Role of Btg2 in the Progression of a PDGF-Induced Oligodendroglioma Model


Abstract
:1. Introduction
2. Results and Discussion
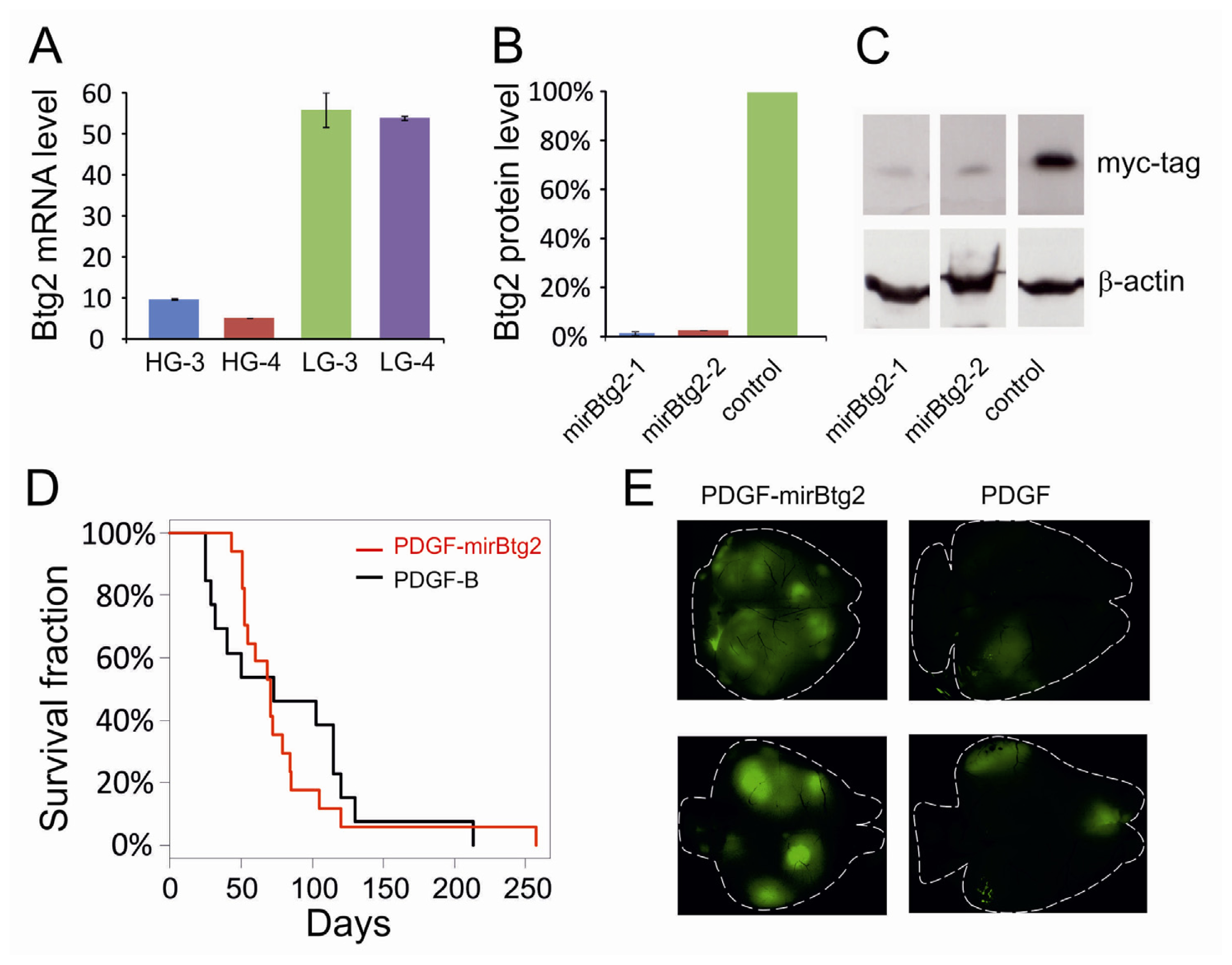
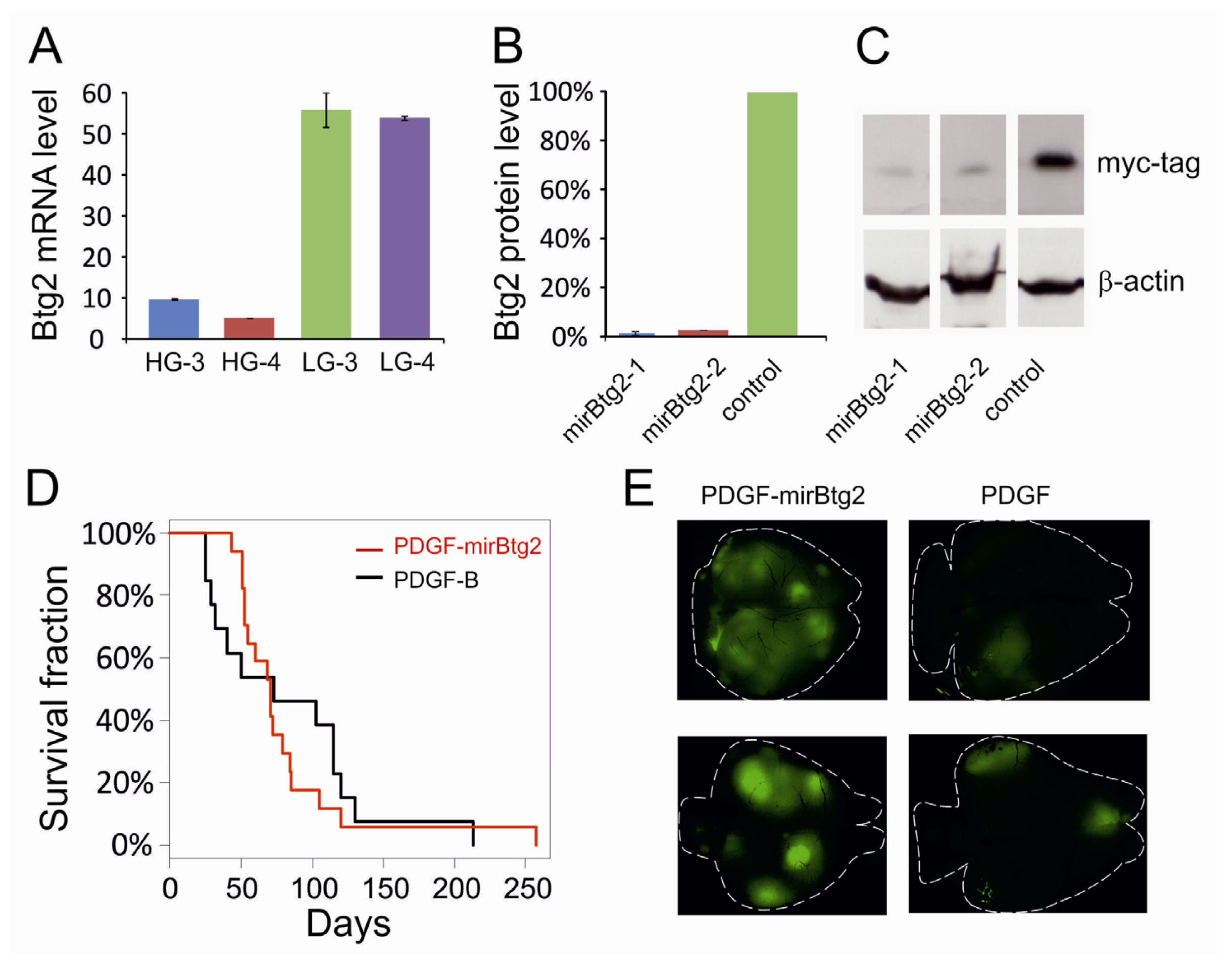
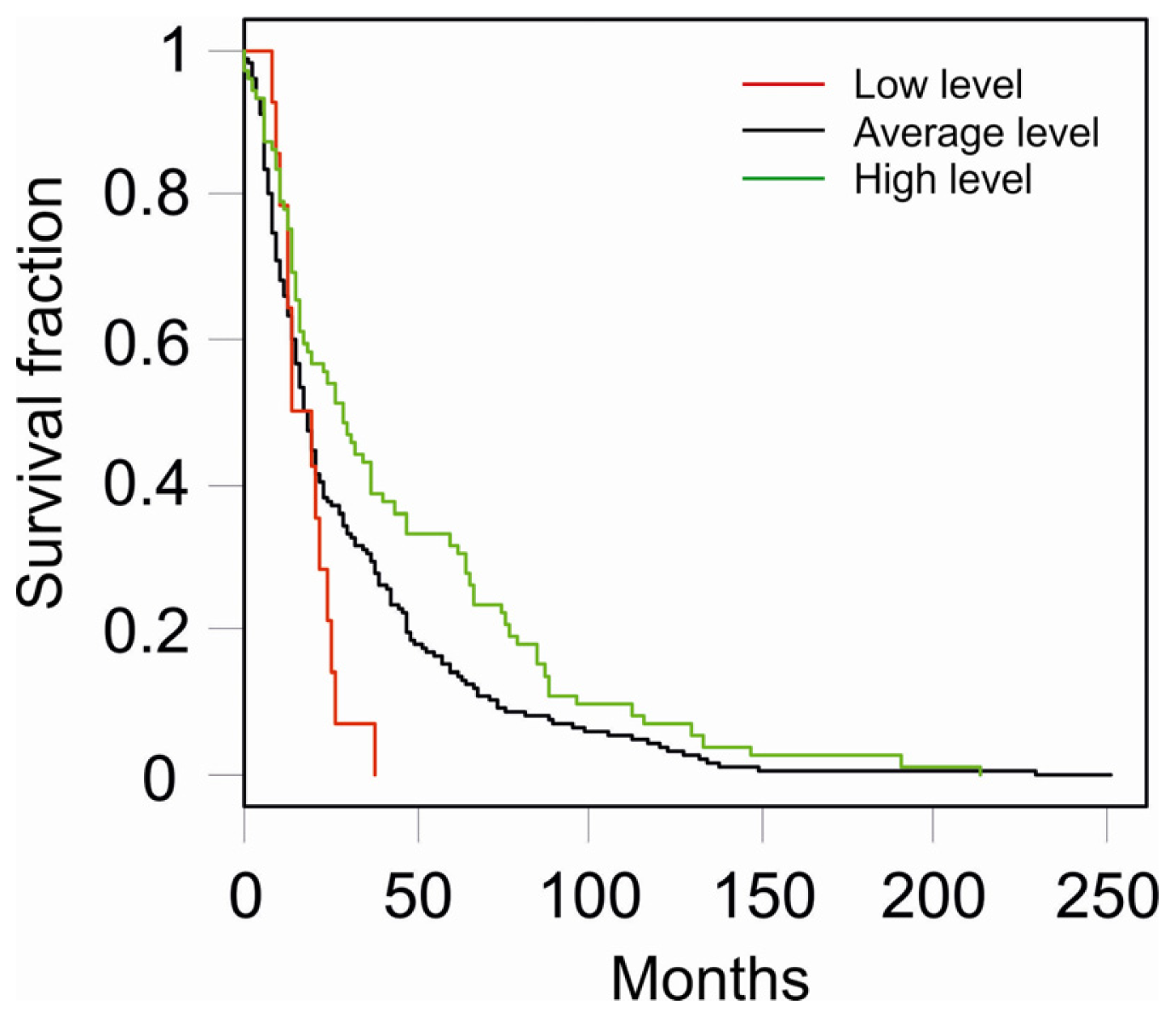
2.1. Downregulation of Btg2
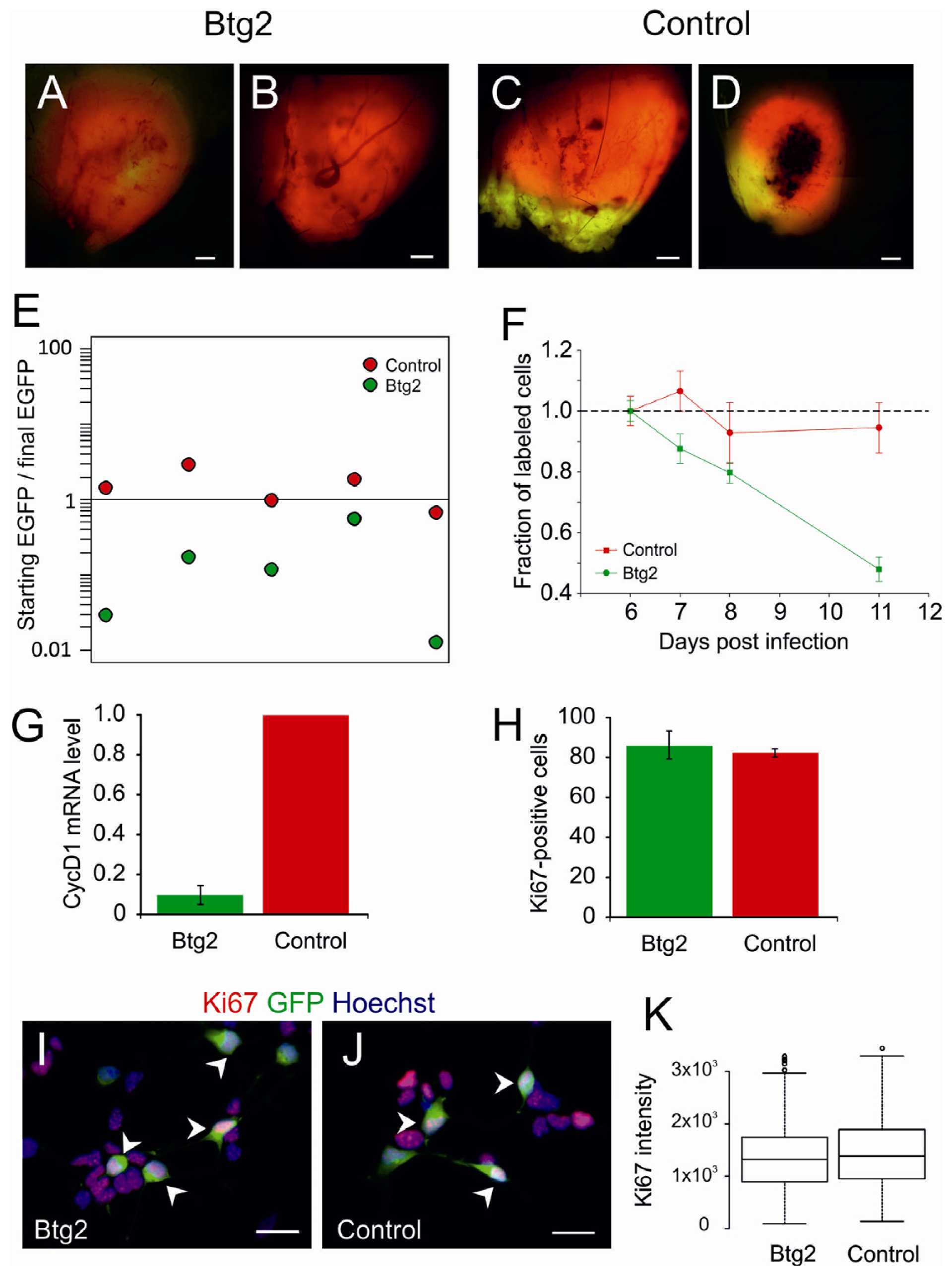
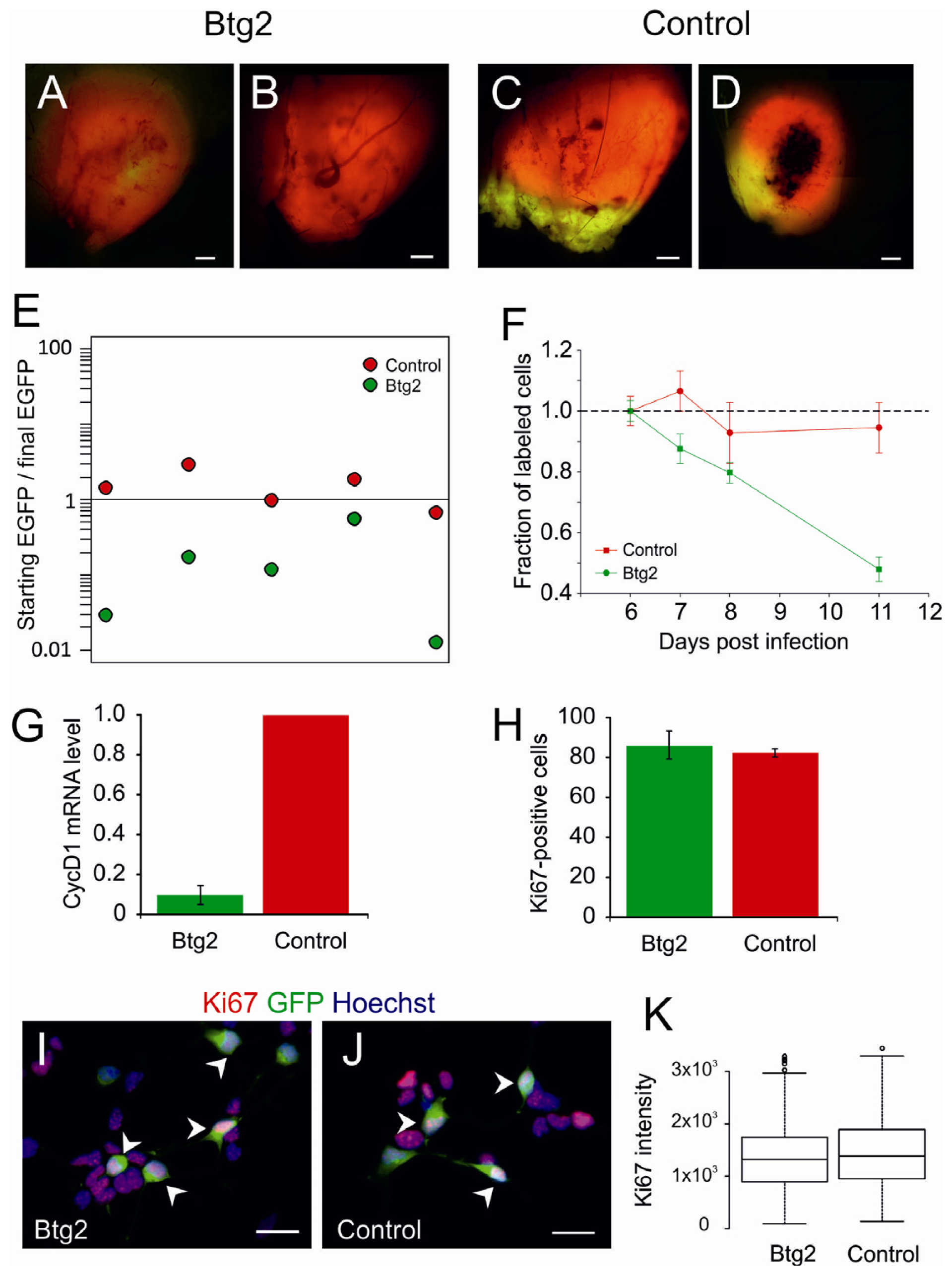
2.2. Upregulation of Btg2
3. Experimental Section
3.1. Animal Procedures
3.3. Cell Cultures and Immunostainings
3.4. Microarray and Real-Time PCR
3.5. Western Blot
4. Conclusions
Supplementary Materials
ijms-13-14667-s001.pdfAcknowledgments
References
- Appolloni, I.; Calzolari, F.; Tutucci, E.; Caviglia, S.; Terrile, M.; Corte, G.; Malatesta, P. PDGF-B induces a homogeneous class of oligodendrogliomas from embryonic neural progenitors. Int. J. Cancer 2009, 124, 2251–2259. [Google Scholar]
- Calzolari, F.; Appolloni, I.; Tutucci, E.; Caviglia, S.; Terrile, M.; Corte, G.; Malatesta, P. Tumor progression and oncogene addiction in a PDGF-B-induced model of gliomagenesis. Neoplasia 2008, 10, 1373–1382. [Google Scholar]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev 2007, 21, 2683–2710. [Google Scholar]
- Yin, D.; Ogawa, S.; Kawamata, N.; Tunici, P.; Finocchiaro, G.; Eoli, M.; Ruckert, C.; Huynh, T.; Liu, G.; Kato, M.; et al. High-resolution genomic copy number profiling of glioblastoma multiforme by single nucleotide polymorphism DNA microarray. Mol. Cancer Res 2009, 7, 665–677. [Google Scholar]
- Zheng, H.; Ying, H.; Yan, H.; Kimmelman, A.C.; Hiller, D.J.; Chen, A.J.; Perry, S.R.; Tonon, G.; Chu, G.C.; Ding, Z.; et al. p53 and Pten control neural and glioma stem/progenitor cell renewal and differentiation. Nature 2008, 455, 1129–1133. [Google Scholar]
- Malatesta, P.; Gotz, M.; Barsacchi, G.; Price, J.; Zoncu, R.; Cremisi, F. PC3 overexpression affects the pattern of cell division of rat cortical precursors. Mech. Dev 2000, 90, 17–28. [Google Scholar]
- Tirone, F. The gene PC3 (TIS21/BTG2), prototype member of the PC3/BTG/TOB family: Regulator in control of cell growth, differentiation, and DNA repair? J. Cell Physiol 2001, 187, 155–165. [Google Scholar]
- Coppola, V.; Musumeci, M.; Patrizii, M.; Cannistraci, A.; Addario, A.; Maugeri-Sacca, M.; Biffoni, M.; Francescangeli, F.; Cordenonsi, M.; Piccolo, S.; et al. BTG2 loss and miR-21 upregulation contribute to prostate cell transformation by inducing luminal markers expression and epithelial-mesenchymal transition. Oncogene 2012. [Google Scholar] [CrossRef]
- Wei, S.; Hao, C.; Li, X.; Zhao, H.; Chen, J.; Zhou, Q. Effects of BTG2 on proliferation inhibition and anti-invasion in human lung cancer cells. Tumour Biol 2012, 33, 1223–1230. [Google Scholar]
- Takahashi, F.; Chiba, N.; Tajima, K.; Hayashida, T.; Shimada, T.; Takahashi, M.; Moriyama, H.; Brachtel, E.; Edelman, E.J.; Ramaswamy, S.; et al. Breast tumor progression induced by loss of BTG2 expression is inhibited by targeted therapy with the ErbB/HER inhibitor lapatinib. Oncogene 2011, 30, 3084–3095. [Google Scholar]
- Farioli-Vecchioli, S.; Tanori, M.; Micheli, L.; Mancuso, M.; Leonardi, L.; Saran, A.; Ciotti, M.T.; Ferretti, E.; Gulino, A.; Pazzaglia, S.; et al. Inhibition of medulloblastoma tumorigenesis by the antiproliferative and pro-differentiative gene PC3. Faseb. J 2007, 21, 2215–2225. [Google Scholar]
- Riley, T.; Sontag, E.; Chen, P.; Levine, A. Transcriptional control of human p53-regulated genes. Nat. Rev. Mol. Cell Biol 2008, 9, 402–412. [Google Scholar]
- Appolloni, I.; Calzolari, F.; Barilari, M.; Terrile, M.; Daga, A.; Malatesta, P. Antagonistic modulation of gliomagenesis by Pax6 and Olig2 in PDGF-induced oligodendroglioma. Int. J. Cancer 2012, 131, E1078–E1087. [Google Scholar]
- Rasband, W.S. ImageJ Home Page; Available online: http://rsb.info.nih.gov/ij/ accessed on 6 November 2012U.S. National Institutes of 13 Health: Bethesda, MD, USA.
- Lange, C.; Huttner, W.B.; Calegari, F. Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 2009, 5, 320–331. [Google Scholar]
- Quelle, D.E.; Ashmun, R.A.; Shurtleff, S.A.; Kato, J.Y.; Bar-Sagi, D.; Roussel, M.F.; Sherr, C.J. Overexpression of mouse d-type cyclins accelerates G1 phase in rodent fibroblasts. Genes Dev 1993, 7, 1559–1571. [Google Scholar]
- National Cancer Institute. REMBRANDT Home Page. 2005. Available online: http://rembrandt.nci.nih.gov accessed on 26 September 2012.
- Gambini, E.; Reisoli, E.; Appolloni, I.; Gatta, V.; Campadelli-Fiume, G.; Menotti, L.; Malatesta, P. Replication-competent herpes simplex virus retargeted to HER2 as therapy for high-grade glioma. Mol. Ther 2012, 20, 994–1001. [Google Scholar]
- Terrile, M.; Appolloni, I.; Calzolari, F.; Perris, R.; Tutucci, E.; Malatesta, P. PDGF-B-driven gliomagenesis can occur in the absence of the proteoglycan NG2. BMC Cancer 2010, 10, 550. [Google Scholar]
- Gavazzo, P.; Vella, S.; Marchetti, C.; Nizzari, M.; Cancedda, R.; Pagano, A. Acquisition of neuron-like electrophysiological properties in neuroblastoma cells by controlled expression of NDM29 ncRNA. J. Neurochem 2011, 119, 989–1001. [Google Scholar]
- Gentleman, R.C.; Carey, V.J.; Bates, D.M.; Bolstad, B.; Dettling, M.; Dudoit, S.; Ellis, B.; Gautier, L.; Ge, Y.; Gentry, J.; et al. Bioconductor: Open software development for computational biology and bioinformatics. Genome Biol 2004, 5, R80. [Google Scholar]
- Irizarry, R.A.; Bolstad, B.M.; Collin, F.; Cope, L.M.; Hobbs, B.; Speed, T.P. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res 2003, 31, e15. [Google Scholar]
- Massone, S.; Ciarlo, E.; Vella, S.; Nizzari, M.; Florio, T.; Russo, C.; Cancedda, R.; Pagano, A. NDM29, a RNA polymerase III-dependent non coding RNA, promotes amyloidogenic processing of APP and amyloid beta secretion. Biochim. Biophys. Acta 2012, 1823, 1170–1177. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
| Symb | L1 | L2 | H1 | H1 | p |
|---|---|---|---|---|---|
| Btg2 | 9.48 | 9.54 | 8.67 | 8.26 | 0.037 |
| Trpm2 | 7.05 | 6.78 | 5.80 | 6.11 | 0.042 |
| Ddb2 | 7.60 | 7.93 | 7.00 | 6.86 | 0.045 |
| Vdr | 5.55 | 5.51 | 5.89 | 6.12 | >0.05 |
| Igfbp3 | 10.46 | 10.40 | 10.89 | 11.27 | >0.05 |
| Pcna | 11.82 | 11.51 | 11.98 | 12.09 | >0.05 |
| Cdkn1a | 10.91 | 9.89 | 9.03 | 9.36 | >0.05 |
| Sh2d1a | 4.71 | 4.63 | 4.85 | 4.74 | >0.05 |
| Tyrp1 | 5.01 | 4.90 | 4.95 | 5.20 | >0.05 |
| Hras1 | 8.77 | 8.65 | 8.70 | 8.81 | >0.05 |
| Mdm2 | 10.05 | 9.74 | 9.81 | 10.03 | >0.05 |
| Gml | 5.00 | 5.37 | 5.39 | 4.96 | >0.05 |
| Trp53 | 9.47 | 9.53 | 8.45 | 10.40 | >0.05 |
| Animal | Latency (days) | Secondary tumors |
|---|---|---|
| 627B | 51 | 0 |
| 627C | 51 | 0 |
| 627D | 52 | 0 |
| 630A | 52 | 0 |
| 627E | 55 | 0 |
| 645A | 71 | 0 |
| 645B | 71 | 0 |
| 630D | 72 | 0 |
| 630E | 84 | 0 |
| 627F | 85 | 0 |
| 627G | 105 | 1 |
| 646C | 257 | 3 |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Appolloni, I.; Curreli, S.; Caviglia, S.; Barilari, M.; Gambini, E.; Pagano, A.; Malatesta, P. Role of Btg2 in the Progression of a PDGF-Induced Oligodendroglioma Model. Int. J. Mol. Sci. 2012, 13, 14667-14678. https://doi.org/10.3390/ijms131114667
Appolloni I, Curreli S, Caviglia S, Barilari M, Gambini E, Pagano A, Malatesta P. Role of Btg2 in the Progression of a PDGF-Induced Oligodendroglioma Model. International Journal of Molecular Sciences. 2012; 13(11):14667-14678. https://doi.org/10.3390/ijms131114667
Chicago/Turabian StyleAppolloni, Irene, Sebastiano Curreli, Sara Caviglia, Manuela Barilari, Eleonora Gambini, Aldo Pagano, and Paolo Malatesta. 2012. "Role of Btg2 in the Progression of a PDGF-Induced Oligodendroglioma Model" International Journal of Molecular Sciences 13, no. 11: 14667-14678. https://doi.org/10.3390/ijms131114667