Structure Prediction, Molecular Dynamics Simulation and Docking Studies of D-Specific Dehalogenase from Rhizobium sp. RC1


Abstract
:1. Introduction
2. Results and Discussion
2.1. The Primary and Secondary Structure of DehD
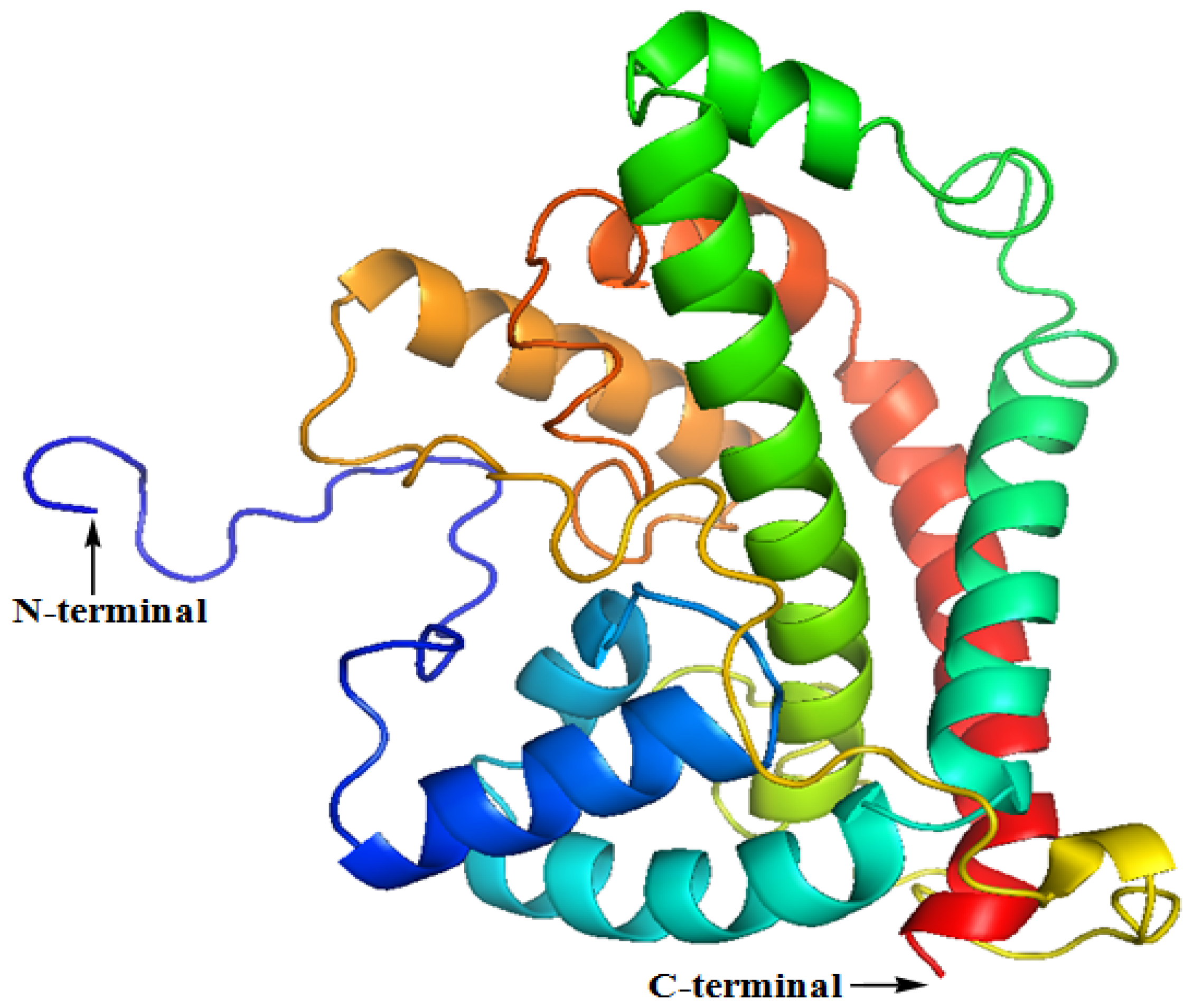
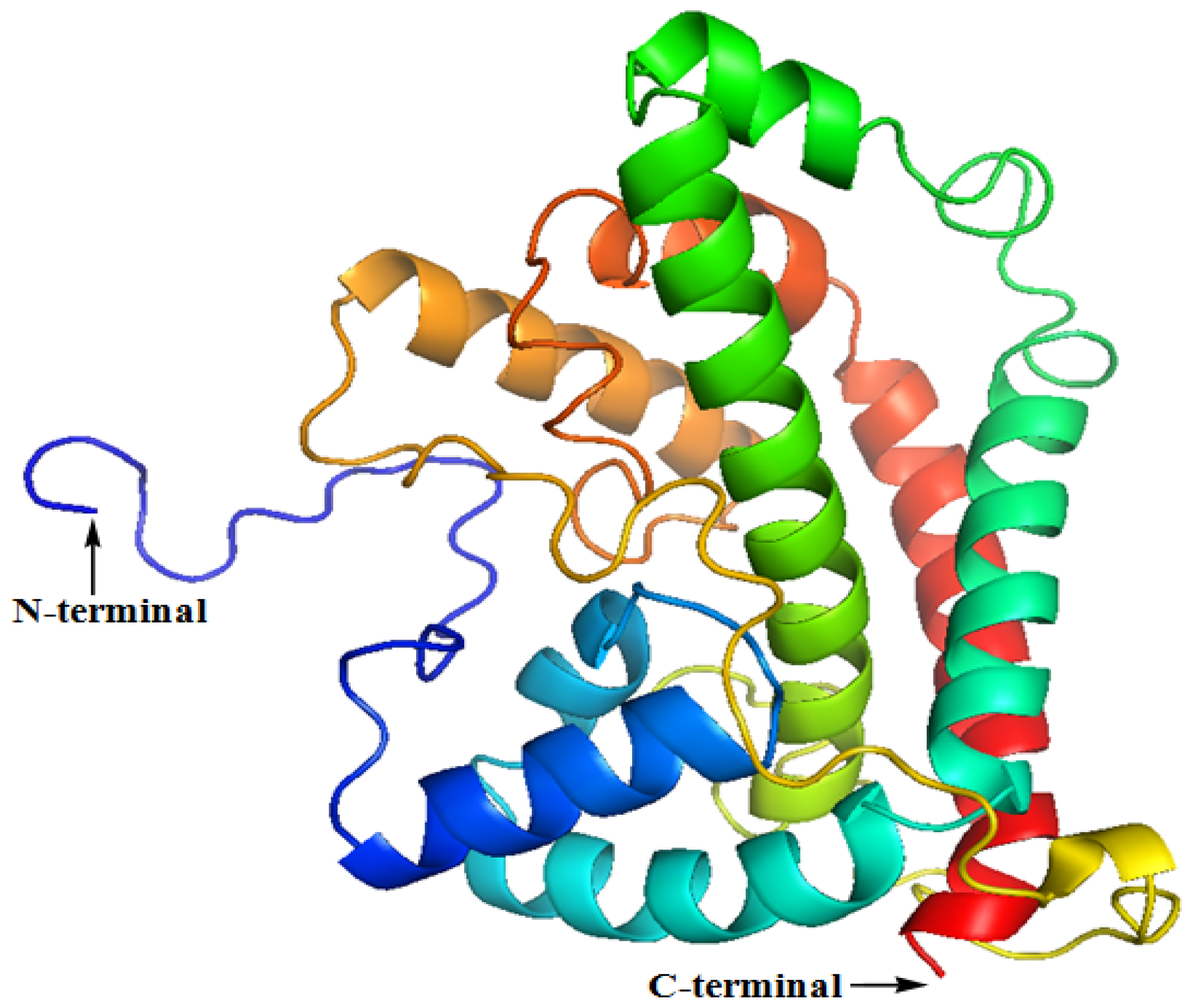
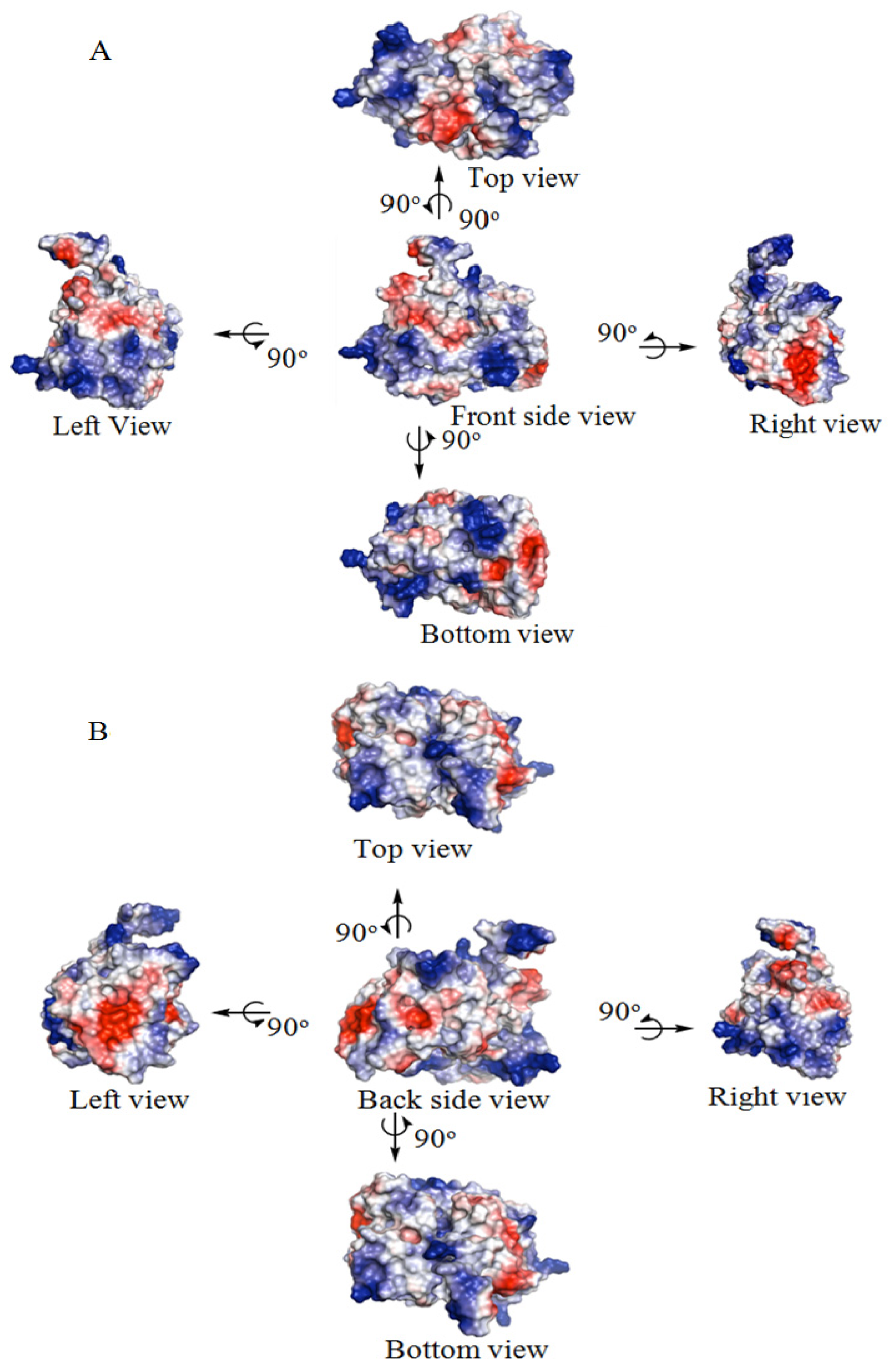
2.2. The Three-Dimensional (3D) Structure of DehD
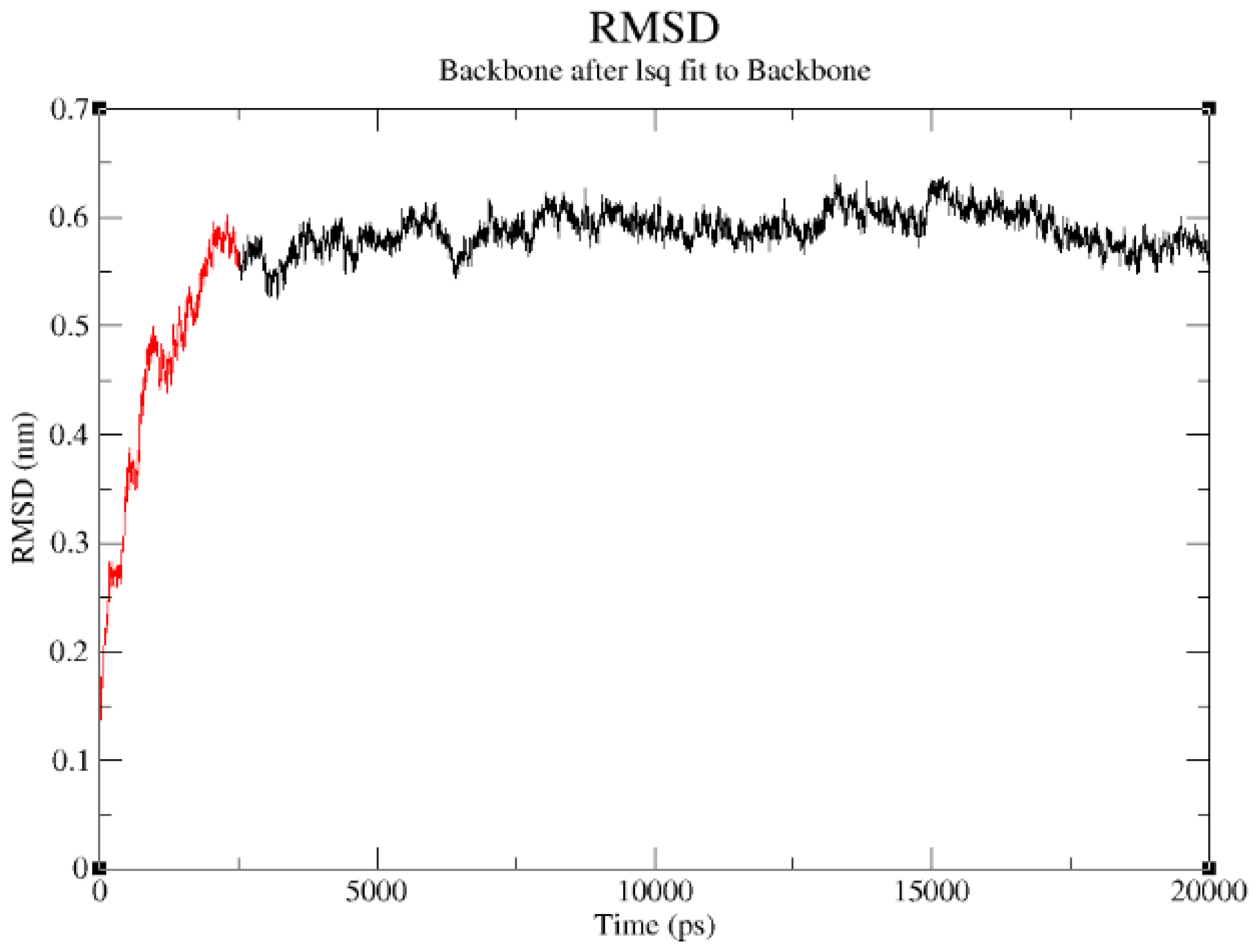
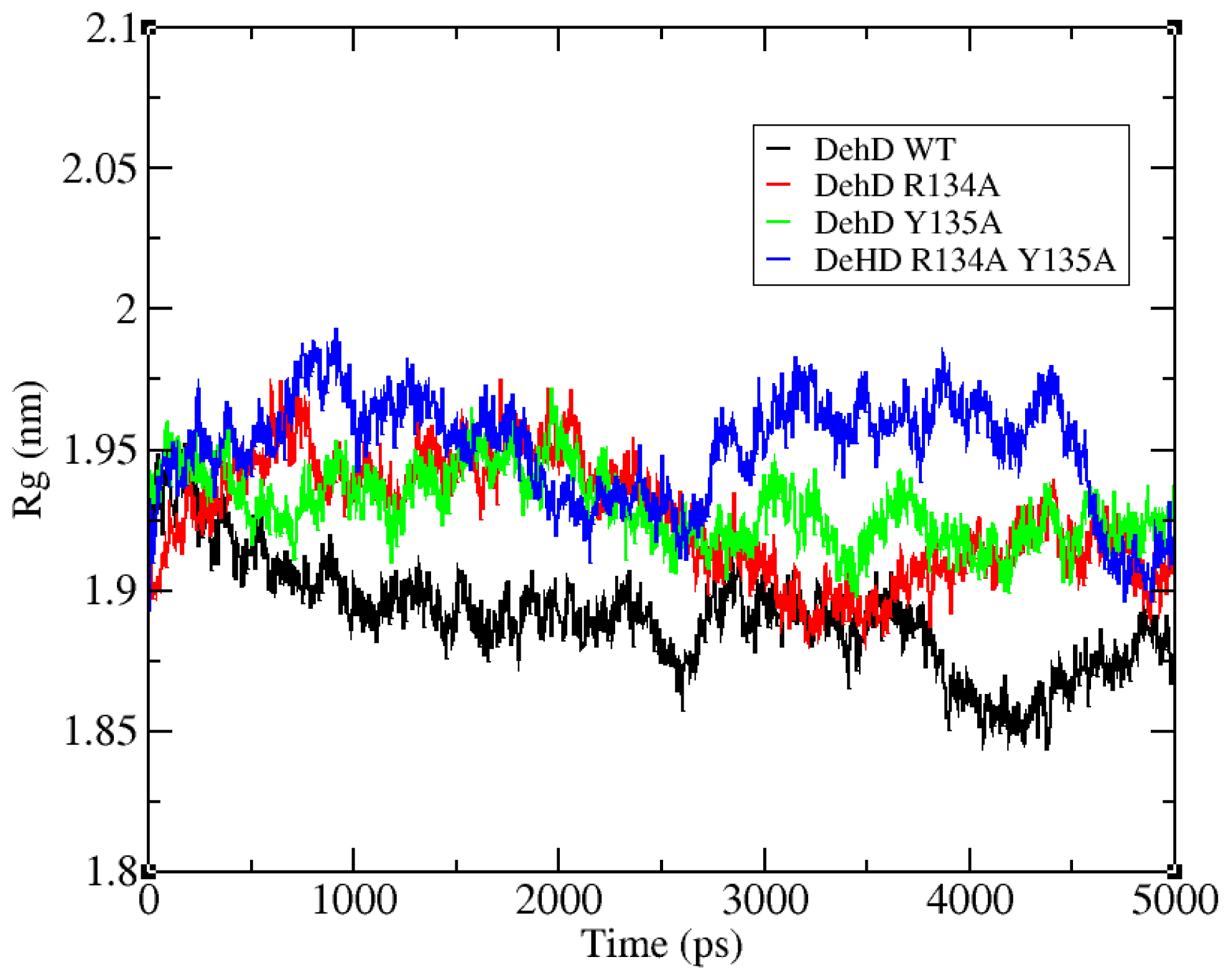
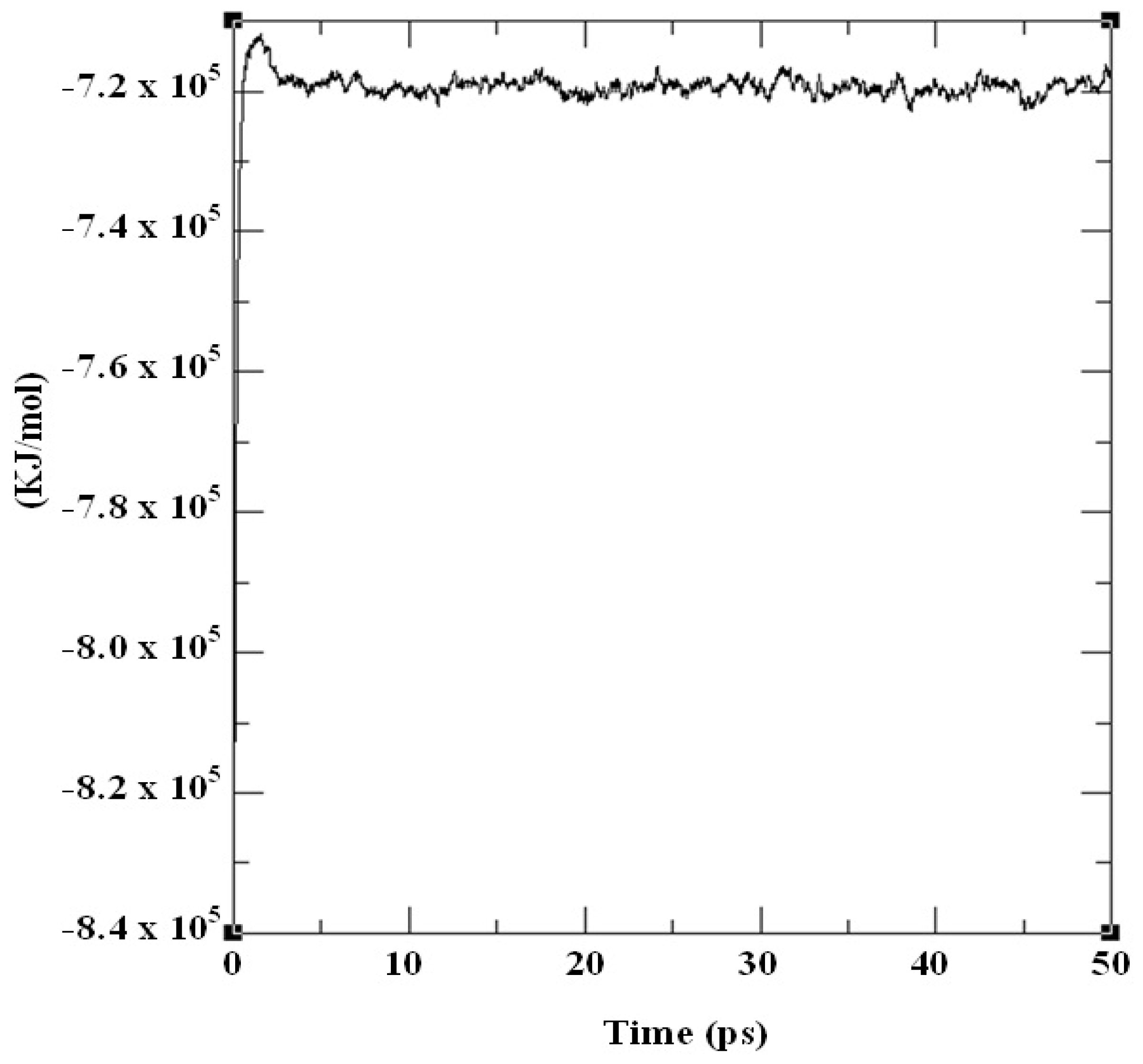
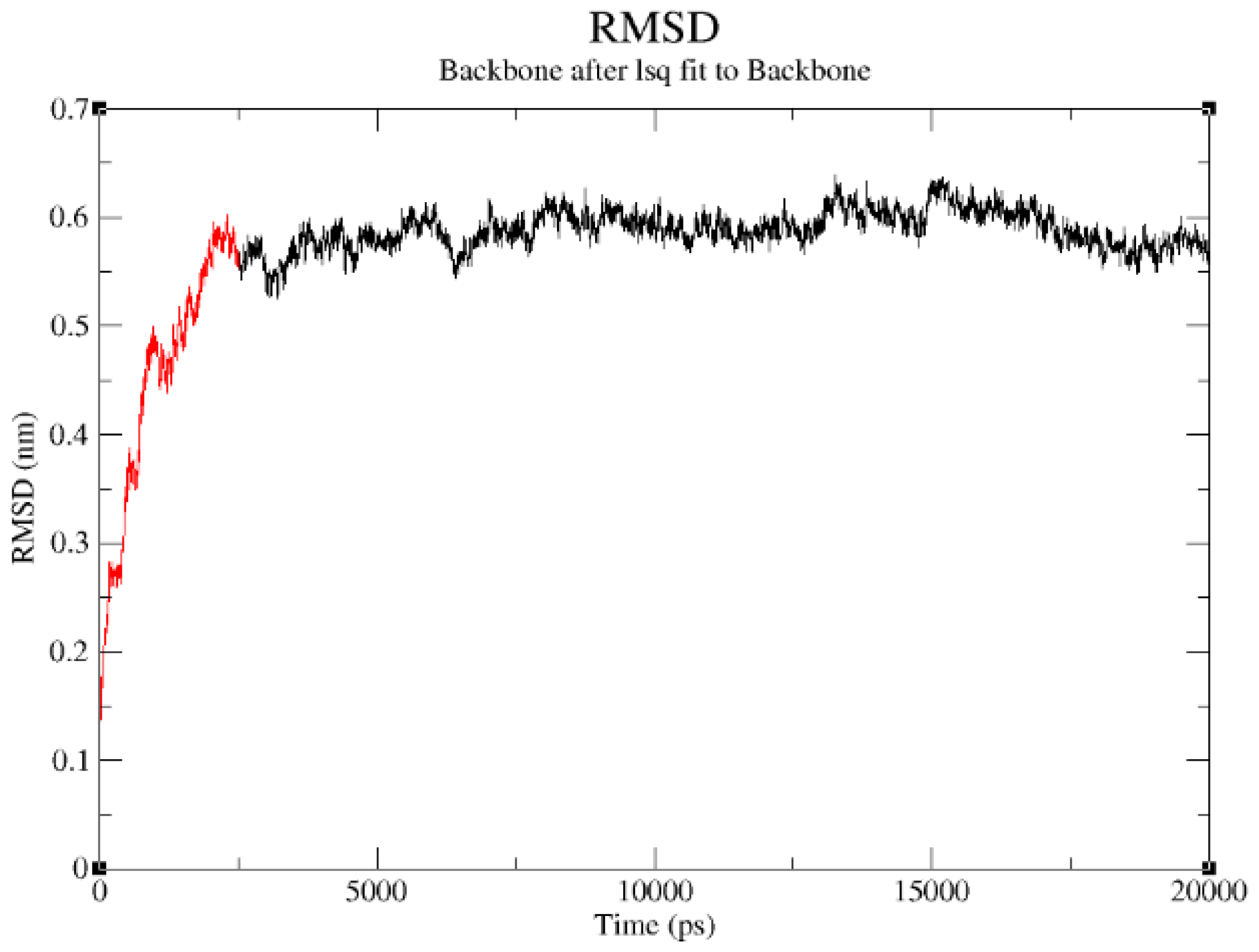
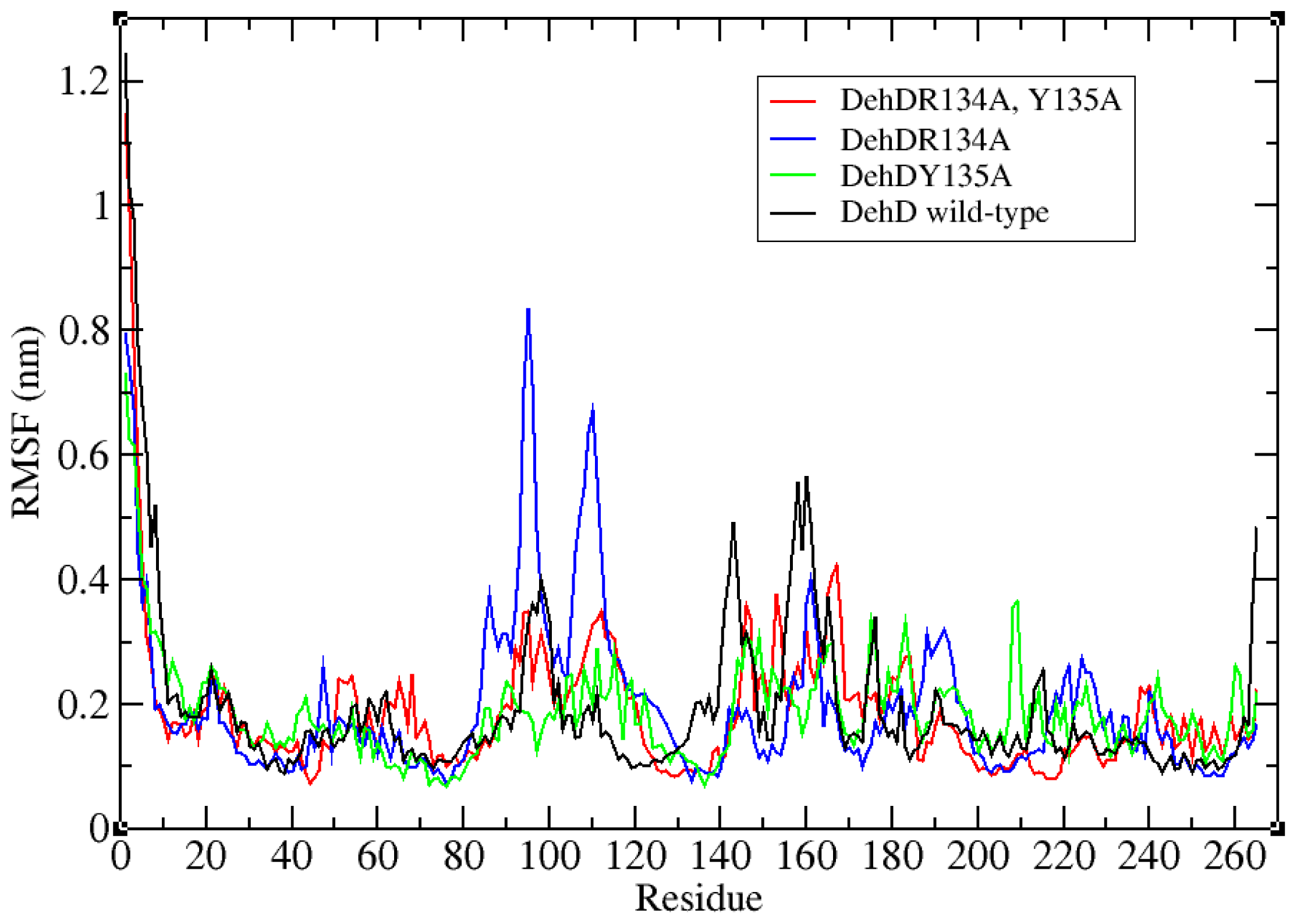
2.3. Structural Model Refinement
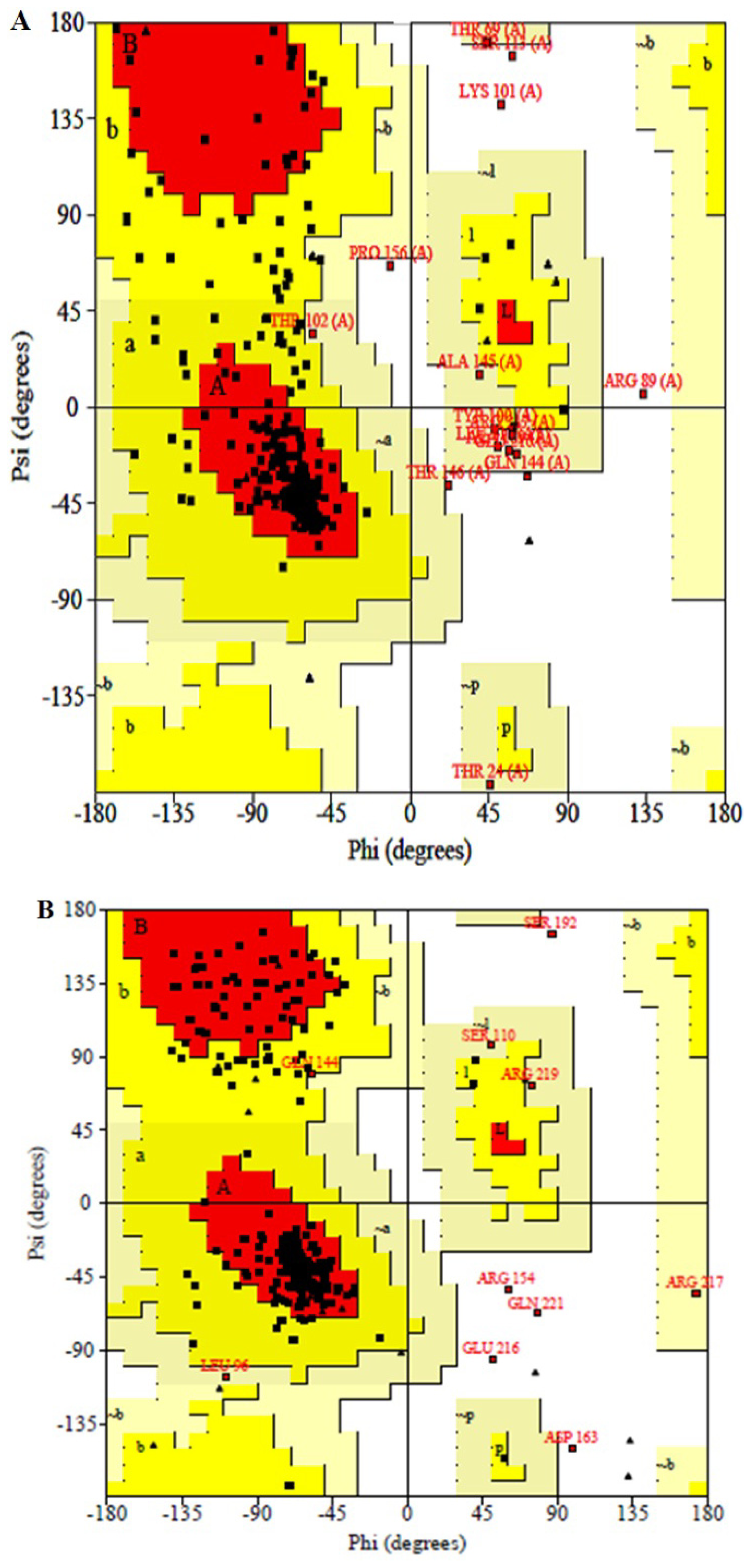
2.4. Structural Model Validation
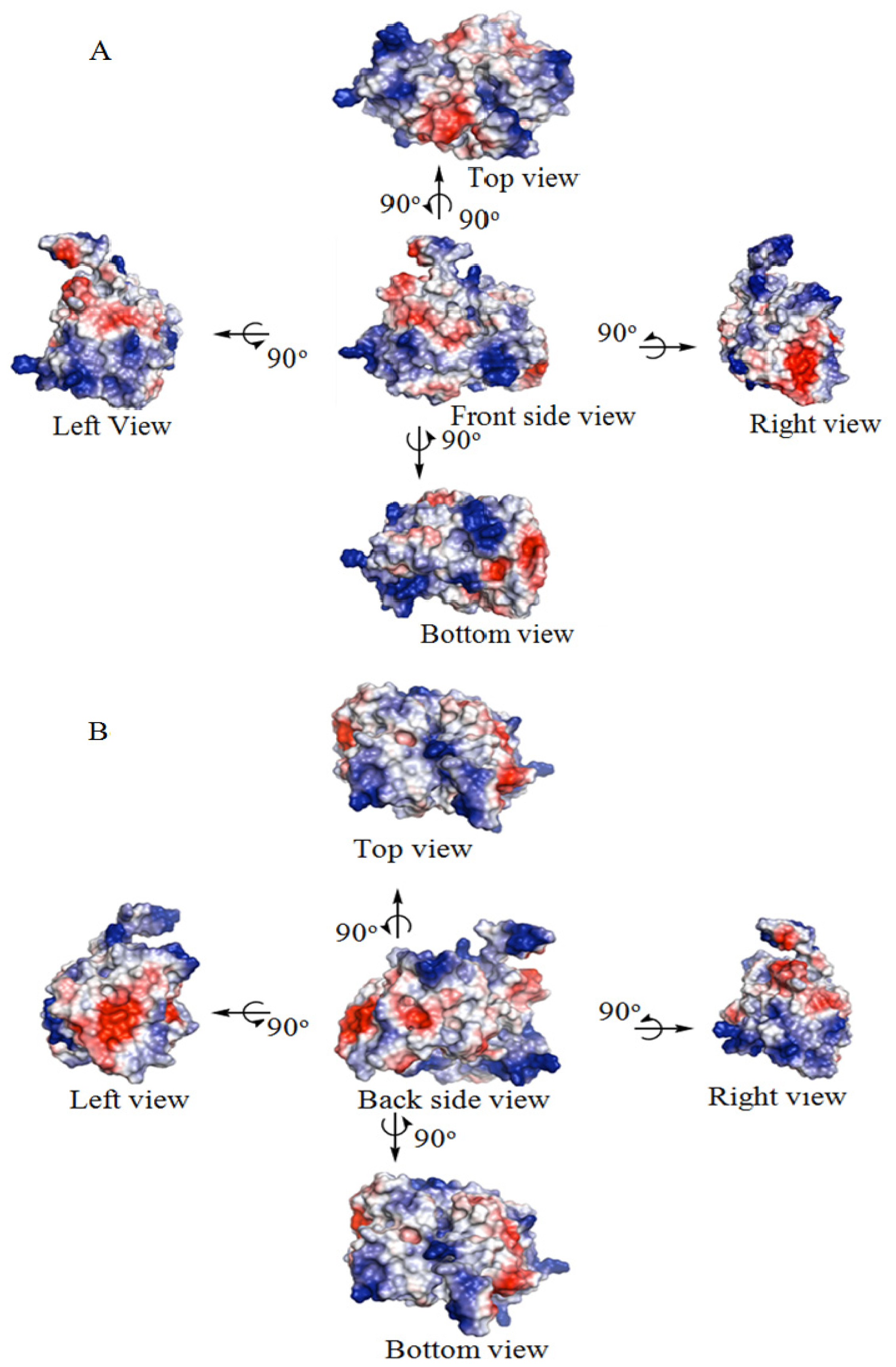
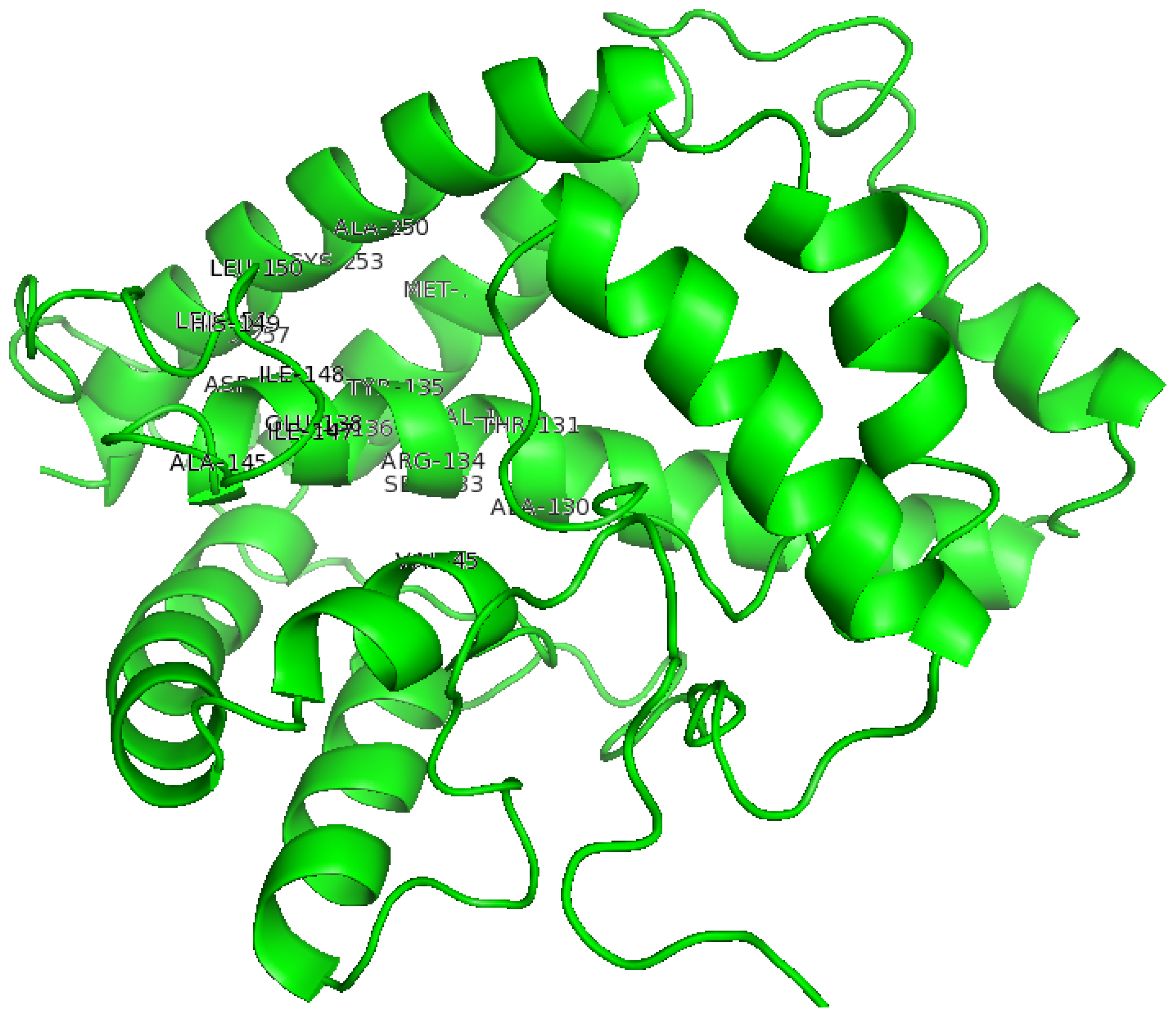
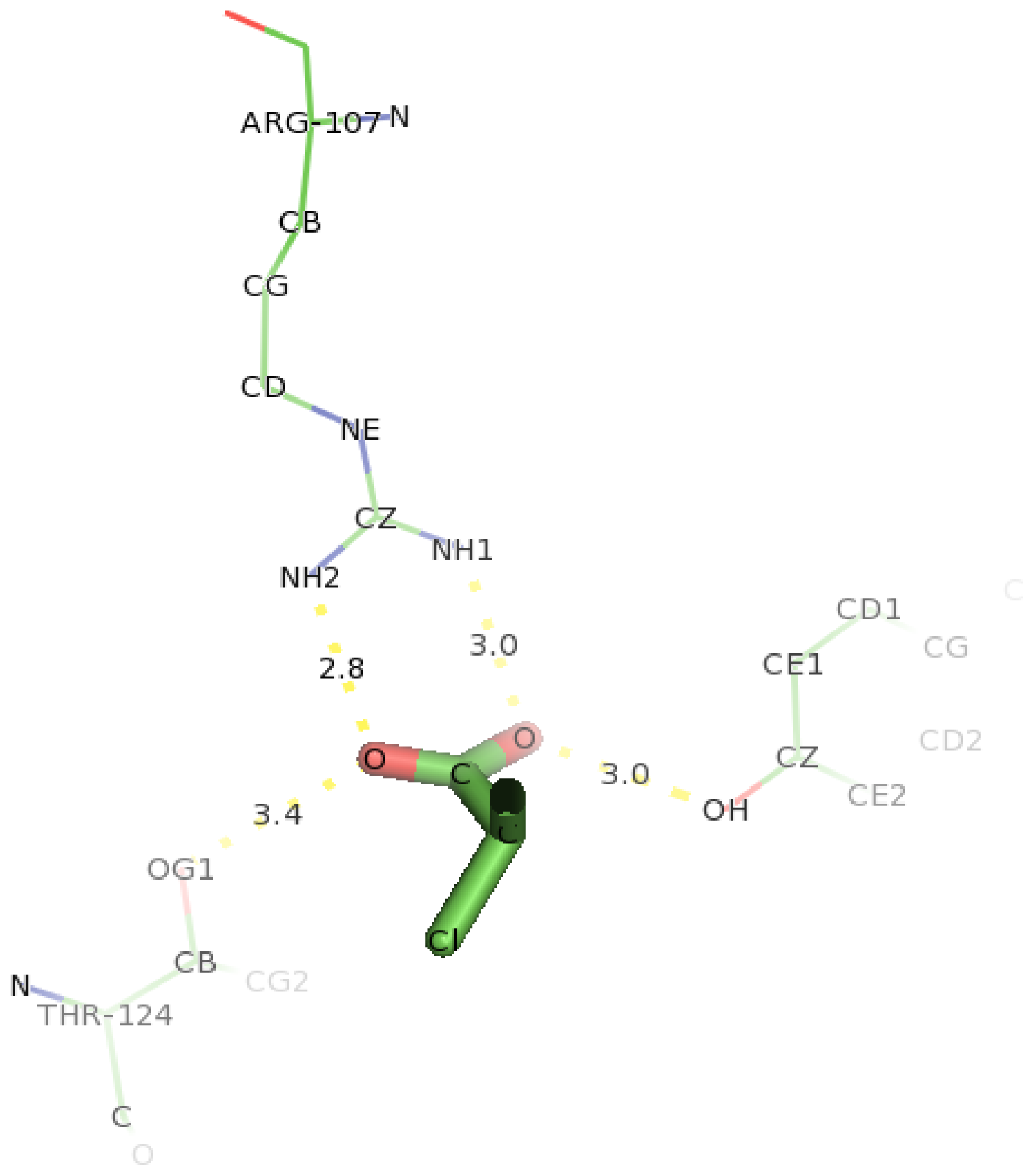
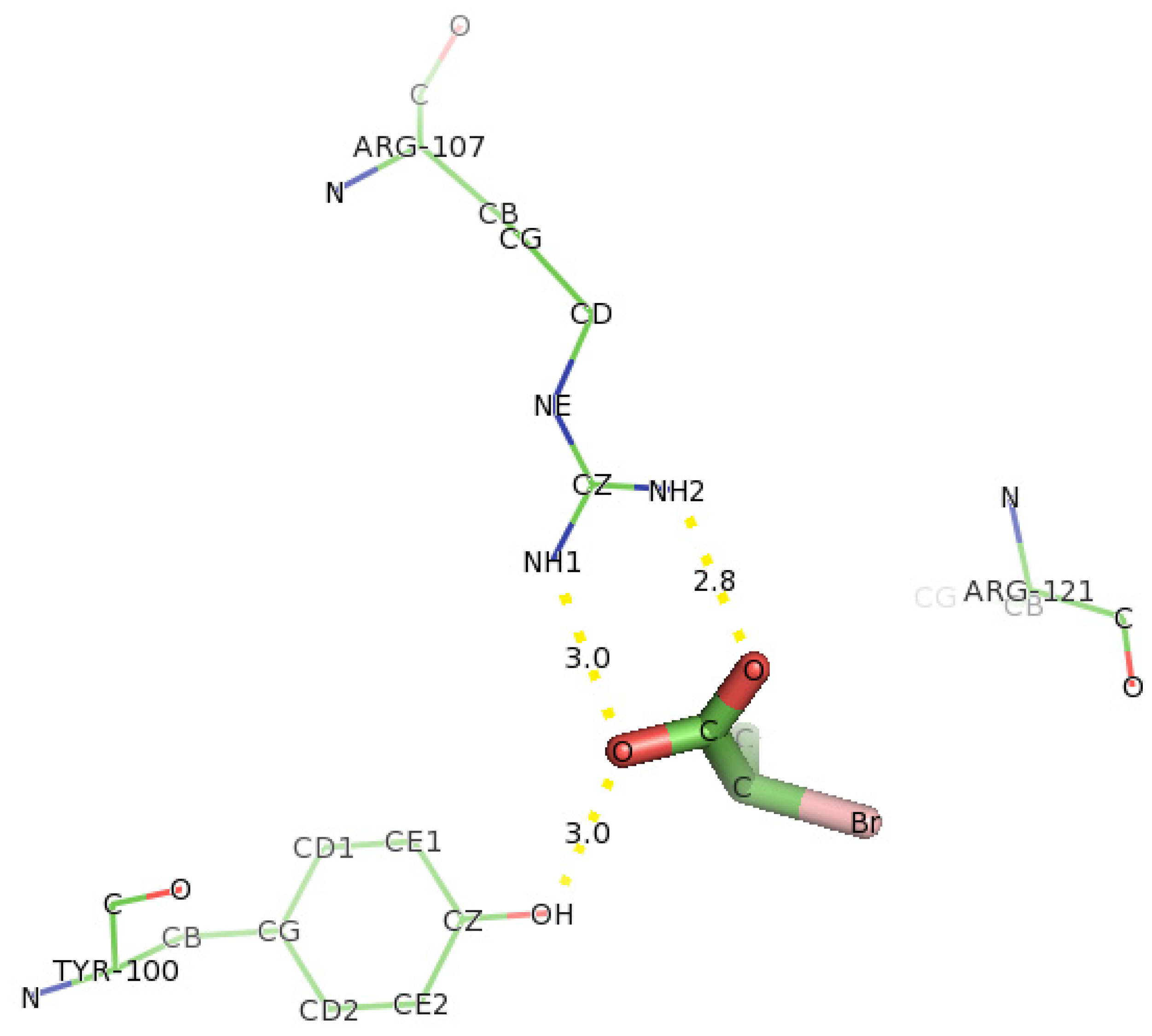
2.5. Identification of the Binding Site and Catalytic Residues Lining the Active Site of DehD
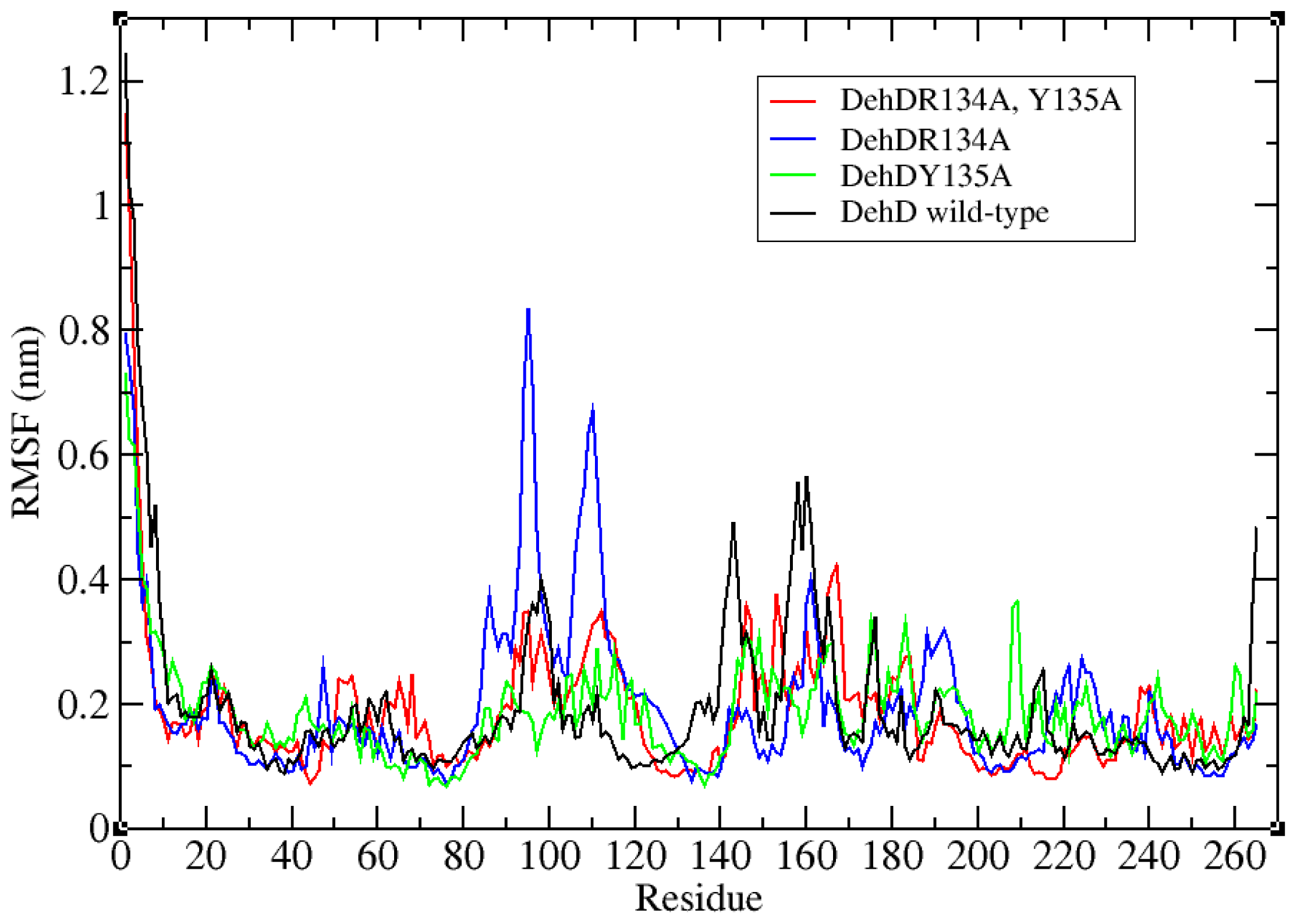
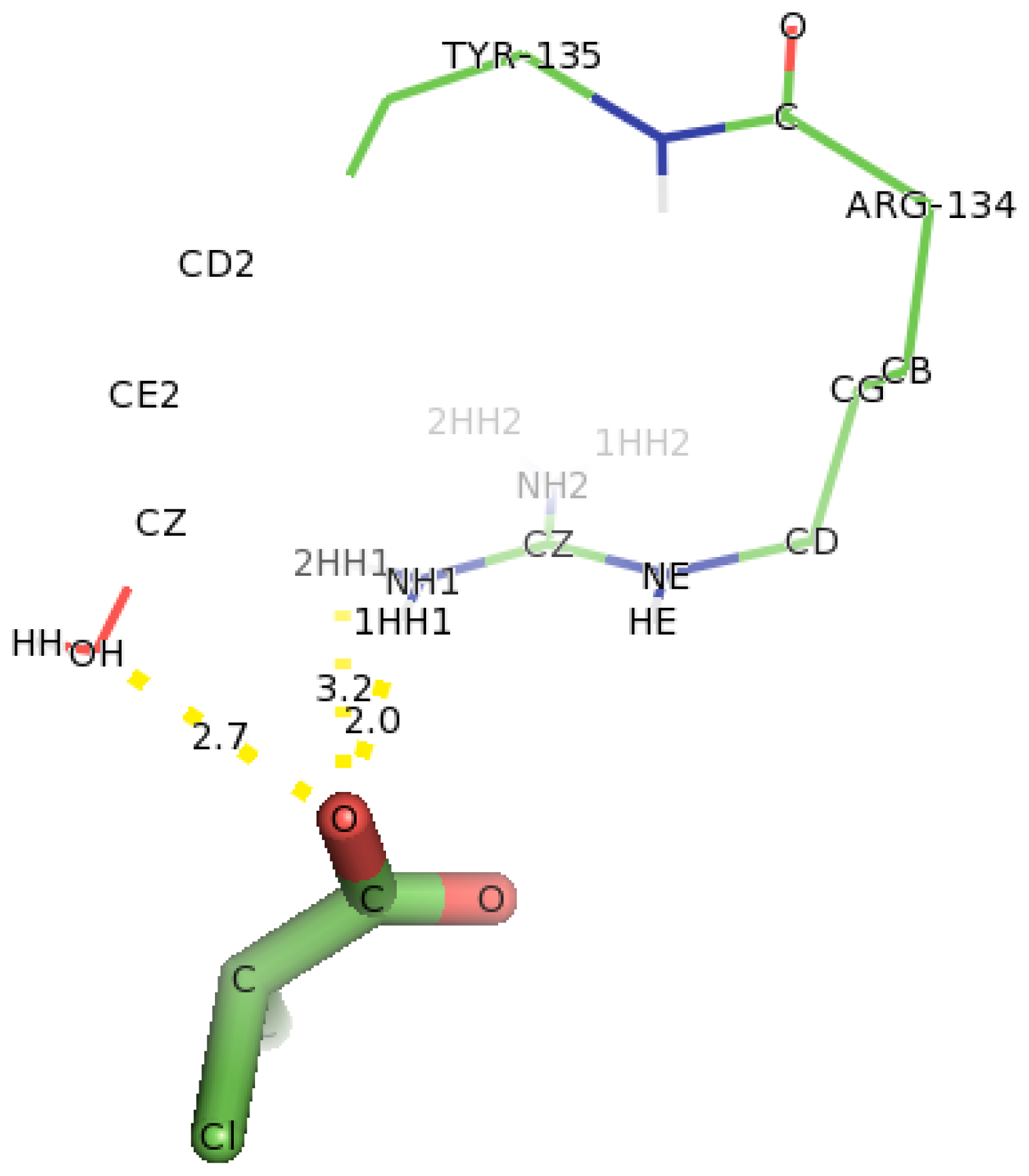
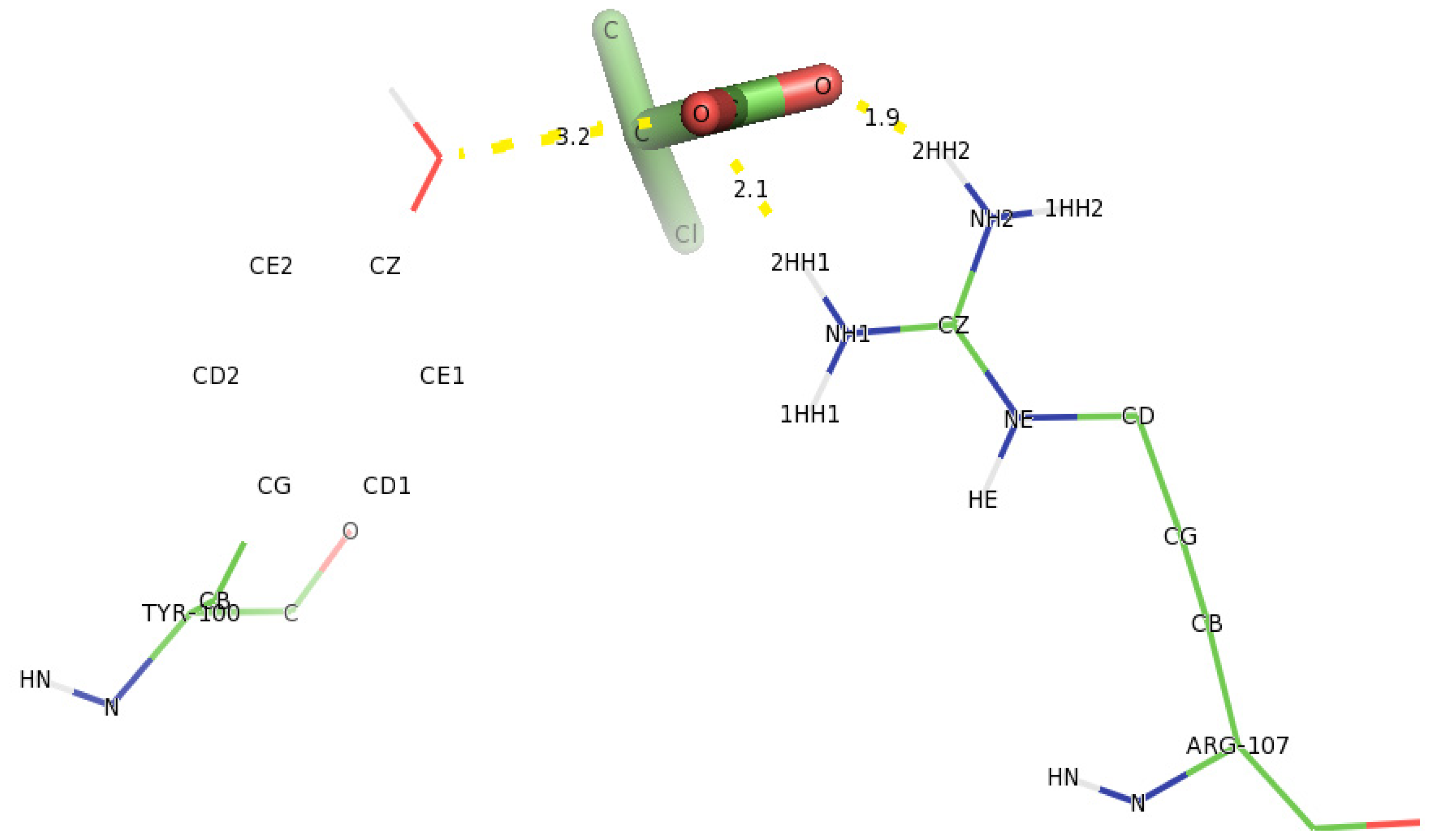
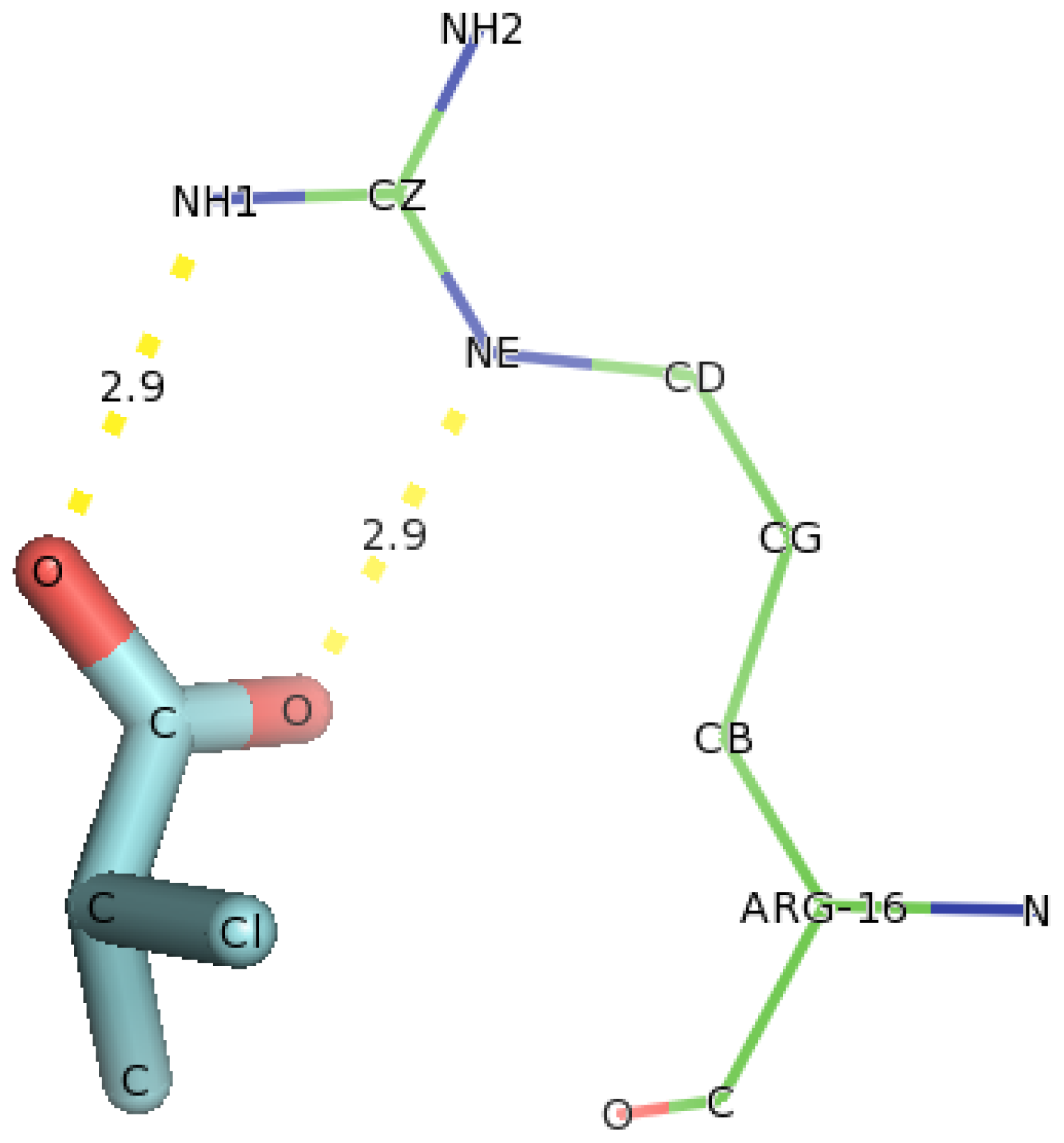
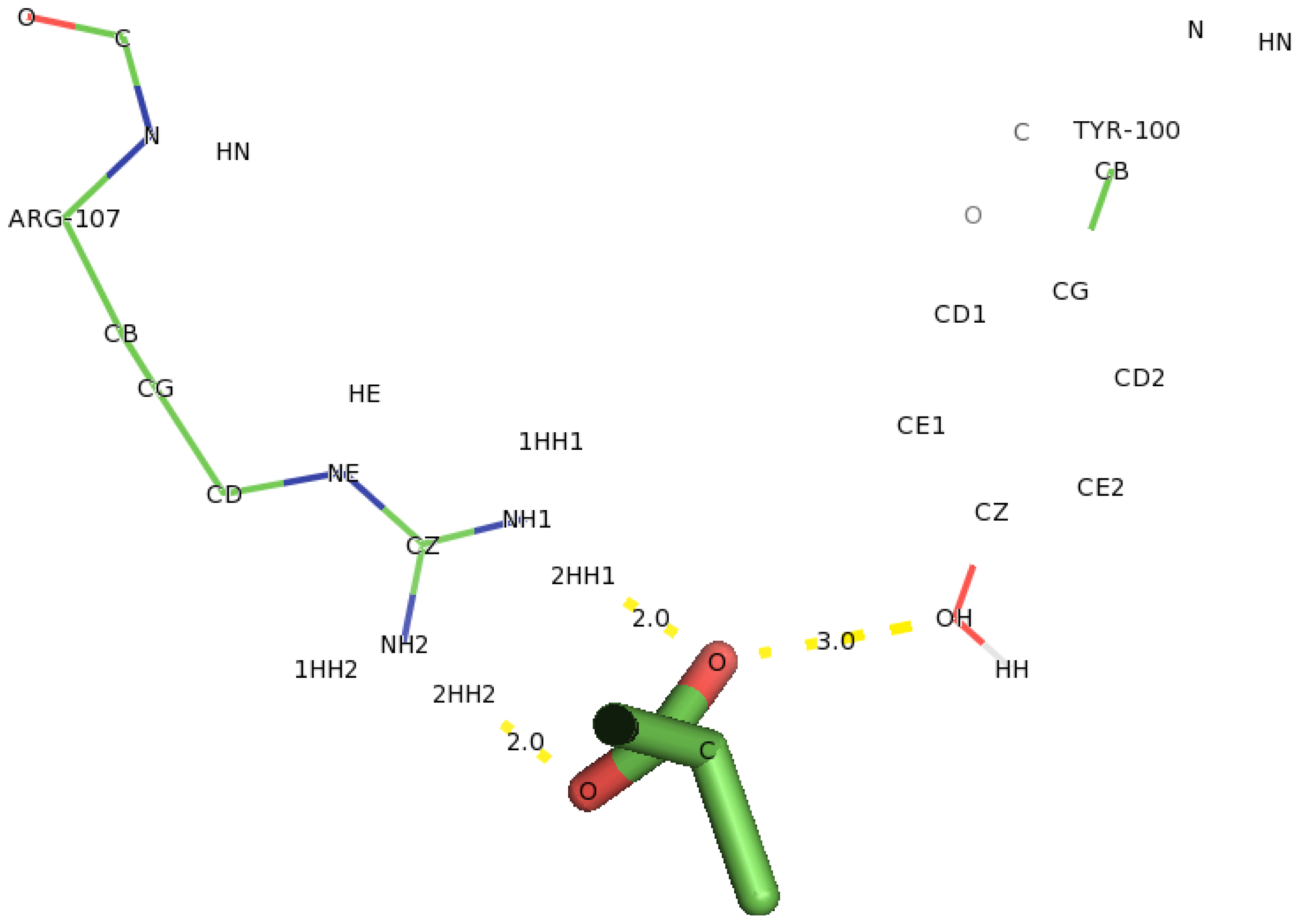
2.6. Identification of the Key Catalytic Residues of DehD by Docking
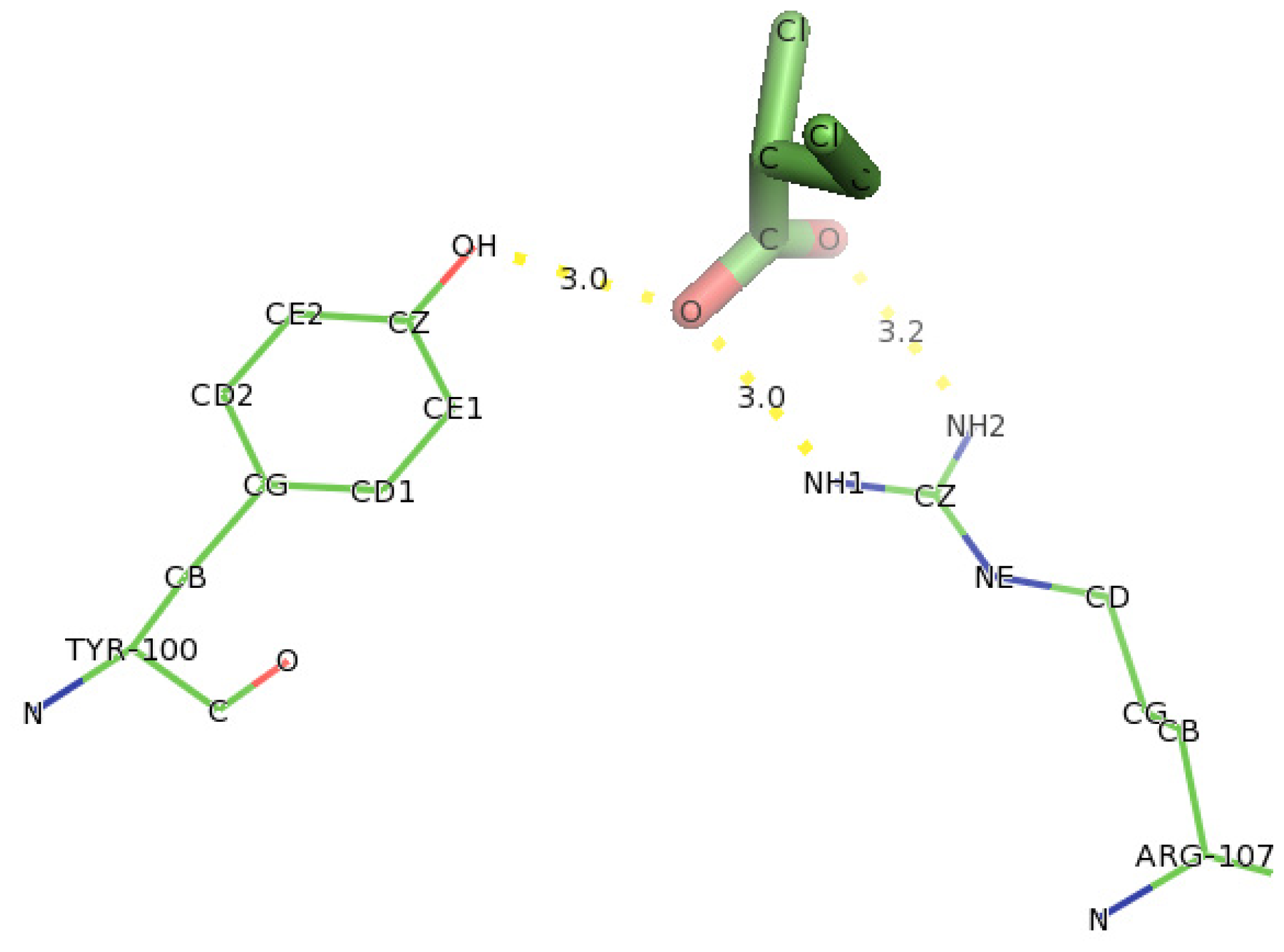
2.7. Docking of Other Substrates into DehD Active Site
3. Experimental Section
3.1. The Primary and Secondary Structure of DehD
3.2. Three-Dimensional (3D) Model of DehD
3.3. Structure Refinement and Validation
3.4. Identification of Catalytic Residues and Substrate Docking into the Active Site of DehD
4. Conclusions
Supplementary Information
ijms-13-15724-s001.pdfAcknowledgments
- Conflict of InterestThe authors declare that they have no conflict of interest and are responsible for the content of the paper.
References
- Slater, J.H.; Bull, A.T.; Hardman, D.J. Microbial dehalogenation of halogenated alkanoic acids, alcohols and alkanes. Adv. Microb. Physiol 1997, 38, 133–176. [Google Scholar]
- Weightman, A.J.; Topping, A.W.; Hill, K.E.; Lee, L.L.; Sakai, K.; Slater, J.H.; Thomas, A.W. Transposition of DEH, a broad-host-range transposon flanked by ISPpu12, in Pseudomonas putida is associated with genomic rearrangements and dehalogenase gene silencing. J. Bacteriol 2002, 184, 6581–6591. [Google Scholar]
- Janssen, D.B.; Oppentocht, J.E.; Poelarends, G.J. Microbial dehalogenation. Curr. Opin. Biotechnol 2001, 12, 254–258. [Google Scholar]
- Ridder, I.S.; Rozeboom, H.J.; Kalk, K.H.; Dijkstra, B.W. Crystal structures of intermediates in the dehalogenation of haloalkanoates by L-2-haloacid dehalogenase. J. Biol. Chem 1999, 274, 30672–30678. [Google Scholar]
- Li, Y.-F.; Hata, Y.; Fujii, T.; Hisano, T.; Nishihara, M.; Kurihara, T.; Esaki, N. Crystal structures of reaction intermediates of L-2-haloacid dehalogenase and implications for the reaction mechanism. J. Biol. Chem 1998, 273, 15035–15044. [Google Scholar]
- Leigh, J.A.; Skinner, A.J.; Cooper, R.A. Partial purification, stereospecificity and stoichiometry of three dehalogenases from a Rhizobium species. FEMS Microbiol. Lett 1988, 49, 353–356. [Google Scholar]
- Swanson, P.E. Dehalogenases applied to industrial-scale biocatalysis. Curr. Opin. Biotechnol 1999, 10, 365–369. [Google Scholar]
- Hill, K.E.; Marchesi, J.R.; Weightman, A.J. Investigation of two evolutionarily unrelated halocarboxylic acid dehalogenase gene families. J. Bacteriol 1999, 181, 2535–2547. [Google Scholar]
- Berry, E.K.M.; Allison, N.; Skinner, A.J.; Cooper, R.A. Degradation of the selective herbicide 2,2-dichloropropionate (Dalapon) by a soil bacterium. J. Gen. Microbiol 1979, 110, 39–45. [Google Scholar]
- Smith, J.M.; Harrison, K.; Colby, J.; Taylor, S.C. Determination of D-2-halopropionate dehalogenase activity from Pseudomonas putida strain AJ1/23 by ion chromatography. FEMS Microbiol. Lett 1989, 57, 71–74. [Google Scholar]
- Smith, J.M.; Harrison, K.; Colby, J. Purification and characterization of D-2-haloacid dehalogenase from Pseudomonas putida strain AJ1/23. J. Gen. Microbiol 1990, 136, 881–886. [Google Scholar]
- Barth, P.T.; Bolton, L.; Thomson, J.C. Cloning and partial sequencing of an operon encoding two Pseudomonas putida haloalkanoate dehalogenases of opposite stereospecificity. J. Bacteriol 1992, 174, 2612–2619. [Google Scholar]
- Schmidberger, J.W.; Wilce, J.A.; Weightman, A.J.; Whisstock, J.C.; Wilce, M.C.J. The Crystal structure of dehI reveals a new α-haloacid dehalogenase fold and active-site mechanism. J. Mol. Biol 2008, 378, 284–294. [Google Scholar]
- Stringfellow, J.M.; Cairns, S.S.; Cornish, A.; Cooper, R.A. Haloalkanoate dehalogenase II (dehE) of a Rhizobium sp: Molecular analysis of the gene and formation of carbon monoxide from trihaloacetate by the enzyme. Eur. J. Biochem 1997, 250, 789–79. [Google Scholar]
- Mikos, A.G.; Lyman, M.D.; Freed, L.E.; Langer, R. Wetting of poly(L-lactic acid) and poly(DL-lactic-co-glycolic acid) foams for tissue culture. Biomaterials 1994, 15, 55–58. [Google Scholar]
- Mooney, D.J.; Baldwin, D.F.; Suh, N.P.; Vacanti, J.P.; Langer, R. Novel approach to fabricate porous sponges of poly(D,L-lactic-co-glycolic acid) without the use of organic solvents. Biomaterials 1996, 17, 1417–1422. [Google Scholar]
- Tamai, H.; Igaki, K.; Kyo, E.; Kosuga, K.; Kawashima, A.; Matsui, S.; Komori, H.; Tsuji, T.; Motohara, S.; Uehata, H. Initial and 6-month results of biodegradable poly-L-lactic acid coronary stents in humans. Circulation 2000, 102, 399–404. [Google Scholar]
- Vochelle, D. The use of poly-L-lactic acid in the management of soft-tissue augmentation: A five-year experience. Sem. Cut. Med. Surg 2004, 23, 223–226. [Google Scholar]
- Burgess, C.M.; Quiroga, R.M. Assessment of the safety and efficacy of poly-L-lactic acid for the treatment of HIV-associated facial lipoatrophy. J. Am. Acad. Dermatol 2005, 52, 233–239. [Google Scholar]
- Middleton, J.C.; Tipton, A.J. Synthetic biodegradable polymers as orthopedic devices. Biomaterials 2000, 21, 2335–2346. [Google Scholar]
- Fiore, G.L.; Jing, F.; Young, J.V.G.; Cramer, C.J.; Hillmyer, M.A. High Tg aliphatic polyesters by the polymerization of spirolactide derivatives. Polym. Chem 2010, 1, 870–877. [Google Scholar]
- Sadove, R. Injectable poly-L-lactic acid: A novel sculpting agent for the treatment of dermal fat atrophy after severe acne. Aesth. Plast. Surg 2009, 33, 113–116. [Google Scholar]
- Bryceland, A.; Jones, R. L-Lactic Acid. In Biopesticides Registration Action Document; United States Environmental Protection Agency: Washington, DC, USA, 2009. [Google Scholar]
- Taylor, S.C. S-2-Chloropropionic Acid by Biotransformations. In Opportunities in Biotransformations; Copping, L.G., Martin, R., Pickett, J.A., Bucke, C., Bunch, A.W., Eds.; Elsevier: London, UK, 1990; pp. 170–76. [Google Scholar]
- Leigh, J.A.; Skinner, A.J.; Cooper, R.A. Isolation and partial characterisation of dehalogenase-deficient mutants of a Rhizobium sp. FEMS Microbiol. Lett 1986, 36, 163–166. [Google Scholar]
- Cairns, S.S.; Cornish, A.; Cooper, R.A. Cloning, sequencing and expression in Escherichia coli of two Rhizobium sp. genes encoding haloalkanoate dehalogenases of opposite stereospecificity. Eur. J. Biochem 1996, 235, 744–749. [Google Scholar]
- Lin, C.; Yang, L.; Xu, G.; Wu, J. Biodegradation and metabolic pathway of β-chlorinated aliphatic acid in Bacillus sp. CGMCC no. 4196. Appl. Microbiol. Biotechnol 2011, 90, 689–96. [Google Scholar]
- Huyop, F.; Sudi, I.Y. D-specific dehalogenases, a review. Biotechnol. Biotechnol. Equip 2012, 26, 2817–2822. [Google Scholar]
- Huyop, F.Z.; Ronald, A.C. A potential use of dehalogenase D (DehD) from Rhizobium sp. for industrial process. J. Teknologi 2003, 38, 69–75. [Google Scholar]
- Mesri, S.; Wahab, R.; Huyop, F. Degradation of 3-chloropropionic acid (3CP) by Pseudomonas sp. B6P isolated from a rice paddy field. Ann. Microbiol 2009, 59, 447–451. [Google Scholar]
- Torrance, J.W.; Thornton, J.M. Structure-Based Prediction of Enzymes and Their Active Sites. In Prediction of Protein Structures, Functions, and Interactions; Bujnicki, J.M., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2008; pp. 187–209. [Google Scholar]
- Tress, M.; Bujnicki, J.M.; Lopez, G.; Valencia, A. Integrating Prediction of Structure, Function, and Interactions. In Prediction of Protein Structures, Functions, and Interactions; Bujnicki, J.M., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2008; pp. 259–279. [Google Scholar]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucl. Acids Res 1997, 25, 3389–3402. [Google Scholar]
- Fitch, W.M. Random sequences. J. Mol. Biol 1983, 163, 171–176. [Google Scholar]
- Corpet, F. Multiple sequence alignment with hierarchical clustering. Nucl. Acids Res 1988, 16, 10881–10890. [Google Scholar]
- Majorek, K.; Kozłowski, Ł.; Jąkalski, M.; Bujnicki, J.M. First Steps of Protein Structure Prediction. In Prediction of Protein Structures, Functions, and Interactions; Bujnicki, J., Ed.; John Wiley & Sons, Ltd: Chichester, UK, 2008; pp. 39–62. [Google Scholar]
- Hutchinson, E.G.; Thornton, J.M. PROMOTIF—A program to identify and analyze structural motifs in proteins. Prot. Sci 1996, 5, 212–220. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of protein secondary structure: Pattern recognition of hydrogen-bonded and geometrical features. Biopolymers 1983, 22, 2577–2637. [Google Scholar]
- Laskowski, R.A.; MacArthur, M.W.; Moss, D.S.; Thornton, J.M. PROCHECK: A program to check the stereochemical quality of protein structures. J. Appl. Cryst 1993, 26, 283–291. [Google Scholar]
- Ramachandran, G.N.; Ramakrishnan, C.; Sasisekharan, V. Stereochemistry of polypeptide chain configurations. J. Mol. Biol 1963, 7, 95–99. [Google Scholar]
- Raushel, F.M.; Thoden, J.B.; Holden, H.M. Enzymes with molecular tunnels. Acc. Chem. Res 2003, 36, 539–548. [Google Scholar]
- Zarina, S.; Zaidi, Z.H. Homology modeling of rho-crystallin from bullfrog (Rana catesbeiana) lens. J. Mol. Graphics Modell 2004, 22, 285–291. [Google Scholar]
- Tumbale, P.; Jamaluddin, H.; Thiyagarajan, N.; Brew, K.; Acharya, K.R. Structural basis of UDP-galactose binding by alpha-1,3-galactosyltransferase (alpha3GT): Role of negative charge on aspartic acid 316 in structure and activity. CORD Conf. Proc 2008, 47, 8711–8718. [Google Scholar]
- Kurihara, T.; Esaki, N.; Soda, K. Bacterial 2-haloacid dehalogenases: Structures and reaction mechanisms. J. Mol. Catal. B 2000, 10, 57–65. [Google Scholar]
- Thallapally, P.K.; Nangia, A. A Cambridge structural database analysis of the C-HCl interaction: C-HCl and C-HCl-M often behave as hydrogen bonds but C-HCl-C is generally a Van der Waals interaction. CrystEngComm 2001, 3, 114–119. [Google Scholar]
- Mowafy, A.M.; Kurihara, T.; Kurata, A.; Uemura, T.; Esaki, N. 2-Haloacrylate hydratase, a new class of flavoenzyme that catalyzes the addition of water to the substrate for dehalogenation. Appl. Environ. Microbiol 2010, 76, 6032–6037. [Google Scholar]
- Weightman, A.J.; Weightman, A.L.; Slater, J.H. Stereospecificity of 2-monochloropropionate dehalogenation by the two dehalogenases of Pseudomonas putida PP3: Evidence for two different dehalogenation mechanisms. J. Gen. Microbiol 1982, 128, 1755–1762. [Google Scholar]
- Bajaj, C.; Park, S.; Thane, A.G. Parallel Multi-PC Volume Rendering System. In CS and TICAM Technical Report; Park, S., Bajaj, C., Eds.; University of Texas, Austin: Austin, TX, USA; p. 2002.
- Bajaj, C.L.; Pascucci, V.; Schikore, D.R. Fast Isocontouring for Improved Interactivity. In Proceedings of the 1996 Symposium on Volume Visualization; IEEE Press: San Francisco, CA, USA, 1996; pp. 39–46. [Google Scholar]
- Sanner, M.F. Python: A programming language for software integration and development. J. Mol. Graphics Modell 1999, 17, 57–61. [Google Scholar]
- Sanner, M.F.; Olson, A.J.; Spehner, J.C. Reduced surface: An efficient way to compute molecular surfaces. Biopolymers 1996, 38, 305–320. [Google Scholar]
- Sanner, M.F.; Stoffler, D.; Olson, A.J. ViPEr, a Visual Programming Environment for Python. Proceedings of the 10th International Python Conference, Alexandria, VA, USA, 4–7 February 2002; pp. 103–115.
- Nardi-Dei, V.; Kurihara, T.; Park, C.; Miyagi, M.; Tsunasawa, S.; Soda, K.; Esaki, N. D,L-2-haloacid dehalogenase from Pseudomonas sp. 113 is a new class of dehalogenase catalyzing hydrolytic dehalogenation not involving enzyme-substrate ester intermediate. J. Biol. Chem 1999, 274, 20977–20981. [Google Scholar]
- Jing, N.H.; Ab. Wahab, R.; Cooper, R.A.; Huyop, F. Degradation of Herbicide (3-Chloropropionic Acid) by Bacterial Dehalogenases. Proceedings of KUSTEM 4th Annual Seminar, Kuala Terengganu, Malaysia, 2–5 May 2005; pp. 586–590.
- Cheng, J. DOMAC: An accurate, hybrid protein domain prediction server. Nucl. Acids Res 2007, 35, 354–356. [Google Scholar]
- Cheng, J.; Sweredoski, M.; Baldi, P. DOMpro: Protein domain prediction using profiles, secondary structure, relative solvent accessibility, and recursive neural networks. Data Min. Knowl. Discov 2006, 13, 1–10. [Google Scholar]
- Larsen, L.S.Z.; Zhang, M.; Beliakova-Bethell, N.; Bilanchone, V.; Lamsa, A.; Nagashima, K.; Najdi, R.; Kosaka, K.; Kovacevic, V.; Cheng, J.; et al. Ty3 capsid mutations reveal early and late functions of the amino-terminal domain. J. Virol 2007, 81, 6957–6972. [Google Scholar]
- Tress, M.; Cheng, J.; Baldi, P.; Joo, K.; Lee, J.; Seo, J.H.; Lee, J.; Baker, D.; Chivian, D.; Kim, D.; et al. Assessment of predictions submitted for the CASP7 domain prediction category. Proteins 2007, 69, 137–151. [Google Scholar]
- Cheng, J.; Randall, A.Z.; Sweredoski, M.J.; Baldi, P. SCRATCH: A protein structure and structural feature prediction server. Nucl. Acids Res 2005, 33, 72–76. [Google Scholar]
- Cuff, J.A.; Barton, G.J. Application of multiple sequence alignment profiles to improve protein secondary structure prediction. Proteins 2000, 40, 502–511. [Google Scholar]
- Jones, D.T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol 1999, 292, 195–202. [Google Scholar]
- Kelley, L.A.; Sternberg, M.J.E. Protein structure prediction on the Web: A case study using the Phyre server. Nat. Protoc 2009, 4, 363–371. [Google Scholar]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protoc 2010, 5, 725–738. [Google Scholar]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinf 2008, 9, 40. [Google Scholar]
- Zhang, Y. I-TASSER: Fully automated protein structure prediction in CASP8. Proteins 2009, 77, 100–113. [Google Scholar]
- Delano, W.L. The PyMOL molecular graphics system, 2002. Available online: http://www.pymol.org accessed on 28 April 2012.
- Van Der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, flexible, and free. J. Comput. Chem 2005, 26, 1701–1718. [Google Scholar]
- Shroll, R.M.; Straatsma, T.P. Molecular structure of the outer bacterial membrane of Pseudomonas aeruginosa via classical simulation. Biopolymers 2002, 65, 395–407. [Google Scholar]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem 1997, 18, 1463–1472. [Google Scholar]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys 1995, 103, 8577–8593. [Google Scholar]
- Berendsen, H. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun 1995, 91, 43–56. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Fellinger, K.; Leonhardt, H.; Spada, F. A mutagenesis strategy combining systematic alanine scanning with larger mutations to study protein interactions. Anal. Biochem 2008, 373, 176–178. [Google Scholar]
- Bolton, E.; Wang, Y.; Thiessen, P.A.; Bryant, S.H. PubChem: Integrated Platform of Small Molecules and Biological Activities. In Annual Reports in Computational Chemistry; American Chemical Society: Washington, DC, USA, 2008; Volume 4. [Google Scholar]
- Li, T.; Froeyen, M.; Herdewijn, P. Insight into ligand selectivity in HCV NS5B polymerase: Molecular dynamics simulations, free energy decomposition and docking. J. Mol. Model 2010, 16, 49–59. [Google Scholar]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem 1998, 19, 1639–1662. [Google Scholar]
- Bromberg, Y.; Rost, B. SNAP: Predict effect of non-synonymous polymorphisms on function. Nucl. Acids Res 2007, 35, 3823–3835. [Google Scholar]
- Capriotti, E.; Fariselli, P.; Casadio, R. I-Mutant2.0: Predicting stability changes upon mutation from the protein sequence or structure. Nucl. Acids Res 2005, 33, 306–310. [Google Scholar]







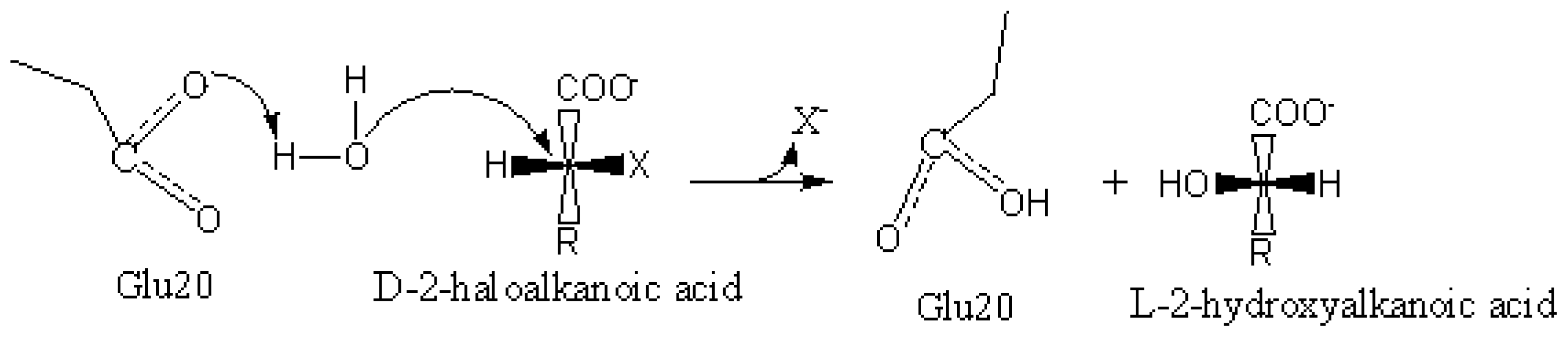

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of amino acid residues in the loop | Loop length | Number of amino acid residues in the helix | Helix length |
|---|---|---|---|
| 24 | M1–T24 | 33 | G5–A37 |
| 5 | F38–W42 | 8 | M43–I50 |
| 2 | P51–I52 | 16 | S53–G68 |
| 1 | T69 | 18 | R70–V87 |
| 14 | E88–K101 | 9 | T102–S110 |
| 3 | G111–S113 | 28 | E114–R141 |
| 25 | G142–G166 | 3 | T167–G169 |
| 24 | F170–D193 | 17 | E194–I210 |
| 9 | L211–R219 | 4 | G220–I223 |
| 4 | S224–V227 | 37 | G228–L264 |
| 1 | P265 | ||
| Total (percentage) | 112 (42.26%) | 173 (65.28%) |
| Stereochemical parameter | Parameter value | Typical value | Bandwidth | Number of bandwidths from mean |
|---|---|---|---|---|
| Percentage residues in favored region | 79.6 | 76.6 | 10.0 | 0.3 |
| Omega angle standard deviation (degrees) | 8.4 | 6.0 | 3.0 | 0.8 |
| Number of bad contacts/100 residues | 0.0 | 10.5 | 10.0 | −1.1 |
| Zeta angle standard deviation (degrees) | 5.6 | 3.1 | 1.6 | 1.6 |
| Hydrogen bond standard deviation | 0.8 | 0.9 | 0.2 | −0.7 |
| Overall G-factor | −0.8 | −0.6 | 0.3 | −0.6 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sudi, I.Y.; Wong, E.L.; Joyce-Tan, K.H.; Shamsir, M.S.; Jamaluddin, H.; Huyop, F. Structure Prediction, Molecular Dynamics Simulation and Docking Studies of D-Specific Dehalogenase from Rhizobium sp. RC1. Int. J. Mol. Sci. 2012, 13, 15724-15754. https://doi.org/10.3390/ijms131215724
Sudi IY, Wong EL, Joyce-Tan KH, Shamsir MS, Jamaluddin H, Huyop F. Structure Prediction, Molecular Dynamics Simulation and Docking Studies of D-Specific Dehalogenase from Rhizobium sp. RC1. International Journal of Molecular Sciences. 2012; 13(12):15724-15754. https://doi.org/10.3390/ijms131215724
Chicago/Turabian StyleSudi, Ismaila Yada, Ee Lin Wong, Kwee Hong Joyce-Tan, Mohd Shahir Shamsir, Haryati Jamaluddin, and Fahrul Huyop. 2012. "Structure Prediction, Molecular Dynamics Simulation and Docking Studies of D-Specific Dehalogenase from Rhizobium sp. RC1" International Journal of Molecular Sciences 13, no. 12: 15724-15754. https://doi.org/10.3390/ijms131215724
APA StyleSudi, I. Y., Wong, E. L., Joyce-Tan, K. H., Shamsir, M. S., Jamaluddin, H., & Huyop, F. (2012). Structure Prediction, Molecular Dynamics Simulation and Docking Studies of D-Specific Dehalogenase from Rhizobium sp. RC1. International Journal of Molecular Sciences, 13(12), 15724-15754. https://doi.org/10.3390/ijms131215724