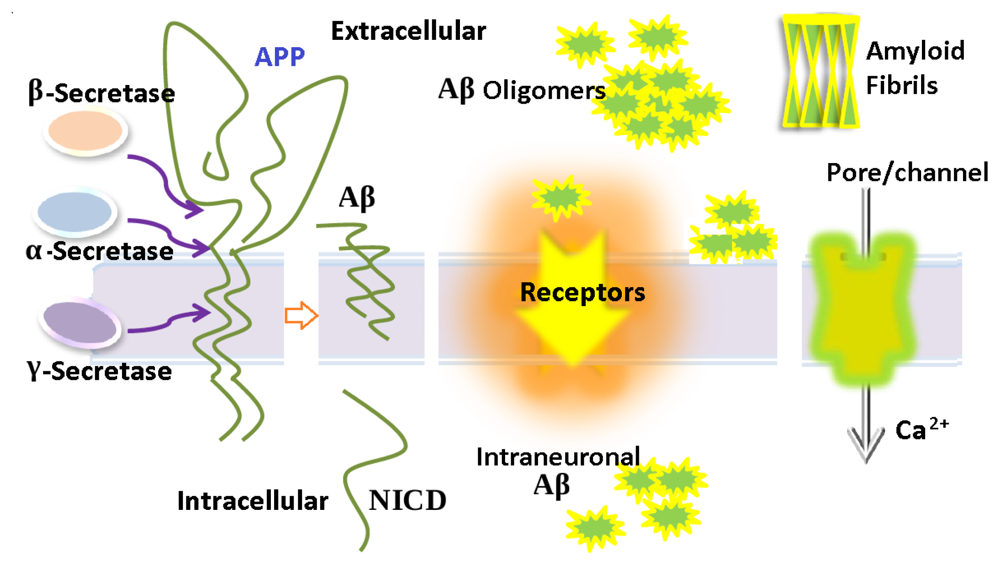
The Toxicity of Amyloid ß Oligomers



Abstract
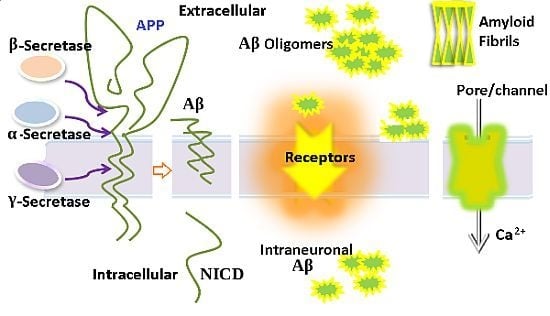
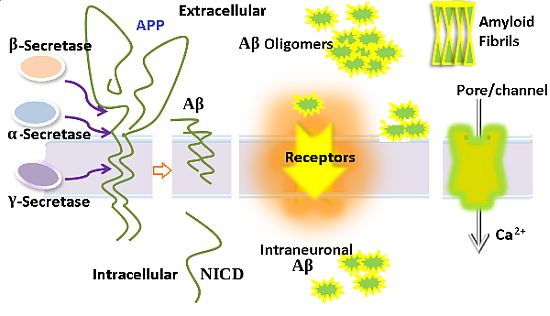
:
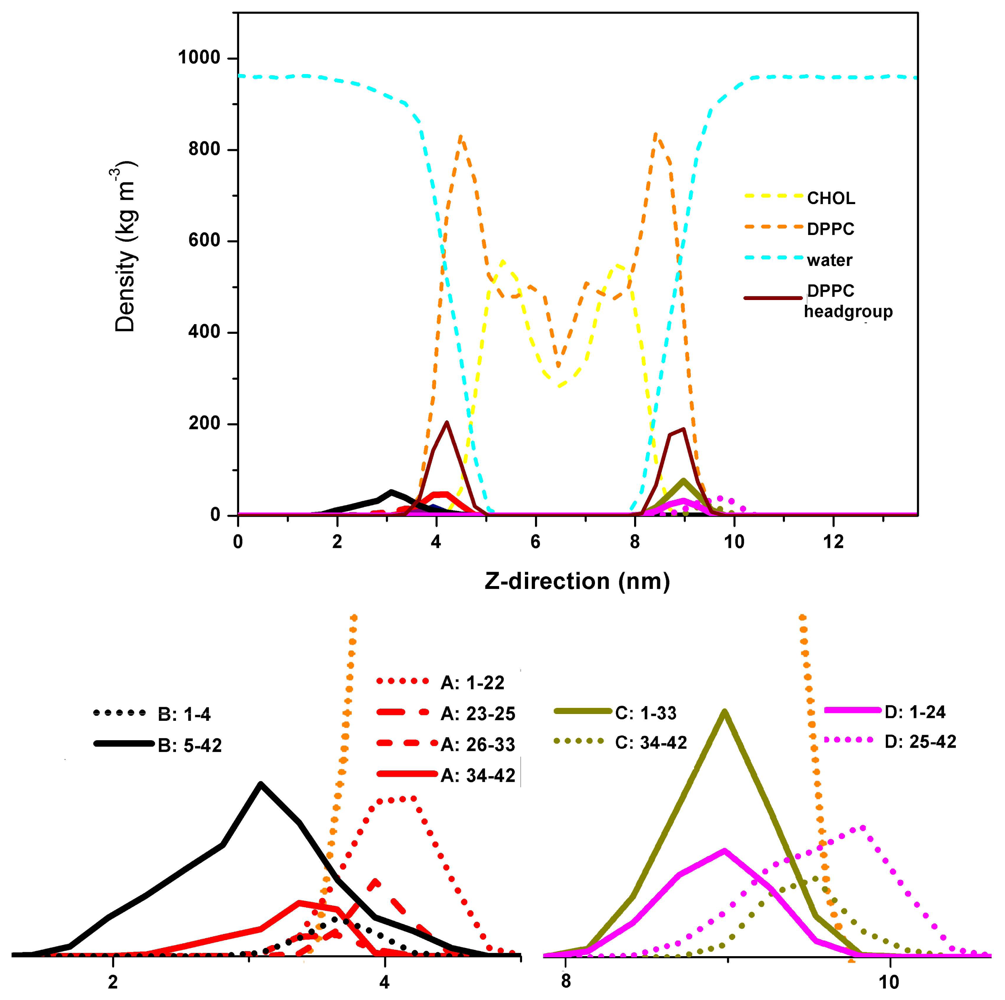
{kind=link}
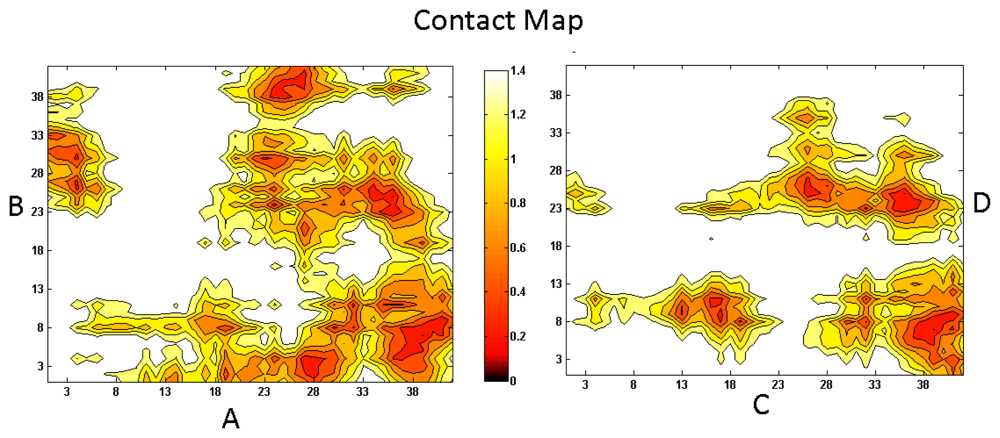
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Aβ Oligomers in Aqueous Environment
3. Aβ Adsorption and Insertion Mechanism
4. Membrane-Mediated Aβ Aggregation Mechanism
5. Intraneuronal Aβ Accumulation Mechanism
6. Ion Channels/Pore Formation by the Incorporation of Aβ into the Cell Membrane
7. The Relationship of Aβ with Different Receptors
8. Oxidative Stress
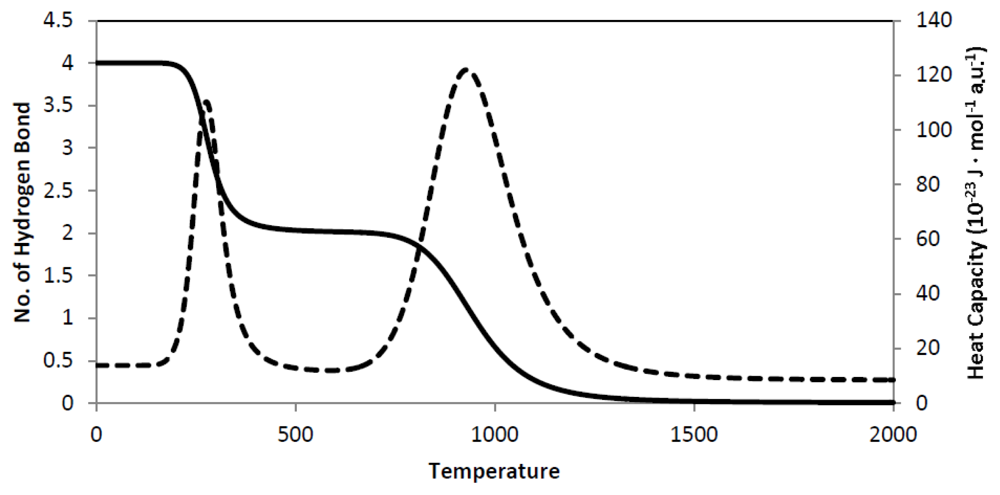
9. Secondary Structure Phase Transition
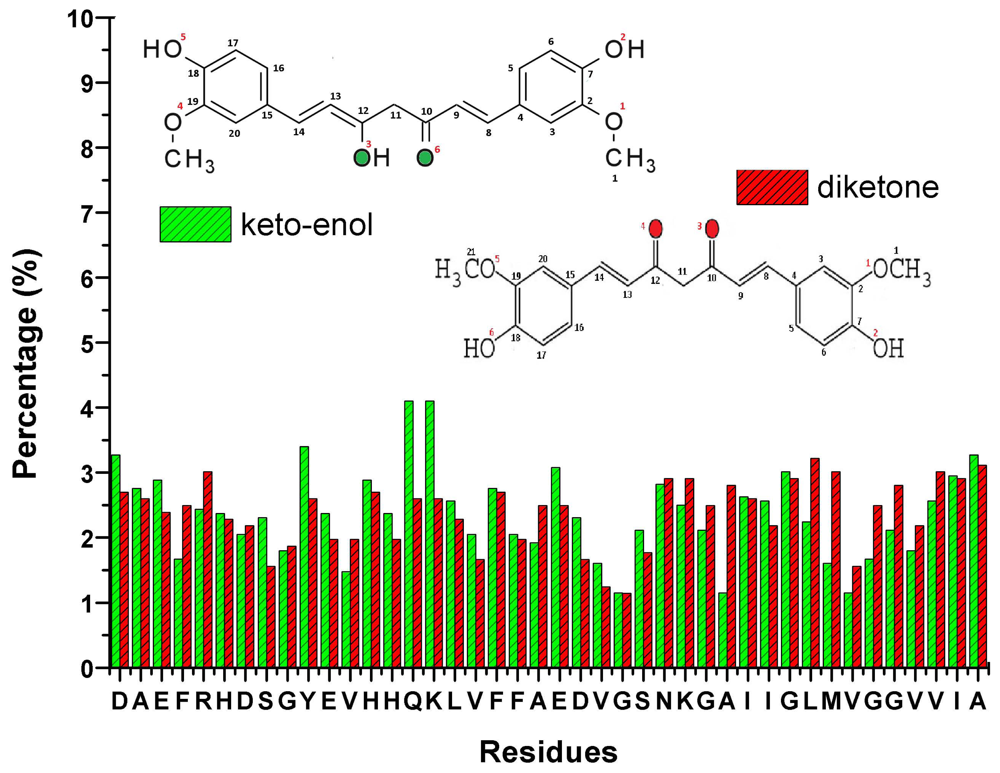
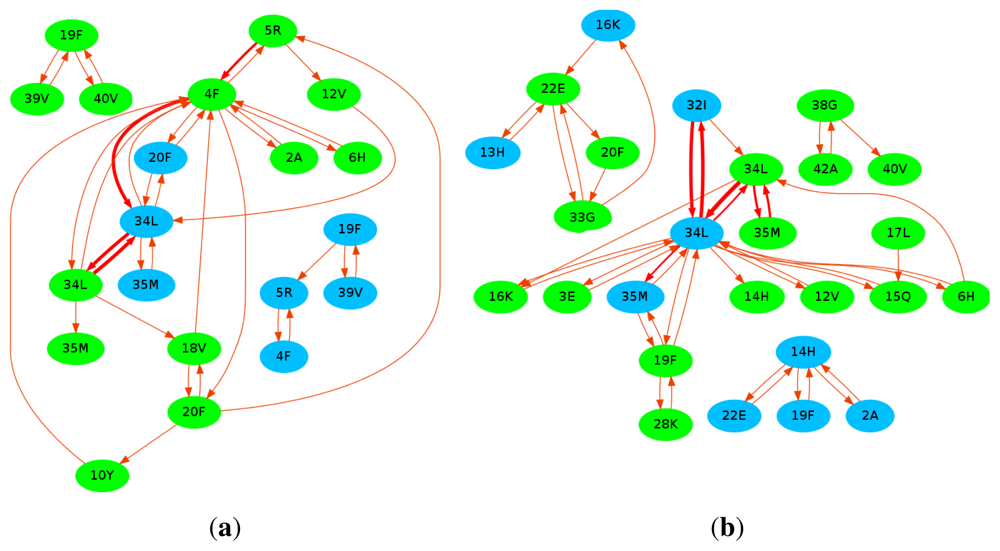
10. Curcumin
11. Conclusions
Acknowledgements
References
- Kirkitadze, M.D.; Bitan, G.; Teplow, D.B. Paradigm shifts in Alzheimer’s disease and other neurodegenerative disorders: The emerging role of oligomeric assemblies. J. Neurosci. Res 2002, 69, 567–577. [Google Scholar]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar]
- Verdier, Y.; Penke, B. Binding sites of amyloid beta-peptide in cell plasma membrane and implications for Alzheimer’s disease. Curr. Protein Pept. Sci 2004, 5, 19–31. [Google Scholar]
- Mutisya, E.M.; Bowling, A.C.; Beal, M.F. Cortical cytochrome oxidase activity is reduced in Alzheimer’s disease. J. Neurochem 1994, 63, 2179–2184. [Google Scholar]
- Huang, H.C.; Lin, C.J.; Liu, W.J.; Jiang, R.R.; Jiang, Z.F. Dual effects of curcumin on neuronal oxidative stress in the presence of Cu(II). Food Chem. Toxicol 2011, 49, 1578–1583. [Google Scholar]
- Atamna, H.; Boyle, K. Amyloid-β peptide binds with heme to form a peroxidase: Relationship to the cytopathologies of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2006, 103, 3381–3386. [Google Scholar]
- Wang, Q.; Walsh, D.M.; Rowan, M.J.; Selkoe, D.J.; Anwyl, R. Block of long-term potentiation by naturally secreted and synthetic amyloid beta-peptide in hippocampal slices is mediated via activation of the kinases c-Jun N-terminal kinase, cyclin-dependent kinase 5, and p38 mitogen-activated protein kinase as well as metabotropic glutamate receptor type 5. J. Neurosci 2004, 24, 3370–3378. [Google Scholar]
- Jang, H.; Zheng, J.; Lal, R.; Nussinov, R. New structures help the modeling of toxic amyloidβ ion channels. Trends Biochem. Sci 2008, 33, 91–100. [Google Scholar]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Lal, R.; Nussinov, R. Structural convergence among diverse, toxic beta-sheet ion channels. J. Phys. Chem. B 2010, 114, 9445–9451. [Google Scholar]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Azimova, R.; Kagan, B.L.; Nussinov, R.; Lal, R. Truncated β-amyloid peptide channels provide an alternative mechanism for Alzheimer’s disease and Down syndrome. Proc. Natl. Acad. Sci. USA 2010, 107, 6538–6543. [Google Scholar]
- Jang, H.; Arce, F.T.; Ramachandran, S.; Capone, R.; Lal, R.; Nussinov, R. β-Barrel Topology of Alzheimer’s β-Amyloid Ion Channels. J. Mol. Biol 2010, 404, 917–934. [Google Scholar]
- Shafrir, Y.; Durell, S.; Arispe, N.; Guy, H.R. Models of membrane-bound Alzheimer’s Abeta peptide assemblies. Proteins Struct. Funct. Bioinforma 2010, 78, 3473–3487. [Google Scholar]
- Sepulveda, F.J.; Parodi, J.; Peoples, R.W.; Opazo, C.; Aguayo, L.G. Synaptotoxicity of Alzheimer beta amyloid can be explained by its membrane perforating property. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Geng, J.; Zhao, C.; Ren, J.; Qu, X. Alzheimer’s disease Amyloid beta converting left-handed Z-DNAs back to right-handed B-form. Chem. Commun 2010, 46, 7187–7189. [Google Scholar]
- Lührs, T.; Ritter, C.; Adrian, M.; RiekLoher, D.; Bohrmann, B.; Dbeli, H.; Schubert, D.; Riek, R. 3D structure of Alzheimer’s amyloid-β (1–42) fibrils. Proc. Natl. Acad. Sci. USA 2005, 102, 17342–17347. [Google Scholar]
- Takahashi, T.; Mihara, H. Peptide and protein mimetics inhibiting amyloid beta-peptide aggregation. Acc. Chem. Res 2008, 41, 1309–1318. [Google Scholar]
- Kirkitadze, M.D.; Kowalska, A. Molecular mechanisms initiating amyloid beta-fibril formation in Alzheimer’s disease. Acta Biochim. Pol 2005, 52, 417–423. [Google Scholar]
- Zhao, L.N.; Chiu, S.W.; Benoit, J.; Chew, L.Y.; Mu, Y. Amyloid β peptide aggregation in a mixed membrane bilayer: A molecule dynamic study. J. Phys. Chem. B 2011, 115, 12247–12256. [Google Scholar]
- Zhao, L.N.; Chiu, S.W.; Benoit, J.; Chew, L.Y.; Mu, Y. The effect of curcumin on the stability of Aβ dimers. J. Phys. Chem. B 2012. (in Press). [Google Scholar]
- Jang, S.; Shin, S. Amyloid β-peptide oligomerization in silico: Dimer and trimer. J. Phys. Chem. B 2006, 110, 1955–1958. [Google Scholar]
- Urbanc, B.; Cruz, L.; Ding, F.; Sammond, D.; Khare, S.; Buldyrev, S.V.; Stanley, H.E.; Dokholyan, N.V. Molecular dynamics simulation of amyloid β dimer formation. Biophys. J 2004, 87, 2310–2321. [Google Scholar]
- Barz, B.; Urbanc, B. Dimer formation enhances structural differences between amyloid β-protein (1–40) and (1–42): An explicit-solvent molecular dynamics study. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Côté, S.; Laghaei, R.; Derreumaux, P.; Mousseau, N. Distinct dimerization for various alloforms of the amyloid-beta protein: Aβ1–40, Aβ1–42, and Aβ1–40(D23N). J. Phys. Chem. B 2012, 116, 4043–4055. [Google Scholar]
- Mitternacht, S.; Staneva, I.; Härd, T.; Irbäck, A. Monte carlo study of the formation and conformational properties of dimers of Aβ 42 variants. J. Mol. Biol 2011, 410, 357–367. [Google Scholar]
- Williams, T.L.; Serpell, L.C. Membrane and surface interactions of Alzheimer’s Aβ peptide-insights into the mechanism of cytotoxicity. FEBS J 2011, 278, 3905–3917. [Google Scholar]
- Zhao, J.; Wang, Q.M.; Liang, G.Z.; Zheng, J. Molecular dynamics simulations of low-ordered Alzheimer beta-amyloid oligomers from dimer to hexamer on self-assembled monolayers. Langmuir 2011, 27, 14876–14887. [Google Scholar]
- Wang, Q.; Zhao, J.; Yu, X.; Zhao, C.; Li, L.; Zheng, J. Alzheimer Abeta(1–42) monomer adsorbed on the self-assembled monolayers. Langmuir 2010, 26, 12722–12732. [Google Scholar]
- Tsai, H.H.; Lee, J.B.; Tseng, S.S.; Pan, X.A.; Shih, Y.C. Folding and membrane insertion of amyloid-beta (25–35) peptide and its mutants: Implications for aggregation and neurotoxicity. Protein 2010, 78, 1909–1925. [Google Scholar]
- Maltseva, E.; Brezesinski, G. Adsorption of amyloid beta (1–40) peptide to phosphatidyl-ethanolamine monolayers. ChemPhysChem 2004, 5, 1185–1190. [Google Scholar]
- Ionov, M.; Klajnert, B.; Gardikis, K.; Hatziantoniou, S.; Palecz, B.; Salakhutdinov, B.; Cladera, J.; Zamaraeva, M.; Demetzos, C.; Bryszewska, M. Effect of amyloid beta peptides A β1–28 and Aβ25–40 on model lipid membranes. J. Therm. Anal. Calorim 2010, 99, 741–747. [Google Scholar]
- Sgourakis, N.G.; Yan, Y.; McCallum, S.A.; Wang, C.; Garcia, A.E. The Alzheimer’s peptides Aβ 40 and 42 adopt distinct conformations in water: A combined MD/NMR study. J. Mol. Biol 2007, 368, 1448–1457. [Google Scholar]
- Krone, M.G.; Hua, L.; Soto, P.; Zhou, R.; Berne, B.J.; Shea, J.E. Role of water in mediating the assembly of Alzheimer amyloid Aβ 16–22 protofilaments. J. Am. Chem. Soc 2008, 130, 11066–11072. [Google Scholar]
- Glabe, C.C. Amyloid accumulation and pathogensis of Alzheimer’s Disease: Significance of monomeric, oligomeric and fibrillar Aβ. Subcell. Biochem 2005, 28, 167–177. [Google Scholar]
- Chiu, S.; Pandti, S.; Scott, H.L.; Jakobsson, E. An improved united atom force field for simulation of mixed lipid bilayers. J. Phys. Chem. B 2009, 113, 2748–2763. [Google Scholar]
- Pandit, S.A.; Chiu, S.W.; Jakobsson, E.; Grama, A.; Scott, H.L. Cholesterol packing around lipids with saturated and unsaturated chains: A simulation study. Langmuir 2008, 24, 6858–6865. [Google Scholar]
- Pandit, S.A.; Chiu, S.W.; Jakobsson, E.; Grama, A.; Scott, H.L. Cholesterol Surrogates: A comparison of cholesterol and 16:0 ceramide in POPC Bilayers. Biophys. J 2007, 92, 920–927. [Google Scholar]
- Pandit, S.A.; Vasudevan, S.; Chiu, S.W.; Jakobsson, E.; Scott, H.L. Sphingomyelin-cholesterol domains in phospholipid membranes: Atomistic simulation. Biophys. J 2004, 87, 1092–1100. [Google Scholar]
- Chiu, S.W.; Vasudevan, S.; Jakobsson, E.; Mashl, R.J.; Scott, H.L. Structure of sphingomyelin bilayers: A simulation study. Biophys. J 2003, 85, 3624–3635. [Google Scholar]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys 1984, 55, 255–268. [Google Scholar]
- Hoover, W.G. Canonical dynamics: Equilibrium phase space distributions. Phys. Rev. A 1985, 31, 1695–1697. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 113, 1463–1472. [Google Scholar]
- Miyashita, N.; Straub, J.E.; Thirumalai, D.; Sugita, Y. Transmembrane structures of amyloid precursor protein dimer predicted by replica-exchange molecular dynamics simulations. J. Am. Chem. Soc 2009, 131, 3438–3439. [Google Scholar]
- Leoni, V.; Solomon, A.; Kivipelto, M. Links between ApoE brain cholesterol metabolism, tau and amyloid β-peptide in patients with cognitive impairment. Biochem. Soc. Trans 2010, 38, 1021–1025. [Google Scholar]
- Okada, T.; Wakabayashi, M.; Ikeda, K.; Matsuzaki, K. Formation of toxic fibrils of Alzheimer’s amyloid beta-protein-(1–40) by monosialoganglioside GM1, a neuronal membrane component. J. Mol.Biol 2007, 371, 481–489. [Google Scholar]
- Molander-Melin, M.; Blennow, K.; Bogdanovic, N.; Dellheden, B.; Månsson, J.E.; Fredman, P. Structural membrane alterations in Alzheimer brains found to be associated with regional disease development: Increased density of gangliosides GM1 and GM2 and loss of cholesterol in detergent-resistant membrane domains. J. Neurochem 2005, 92, 171–182. [Google Scholar]
- Wakabayashi, M.; Matsuzaki, K. Ganglioside-induced amyloid formation by human islet amyloid polypeptide in lipid rafts. FEBS Lett 2009, 583, 2854–2858. [Google Scholar]
- Kakio, A.; Nishimoto, S.I.; Kozotsumi, Y.; Matsuzaki, K. Formation of a membrane-active form of amyloid β-protein in raft-like model membranes. Biochem. Biophys. Res. Commun 2003, 303, 514–518. [Google Scholar]
- Yahi, N.; Aulas, A.; Fantini, J. How cholesterol constrains glycolipid conformation for optimal recognition of Alzheimer’s β amyloid peptide (Aβ1-40). PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Yu, X.; Zheng, J. Cholesterol promotes the interaction of Alzheimer β-amyloid monomer with lipid bilayer. J. Mol. Biol 2011. (in Press). [Google Scholar]
- Wakabayashi, M.; Okada, T.; Kozutsumi, Y.; Matsuzaki, K. GM1 ganglioside-mediated accumulation of amyloid beta-protein on cell membranes. Biochem. Biophys. Res. Commun 2005, 328, 1019–1023. [Google Scholar]
- Kakio, A.; ichi Nishimoto, S.; Yanagisawa, K.; Kozutsumi, Y.; Matsuzaki, K. Interactions of amyloid β-protein with various gangliosides in raft-like membranes: Importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid? Biochemistry 2002, 41, 7385–7390. [Google Scholar]
- Abramov, A.Y.; Ionov, M.; Pavlov, E.; Duchen, M.R. Membrane cholesterol content plays a key role in the neurotoxicity of β-amyloid: Implications for Alzheimer’s disease. Aging Cell 2011, 10, 595–603. [Google Scholar]
- Fantini, J.; Yahi, N. Molecular insights into amyloid regulation by membrane cholesterol and sphingolipids: Common mechanisms in neurodegenerative diseases. Expert Rev. Mol. Med 2010, 12. [Google Scholar] [CrossRef]
- Gouras, G.K.; Tsai, J.; Naslund, J.; Vincent, B.; Edgar, M.; Checler, F.; Greenfield, J.P.; Haroutunian, V.; Buxbaum, J.D.; Xu, H.; et al. Intraneuronal Aβ42 accumulation in human brain. Am. J. Pathol 2000, 156, 15–20. [Google Scholar]
- Wegiel, J.; Kuchna, I.; Nowicki, K.; Frackowiak, J.; Mazur-Kolecka, B.; Imaki, H.; Wegiel, J.; Mehta, P.D.; Silverman, W.P.; Reisberg, B.; et al. Intraneuronal Aβ immunoreactivity is not a predictor of brain amyloidosis-β or neurofibrillary degeneration. Acta Neuropathol 2007, 113, 389–420. [Google Scholar]
- Bayer, T.A.; Wirths, O. Intracellular accumulation of amyloid-beta—A predictor for synaptic dysfunction and neuron loss in Alzheimer’s disease. Front. Aging Neurosci 2010, 2, 1–9. [Google Scholar]
- Cuello, A.C. Intracellular and extracellular Abeta, a tale of two neuropathologies. Brain Pathol 2005, 15, 66–71. [Google Scholar]
- Nagele, R.G.; DÁndrea, M.R.; Anderson, W.J.; Wang, H.Y. Intracellular accumulation of beta-amyloid (1-42) in neurons is facilitated by the alpha 7 nicotinic acetylcholine receptor in Alzheimer’s disease. Neuroscience 2002, 110, 199–211. [Google Scholar]
- DÁndrea, M.R.; Nagele, R.G. Targeting the alpha 7 nicotinic acetylcholine receptor to reduce amyloid accumulation in Alzheimer’s disease pyramidal neurons. Curr. Pharm. Des 2006, 12, 677–684. [Google Scholar]
- Fuentealba, R.A.; Liu, Q.; Zhang, J.; Kanekiyo, T.; Hu, X.; Lee, J.M.; LaDu, M.; Bu, G. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Aβ42 uptake and lysosomal trafficking. PLoS One 2010, 5. [Google Scholar] [CrossRef]
- Takuma, K.; Fang, F.; Zhang, W.; Yan, S.; Fukuzaki, E.; Du, H.; Sosunov, A.; McKhann, G.; Funatsu, Y.; Nakamichi, N.; et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proc. Natl. Acad. Sci. USA 2009, 106, 20021–20026. [Google Scholar]
- LaFerla1, F.M.; Green1, K.N.; Oddo, S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci 2007, 8, 499–509. [Google Scholar]
- Lai, A.Y.; McLaurin, J. Mechanisms of amyloid-beta peptide uptake by neurons: The role of lipid rafts and lipid raft-associated proteins. Int. J. Alzheimer’s Dis 2011, 2011. [Google Scholar] [CrossRef]
- Christensen, D.Z.; Schneider-Axmann, T.; Lucassen, P.J.; Bayer, T.A.; Wirths, O. Accumulation of intraneuronal Aβ correlates with ApoE4 genotype. Acta Neuropathol 2010, 119, 555–566. [Google Scholar]
- Pigino, G.; Morfini, G.; Atagi, Y.; Deshpande, A.; Yu, C.; Jungbauer, L.; LaDu, M.; Busciglio, J.; Brady, S. Disruption of fast axonal transport is a pathogenic mechanism for intraneuronal amyloid beta. Proc. Natl. Acad. Sci. USA 2009, 106, 5907–5912. [Google Scholar]
- Christensen, D.Z.; Bayer, T.A.; Wirths, O. Intracellular Aβ triggers neuron loss in the cholinergic system of the APP/PS1KI mouse model of Alzheimer’s disease. Neurobiol. Aging 2010, 31, 1153–1163. [Google Scholar]
- Christensen, D.Z.; Kraus, S.L.; Flohr, A.; Cotel, M.C.; Wirths, O.; Bayer, T.A. Transient intraneuronal Aβ rather than extracellular plaque pathology correlates with neuron loss in the frontal cortex of APP/PS1KI mice. Acta Neuropathol 2008, 116, 647–655. [Google Scholar]
- Bayer, T.A.; Wirths, O. Review on the APP/PS1KI mouse model: Intraneuronal Aβ accumulation triggers axonopathy, neuron loss and working memory impairment. Genes Brain Behav 2008, 7, 6–11. [Google Scholar]
- Breyhan, H.; Wirths, O.; Duan, K.; Marcello, A.; Rettig, J.; Bayer, T.A. APP/PS1KI bigenic mice develop early synaptic deficits and hippocampus atrophy. Acta Neuropathol 2009, 117, 677–685. [Google Scholar]
- Gouras, G.K.; Tampellini, D.; Takahashi, R.H.; Capetillo-Zarate, E. Intraneuronal β-amyloid accumulation and synapse pathology in Alzheimer’s disease. Acta Neuropathol 2010, 119, 523–541. [Google Scholar]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar]
- Arispe, N.; Rojas, E.; Pollard, H.B. Alzheimer disease amyloid β protein forms calcium channels in bilayer membranes: Blockade by tromethamine and aluminum. Proc. Natl. Acad. Sci. USA 1993, 90, 567–571. [Google Scholar]
- Wong, P.T.; Schauerte, J.A.; Wisser, K.C.; Ding, H.; Lee, E.L.; Steel, D.G.; Gafni, A. Amyloid-beta membrane binding and permeabilization are distinct processes influenced separately by membrane charge and fluidity. J. Mol. Biol 2009, 386, 81–96. [Google Scholar]
- Strodel, B.; Lee, J.W.L.; Whittleston, C.S.; Wales, D.J. Transmembrane structures for Alzheimer’s Aβ1–42 oligomers. J. Am. Chem. Soc 2010, 132, 13300–13312. [Google Scholar]
- Lin, H.; Bhatia, R.; Lal, R. Amyloid beta protein forms ion channels: Implications for Alzheimer’s disease pathophysiology. FASEB J 2001, 15, 2433–2444. [Google Scholar]
- Lin, H.; Zhu, Y.J.; Lal, R. Amyloid β protein (1-40) forms calcium-permeable, Zn2+-sensitive channel in reconstituted lipid vesicles. Biochemistry 1999, 38, 11189–11196. [Google Scholar]
- Kagan, B.L.; Hirakura, Y.; Azimov, R.; Azimova, R.; Lin, M.C. The channel hypothesis of Alzheimer’s disease: Current status. Peptides 2002, 23, 1311–1315. [Google Scholar]
- Diaz, J.C.; Simakova, O.; Jacobson, K.A.; Arispe, N.; Pollard, H.B. Small molecule blockers of the Alzheimer Aβ calcium channel potently protect neurons from Aβ cytotoxicity. Proc. Natl. Acad. Sci USA 2009, 106, 3348–3353. [Google Scholar]
- Felice, F.G.D.; Vieira, M.N.N.; Bomfim, T.R.; Decker, H.; Velasco, P.T.; Lambert, M.P.; Viola, K.L.; Zhao, W.Q.; Ferreira, S.T.; Klein, W.L. Protection of synapses against Alzheimer’s-linked toxins: Insulin signaling prevents the pathogenic binding of Aβ oligomers. Neuroscience 2009, 106, 1971–1976. [Google Scholar]
- Lacor, P.N.; Buniel, M.C.; Furlow, P.W.; Clemente, A.S.; Velasco, P.T.; Wood, M.; Viola, K.L.; Kleinaalina, W.L. Aβ oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. J. Neurosci 2007, 27, 796–807. [Google Scholar]
- Zhao, W.Q.; Felice, F.G.D.; Fernandez, S.; Chen, H.; Lambert, M.P.; Quon, M.J.; Krafft, G.A.; Klein, W.L. Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 2008, 22, 246–260. [Google Scholar]
- Snyder, E.M.; Nong, Y.; Almeida, C.G.; Paul, S.; Moran, T.; Choi, E.Y.; Nairn, A.C.; Salter, M.W.; Lombroso, P.J.; Greengard, G.K.G.P. Regulation of NMDA receptor trafficking by amyloid-β. Nat. Neurosci 2005, 8, 1051–1058. [Google Scholar]
- Yan, S.D.; Chen, X.; Fu, J.; Chen, M.; Zhu, H.; Roher, A.; Slattery, T.; Zhao, L.; Nagashima, M.; Morser, J.; et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature 1996, 382, 685–691. [Google Scholar]
- Parri, R.H.; Dineley, T.K. Nicotinic acetylcholine receptor interaction with beta-amyloid: Molecular, cellular, and physiological consequences. Curr. Alzheimer Res 2010, 7, 27–39. [Google Scholar]
- Diarra, A.; Geetha, T.; Potter, P.; Babu, J.R. Signaling of the neurotrophin receptor p75 in relation to Alzheimer’s disease. Biochem. Biophys. Res. Commun 2009, 390, 352–356. [Google Scholar]
- Alberdi, E.; Sánchez-Gómez, M.V.; Cavaliere, F.; Pérez-Samartín, A.; Zugaza, J.L.; Trullas, R.; Domercq, M.; Matute, C. Amyloid β oligomers induce Ca2+ dysregulation and neuronal death through activation of ionotropic glutamate receptors. Cell Calcium 2010, 47, 264–272. [Google Scholar]
- Ma, Q.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. β-Amyloid oligomers induce phosphorylation of Tau and inactivation of insulin receptor substrate via c-Jun N-Terminal kinase signaling: Suppression by omega-3 fatty acids and curcumin. J. Neurosci 2009, 29, 9078–9089. [Google Scholar]
- Malinow, R. New developments on the role of NMDA receptors in Alzheimer’s disease. Curr. Opi. Neurobiol 2011. (in Press). [Google Scholar]
- Li, S.; Jin, M.; Koeglsperger, T.; Shepardson, N.E.; Shankar, G.M.; Selkoe, D.J. Soluble Aβ oligomers inhibit long-term potentiation through a mechanism involving excessive activation of extrasynaptic NR2B-containing NMDA receptors. J. Neurosci 2011, 31, 6627–6638. [Google Scholar]
- Texido, L.; Martin-Satue, M.; Alberdi, E.; Solsona, C.; Matute, C. Amyloid β peptide oligomers directly activate NMDA receptors. Cell Calcium 2011, 49, 184–190. [Google Scholar]
- Verges, D.K.; Restivo, J.L.; Goebel, W.D.; Holtzman, D.M.; Cirrito, J.R. Opposing synaptic regulation of amyloid-β metabolism by NMDA receptors in vivo. J. Neurosci 2011, 31, 11328–11337. [Google Scholar]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-β production. J. Neurosci 2010, 30, 15927–15942. [Google Scholar]
- Rönicke, R.; Mikhaylova, M.; Rönicke, S.; Meinhardt, J.; Schröder, U.H.; Fändrich, M.; Reiser, G.; Kreutz, M.R.; Reymann, K.G. Early neuronal dysfunction by amyloid β oligomers depends on activation of NR2B-containing NMDA receptors. Neurobiol. Aging 2011, 32, 2219–2228. [Google Scholar]
- Bicca, M.A.; Figueiredo, C.P.; Piermartiri, T.C.; Meotti, F.C.; Bouzon, Z.L.; Tasca, C.I.; Medeiros, R.; Calixto, J.B. The selective and competitive N-methyl-D-aspartate receptor antagonist, (−)-6-phosphonomethyl-deca-hydroisoquinoline-3-carboxylic acid, prevents synaptic toxicity induced by amyloid-β in mice. Neuroscience 2011, 192, 631–641. [Google Scholar]
- Maglio, L.E.; Martins, V.R.; Izquierdo, I.; Ramirez, O.A. Role of cellular prion protein on LTP expression in aged mice. Brain Res 2006, 1097, 11–18. [Google Scholar]
- Criado, J.R.; Sanchez-Alavez, M.; Conti, B.; Giacchino, J.L.; Wills, D.N.; Henriksen, S.J.; Race, R.; Manson, J.C.; Chesebro, B.; Oldstone, M.B. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of PrP in neurons. Neurobiol. Dis 2005, 19, 255–265. [Google Scholar]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol 2008, 181, 551–565. [Google Scholar]
- Vassallo, N.; Herms, J. Cellular prion protein function in copper homeostasis and redox signalling at the synapse. J. Neurochem 2003, 86, 538–544. [Google Scholar]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol 2004, 165, 227–235. [Google Scholar]
- Shyu, W.C.; Lin, S.Z.; Chiang, M.F.; Ding, D.C.; Li, K.W.; Chen, S.F.; Yang, H.I.; Li, H. Overexpression of PrPC by adenovirus-mediated gene targeting reduces ischemic injury in a stroke rat model. J. Neurosci 2005, 25, 8967–8977. [Google Scholar]
- Santuccione, A.; Sytnyk, V.; Leshchyns’ka, I.; Schachner, M. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol 2005, 169, 341–354. [Google Scholar]
- Laurén, J.; Gimbel, D.A.; Nygaard, H.B.; Gilbert, J.W.; Strittmatter, S.M. Cellular prion protein mediates impairment of synaptic plasticity by amyloid-β oligomers. Nature 2009, 457, 1128–1132. [Google Scholar]
- Kessels, H.W.; Nguyen, L.N.; Nabavi, S.; Malinow, R. The prion protein as a receptor for amyloid-β. Nature 2010, 457, 1128–1132. [Google Scholar]
- Calella, A.M.; Farinelli, M.; Nuvolone, M.; Mirante, O.; Moos, R.; Falsig, J.; Mansuy, I.M.; Aguzzi, A. Prion protein and Aβ-related synaptic toxicity impairment. EMBO Mol. Med 2010, 2, 306–314. [Google Scholar]
- Wang, H.Y.; Lee, D.H.S.; D’Andrea, M.R.; Peterson, P.A.; Shank, R.P.; Reitz, A.B. β-Amyloid(1-42) binds to α7 nicotinic acetylcholine receptor with high affinity: Implications for Alzheimer’s disease pathology. J. Biol. Chem 2000, 275, 5626–5632. [Google Scholar]
- Pettit, D.L.; Shao, Z.; Yakel, J.L. β-amyloid 1-42 peptide directly modulates nicotinic receptors in the rat hippocampal slice. J. Neurosci 2001, 21, RC120–RC125. [Google Scholar]
- Liu, Q.S.; Kawai, H.; Berg, D.K. Beta-amyloid peptide blocks the response of alpha7-containing nicotinic receptors on hippocampal neurons. Proc. Natl. Acad. Sci. USA 2001, 48, 4734–4739. [Google Scholar]
- Dougherty, J.J.; Wu, J.; Nichols, R.A. β-Amyloid regulation of presynaptic nicotinic receptors in rat hippocampus and neocortex. J. Neurosci 2003, 23, 6740–6747. [Google Scholar]
- Hernandez, C.M.; Kayed, R.; Zheng, H.; Sweatt, J.D.; Dineley, K.T. Loss of α7 nicotinic receptors enhances β-amyloid oligomer accumulation, exacerbating early-stage cognitive decline and Septohippocampal pathology in a mouse model of Alzheimer’s disease. J. Neurosci 2010, 30, 2442–2453. [Google Scholar]
- Wang, H.Y.; Bakshi, K.; Shen, C.; Frankfurt, M.; Trocmé-Thibíerge, C.; Morain, P. S24795 limits β-amyloid-α7 nicotinic receptor interaction and reduces Alzheimer’s disease-like pathologies. Biol. Psychiatry 2010, 67, 522–530. [Google Scholar]
- Lagostena, L.; Trocme-Thibierge, C.; Morain, P.; Cherubini, E. The partial α7 nicotine acetylcholine receptor agonist S 24795 enhances long-term potentiation at CA3-CA1 synapses in the adult mouse hippocampus. Neuropharmacology 2008, 54, 676–685. [Google Scholar]
- Beracochea, D.; Boucard, A.; Trocme-Thibierge, C.; Morain, P. Improvement of contextual memory by S24795 in aged mice: Comparison with memantine. Psychopharmacology 2008, 196, 555–564. [Google Scholar]
- Small, D.H.; Maksel, D.; Kerr, M.L.; Ng, J.; Hou, X.; Chu, C.; Mehrani, H.; Unabia, S.; Azari, M.F.; Loiacono, R.; et al. The beta-amyloid protein of Alzheimer’s disease binds to membrane lipids but does not bind to the alpha7 nicotinic acetylcholine receptor. J. Neurochem 2007, 101, 1527–1538. [Google Scholar]
- Knowles, J.K.; Rajadas, J.; Nguyen, T.V.V.; Yang, T.; LeMieux, M.C.; Griend, L.V.; Ishikawa, C.; Massa, S.M.; Wyss-Coray, T.; Longo, F.M. The p75 neurotrophin receptor promotes amyloid-β (1-42)-induced neuritic dystrophy in vitro and in vivo. J. Neurosci 2009, 29, 10627–10637. [Google Scholar]
- Bengoechea, T.G.; Chen, Z.; O’Learya, D.A.; Masliahb, E.; Lee, K.F. p75 reduces β-amyloid-induced sympathetic innervation deficits in an Alzheimer’s disease mouse model. Proc. Natl. Acad. Sci. USA 2009, 106, 7870–7675. [Google Scholar]
- Boland, K.; Manias, K.; Perlmutter, D.H. Specificity in recognition of amyloid-beta peptide by the serpin-enzyme complex receptor in hepatoma cells and neuronal cells. J. Biol. Chem 1995, 270, 28022–28028. [Google Scholar]
- Joslin, G.; Griffin, G.L.; August, A.M.; Adams, S.; Fallon, R.J.; Senior, R.M.; Perlmutter, D.H. The serpin-enzyme complex (SEC) receptor mediates the neutrophil chemotactic effect of alpha-1 antitrypsin-elastase complexes and amyloid-beta peptide. J. Clin. Invest 1992, 90, 1150–1154. [Google Scholar]
- Schmidt, A.M.; Sahagan, B.; Nelson, R.B.; Selmer, J.; Rothlein, R.; Bell, J.M. The role of RAGE in amyloid-beta peptide-mediated pathology in Alzheimer’s disease. Curr. Opin. Investig. Drugs 2009, 10, 672–680. [Google Scholar]
- Baiguera, S.; Fioravanzo, L.; Grandi, C.; Liddo, R.D.; Parnigotto, P.P.; Folin, M. Involvement of the receptor for advanced glycation-end products (RAGE) in beta-amyloid-induced toxic effects in rat cerebromicrovascular endothelial cells cultured in vitro. Int. J. Mol. Med 2009, 24, 9–15. [Google Scholar]
- Prk, L.; Wng, G.; Zhou, P.; Zhou, J.; Pitstick, R.; Previtic, M.L.; Younkind, L.; Younkind, S.G.; Nostrndc, W.E.V.; Choe, S.; et al. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-β. Proc. Natl. Acad. Sci. USA 2011, 108, 5063–5068. [Google Scholar]
- Silverstein, R.L.; Febbraio, M. CD36, a scavenger receptor involved in immunity, metabolism, angiogenesis, and behavior. Sci. Signal 2009, 2, 1–8. [Google Scholar]
- Coraci, I.S.; Husemann, J.; Berman, J.W.; Hulette, C.; Dufour, J.H.; Campanella, G.K.; Luster, A.D.; Silverstein, S.C.; El-Khoury, J.B. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol 2002, 160, 101–112. [Google Scholar]
- Varadarajan, S.; Yatin, S.; Aksenova, M.; Butterfield, D.A. Review: Alzheimer’s amyloid β-peptide-associated free radical oxidative stress and neurotoxicity. J. Struct. Biol 2000, 130, 184–208. [Google Scholar]
- Butterfield, D.A.; Reed, T.; Newman, S.F.; Sultana, R. Roles of amyloid β-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med 2007, 43, 658–677. [Google Scholar]
- Jo, D.G.; Arumugam, T.V.; Woo, H.N.; Park, J.S.; Tang, S.C.; Mughal, M.; Hyun, D.H.; Park, J.H.; Choi, Y.H.; Gwon, A.R.; et al. Evidence that γ-secretase mediates oxidative stress-induced β-secretase expression in Alzheimer’s disease. Neurobiol. Aging 2010, 31, 917–925. [Google Scholar]
- Zheng, L.; Kågedal, K.; Dehvari, N.; Benedikz, E.; Cowburn, R.; Marcusson, J.; Terman, A. Oxidative stress induces macroautophagy of amyloid β-protein and ensuing apoptosis. Free Radic. Biol. Med 2009, 46, 422–429. [Google Scholar]
- Abdul, H.M.; Butterfield, D.A. Involvement of PI3K/PKG/ERK1/2 signaling pathways in cortical neurons to trigger protection by cotreatment of acetyl-l-carnitine and α-lipoic acid against HNE-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. Free Radic. Biol. Med 2007, 42, 371–384. [Google Scholar]
- Naoi, M.; Maruyama, W.; Shamoto-Nagai, M.; Yi, H.; Akao, Y.; Tanaka, M. Oxidative stress in mitochondria: Decision to survive and death of neurons in neurodegenerative disorder. Mol. Neurobiol 2005, 31, 81–93. [Google Scholar]
- Castegna, A.; Aksenov, M.; Aksenova, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part I. Creatine kinase BB, glutamine synthase, and ubiquitin carboxy-terminal hydrolase L-1. Free Radic. Biol. Med 2002, 33, 562–571. [Google Scholar]
- Castegna, A.; Aksenov, M.; Thongboonkerd, V.; Klein, J.B.; Pierce, W.M.; Booze, R.; Markesbery, W.R.; Butterfield, D.A. Proteomic identification of oxidatively modified proteins in Alzheimer’s disease brain. Part II. Dihydropyrimidinase-related protein 2, alpha-enolase and heat shock cognate 71. J. Neurochem 2002, 82, 1524–1532. [Google Scholar]
- Sultana, R.; Perluigi, M.; Butterfield, D.A. Redox proteomics identification of oxidatively modified proteins in Alzheimer’s disease brain and in vivo and in vitro models of AD centered around Abeta(1-42). J. Chromatogr. B Anal. Technol. Biomed. Life Sci 2006, 833, 3–11. [Google Scholar]
- Butterfield, D.A.; Boyd-Kimball, D. The critical role of methionine 35 in Alzheimer’s amyloid β- peptide (1-42)-induced oxidative stress and neurotoxicity. Biochimi. Biophys. Acta (BBA)-Protein Proteomics 2005, 1703, 149–156. [Google Scholar]
- Butterfield, D.A.; Galvan, V.; Lange, M.B.; Tang, H.; Sowell, R.A.; Spilman, P.; Fombonne, J.; Gorostiza, O.; Zhang, J.; Sultana, R.; et al. In vivo oxidative stress in brain of Alzheimer disease transgenic mice: Requirement for methionine 35 in amyloid β-peptide of APP. Free Radic. Biol. Med 2010, 48, 136–144. [Google Scholar]
- Muthaiyah, B.; Essa, M.M.; Chauhan, V.; Chauhan, A. Protective effects of walnut extract against amyloid beta peptide-induced cell death and oxidative stress in PC12 cells. Biomed. Life Sci 2011, 36, 2096–2103. [Google Scholar]
- Barten, D.M.; Albright, C.F. Therapeutic strategies for Alzheimer’s disease. Mol. Neurobiol 2008, 37, 171–186. [Google Scholar]
- Tomaselli, S.; Esposito, V.; Vangone, P.; van Nuland, N.A.J.; Bonvin, A.M.J.J.; Guerrini, R.; Tancredi, T.; Temussi, P.A.; Picone, D. The α-to-β conformational transition of Alzheimer’s Aβ-(1-42) peptide in aqueous media is reversible: A step by step conformational analysis suggests the location of β conformation seeding. ChemBioChem 2006, 7, 257–267. [Google Scholar]
- Hayat, S.; Helms, V. Towards understanding the early events in the conformational transition of amyloid beta peptides. Comput. Biophys. Syst. Biol. (CBSB08) 2008, 40, 227–230. [Google Scholar]
- Xu, Y.; Shen, J.; Luo, X.; Zhu, W.; Chen, K.; Ma, J.; Jiang, H. Conformational transition of amyloid β-peptide. Proc. Natl. Acad. Sci. USA 2005, 102, 5403–5407. [Google Scholar]
- Creighton, T.E. Protein Folding; W.H. Freeman & Company: New York, NY, USA, 1992. [Google Scholar]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar]
- Zimm, H.; Bragg, J.K. Theory of the phase transition between helix and random coil in polypeptide chains. J. Chem. Phys 1959, 31, 526–535. [Google Scholar]
- Lifson, S.; Roig, A. On the theory of helix-coil transition in polypeptides. J. Chem. Phys 1961, 34, 1963. [Google Scholar]
- Kao, Y.M.; Jiang, T.F. Transition temperatures of the trapped ideal spinor Bose gas. Eur. Phys. J. D 2006, 40, 363–269. [Google Scholar]
- Yakubovich, A.V.; Solov’yov, I.A.; Solov’yov, A.V.; Greiner, W. Ab initio theory of helix-coil phase transition. Eur. Phys. J. D 2008, 46. [Google Scholar] [CrossRef]
- Solovyov, I.A.; Yakubovich, A.V.; Solovyov, A.V.; Greiner, W. α-helix ↔ random coil phase transition: Analysis of ab initio theory predictions. Eur. Phys. J. D 2008, 46, 227–240. [Google Scholar]
- Ding, F.; Borreguero, J.M.; Buldyrey, S.V.; Stanley, H.E.; Dokholyan, N.V. Mechanism for the alpha-helix to beta-hairpin transition. Proteins 2003, 53, 220–228. [Google Scholar]
- Hong, L.; Lei, J. Statistical mechanical model for helix-sheet-coil transitions in homopolypeptides. Phys. Rev. E 2008, 78, 051904. [Google Scholar]
- Yasar, F.; Demir, K. The study of helix-coil transition of polyalanine with single histogram method. Comput. Phys. Commun 2006, 175, 604–611. [Google Scholar]
- Gibbs, H.; Dimarzio, E.A. Statistical mechanics of helix-coil transitions in biological macromolecules. J. Chem. Phys 1959, 30, 271–282. [Google Scholar]
- Huang, K. Protein folding as a physical stochastic process. Biophys. Rev. Lett 2008, 3, 1–2. [Google Scholar]
- Huang, K. Conditioned self-avoiding walk (csaw): Stochastic approach to protein folding. Biophys. Rev. Lett 2007, 139–154. [Google Scholar]
- Leong, H.W.; Chew, L.Y.; Huang, K. Normal modes and phase transition of the protein chain based on the Hamiltonian formalism. Phys. Rev. E 2010, 82. [Google Scholar] [CrossRef]
- Goh, B.C.; Leong, H.W.; Qu, X.; Chew, L.Y. The mechanism of antiparallel β-sheet formation based on conditioned self-avoiding walk. Eur. Phys. J. E 2012, 35. [Google Scholar] [CrossRef]
- Jiang, P.; Li, W.F.; Shea, J.E.; Mu, Y. Resveratrol inhibits the formation of multiple-layered β-sheet oligomers of the human islet amyloid polypeptide segment 22–27. Biophys. J 2011, 100, 1550–1558. [Google Scholar]
- Qin, X.R.; Abe, H.; Nakanishi, H. NMR and CD studies on the interaction of Alzheimer β-amyloid peptide (12-28) with β-cyclodextrin. Biochem. Biophys. Res. Commun 2002, 297, 1011–1015. [Google Scholar]
- Camilleri, P.; Haskins, N.J.; Hewlett, D.R. β-Cyclodextrin interacts with the Alzheimer amyloid β-A4 peptide. FEBS Lett 1994, 341, 256–258. [Google Scholar]
- Colombo, R.; Carotti, A.; Catto, M.; Racchi, M.; Lanni, C.; Verga, L.; Caccialanza, G.; Lorenzi, E.D. CE can identify small molecules that selectively target soluble oligomers of amyloid β protein and display antifibrillogenic activity. Electrophoresis 2009, 30, 1418–1429. [Google Scholar]
- Cohen, A.D.; Ikonomovic, M.D.; Abrahamson, E.E.; Paljug, W.R.; DeKosky, S.T.; Lefterov, I.M.; Koldamova, R.P.; Shao, L.; Debnath, M.L.; Mason, N.S.; et al. Anti-amyloid effects of small molecule Aβ-binding agents in PS1/APP mice. Lett. Drug. Des. Discov 2009, 6, 437. [Google Scholar]
- Yang, F.; Lim, G.P.; Begum, A.N.; Ubeda, O.J.; Simmons, M.R.; Ambegaokar, S.S.; Chen, P.P.; Kayed, R.; Glabe, C.G.; Frautschy, S.A.; et al. Curcumin inhibits formation of amyloid β oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J. Biol. Chem 2005, 280, 5892–5901. [Google Scholar]
- Inbar, P.; Bautista, M.R.; Takayama, S.A.; Yang, J. Assay to screen for molecules that associate with Alzheimer’s related beta-amyloid fibrils. Anal. Chem 2008, 80, 3502–3506. [Google Scholar]
- Garcia-Alloza, M.; Borrelli, L.A.; Rozkalne, A.; Hyman, B.T.; Bacskai, B.J. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J. Neurochem 2007, 102, 1095–1104. [Google Scholar]
- Caesar, I.; Jonson, M.; Nilsson, K.P.R.; Thor, S.; Hammarström, P. Curcumin promotes A-beta fibrillation and reduces neurotoxicity in transgenic drosophila. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Park, S.Y.; Kim, H.S.; Cho, E.K.; Kwon, B.Y.; Phark, S.; Hwang, K.W.; Sula, D. Curcumin protected PC12 cells against beta-amyloid-induced toxicity through the inhibition of oxidative damage and tau hyperphosphorylation. Food Chem. Toxicol 2008, 46, 2881–2887. [Google Scholar]
- Masuda, Y.; Fukuchi, M.; Yatagawa, T.; Tada, M.; Takeda, K.; Irie, K.; ichi Akagi, K.; Monobe, Y.; Imazawa, T.; Takegoshi, K. Solid-state NMR analysis of interaction sites of curcumin and 42-residue amyloid β-protein fibrils. Bioorg. Med. Chem 2011, 19, 5967–5974. [Google Scholar]
- Ellson, J.; Gansner, E.; Koutsofios, L.; North, S.; Woodhull, G.; Description, S.; Technologies, L. Graphviz-Open Source Graph Drawing Tools. In Lecture Notes in Computer Science; Springer-Verlag: Heidelberger/Berlin, Germany, 2001; pp. 483–484. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhao, L.N.; Long, H.W.; Mu, Y.; Chew, L.Y. The Toxicity of Amyloid ß Oligomers. Int. J. Mol. Sci. 2012, 13, 7303-7327. https://doi.org/10.3390/ijms13067303
Zhao LN, Long HW, Mu Y, Chew LY. The Toxicity of Amyloid ß Oligomers. International Journal of Molecular Sciences. 2012; 13(6):7303-7327. https://doi.org/10.3390/ijms13067303
Chicago/Turabian StyleZhao, Li Na, Hon Wai Long, Yuguang Mu, and Lock Yue Chew. 2012. "The Toxicity of Amyloid ß Oligomers" International Journal of Molecular Sciences 13, no. 6: 7303-7327. https://doi.org/10.3390/ijms13067303