MiR-218 Impairs Tumor Growth and Increases Chemo-Sensitivity to Cisplatin in Cervical Cancer

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
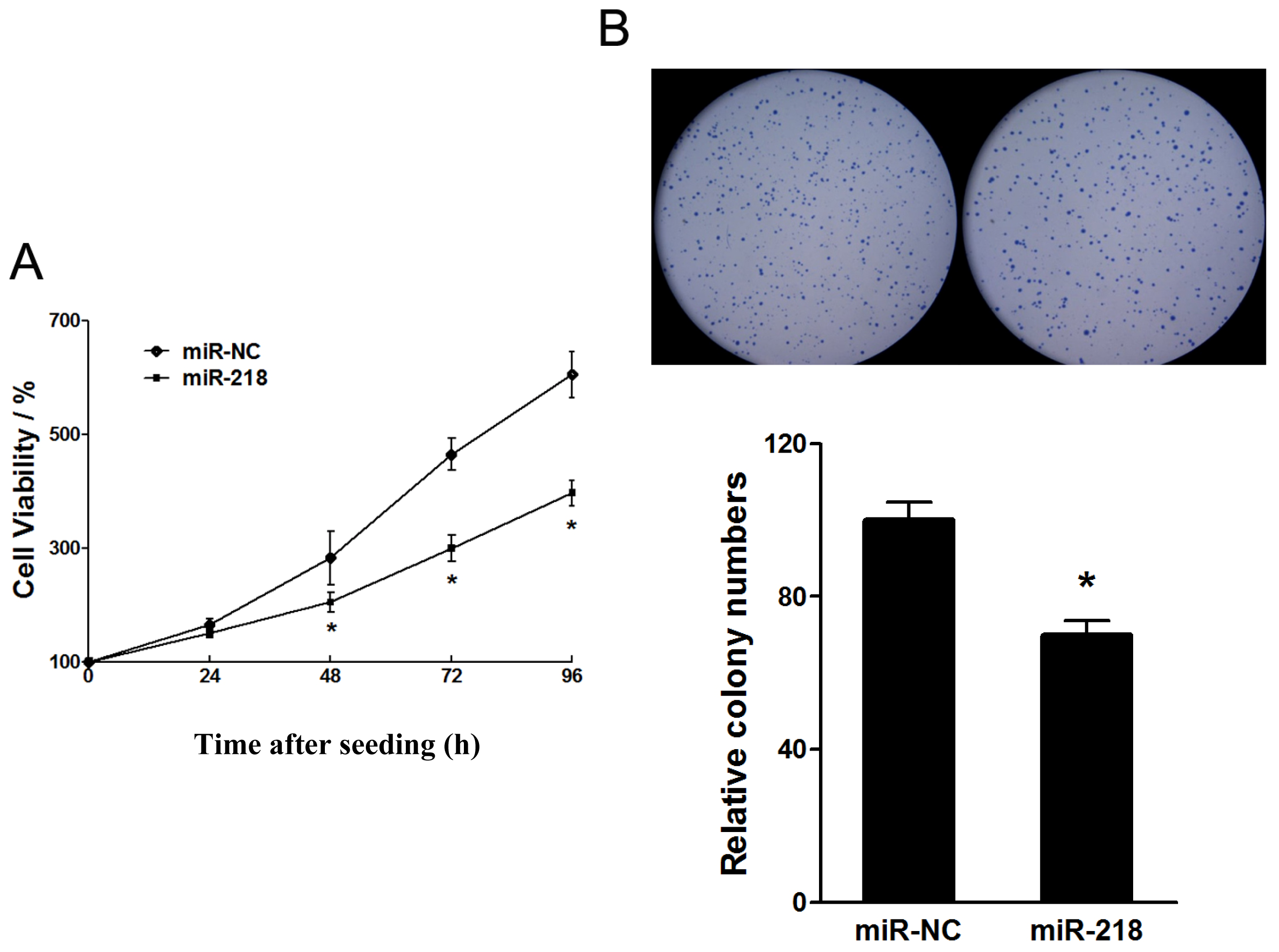
2.1.1. Tumor Suppressive Effect of miR-218 in the Proliferation of Cervical Cancer Cell Growth
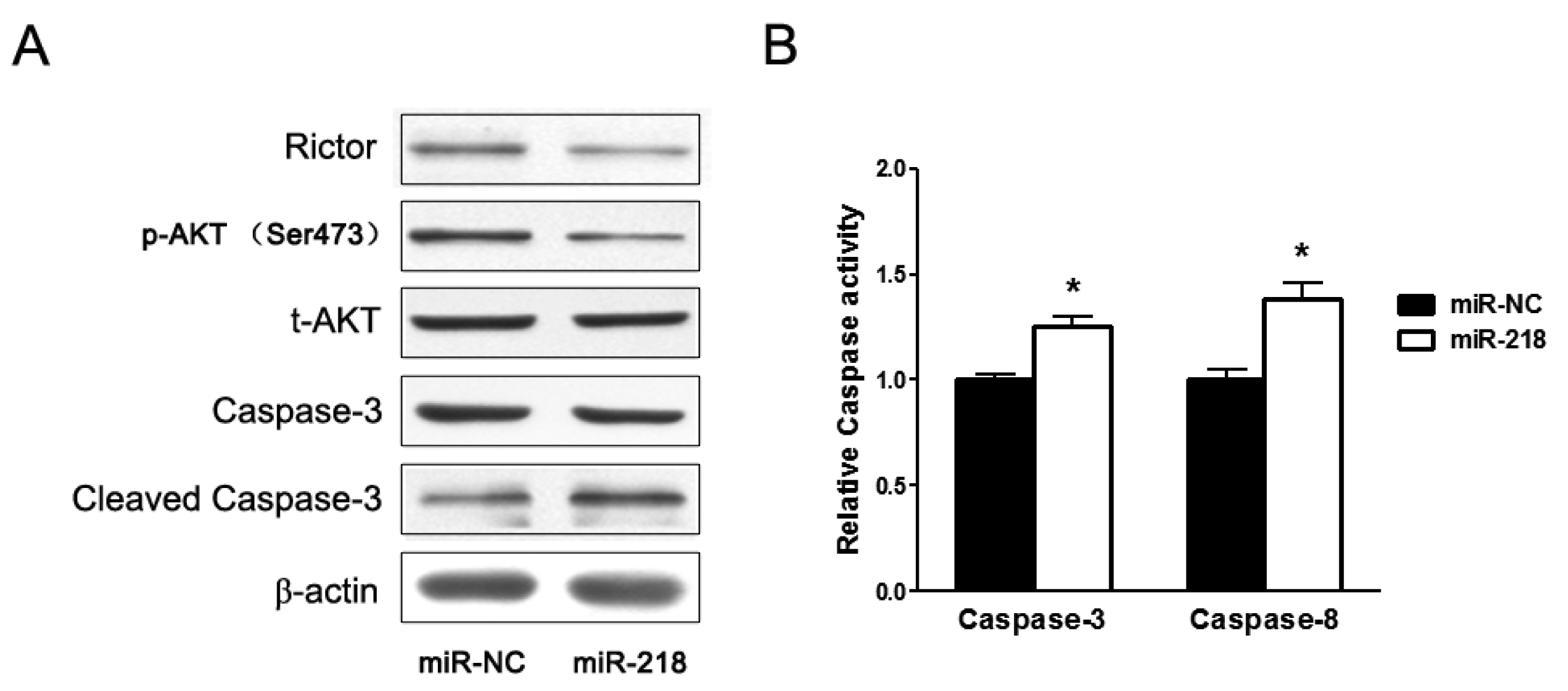
2.1.2. Overexpression of miR-218 Inhibited Expression of Rictor, an mTOR Component, and Its Downstream Pathway
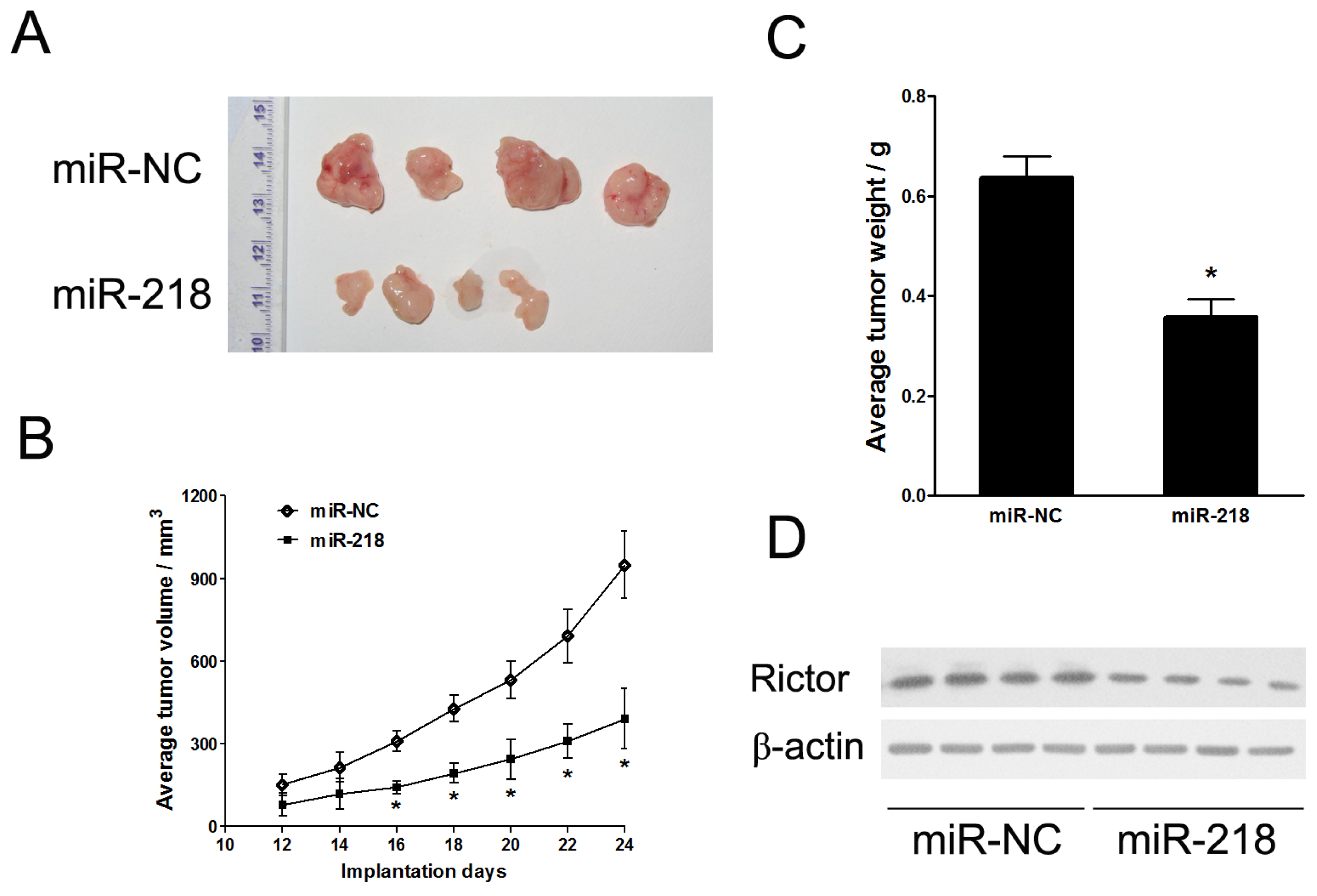
2.1.3. MiR-218 Impaired In Vivo Tumor Growth
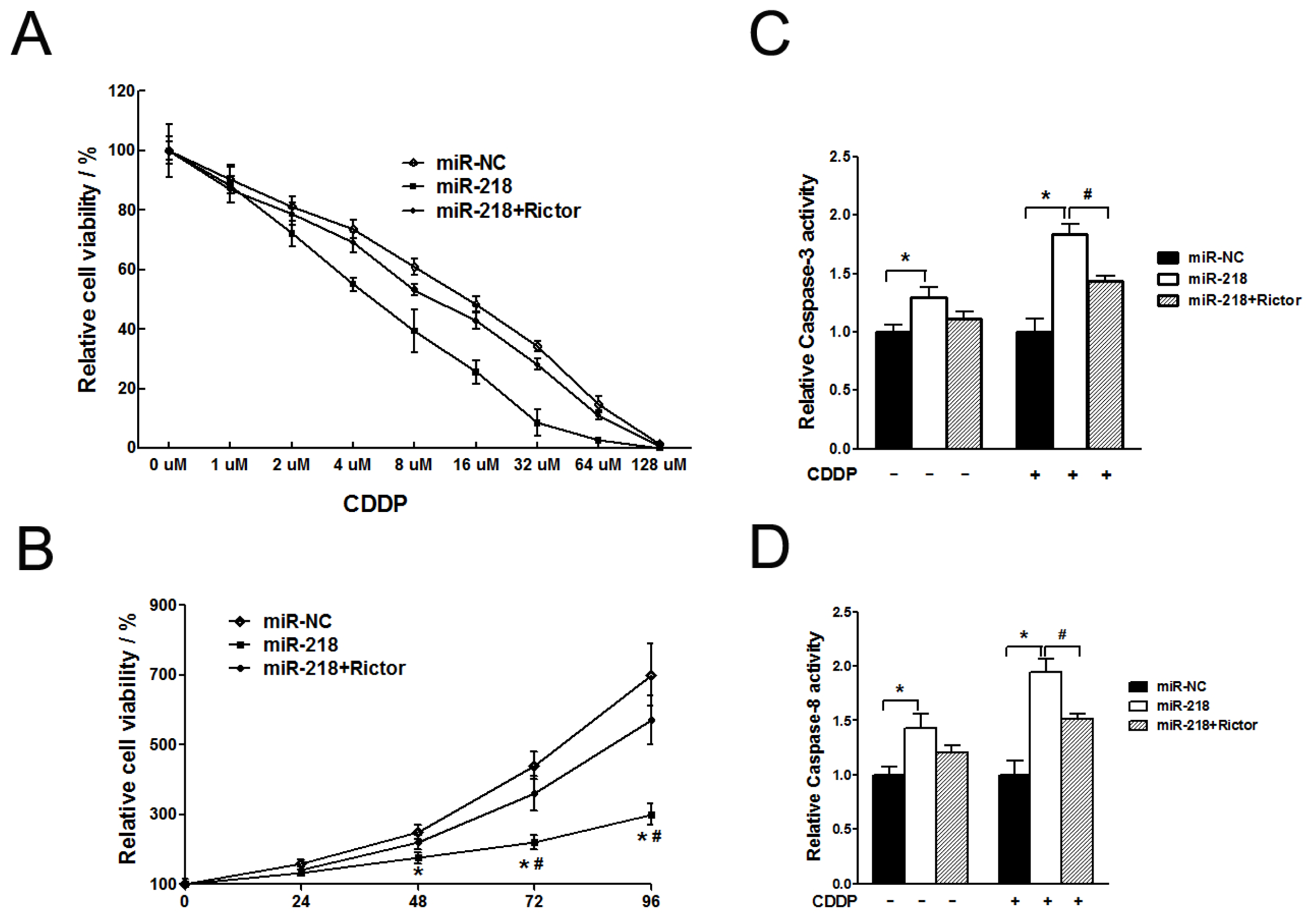
2.1.4. MiR-218 Increased Chemosensitivity of Cervical Cancer Cells to Cisplatin via Its Target Rictor
2.2. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Generation of HeLa Cells with Stable Expression of miR-218
3.3. Cell Proliferation Assay
3.4. Anchorage-Independent Colony Formation Assay
3.5. Western Blotting Assay
3.6. Caspase Activity
3.7. In Vivo Tumorigenesis Study
3.8. Adenovirus Preparation
3.9. In Vitro Assay of Chemosensitivity to Cisplatin (CDDP)
3.10. CDDP Treatment of Cancer Cell in Vitro
3.11. Statistical Analysis
4. Conclusions
Supplementary Information
ijms-13-16053-s001.pdfAcknowledgements
- Conflict of InterestAll authors declare that there are no conflicts of interest and agree with the contents of the manuscript for publication and support open access publishing to allow unlimited access and high publicity of my published paper.
References
- Shi, J.F.; Chen, J.F.; Canfell, K.; Feng, X.X.; Ma, J.F.; Zhang, Y.Z.; Zhao, F.H.; Li, R.; Ma, L.; Li, Z.F.; et al. Estimation of the costs of cervical cancer screening, diagnosis and treatment in rural Shanxi Province, China: A micro-costing study. BMC Health Serv. Res 2012, 12, 123. [Google Scholar]
- Lea, J.S.; Sunaga, N.; Sato, M.; Kalahasti, G.; Miller, D.S.; Minna, J.D.; Muller, C.Y. Silencing of HPV 18 oncoproteins With RNA interference causes growth inhibition of cervical cancer cells. Reprod. Sci 2007, 14, 20–28. [Google Scholar]
- Ji, J.; Zheng, P.S. Activation of mTOR signaling pathway contributes to survival of cervical cancer cells. Gynecol. Oncol 2010, 117, 103–108. [Google Scholar]
- Noh, W.C.; Kim, Y.H.; Kim, M.S.; Koh, J.S.; Kim, H.A.; Moon, N.M.; Paik, N.S. Activation of the mTOR signaling pathway in breast cancer and its correlation with the clinicopathologic variables. Breast Cancer Res. Treat 2008, 110, 477–483. [Google Scholar]
- Marinov, M.; Ziogas, A.; Pardo, O.E.; Tan, L.T.; Dhillon, T.; Mauri, F.A.; Lane, H.A.; Lemoine, N.R.; Zangemeister-Wittke, U.; Seckl, M.J.; et al. AKT/mTOR pathway activation and BCL-2 family proteins modulate the sensitivity of human small cell lung cancer cells to RAD001. Clin. Cancer Res 2009, 15, 1277–1287. [Google Scholar]
- Babcock, J.T.; Quilliam, L.A. Rheb/mTOR activation and regulation in cancer: Novel treatment strategies beyond rapamycin. Curr. Drug Targets 2011, 12, 1223–1231. [Google Scholar]
- Yanokura, M.; Banno, K.; Kobayashi, Y.; Kisu, I.; Ueki, A.; Ono, A.; Masuda, K.; Nomura, H.; Hirasawa, A.; Susumu, N.; et al. MicroRNA and endometrial cancer: Roles of small RNAs in human tumors and clinical applications (Review). Oncol. Lett 2010, 1, 935–940. [Google Scholar]
- Jiang, Y.W.; Chen, L.A. microRNAs as tumor inhibitors, oncogenes, biomarkers for drug efficacy and outcome predictors in lung cancer (review). Mol. Med. Report 2012, 5, 890–894. [Google Scholar]
- Kelly, B.D.; Miller, N.; Healy, N.A.; Walsh, K.; Kerin, M.J. A review of expression profiling of circulating microRNAs in men with prostate cancer. BJU Int. 2012. [Google Scholar] [CrossRef]
- Nair, V.S.; Maeda, L.S.; Ioannidis, J.P. Clinical outcome prediction by microRNAs in human cancer: A systematic review. J. Natl. Cancer Inst 2012, 104, 528–540. [Google Scholar]
- Lujambio, A.; Calin, G.A.; Villanueva, A.; Ropero, S.; Sanchez-Cespedes, M.; Blanco, D.; Montuenga, L.M.; Rossi, S.; Nicoloso, M.S.; Faller, W.J.; et al. A microRNA DNA methylation signature for human cancer metastasis. Proc. Natl. Acad. Sci. USA 2008, 105, 13556–13561. [Google Scholar]
- Lages, E.; Ipas, H.; Guttin, A.; Nesr, H.; Berger, F.; Issartel, J.P. MicroRNAs: Molecular features and role in cancer. Front. Biosci 2012, 17, 2508–2540. [Google Scholar]
- Uesugi, A.; Kozaki, K.; Tsuruta, T.; Furuta, M.; Morita, K.; Imoto, I.; Omura, K.; Inazawa, J. The tumor suppressive microRNA miR-218 targets the mTOR component Rictor and inhibits AKT phosphorylation in oral cancer. Cancer Res 2011, 71, 5765–5778. [Google Scholar]
- Martinez, I.; Gardiner, A.S.; Board, K.F.; Monzon, F.A.; Edwards, R.P.; Khan, S.A. Human papillomavirus type 16 reduces the expression of microRNA-218 in cervical carcinoma cells. Oncogene 2008, 27, 2575–2582. [Google Scholar]
- Zhou, X.; Chen, X.; Hu, L.; Han, S.; Qiang, F.; Wu, Y.; Pan, L.; Shen, H.; Li, Y.; Hu, Z. Polymorphisms involved in the miR-218-LAMB3 pathway and susceptibility of cervical cancer, a case-control study in Chinese women. Gynecol. Oncol 2010, 117, 287–290. [Google Scholar]
- Rostaing, L.; Kamar, N. mTOR inhibitor/proliferation signal inhibitors: Eentering or leaving the field? J. Nephrol 2010, 23, 133–142. [Google Scholar]
- Morgensztern, D.; McLeod, H.L. PI3K/Akt/mTOR pathway as a target for cancer therapy. Anticancer Drugs 2005, 16, 797–803. [Google Scholar]
- Guo, C.; Gasparian, A.V.; Zhuang, Z.; Bosykh, D.A.; Komar, A.A.; Gudkov, A.V.; Gurova, K.V. 9-Aminoacridine-based anticancer drugs target the PI3K/AKT/mTOR, NF-kappaB and p53 pathways. Oncogene 2009, 28, 1151–1161. [Google Scholar]
- Breuleux, M.; Klopfenstein, M.; Stephan, C.; Doughty, C.A.; Barys, L.; Maira, S.M.; Kwiatkowski, D.; Lane, H.A. Increased AKT S473 phosphorylation after mTORC1 inhibition is rictor dependent and does not predict tumor cell response to PI3K/mTOR inhibition. Mol. Cancer Ther 2009, 8, 742–753. [Google Scholar]
- Brewer, C.A.; Blessing, J.A.; Nagourney, R.A.; McMeekin, D.S.; Lele, S.; Zweizig, S.L. Cisplatin plus gemcitabine in previously treated squamous cell carcinoma of the cervix: A phase II study of the Gynecologic Oncology Group. Gynecol. Oncol 2006, 100, 385–388. [Google Scholar]
- Long, H.J., III; Monk, B.J.; Huang, H.Q.; Grendys, E.C., Jr; McMeekin, D.S.; Sorosky, J.; Miller, D.S.; Eaton, L.A.; Fiorica, J.V. Clinical results and quality of life analysis for the MVAC combination (methotrexate, vinblastine, doxorubicin, and cisplatin) in carcinoma of the uterine cervix: A Gynecologic Oncology Group study. Gynecol. Oncol. 2006, 100, 537–543. [Google Scholar]
- Anders, J.C.; Grigsby, P.W.; Singh, A.K. Cisplatin chemotherapy (without erythropoietin) and risk of life-threatening thromboembolic events in carcinoma of the uterine cervix: The tip of the iceberg? A review of the literature. Radiat. Oncol 2006, 1, 14. [Google Scholar]
- Coleman, R.E.; Clarke, J.M.; Slevin, M.L.; Sweetenham, J.; Williams, C.J.; Blake, P.; Calman, F.; Wiltshaw, E.; Harper, P.G. A phase II study of ifosfamide and cisplatin chemotherapy for metastatic or relapsed carcinoma of the cervix. Cancer Chemother. Pharmacol 1990, 27, 52–54. [Google Scholar]
- Nagai, Y.; Toita, T.; Wakayama, A.; Nakamoto, T.; Ooyama, T.; Tokura, A.; Inamine, M.; Kudaka, W.; Murayama, S.; Aoki, Y. Concurrent chemoradiotherapy with paclitaxel and cisplatin for adenocarcinoma of the cervix. Anticancer Res 2012, 32, 1475–1479. [Google Scholar]
- Nakada, S.; Aoki, D.; Ohie, S.; Horiuchi, M.; Suzuki, N.; Kanasugi, M.; Susumu, N.; Udagawa, Y.; Nozawa, S. Chemosensitivity testing of ovarian cancer using the histoculture drug response assay: Sensitivity to cisplatin and clinical response. Int. J. Gynecol. Cancer 2005, 15, 445–452. [Google Scholar]
- Calabro, A.; Singletary, S.E.; Tucker, S.; Boddie, A.; Spitzer, G.; Cavaliere, R. In vitro thermo-chemosensitivity screening of spontaneous human tumors: Significant potentiation for cisplatin but not adriamycin. Int. J. Cancer 1989, 43, 385–390. [Google Scholar]
- Peng, D.J.; Wang, J.; Zhou, J.Y.; Wu, G.S. Role of the Akt/mTOR survival pathway in cisplatin resistance in ovarian cancer cells. Biochem. Biophys. Res. Commun 2010, 394, 600–605. [Google Scholar]
- Wangpaichitr, M.; Wu, C.; You, M.; Kuo, M.T.; Feun, L.; Lampidis, T.; Savaraj, N. Inhibition of mTOR restores cisplatin sensitivity through down-regulation of growth and anti-apoptotic proteins. Eur. J. Pharmacol 2008, 591, 124–127. [Google Scholar]
- Tie, J.; Pan, Y.; Zhao, L.; Wu, K.; Liu, J.; Sun, S.; Guo, X.; Wang, B.; Gang, Y.; Zhang, Y.; et al. MiR-218 inhibits invasion and metastasis of gastric cancer by targeting the Robo1 receptor. PLoS Genet 2010, 6, e1000879. [Google Scholar]
- Boeri, M.; Pastorino, U.; Sozzi, G. Role of MicroRNAs in Lung Cancer: MicroRNA Signatures in Cancer Prognosis. Cancer J 2012, 18, 268–274. [Google Scholar]
- Li, X.; Wang, Q.; Zheng, Y.; Lv, S.; Ning, S.; Sun, J.; Huang, T.; Zheng, Q.; Ren, H.; Xu, J.; et al. Prioritizing human cancer microRNAs based on genes’ functional consistency between microRNA and cancer. Nucleic Acids Res 2011, 39, e153. [Google Scholar]
- Brenner, B.; Hoshen, M.B.; Purim, O.; David, M.B.; Ashkenazi, K.; Marshak, G.; Kundel, Y.; Brenner, R.; Morgenstern, S.; Halpern, M.; et al. MicroRNAs as a potential prognostic factor in gastric cancer. World J. Gastroenterol 2011, 17, 3976–3985. [Google Scholar]
- Borralho, P.M.; Kren, B.T.; Castro, R.E.; da Silva, I.B.; Steer, C.J.; Rodrigues, C.M. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J 2009, 276, 6689–6700. [Google Scholar]
- Luo, J.; Deng, Z.L.; Luo, X.; Tang, N.; Song, W.X.; Chen, J.; Sharff, K.A.; Luu, H.H.; Haydon, R.C.; Kinzler, K.W.; et al. A protocol for rapid generation of recombinant adenoviruses using the AdEasy system. Nat. Protocols 2007, 2, 1236–1247. [Google Scholar]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Stable Cell Line | IC50 (μM) of Cells Treated by CDDP |
|---|---|
| HeLa/miR-NC | 15.85 ± 1.21 |
| HeLa/miR-218 | 5.96 ± 0.57 *,# |
| HeLa/miR-218 + Rictor | 11.88 ± 0.94 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, J.; Ping, Z.; Ning, H. MiR-218 Impairs Tumor Growth and Increases Chemo-Sensitivity to Cisplatin in Cervical Cancer. Int. J. Mol. Sci. 2012, 13, 16053-16064. https://doi.org/10.3390/ijms131216053
Li J, Ping Z, Ning H. MiR-218 Impairs Tumor Growth and Increases Chemo-Sensitivity to Cisplatin in Cervical Cancer. International Journal of Molecular Sciences. 2012; 13(12):16053-16064. https://doi.org/10.3390/ijms131216053
Chicago/Turabian StyleLi, Jiarui, Zhang Ping, and Hui Ning. 2012. "MiR-218 Impairs Tumor Growth and Increases Chemo-Sensitivity to Cisplatin in Cervical Cancer" International Journal of Molecular Sciences 13, no. 12: 16053-16064. https://doi.org/10.3390/ijms131216053