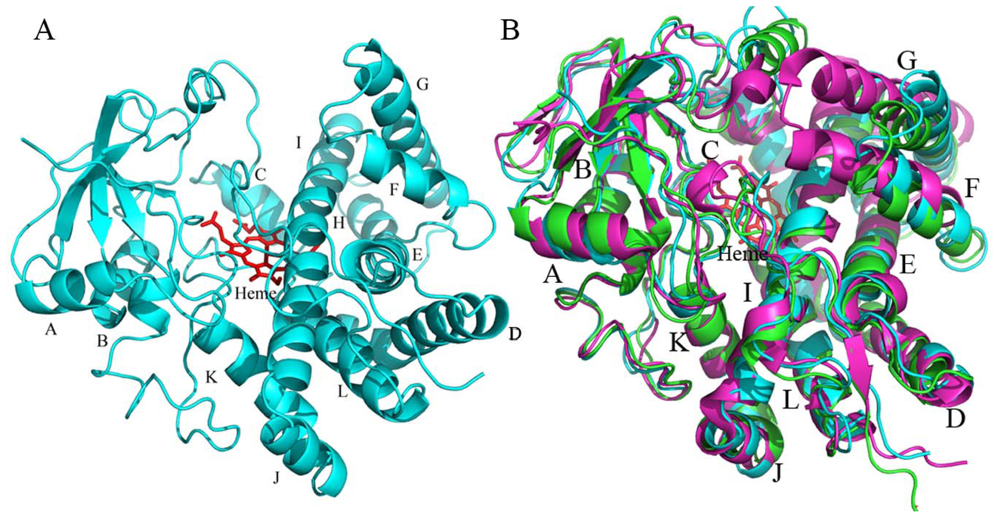
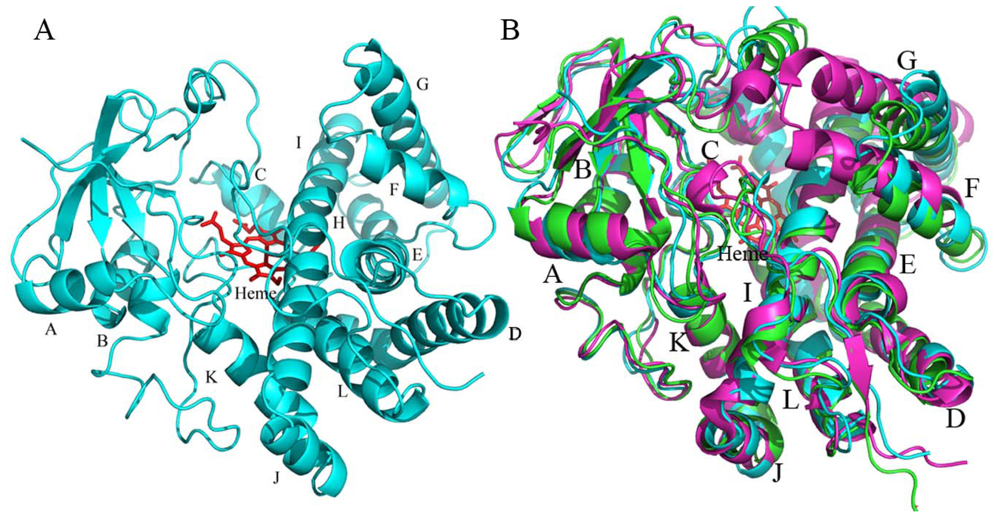
2.4.1. The Overall Structure of CYP105N1
2.4.2. The Heme-Binding Pocket
2.4.3. Comparison of CYP105N1 with Other CYP105s
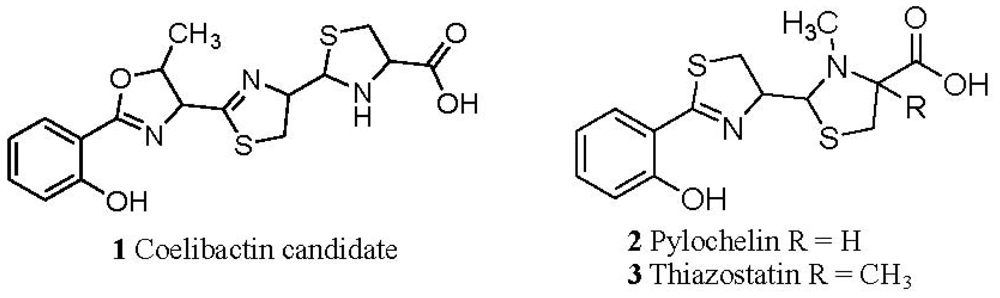
2.5. Conjecture on the Enzymatic Role(s) of CYP105N1 in Production of an Active Siderophore
3. Experimental Section
3.1. Cloning, Heterologous Expression and Purification of Streptomyces coelicolor A3(2) CYP105N1
3.2. Crystallization, Data collection and Structure Determination of CYP105N1
3.3. General Methods
4. Conclusions
Acknowledgments
References
- Nelson, D.R. The cytochrome P450 homepage. Available online: http://drnelson.uthsc.edu/CytochromeP450.html accessed on 3 July 2012.
- De Montellano, P.R.O. Cytochrome P450: Structure, Mechanism and Biochemistry, 3rd ed; Kluwer Academic: New York, NY, USA, 2005; pp. 1–45. [Google Scholar]
- Omura, T. Structural diversity of cytochrome P450 enzyme system. J. Biochem 2010, 147, 297–306. [Google Scholar]
- Rittle, J.; Green, M.T. Cytochrome P450 compound I: Capture, characterization and C–H bond activation kinetics. Science 2010, 330, 933–937. [Google Scholar]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.M.; Maves, S.A.; Benson, D.E.; Sweet, R.M.; Ringe, D.; Petsko, G.A.; Sligar, S.G. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar]
- Guengerich, F.P. Common and uncommon cytochrome P450 reactions related to metabolism and toxicity. Chem. Res. Toxicol 2001, 14, 611–650. [Google Scholar]
- Paine, M.J.I.; Scrutton, N.S.; Munro, A.W.; Guiterrez, A.; Roberts, G.C.K.; Wolf, C.R. Electron Transfer Partners of Cytochrome P450. In Cytochrome P450, Structure, Mechanism and Biochemistry, 3rd ed; de Montellano, P.R.O., Ed.; Kluwer Academic: New York, NY, USA, 2005; pp. 115–138. [Google Scholar]
- Lamb, D.C.; Zhao, B.; Guengerich, F.P.; Kelly, S.L.; Waterman, M.R. Genomics of Streptomyces Cytochromes P450. In Streptomyces: Molecular Biology and Biotechnology; Dyson, P.J., Ed.; Caister Academic Press: Norfolk, UK, 2011; pp. 233–253. [Google Scholar]
- Bentley, S.D.; Chater, K.F.; Cerdeńo-Tárraga, A.M.; Challis, G.L.; Thomson, N.R.; James, K.D.; Harris, D.E.; Quail, M.A.; Kieser, H.; Harper, D.; et al. Complete genome sequence of the model actinomycete Streptomyces coeicolor A3(2). Nature 2002, 417, 141–147. [Google Scholar]
- Hopwood, D.A. Forty years of genetics with Streptomyces, from in vivo through in vitro to in silico. Microbiology 1999, 145, 2183–2202. [Google Scholar]
- Lamb, D.C.; Guengerich, F.P.; Kelly, S.L.; Waterman, M.R. Exploiting Streptomyces coelicolor A3(2) P450s as a model for application in drug discovery. Expert Opin. Drug Metab. Toxicol 2006, 2, 27–40. [Google Scholar]
- Shafiee, A.; Hutchinson, C.R. Purification and reconstitution of the electron transport components for 6-deoxyerythronolide B hydroxylase, a cytochrome P-450 enzyme of macrolide antibiotic (erythromycin) biosynthesis. J. Bacteriol 2007, 170, 1548–1553. [Google Scholar]
- Betlach, M.C.; Kealey, J.T.; Ashley, G.W.; McDaniel, R. Characterization of the macrolide P-450 hydroxylase from Streptomyces venezuelae which converts narbomycin to picromycin. Biochemistry 1998, 37, 14937–14942. [Google Scholar]
- Trower, M.K.; Sariaslani, F.S.; O’Keefe, D.P. Purification and characterization of a soybean flour-induced cytochrome P-450 from Streptomyces griseus. J. Bacteriol 1989, 171, 1781–1787. [Google Scholar]
- Xu, L.H.; Fushinobu, S.; Takamatsu, S.; Wakagi, T.; Ikeda, H.; Shoun, H. Regio- and stereospecificity of filipin hydroxylation sites revealed by crystal structures of cytochrome P450 105P1 and 105D6 from Streptomyces avermitilis. J. Biol. Chem 2010, 285, 16844–16853. [Google Scholar]
- Lamb, D.C.; Skaug, T.; Song, H.L.; Jackson, C.J.; Podust, L.M.; Waterman, M.R.; Kell, D.B.; Kelly, D.E.; Kelly, S.L. The cytochrome P450 complement (CYPome) of Streptomyces coelicolor A3(2). J. Biol. Chem 2002, 277, 24000–24005. [Google Scholar]
- Zhao, B.; Guengerich, F.P.; Bellamine, A.; Lamb, D.C.; Izumikawa, M.; Lei, L.; Podust, L.M.; Sundaramoorthy, M.; Kalaitzis, J.A.; Reddy, L.M.; et al. Binding of two flaviolin substrate molecules, oxidative coupling, and crystal structure of Streptomyces coelicolor A3(2) cytochrome P450 158A2. J. Biol. Chem 2005, 280, 11599–11607. [Google Scholar]
- Zhao, B.; Lin, X.; Lei, L.; Lamb, D.C.; Kelly, S.L.; Waterman, M.R.; Cane, D.E. Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor A3(2). J. Biol. Chem 2008, 283, 8183–8189. [Google Scholar]
- Zhao, B.; Lei, L.; Vassylyev, D.G.; Lin, X.; Cane, D.E.; Kelly, S.L.; Yuan, H.; Lamb, D.C.; Waterman, M.R. Crystal structure of albaflavenone monooxygenase containing a moonlighting terpene synthase active site. J. Biol. Chem 2009, 284, 36711–36719. [Google Scholar]
- Hesketh, A.; Kock, H.; Mootien, S.; Bibb, M. The role of absC, a novel regulatory gene for secondary metabolism, in zinc-dependent antibiotic production in Streptomyces coelicolor A3(2). Mol. Microbiol 2009, 74, 1427–1444. [Google Scholar]
- Kallifidas, D.; Pascoe, B.; Owen, G.A.; Strain-Damerell, C.M.; Hong, H.J.; Paget, M.S. The zinc-responsive regulator Zur controls expression of the coelibactin gene cluster in Streptomyces coelicolor. J. Bacteriol 2010, 192, 608–611. [Google Scholar]
- Matsuoka, T.; Miyakoshi, S.; Tanzawa, K.; Nakahara, K.; Hosobuchi, M.; Serizawa, N. Purification and characterization of cytochrome P-450sca from Streptomyces carbophilus. Eur. J. Biochem 1989, 184, 707–713. [Google Scholar]
- Sugimoto, H.; Shinkyo, R.; Hayashi, K.; Yoneda, S.; Yamada, M.; Kamakura, M.; Ikushiro, S.; Shiro, Y.; Sakaki, T. Crystal structure of CYP105A1 (P450SU-1) in complex with 1alpha, 25-dihydroxyvitamin D3. Biochemistry 2008, 47, 4017–4027. [Google Scholar]
- Ito, S.; Matsuoka, T.; Watanabe, I.; Kagasaki, T.; Serizawa, N.; Hata, T. Crystallization and preliminary X-ray diffraction analysis of cytochrome P450sca-2 from Streptomyces carbophilus involved in production of pravastatin sodium, a tissue-selective inhibitor of HMG-CoA reductase. Acta Crystallogr. D 1999, 55, 1209–1211. [Google Scholar]
- Yasutake, Y.; Imoto, N.; Fujii, Y.; Fujii, T.; Arisawa, A.; Tamura, T. Crystal structure of cytochrome P450 MoxA from Nonomuraea recticatena (CYP105). Biochem. Biophys. Res. Commun 2007, 361, 876–882. [Google Scholar]
- Arase, M.; Waterman, M.R.; Kawaga, N. Purification and characterization of bovine steroid 21-hydroxylase (P450c21) efficiently expressed in Escherichia coli. Biochem. Biophys. Res. Commun 2006, 344, 400–405. [Google Scholar]
- Cytochrome P450 Homepage. Available online: http://drnelson.uthsc.edu/bacterial.P450s.2011.htm accessed on 3 July 2012.
- Hider, R.C.; Kong, X. Chemistry and biology of siderophores. Nat. Prod. Rep 2010, 27, 637–657. [Google Scholar]
- Cox, C.D.; Rinehart, K.L.; Moore, M.L.; Cook, J.C. Pyochelin: Novel structure of an iron-chelating growth promoter for Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 1981, 78, 4256–4260. [Google Scholar]
- Shindo, K.; Takenaka, A.; Noguchi, T.; Hayakawa, Y.; Seto, H. Thiazostatin A and thiazostatin B, new antioxidants produced by Streptomyces tolurosus. J. Antibiot 1989, 42, 1526–1529. [Google Scholar]
- Otwinowski, Z.; Borek, D.; Majewski, W.; Minor, W. Multiparametric scaling of diffraction intensities. Acta Crystallogr. A 2003, 59, 228–234. [Google Scholar]
- Storoni, L.C.; McCoy, A.J.; Read, R.J. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D 2004, 60, 432–438. [Google Scholar]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of Coot. Acta Crystallogr. D 2010, 66, 486–501. [Google Scholar]
- Brunger, A.T.; Adams, P.D.; Clore, G.M.; DeLano, W.L.; Gros, P.; Grosse-Kunstleve, R.W.; Jiang, J.S.; Kuszewski, J.; Nilges, M.; Pannu, N.S.; et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D 1998, 54, 905–921. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.2r3pre; Schrödinger LLC: New York, NY, USA.
- Omura, T.; Sato, R. The carbon monoxide binding pigment of liver microsomes. I. Evidence for its hemoprotein in nature. J. Biol. Chem 1964, 239, 2370–2378. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Collection Statistics | Substrate-free |
|---|---|
| Space group | P61 |
| Unit Cell (Å) | a = b = 134.537 c = 230.58 |
| Molecules/asymmetric unit | 4 |
| Data resolution (Å) | 2.9 |
| Redundancya | 16.3 (9.9) |
| Completeness%a | 99.8 (99.8) |
| I/σ(I)a | 26.8 (6.1) |
| Rmerge%a | 7.1 (54.2) |
| Refinement statistics | |
| No. of reflections used in refinement | 51760 |
| No. of water molecules | 100 |
| Protein atoms | 12366 |
| Heme atoms | 172 |
| Ligand atoms | 0 |
| Rwork% | 28.21 |
| Rfree% | 30.05 |
| Rmsdb in bond lengths (Å) | 0.009 |
| Rmsd in bond angles (°) | 1.2 |
| Ramachandran statistics | |
| Favored regions (%) | 94.9 |
| Allowed regions (%) | 3.2 |
| Outerliers (%) | 1.9 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhao, B.; Moody, S.C.; Hider, R.C.; Lei, L.; Kelly, S.L.; Waterman, M.R.; Lamb, D.C. Structural Analysis of Cytochrome P450 105N1 Involved in the Biosynthesis of the Zincophore, Coelibactin. Int. J. Mol. Sci. 2012, 13, 8500-8513. https://doi.org/10.3390/ijms13078500
Zhao B, Moody SC, Hider RC, Lei L, Kelly SL, Waterman MR, Lamb DC. Structural Analysis of Cytochrome P450 105N1 Involved in the Biosynthesis of the Zincophore, Coelibactin. International Journal of Molecular Sciences. 2012; 13(7):8500-8513. https://doi.org/10.3390/ijms13078500
Chicago/Turabian StyleZhao, Bin, Suzy C. Moody, Robert C. Hider, Li Lei, Steven L. Kelly, Michael R. Waterman, and David C. Lamb. 2012. "Structural Analysis of Cytochrome P450 105N1 Involved in the Biosynthesis of the Zincophore, Coelibactin" International Journal of Molecular Sciences 13, no. 7: 8500-8513. https://doi.org/10.3390/ijms13078500