Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine soja Sieb. et Zucc) Based on Nuclear and Chloroplast Microsatellite Variation

Abstract
:
1. Introduction
2. Results
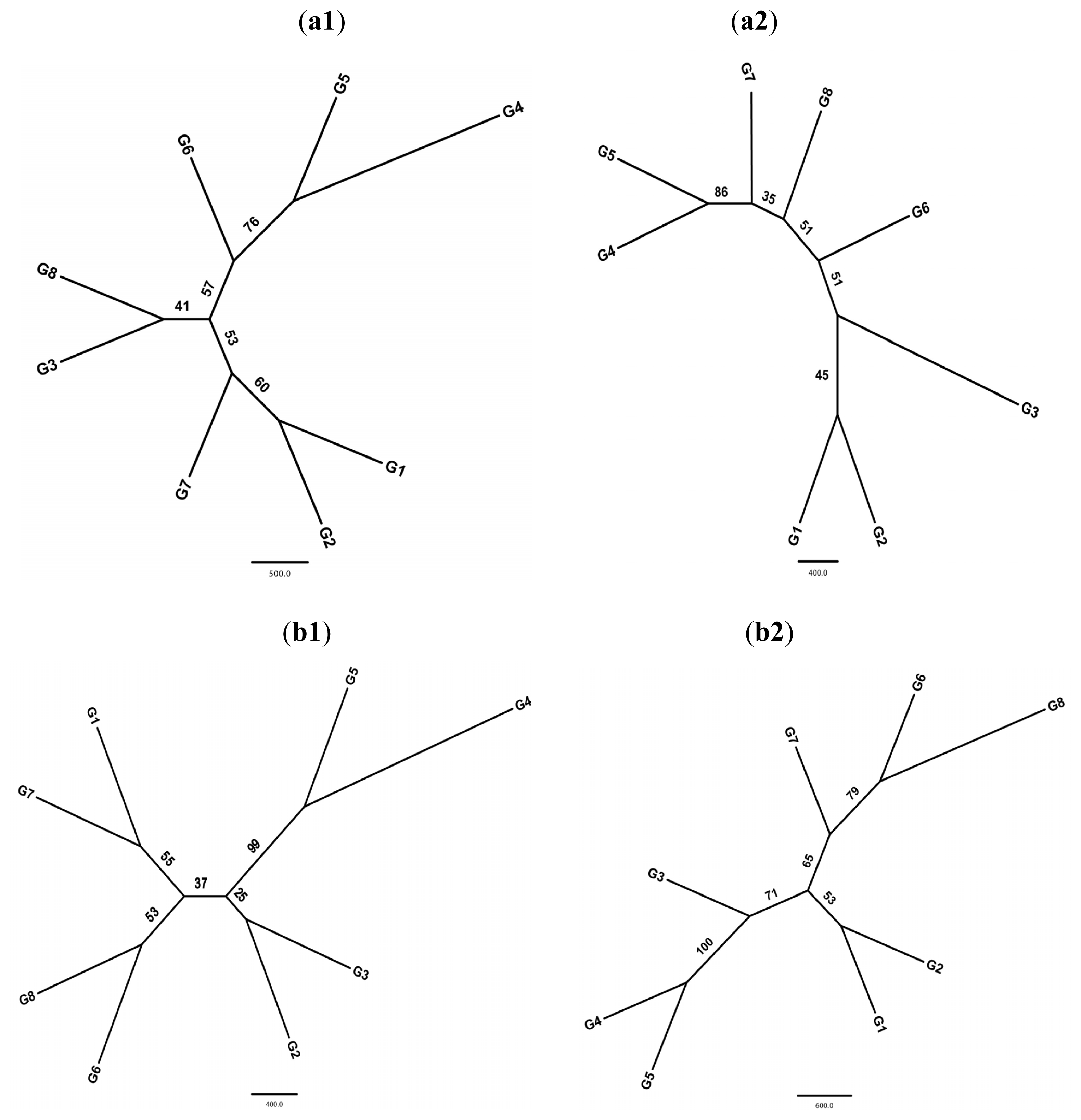
2.1. Equilibrium Test and Genetic Diversity
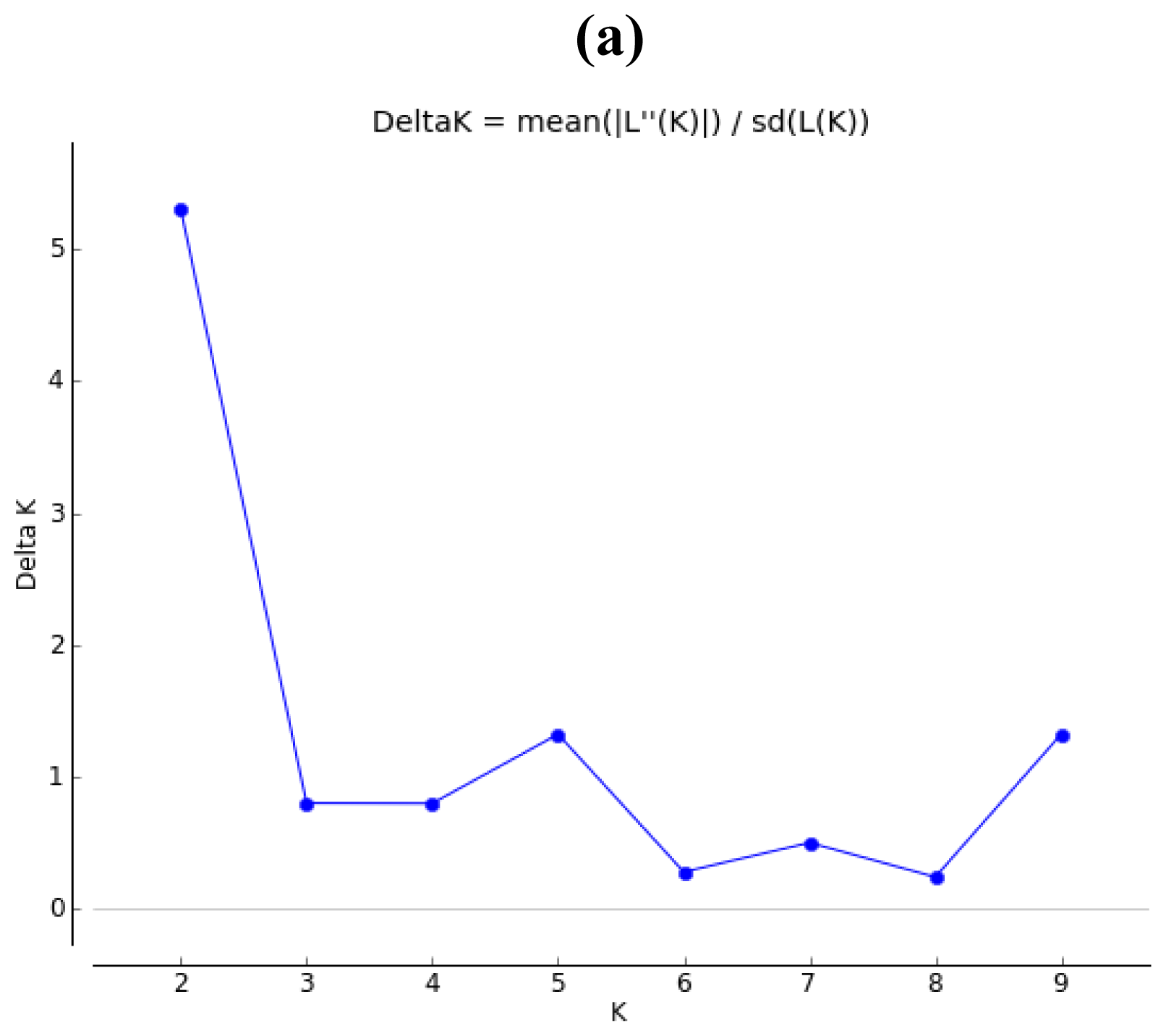
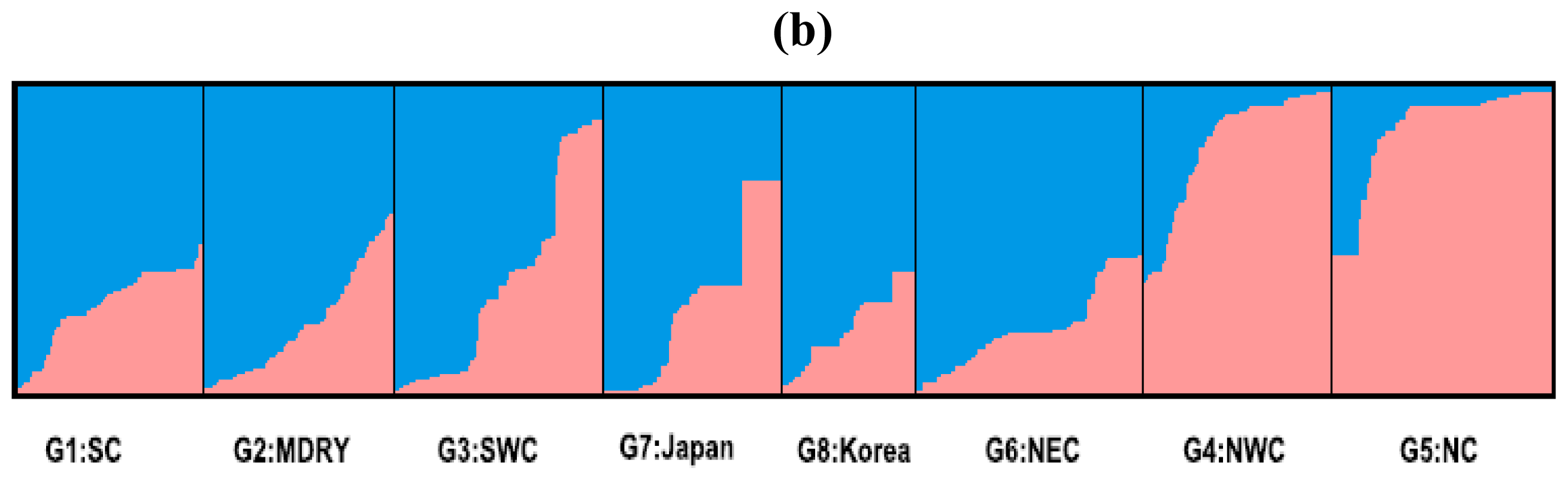
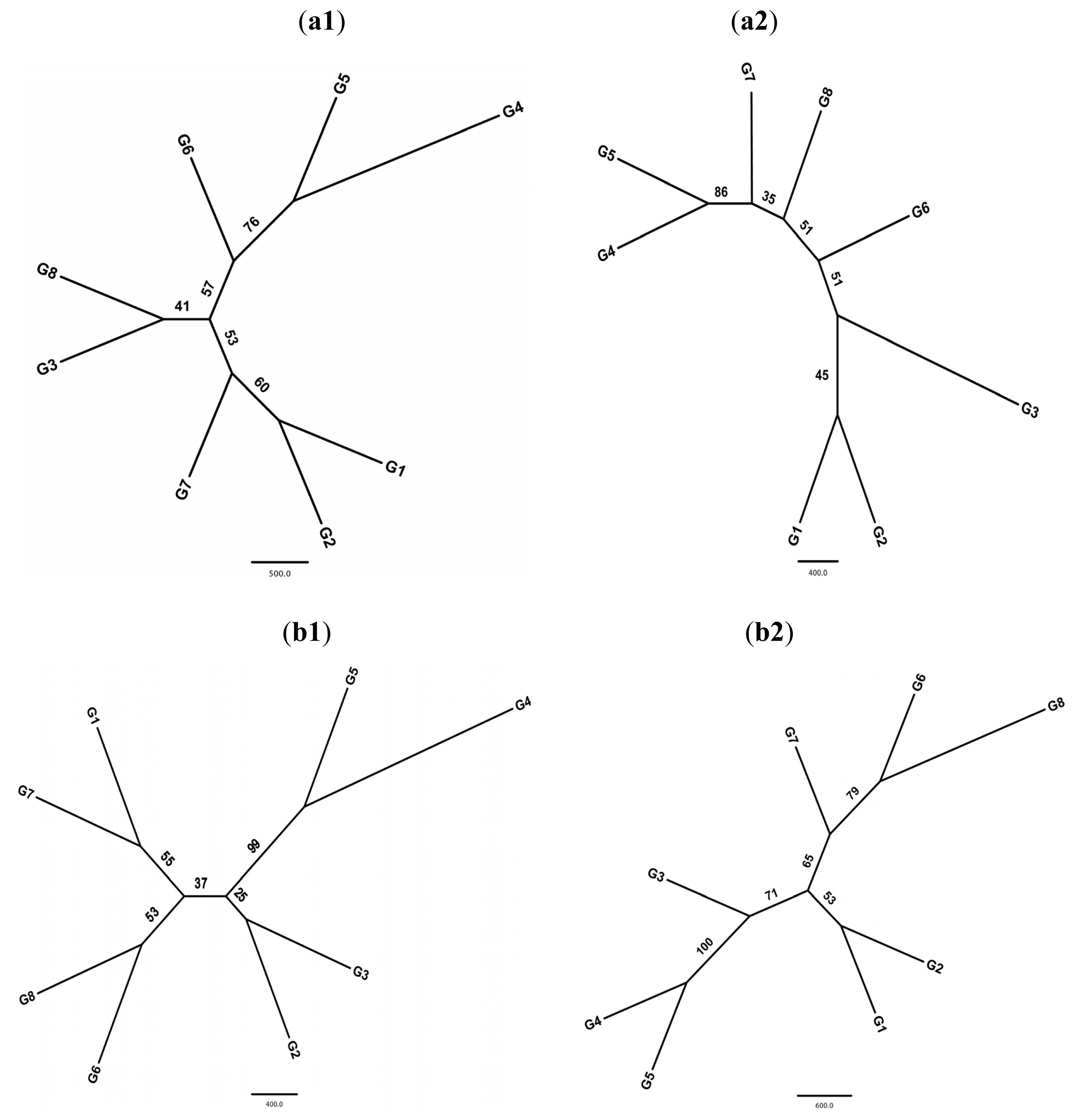
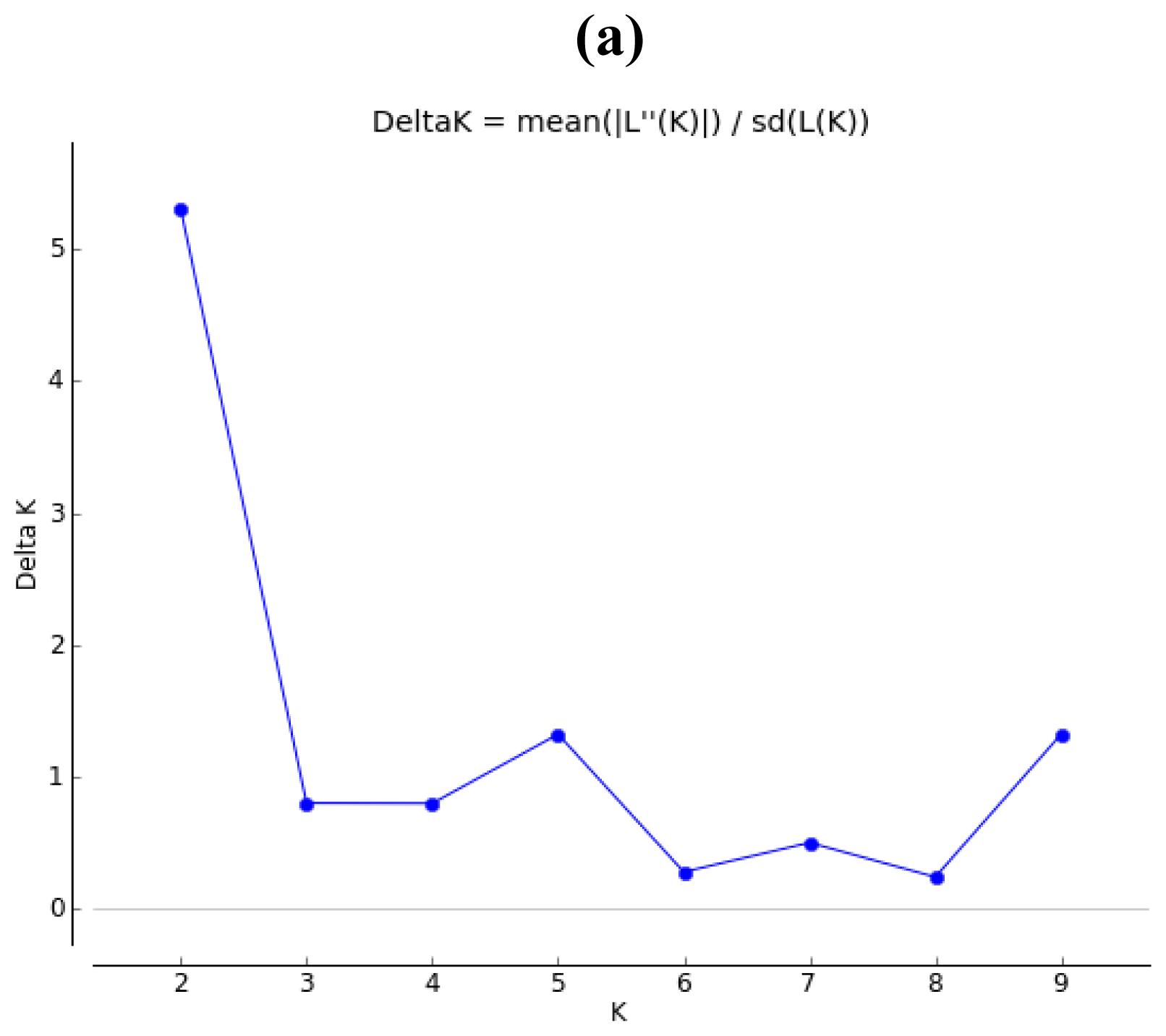
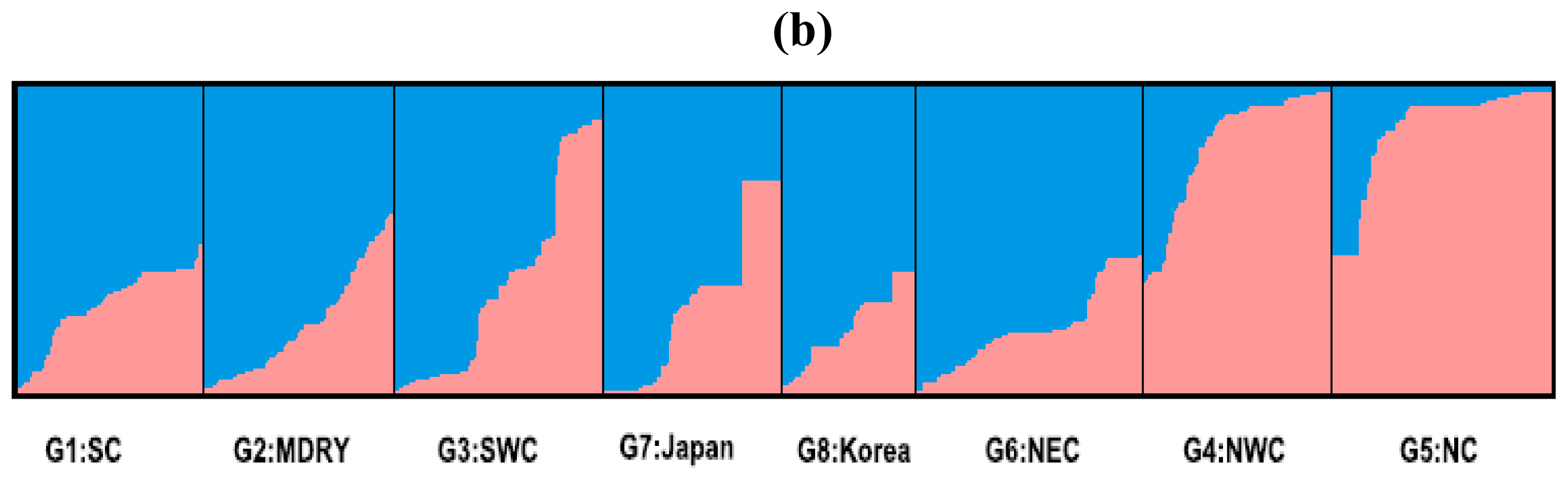
2.2. Population Structure
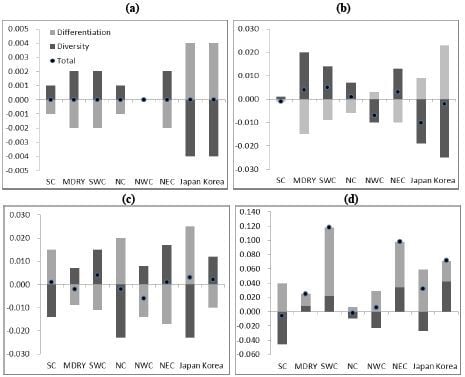
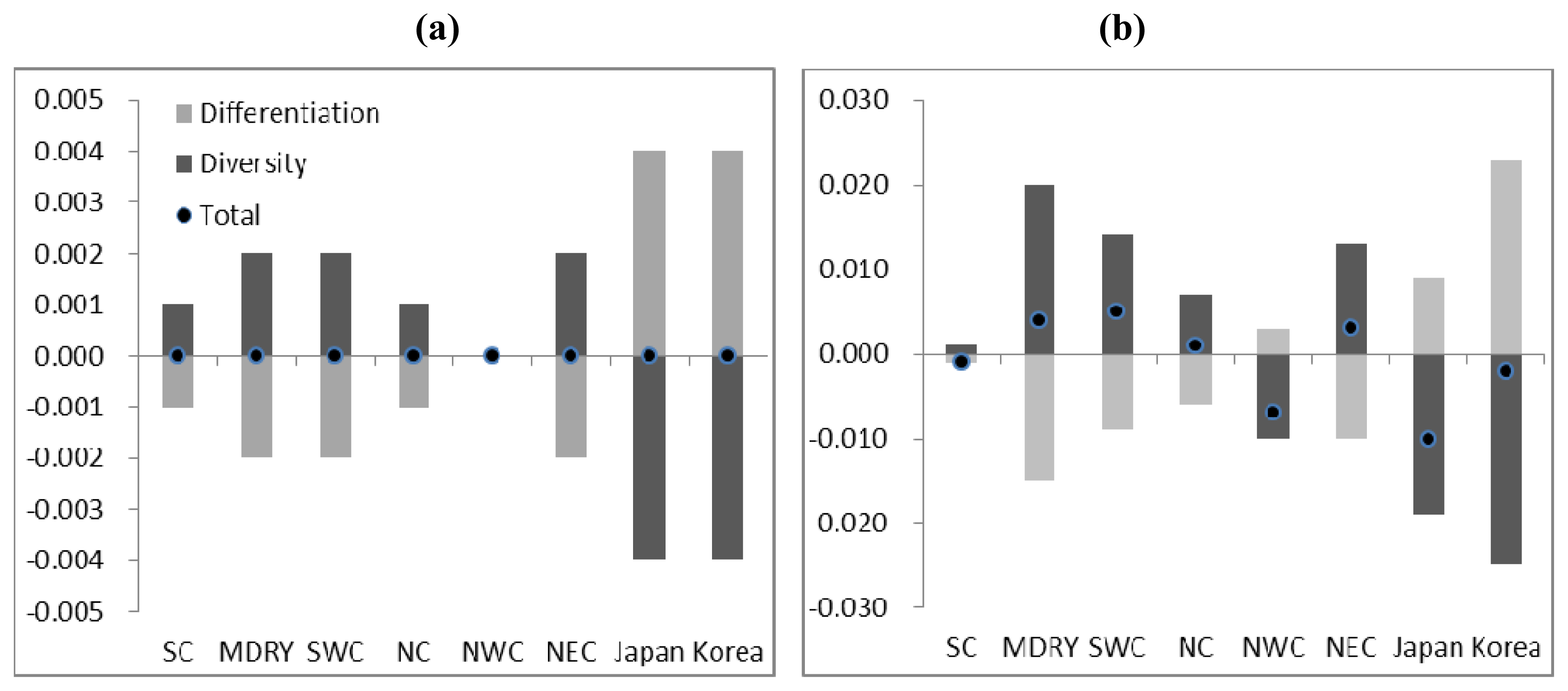
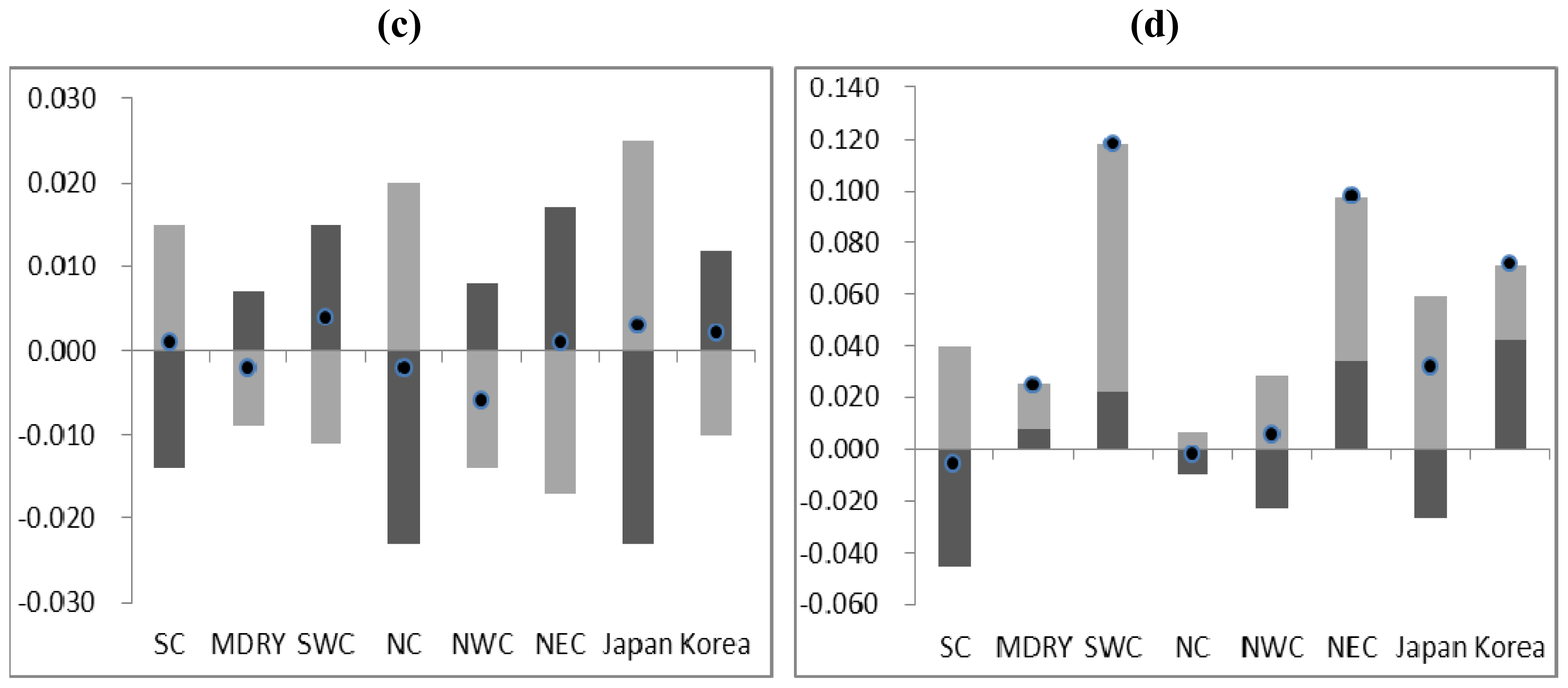
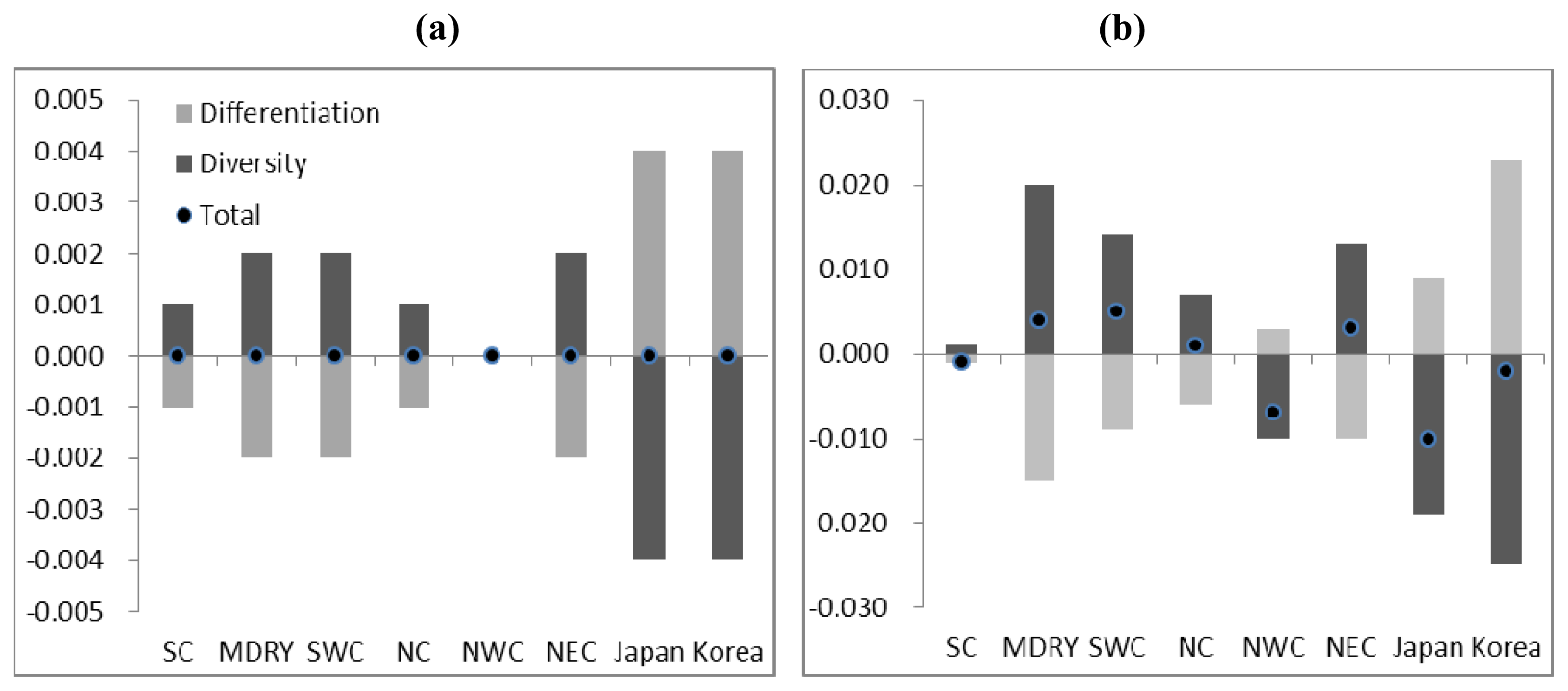
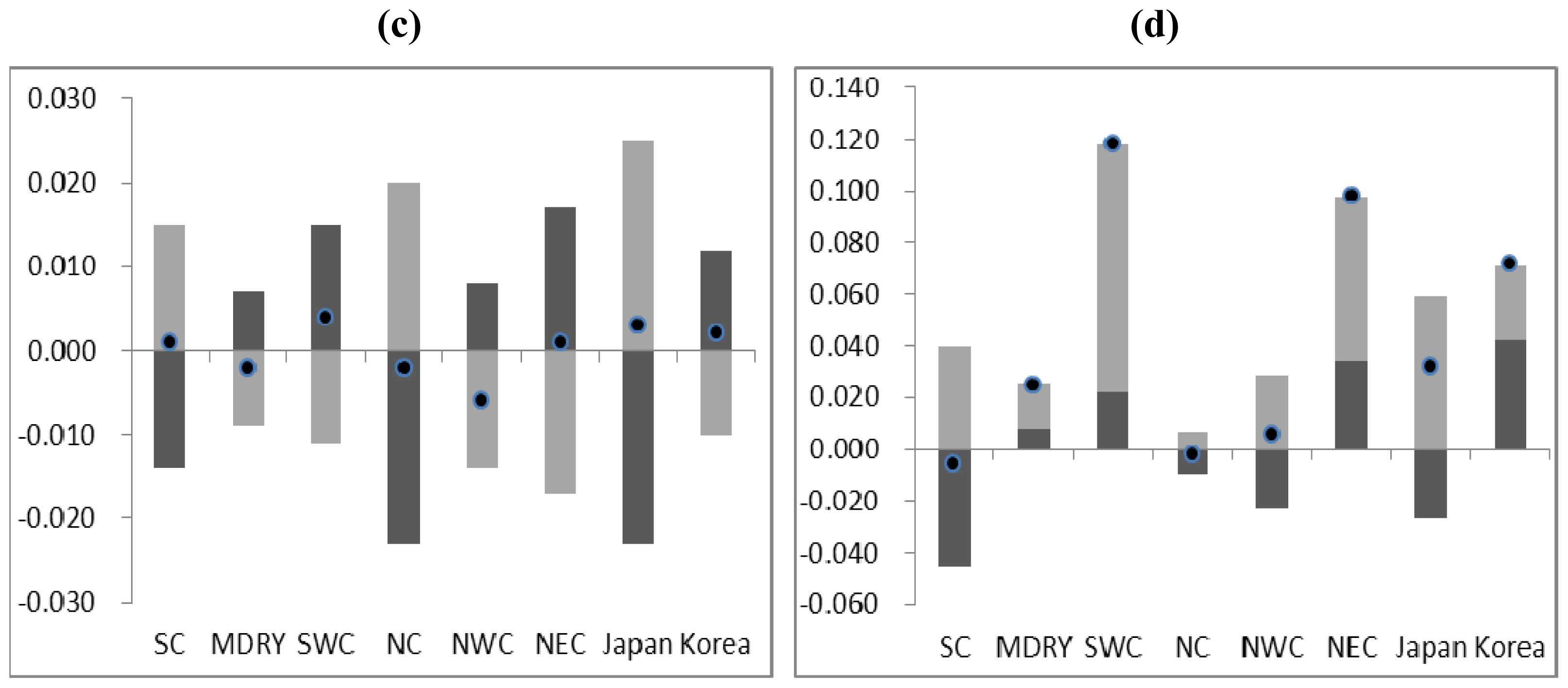
2.3. Demographic History
3. Discussion
3.1. Genetic Diversity in Wild Soybean
3.2. Genetic Structure of Wild Soybean
3.3. Conservation Implications
4. Experimental Section
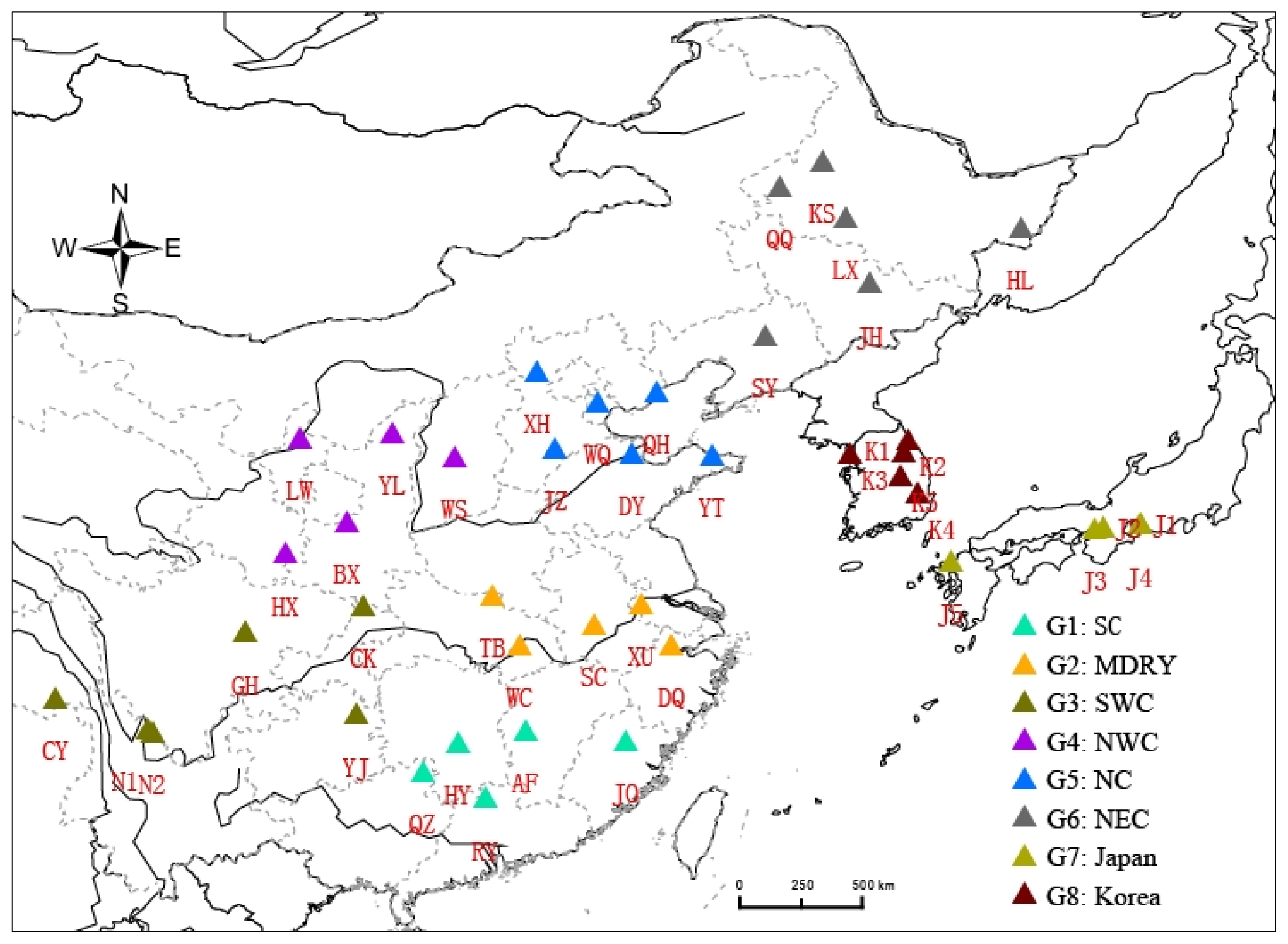
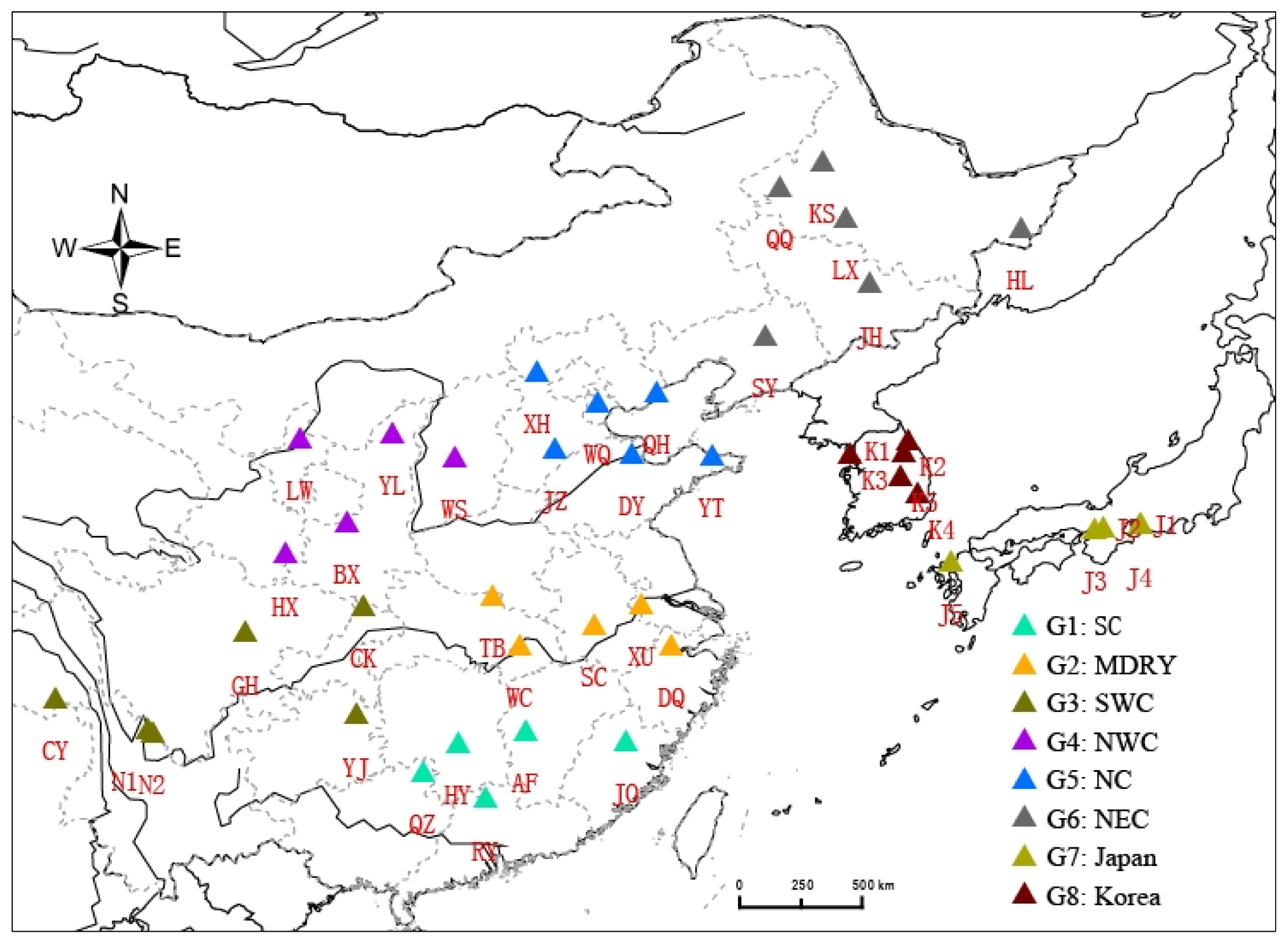
4.1. Samples Collection, DNA Extraction and Microsatellite Genotyping
4.2. Microsatellite Validation and Diversity
4.3. Population Spatial Structure
4.4. Demographic History
5. Conclusions
Acknowledgements
References
- Smil, V. Magic beans. Nature 2000, 407, 567–567. [Google Scholar]
- Lam, H.M.; Xu, X.; Liu, X.; Chen, W.B.; Yang, G.H.; Wong, F.L.; Li, M.W.; He, W.M.; Qin, N.; Wang, B.; et al. Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet 2010, 42, 1053–1061. [Google Scholar]
- Dong, Y.S.; Zhuang, B.C.; Zhao, L.M.; Sun, H.; He, M.Y. The genetic diversity of annual wild soybeans grown in China. Theor. Appl. Genet 2001, 103, 98–103. [Google Scholar]
- Li, F.S. Studies on the ecological and geographical distribution of the Chinese resources of wild soybean. Sci. Agric. Sin 1993, 26, 47–55. [Google Scholar]
- Lu, B.R. Conserving biodiversity of soybean gene pool in the biotechnology era. Plant. Species Biol 2004, 19, 115–125. [Google Scholar]
- Jin, Y.; He, T.H.; Lu, B.R. Fine scale genetic structure in a wild soybean (Glycine soja) population and the implications for conservation. New Phytol 2003, 159, 513–519. [Google Scholar]
- Dong, Y.S. Advances of research on wild soybean in China. J. Jilin Agric. Univers 2008, 30, 394–400. [Google Scholar]
- Yu, Y.F. China’s wild plants protection work milestone_National wild plants list for protection (I). Plant J 1999, 5, 3–11. [Google Scholar]
- Wang, K.J.; Li, X.H.; Li, F.S. Phenotypic diversity of the big seed type subcollection of wild soybean (Glycine soja Sieb. et Zucc.) in China. Genet. Resour. Crop Evol 2008, 55, 1335–1346. [Google Scholar]
- Sakamoto, S.I.; Abe, J.; Kanazawa, A.; Shimamoto, Y. Marker-assisted analysis for soybean hard seededness with isozyme and simple sequence repeat loci. Breed. Sci 2004, 54, 133–139. [Google Scholar]
- Abe, J.; Hasegawa, A.; Fukushi, H.; Mikami, T.; Ohara, M.; Shimamoto, Y. Introgression between wild and cultivated soybeans of Japan revealed by RFLP analysis for chloroplast DNAs. Econ. Bot 1999, 53, 285–291. [Google Scholar]
- Shimamoto, Y.; Tozuka, A.; Fukushi, H.; Hirata, T.; Ohara, M.; Kanazawa, A.; Mikami, T.; Abe, J. Composite and clinal distribution of Glycine soja in Japan revealed by RFLP analysis of mitochondrial DNA. Theor. Appl. Genet 1998, 96, 170–176. [Google Scholar]
- Shimamoto, Y.; Fukushi, H.; Abe, J.; Kanazawa, A.; Gai, J.Y.; Gao, Z.; Xu, D.H. RFLPs of chloroplast and mitochondrial DNA in wild soybean, Glycine soja, growing in China. Genet. Resour. Crop Evol 1998, 45, 433–439. [Google Scholar]
- Lee, J.D.; Yu, J.K.; Hwang, Y.H.; Blake, S.; So, Y.S.; Lee, G.J.; Nguyen, H.T.; Shannon, J.G. Genetic diversity of wild soybean (Glycine soja Sieb. and Zucc.) accessions from South Korea and other countries. Crop Sci 2008, 48, 606–616. [Google Scholar]
- Guan, R.X.; Chang, R.Z.; Li, Y.H.; Wang, L.X.; Liu, Z.X.; Qiu, L.J. Genetic diversity comparison between Chinese and Japanese soybeans (Glycine max (L.) Merr.) revealed by nuclear SSRs. Genet. Resour. Crop Evol 2010, 57, 229–242. [Google Scholar]
- Li, Y.H.; Li, W.; Zhang, C.; Yang, L.A.; Chang, R.Z.; Gaut, B.S.; Qiu, L.J. Genetic diversity in domesticated soybean (Glycine max) and its wild progenitor (Glycine soja) for simple sequence repeat and single-nucleotide polymorphism loci. New Phytol 2010, 188, 242–253. [Google Scholar]
- Wang, M.; Li, R.Z.; Yang, W.M.; Du, W.J. Assessing the genetic diversity of cultivars and wild soybeans using SSR markers. Afr. J. Biotechnol 2010, 9, 4857–4866. [Google Scholar]
- Li, X.H.; Wang, K.J.; Jia, J.Z. Genetic diversity and differentiation of Chinese wild soybean germplasm (G. soja Sieb. & Zucc.) in geographical scale revealed by SSR markers. Plant Breed 2009, 128, 658–664. [Google Scholar]
- Kuroda, Y.; Sato, Y.I.; Bounphanousay, C.; Kono, Y.; Tanaka, K. Genetic structure of three Oryza AA genome species (O. rufipogon, O. nivara and O. sativa) as assessed by SSR analysis on the Vientiane Plain of Laos. Conserv. Genet 2007, 8, 149–158. [Google Scholar]
- Kuroda, Y.; Kaga, A.; Tomooka, N.; Vaughan, D.A. Gene flow and genetic structure of wild soybean (Glycine soja) in Japan. Crop Sci 2008, 48, 1071–1079. [Google Scholar]
- Kuroda, Y.; Kaga, A.; Tomooka, N.; Vaughan, D.A. Population genetic structure of Japanese wild soybean (Glycine soja) based on microsatellite variation. Mol. Ecol 2006, 15, 959–974. [Google Scholar]
- Yoshimura, Y.; Mizuguti, A.; Matsuo, K. Analysis of the seed dispersal patterns of wild soybean as a reference for vegetation management around genetically modified soybean fields. Weed Biol. Manag 2011, 11, 210–216. [Google Scholar]
- Shimamoto, Y.; Abe, J.; Gao, Z.; Gai, J.Y.; Thseng, F.S. Characterizing the cytoplasmic diversity and phyletic relationship of Chinese landraces of soybean, Glycine max, based on RFLPs of chloroplast and mitochondrial DNA. Genet. Resour. Crop Evol 2000, 47, 611–617. [Google Scholar]
- Wen, Z.X.; Zhao, T.J.; Ding, Y.L.; Gai, J.Y. Genetic diversity, geographic differentiation and evolutionary relationship among ecotypes of Glycine max and G. soja in China. Chin. Sci. Bull 2009, 54, 4393–4403. [Google Scholar]
- Guo, J.; Wang, Y.S.; Song, C.; Zhou, J.F.; Qiu, L.J.; Huang, H.W.; Wang, Y. A single origin and moderate bottleneck during domestication of soybean (Glycine max): Implications from microsatellites and nucleotide sequences. Ann. Bot 2010, 106, 505–514. [Google Scholar]
- Maughan, P.J.; Maroof, M.A.S.; Buss, G.R. Microsatellite and amplified sequence length polymorphisms in cultivated and wild soybean. Genome 1995, 38, 715–723. [Google Scholar]
- Song, Q.J.; Marek, L.F.; Shoemaker, R.C.; Lark, K.G.; Concibido, V.C.; Delannay, X.; Specht, J.E.; Cregan, P.B. A new integrated genetic linkage map of the soybean. Theor. Appl. Genet 2004, 109, 122–128. [Google Scholar]
- Chapuis, M.P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol 2007, 24, 621–631. [Google Scholar]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software structure: A simulation study. Mol. Ecol 2005, 14, 2611–2620. [Google Scholar]
- Doyle, J.J.; Morgante, M.; Tingey, S.V.; Powell, W. Size homoplasy in chloroplast microsatellites of wild perennial relatives of soybean (Glycine subgenus Glycine). Mol. Biol. Evol 1998, 15, 215–218. [Google Scholar]
- Kuroda, Y.; Tomooka, N.; Kaga, A.; Wanigadeva, S.M.S.W.; Vaughan, D. Genetic diversity of wild soybean (Glycine soja Sieb. et Zucc.) and Japanese cultivated soybeans [G. max (L.) Merr.] based on microsatellite (SSR) analysis and the selection of a core collection. Genet. Resour. Crop Evol 2009, 56, 1045–1055. [Google Scholar]
- Zhao, R.; Xia, H.; Lu, B.R. Fine-scale genetic structure enhances biparental inbreeding by promoting mating events between more related individuals in wild soybean (Glycine soja; Fabaceae) populations. Am. J. Bot 2009, 96, 1138–1147. [Google Scholar]
- Hamrick, J.L.; Godt, M.J.W. Effects of life history traits on genetic diversity in plant species. Philos. T. Roy. Soc. B 1996, 351, 1291–1298. [Google Scholar]
- Luo, M.C.; Yang, Z.L.; You, F.M.; Kawahara, T.; Waines, J.G.; Dvorak, J. The structure of wild and domesticated emmer wheat populations, gene flow between them, and the site of emmer domestication. Theor. Appl. Genet 2007, 114, 947–959. [Google Scholar]
- Ozbek, O.; Millet, E.; Anikster, Y.; Arslan, O.; Feldman, M. Spatio-temporal genetic variation in populations of wild emmer wheat, Triticum turgidum ssp. dicoccoides, as revealed by AFLP analysis. Theor. Appl. Genet 2007, 115, 19–26. [Google Scholar]
- Gao, L.Z. Microsatellite variation within and among populations of Oryza officinalis (Poaceae), an endangered wild rice from China. Mol. Ecol 2005, 14, 4287–4297. [Google Scholar]
- Wen, Z.; Ding, Y.; Zhao, T.; Gai, J. Genetic diversity and peculiarity of annual wild soybean (G. soja Sieb. et Zucc.) from various eco-regions in China. Theor. Appl. Genet 2009, 119, 371–381. [Google Scholar]
- Powell, W.; Morgante, M.; Doyle, J.J.; McNicol, J.W.; Tingey, S.V.; Rafalski, A.J. Genepool variation in genus Glycine subgenus Soja revealed by polymorphic nuclear and chloroplast microsatellites. Genetics 1996, 144, 792–803. [Google Scholar]
- Pakkad, G.; Ueno, S.; Yoshimaru, H. Genetic diversity and differentiation of Quercus semiserrata Roxb. in northern Thailand revealed by nuclear and chloroplast microsatellite markers. Forest Ecol. Manag 2008, 255, 1067–1077. [Google Scholar]
- Robledo-Arnuncio, J.J.; Gil, L. Patterns of pollen dispersal in a small population of Pinus sylvestris L. revealed by total-exclusion paternity analysis. Heredity 2005, 94, 13–22. [Google Scholar]
- Setsuko, S.; Ishida, K.; Ueno, S.; Tsumura, Y.; Tomaru, N. Population differentiation and gene flow within a metapopulation of a threatened tree, Magnolia stellata (Magnoliaceae). Am. J. Bot 2007, 94, 128–136. [Google Scholar]
- Wolfe, K.H.; Li, W.H.; Sharp, P.M. Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. USA 1987, 84, 9054–9058. [Google Scholar]
- Ebert, D.; Peakall, R. Chloroplast simple sequence repeats (cpSSRs): Technical resources and recommendations for expanding cpSSR discovery and applications to a wide array of plant species. Mol. Ecol. Resour 2009, 9, 673–690. [Google Scholar]
- Holtsford, T.P.; Ellstrand, N.C. Variation in outcrossing rate and population genetic-structure of Clarkia tembloriensis (Onagraceae). Theor. Appl. Genet 1989, 78, 480–488. [Google Scholar]
- Wright, S. Systems of mating. II. The effects of inbreeding on the genetic composition of a population. Genetics 1921, 6, 124–143. [Google Scholar]
- Kiang, Y.T.; Chiang, Y.C.; Kaizuma, N. Genetic diversity in natural populations of wild soybean in Iwate prefecture, Japan. J. Hered 1992, 83, 325–329. [Google Scholar]
- Fujita, R.; Ohara, M.; Okazaki, K.; Shimamoto, Y. The extent of natural cross-pollination in wild soybean (Glycine soja). J. Hered 1997, 88, 124–128. [Google Scholar]
- Volis, S.; Zaretsky, M.; Shulgina, I. Fine-scale spatial genetic structure in a predominantly selfing plant: Role of seed and pollen dispersal. Heredity 2010, 105, 384–393. [Google Scholar]
- Li, E.X.; Qiu, Y.X.; Yi, S.; Guo, J.T.; Comes, H.P.; Fu, C.X. Phylogeography of two East Asian species in Croomia (Stemonaceae) inferred from chloroplast DNA and ISSR fingerprinting variation. Mol. Phylogen. Evol 2008, 49, 702–714. [Google Scholar]
- Qiu, Y.X.; Sun, Y.; Zhang, X.P.; Lee, J.; Fu, C.X.; Comes, H.P. Molecular phylogeography of East Asian Kirengeshoma (Hydrangeaceae) in relation to Quaternary climate change and landbridge configurations. New Phytol 2009, 183, 480–495. [Google Scholar]
- Qiu, Y.X.; Qi, X.S.; Jin, X.F.; Tao, X.Y.; Fu, C.X.; Naiki, A.; Comes, H.P. Population genetic structure, phylogeography, and demographic history of Platycrater arguta (Hydrangeaceae) endemic to East China and South Japan, inferred from chloroplast DNA sequence variation. Taxon 2009, 58, 1226–1241. [Google Scholar]
- Qiu, Y.X.; Fu, C.X.; Comes, H.P. Plant molecular phylogeography in China and adjacent regions: Tracing the genetic imprints of Quaternary climate and environmental change in the world’s most diverse temperate flora. Mol. Phylogen. Evol 2011, 59, 225–244. [Google Scholar]
- Takahara, H.; Sugita, S.; Harrison, S.P.; Miyoshi, N.; Morita, Y.; Uchiyama, T. Pollen-based reconstructions of Japanese biomes at 0, 6000 and 18,000 C-14 yr BP. J. Biogeogr 2000, 27, 665–683. [Google Scholar]
- Millien-Parra, V.; Jaeger, J.J. Island biogeography of the Japanese terrestrial mammal assemblages: An example of a relict fauna. J. Biogeogr. 1999, 26, 959–972. [Google Scholar]
- Lee, J.D.; Shannon, J.G.; Vuong, T.D.; Nguyen, H.T. Inheritance of Salt Tolerance in Wild Soybean (Glycine soja Sieb. and Zucc.). J. Hered 2009, 100, 798–801. [Google Scholar]
- Doyle, J.J.; Doyle, J.L. A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull 1987, 19, 11–15. [Google Scholar]
- Xu, D.H.; Abe, J.; Gai, J.Y.; Shimamoto, Y. Diversity of chloroplast DNA SSRs in wild and cultivated soybeans: Evidence for multiple origins of cultivated soybean. Theor. Appl. Genet 2002, 105, 645–653. [Google Scholar]
- Oosterhout, C.V.; Hutchinson, W.F.; Wills, D.P.M.; Shipley, P. micro-checker: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar]
- Rousset, F. GENEPOP’007: A complete re-implementation of the GENEPOP software for Windows and Linux. Mol. Ecol. Resour 2008, 8, 103–106. [Google Scholar]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar]
- Goudet, J. FSTAT: A computer program to calculate F-statistics. J. Hered 2002, 86, 485–486. [Google Scholar]
- Weir, B.S. Genetic Data Analysis: Methods for Discrete Population Genetic Data; Sinauer Associates, Inc: Sunderland, MA, USA; p. 1990.
- Petit, R.J.; Excoffier, L. Gene flow and species delimitation. Trends Ecol. Evol 2009, 24, 386–393. [Google Scholar]
- Petit, R.J.; El Mousadik, A.; Pons, O. Identifying populations for conservation on the basis of genetic markers. Conserv. Biol 1998, 12, 844–855. [Google Scholar]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Falush, D.; Stephens, M.; Pritchard, J.K. Inference of population structure using multilocus genotype data: Linked loci and correlated allele frequencies. Genetics 2003, 164, 1567–1587. [Google Scholar]
- Earl, D.A.; Vonholdt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour 2012, 4, 359–361. [Google Scholar]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar]
- Rosenberg, N.A. Distruct: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar]
- Ruzzante, D.E. A comparison of several measures of genetic distance and population structure with microsatellite data: Bias and sampling variance. Can. J. Fish. Aquat. Sci 1998, 55, 1–14. [Google Scholar]
- Nei, M. Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 1978, 89, 583–590. [Google Scholar]
- Felsenstein, J. PHYLIP—phylogeny inference package (version 3.2). Cladistics 1989, 5, 164–166. [Google Scholar]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar]
- Excoffier, L.; Laval, G.; Schneider, S. Arlequin (version 3.0): An integrated software package for population genetics data analysis. Evol. Bioinform. Online 2005, 1, 47–50. [Google Scholar]
- Hardy, O.J.; Vekemans, X. SPAGEDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618–620. [Google Scholar]
- Slatkin, M. Rare alleles as indicators of gene flow. Evolution 1985, 39, 53–65. [Google Scholar]
- Cornuet, J.M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar]
- Luikart, G.; Allendorf, F.W.; Cornuet, J.M.; Sherwin, W.B. Distortion of allele frequency distributions provides a test for recent population bottlenecks. J. Hered 1998, 89, 238–247. [Google Scholar]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered 1999, 90, 502–503. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| nSSRs | cpSSRs | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Pops | N | A | Na | AR | I | HO | HE | FIS | t (%) | A | Na | AR | I |
| G1:SC | 73 | 199 | 10 | 9.5 | 1.886 | 0.013 | 0.809 | 0.984 | 0.8 | 11 | 2 | 2.2 | 0.421 |
| G1_AF | 15 | 64 | 3 | 1.9 | 0.731 | 0.014 | 0.397 | 0.973 | 1.4 | 6 | 1 | 1.2 | 0.079 |
| G1_HY | 15 | 88 | 4 | 2.4 | 1.177 | 0.014 | 0.605 | 0.979 | 1.1 | 9 | 2 | 1.8 | 0.493 |
| G1_JO | 15 | 45 | 2 | 1.6 | 0.505 | 0.007 | 0.300 | 0.927 | 3.8 | 6 | 1 | 1.2 | 0.079 |
| G1_QZ | 13 | 56 | 3 | 1.9 | 0.751 | 0.015 | 0.456 | 0.969 | 1.6 | 6 | 1 | 1.2 | 0.133 |
| G1_RY | 15 | 36 | 2 | 1.2 | 0.190 | 0.018 | 0.094 | 0.736 | 15.2 | 7 | 1 | 1.3 | 0.098 |
| G2:MDRY | 75 | 292 | 15 | 14.0 | 2.349 | 0.063 | 0.881 | 0.929 | 3.7 | 16 | 3 | 3.1 | 0.742 |
| G2_DQ | 14 | 57 | 3 | 2.0 | 0.792 | 0.051 | 0.468 | 0.904 | 5.0 | 10 | 2 | 2.0 | 0.502 |
| G2_SC | 15 | 114 | 6 | 2.8 | 1.493 | 0.023 | 0.722 | 0.969 | 1.6 | 10 | 2 | 1.9 | 0.369 |
| G2_TB | 15 | 136 | 7 | 3.1 | 1.744 | 0.007 | 0.797 | 0.992 | 0.4 | 14 | 3 | 2.7 | 0.809 |
| G2_WC | 14 | 124 | 6 | 3.0 | 1.616 | 0.053 | 0.762 | 0.933 | 3.5 | 11 | 2 | 2.1 | 0.578 |
| G2_XU | 15 | 114 | 6 | 2.7 | 1.436 | 0.205 | 0.708 | 0.718 | 16.4 | 10 | 2 | 1.9 | 0.402 |
| G3:SWC | 83 | 199 | 10 | 9.3 | 1.884 | 0.035 | 0.803 | 0.956 | 2.2 | 18 | 4 | 3.5 | 0.707 |
| G3_CK | 15 | 57 | 3 | 2.0 | 0.827 | 0.037 | 0.491 | 0.934 | 3.4 | 7 | 1 | 1.3 | 0.149 |
| G3_CY | 12 | 38 | 2 | 1.4 | 0.343 | 0.013 | 0.200 | 0.957 | 2.2 | 7 | 1 | 1.4 | 0.170 |
| G3_GH | 14 | 62 | 3 | 1.9 | 0.761 | 0.048 | 0.426 | 0.759 | 13.7 | 7 | 1 | 1.4 | 0.217 |
| G3_N1 | 12 | 59 | 3 | 1.8 | 0.691 | 0.027 | 0.383 | 0.898 | 5.4 | 12 | 2 | 2.2 | 0.511 |
| G3_N2 | 11 | 52 | 3 | 1.8 | 0.647 | 0.067 | 0.407 | 0.761 | 13.6 | 9 | 2 | 1.8 | 0.375 |
| G3_YJ | 14 | 51 | 3 | 1.8 | 0.663 | 0.014 | 0.414 | 0.914 | 4.5 | 6 | 1 | 1.2 | 0.130 |
| G4:NWC | 75 | 222 | 11 | 10.3 | 1.740 | 0.026 | 0.719 | 0.964 | 1.8 | 13 | 3 | 2.6 | 0.447 |
| G4_BX | 15 | 98 | 5 | 2.4 | 1.162 | 0.028 | 0.570 | 0.962 | 1.9 | 6 | 2 | 2.0 | 0.487 |
| G4_HX | 14 | 97 | 5 | 2.5 | 1.196 | 0.071 | 0.598 | 0.835 | 9.0 | 11 | 2 | 2.1 | 0.475 |
| G4_LW | 15 | 40 | 2 | 1.5 | 0.428 | 0.007 | 0.265 | 0.978 | 1.1 | 11 | 1 | 1.0 | 0.000 |
| G4_WS | 15 | 76 | 4 | 2.2 | 0.974 | 0.017 | 0.520 | 0.968 | 1.6 | 5 | 2 | 1.7 | 0.342 |
| G4_YL | 15 | 48 | 2 | 1.6 | 0.517 | 0.010 | 0.302 | 0.905 | 5.0 | 9 | 2 | 1.6 | 0.284 |
| G5:NC | 86 | 169 | 8 | 8.1 | 1.633 | 0.009 | 0.730 | 0.989 | 0.6 | 11 | 2 | 2.2 | 0.598 |
| G5_DY | 15 | 68 | 3 | 2.2 | 0.934 | 0.018 | 0.526 | 0.973 | 1.4 | 9 | 2 | 2.0 | 0.518 |
| G5_JZ | 15 | 44 | 2 | 1.9 | 0.674 | 0.018 | 0.455 | 0.930 | 3.6 | 10 | 1 | 1.4 | 0.235 |
| G5_QH | 15 | 49 | 2 | 1.4 | 0.403 | 0.010 | 0.212 | 0.975 | 1.3 | 7 | 1 | 1.4 | 0.157 |
| G5_WQ | 15 | 81 | 4 | 2.3 | 1.072 | 0.003 | 0.555 | 0.995 | 0.3 | 7 | 2 | 1.8 | 0.500 |
| G5_XH | 15 | 48 | 2 | 1.7 | 0.603 | 0.000 | 0.367 | 1.000 | 0.0 | 9 | 2 | 1.5 | 0.276 |
| G5_YT | 11 | 23 | 1 | 1.1 | 0.059 | 0.005 | 0.037 | 0.651 | 21.1 | 8 | 1 | 1.0 | 0.000 |
| G6:NEC | 89 | 232 | 12 | 10.7 | 1.932 | 0.035 | 0.802 | 0.956 | 2.2 | 17 | 3 | 3.4 | 0.878 |
| G6_HL | 15 | 101 | 5 | 2.6 | 1.303 | 0.037 | 0.658 | 0.946 | 2.8 | 5 | 2 | 1.8 | 0.411 |
| G6_JH | 15 | 108 | 5 | 2.7 | 1.405 | 0.082 | 0.694 | 0.882 | 6.3 | 9 | 2 | 2.0 | 0.434 |
| G6_KS | 15 | 61 | 3 | 2.3 | 0.979 | 0.003 | 0.582 | 0.995 | 0.3 | 11 | 2 | 1.8 | 0.474 |
| G6_LX | 15 | 68 | 3 | 2.2 | 0.930 | 0.028 | 0.520 | 0.958 | 2.2 | 9 | 2 | 2.1 | 0.571 |
| G6_QQ | 15 | 78 | 4 | 2.1 | 0.946 | 0.003 | 0.504 | 0.995 | 0.3 | 11 | 2 | 1.7 | 0.283 |
| G6_SY | 14 | 26 | 1 | 1.1 | 0.118 | 0.061 | 0.076 | 0.202 | 66.4 | 9 | 1 | 1.2 | 0.120 |
| G7:Japan | 70 | 157 | 8 | 7.6 | 1.651 | 0.023 | 0.759 | 0.969 | 1.6 | 10 | 2 | 2.0 | 0.372 |
| G7_J1 | 15 | 63 | 3 | 2.2 | 0.978 | 0.021 | 0.563 | 0.964 | 1.8 | 23 | 2 | 1.6 | 0.343 |
| G7_J2 | 15 | 23 | 1 | 1.0 | 0.034 | 0.007 | 0.018 | 0.310 | 52.7 | 8 | 1 | 1.0 | 0.000 |
| G7_J3 | 15 | 24 | 1 | 1.1 | 0.100 | 0.003 | 0.064 | 0.957 | 2.2 | 5 | 1 | 1.0 | 0.000 |
| G7_J4 | 15 | 64 | 3 | 2.0 | 0.812 | 0.034 | 0.467 | 0.936 | 3.3 | 5 | 1 | 1.0 | 0.000 |
| G7_J5 | 10 | 57 | 3 | 2.0 | 0.773 | 0.069 | 0.450 | 0.861 | 7.5 | 5 | 1 | 1.2 | 0.065 |
| G8:Korea | 53 | 204 | 10 | 10.0 | 1.774 | 0.039 | 0.770 | 0.949 | 2.6 | 23 | 5 | 4.6 | 0.932 |
| G8_K1 | 10 | 30 | 2 | 1.2 | 0.163 | 0.000 | 0.090 | 1.000 | 0.0 | 5 | 1 | 1.0 | 0.000 |
| G8_K2 | 12 | 25 | 1 | 1.0 | 0.049 | 0.013 | 0.023 | 0.345 | 48.7 | 5 | 1 | 1.0 | 0.000 |
| G8_K3 | 10 | 34 | 2 | 1.3 | 0.259 | 0.000 | 0.153 | 1.000 | 0.0 | 5 | 1 | 1.0 | 0.000 |
| G8_K4 | 12 | 125 | 6 | 2.8 | 1.513 | 0.092 | 0.708 | 0.872 | 6.8 | 16 | 3 | 2.9 | 0.776 |
| G8_K5 | 7 | 90 | 5 | 2.8 | 1.360 | 0.100 | 0.711 | 0.860 | 7.5 | 16 | 3 | 3.2 | 0.908 |
| Mean | 14 | 65 | 3 | 1.9 | 0.793 | 0.031 | 0.426 | 0.913 | 8.1 | 9 | 2 | 1.6 | 0.297 |
| Regions | No. of sample | Cluster 1 | Cluster 2 |
|---|---|---|---|
| G1: SC | 73 | 0.729 | 0.271 |
| G2: MDRY | 75 | 0.771 | 0.229 |
| G3: SWC | 83 | 0.669 | 0.331 |
| G4: NWC | 75 | 0.206 | 0.794 |
| G5: NC | 86 | 0.135 | 0.865 |
| G6: NEC | 89 | 0.812 | 0.188 |
| G7: Japan | 70 | 0.694 | 0.307 |
| G8: Korea | 53 | 0.772 | 0.228 |
| Loci | Source of variation | SS | VC | PV (%) | Fixation indices |
|---|---|---|---|---|---|
| nSSR | Among two clusters | 393.04 | 0.565 | 5.99 | FCT = 0.060 |
| Among populations within clusters | 5106.23 | 4.409 | 46.69 | FST = 0.527 | |
| Within populations | 5050.64 | 4.469 | 47.32 | FSC = 0.497 | |
| cpSSR | Among two clusters | 68.562 | 0.095 | 6.77 | FCT = 0.068 |
| Among populations within clusters | 935.881 | 0.802 | 56.98 | FST = 0.637 | |
| Within populations | 589.753 | 0.510 | 36.25 | FSC = 0.611 |
| Populations. | Standardized difference test | Wilcoxm sign test | ||||
|---|---|---|---|---|---|---|
| TPM | SMM | TPM | SMM | |||
| T2 | P | T2 | P | P | P | |
| G1_AF | −2.655 | 0.0040 | −3.014 | 0.0013 | 0.9914 | 0.9959 |
| G1_HY | 0.713 | 0.2381 | 0.307 | 0.3794 | 0.1387 | 0.3108 |
| G1_JO | −0.121 | 0.4519 | −0.303 | 0.3809 | 0.5699 | 0.6282 |
| G1_QZ | 0.434 | 0.3322 | 0.125 | 0.4504 | 0.0715 | 0.2046 |
| G1_RY | −4.223 | 0.0000 | −4.457 | 0.0000 | 1.0000 | 1.0000 |
| G2_DQ | 1.498 | 0.0670 | 1.288 | 0.0989 | 0.0521 | 0.0668 |
| G2_SC | 0.967 | 0.1668 | 0.420 | 0.3374 | 0.1081 | 0.2729 |
| G2_TB | 3.360 | 0.0004 | 2.991 | 0.0014 | 0.0004 | 0.0004 |
| G2_WC | 2.651 | 0.0040 | 2.235 | 0.0127 | 0.0007 | 0.0018 |
| G2_XU | 0.854 | 0.1965 | 0.308 | 0.3791 | 0.1841 | 0.4492 |
| G3_CK | 2.755 | 0.0029 | 2.577 | 0.0050 | 0.0014 | 0.0027 |
| G3_CY | 0.146 | 0.4421 | −0.018 | 0.4929 | 0.5898 | 0.5898 |
| G3_GH | −0.211 | 0.4165 | −0.451 | 0.3260 | 0.5235 | 0.6603 |
| G3_N1 | −1.945 | 0.0259 | −2.236 | 0.0127 | 0.9893 | 0.9964 |
| G3_N2 | 0.435 | 0.3317 | 0.122 | 0.4514 | 0.7387 | 0.2899 |
| G3_YJ | 1.164 | 0.1223 | 0.923 | 0.1780 | 0.0770 | 0.0982 |
| G4_BX | −1.439 | 0.0751 | −2.067 | 0.0194 | 0.7793 | 0.8533 |
| G4_HX | −0.109 | 0.4567 | −0.624 | 0.2665 | 0.4159 | 0.6802 |
| G4_LW | −0.069 | 0.4726 | −0.201 | 0.4203 | 0.4816 | 0.4816 |
| G4_WS | −0.194 | 0.4233 | −0.643 | 0.2602 | 0.2450 | 0.2839 |
| G4_YL | −0.740 | 0.2295 | −0.965 | 0.1673 | 0.7378 | 0.7378 |
| G5_DY | 0.919 | 0.1790 | 0.557 | 0.2886 | 0.0844 | 0.1127 |
| G5_JZ | 4.755 | 0.0000 | 4.642 | 0.0000 | 0.0000 | 0.0000 |
| G5_QH | −5.224 | 0.0000 | −5.543 | 0.0000 | 1.0000 | 1.0000 |
| G5_WQ | 1.411 | 0.0791 | 1.071 | 0.1421 | 0.0407 | 0.0649 |
| G5_XH | 1.371 | 0.0851 | 1.206 | 0.1139 | 0.0523 | 0.0523 |
| G5_YT | −0.485 | 0.3140 | −0.512 | 0.3043 | 0.8125 | 0.8750 |
| G6_HL | −0.230 | 0.4089 | −0.844 | 0.1993 | 0.5218 | 0.8529 |
| G6_JH | 1.447 | 0.0739 | 0.977 | 0.1642 | 0.0181 | 0.0570 |
| G6_KS | 4.213 | 0.0000 | 4.054 | 0.0000 | 0.0000 | 0.0000 |
| G6_LX | 0.935 | 0.1749 | 0.648 | 0.2584 | 0.0978 | 0.2090 |
| G6_QQ | −3.357 | 0.0004 | −4.032 | 0.0000 | 0.9976 | 0.9994 |
| G6_SY | −0.227 | 0.4102 | −0.378 | 0.3528 | 0.4375 | 0.4375 |
| G7_J1 | 3.089 | 0.0010 | 2.903 | 0.0019 | 0.0001 | 0.0001 |
| G7_J2 | −1.720 | 0.0428 | −1.768 | 0.0385 | 1.0000 | 1.0000 |
| G7_J3 | 0.979 | 0.1637 | 0.910 | 0.1815 | 0.0625 | 0.0625 |
| G7_J4 | −0.924 | 0.1777 | −1.336 | 0.0907 | 0.5938 | 0.7392 |
| G7_J5 | 1.356 | 0.0875 | 1.110 | 0.1335 | 0.0327 | 0.0523 |
| G8_K1 | −2.512 | 0.0060 | −2.579 | 0.0050 | 1.0000 | 1.0000 |
| G8_K2 | −3.161 | 0.0008 | −3.282 | 0.0005 | 1.0000 | 1.0000 |
| G8_K3 | −1.969 | 0.0245 | −2.058 | 0.0198 | 0.9480 | 0.9710 |
| G8_K4 | −0.755 | 0.2253 | −1.373 | 0.0848 | 0.6079 | 0.7848 |
| G8_K5 | 3.511 | 0.0002 | 3.299 | 0.0005 | 0.0004 | 0.0004 |
| Geographical region | Population name | Location of sampling | Longitude | Latitude | Altitude (m) | Habitat |
|---|---|---|---|---|---|---|
| G1: SC | Population AF | Anfu county, Jiangxi province | 27.388 | 114.602 | 85 | Beside road |
| Population JO | Jianou county, Fujian province | 26.962 | 112.153 | 126 | Beside river | |
| Population HY | Hengyang county, Hunan province | 27.024 | 118.293 | 123 | Beside river | |
| Population RY | Ruyuan county, Guangdong province | 25.872 | 110.862 | 510 | Beside road | |
| Population QZ | Quanzhou county, Guangxi province | 24.919 | 113.136 | 722 | Beside road | |
| G2: MDYR | Population WC | Wuchang district, Hubei province | 30.549 | 119.972 | 15 | Beside road |
| Population XU | Xuanwu district, Jiangsu province | 31.314 | 117.128 | Waste land | ||
| Population DQ | Duqing county, Zhejiang province | 32.370 | 113.400 | 15 | Beside canal | |
| Population SC | Shucha county, Anhui province | 30.521 | 114.395 | 45 | Beside road | |
| Population TB | Tongbai county, Henan province | 32.045 | 118.861 | 33 | Beside road | |
| G3: SWC | Population CK | Chengkou county, Chongqing | 31.983 | 108.667 | 805 | Valley |
| Population YJ | Yinjiang county, Guizhou province | 30.996 | 104.349 | 458 | Valley, | |
| Population GH | Guanghan city, Sichuan province | 28.000 | 108.406 | 458 | Beside river | |
| Population CY | Chayu county, Xizang province | 28.600 | 97.400 | 1685 | Unknown | |
| Population NL1 | Ninglang county, Yunnan province | 27.455 | 100.758 | 2600 | Beside filed | |
| Population NL2 | Ninglang county, Yunnan province | 27.340 | 100.954 | 2550 | Beside filed | |
| G4: NWC | Population BX | Bingxian county, Shaanxi province | 35.040 | 108.077 | 835 | Valley, |
| Population HX | Huixian county, Gansu province | 33.893 | 105.826 | 1126 | Canal | |
| Population LW | Lingwu county, Ningxia province | 38.146 | 106.326 | 1103 | Canal | |
| Population WS | Wenshui county, Shanxi province | 37.417 | 112.017 | 759 | Beside canal | |
| Population YL | Yulin city, Shanxi province | 38.281 | 109.738 | 1051 | Along river | |
| G5: NC | Population JZ | Jizhou county, Hebei province | 37.574 | 118.524 | 23 | Beside road |
| Population DY | Dongying city, Shandong province | 37.742 | 115.686 | 6 | Beside ditches | |
| Population WQ | Wuqing district, Tianjing | 39.808 | 119.432 | −6 | Beside ditches | |
| Population XH | Xuanhua county, Hebei province | 39.449 | 117.249 | 601 | Beside river | |
| Population QH | Qinghuangdao city, Hebei province | 40.593 | 115.021 | 18 | Beside river | |
| Population YT | Yantai city, Shandong province | 37.485 | 121.453 | 10 | Wasteland | |
| G6: NEC | Population LX | Lanxi county, Heilongjiang province | 41.893 | 123.411 | 139 | Beside pond |
| Population JH | Jiaohe county, Jinlin province | 45.849 | 132.762 | 126 | Beside river | |
| Population KS | Keshan county, Heilongjaing province | 43.808 | 127.237 | 325 | Aside field | |
| Population QQ | Qiqihaer city, Heilongjiang province | 48.283 | 125.498 | 304 | Beside river | |
| Population HL | Hulin city, Heilongjiang province | 46.218 | 126.338 | 73 | Beside filed | |
| Population SY | Shenyang, Liaoning province | 47.341 | 123.940 | wasteland | ||
| G7: Japan | Population J1 | Kanagawa, Japan | 34.960 | 137.160 | 12 | Wet Land |
| Population J2 | Tokyo, Japan | 34.828 | 135.770 | 35 | Wet Land | |
| Population J3 | Hirakata, Osaka, Japan | 34.810 | 135.480 | 11 | Wet Land | |
| Population J4 | Okazaki, Japan | 34.959 | 137.139 | 37 | Wet Land | |
| Population J5 | Kyushu University, Fukuoka, Japan | 33.597 | 130.215 | Unknown | ||
| Population K1 | Gangwon-do, South Korea | 37.625 | 128.492 | 520 | Wet Land | |
| Population K2 | Gangwon-do, South, Korea | 38.031 | 128.639 | 340 | Wet Land | |
| Population K3 | Incheon, South Korea | 37.533 | 126.497 | 11 | Wet Land | |
| Population K4 | Yeongcheon-si city, Korea | 36.113 | 128.982 | 102 | Along road | |
| Population K5 | Moonkyeong-si city, Korea | 36.721 | 128.358 | 77 | Along road | |
| Primer name | Primer sequence (5′ to 3′) | Repeat motif | Size range | linkage group |
|---|---|---|---|---|
| gmcp1 | F:TCGATTCTATGCCCCTACTT R:AGACTCCCAAGTTTTCAGTCG | (T)12 | 124–126 | TrnT/trnL |
| gmcp3 | F:GCTTCAGAATTGTCCTATTTA R:ATCAAATAACGCCTCATCTA | (A)12CG(T)11 | 103–113 | TrnT/trnL |
| gmcp4 | F:TATCACTGTCAAGATTAAGAG R:CTTTTATATGTATGGCGCAAC | (A)11 | 127–136 | atpB/rbcL |
| RD19 | F:CTAAATATTACAAAATGGAATTCT R:ACCAATTCAAAAAATCGAATA | (A)14 | 149–151 | rps19 |
| SOYCP | F:CATAGATAGGTACCATCCTTTTT R:CGCCGTATGAAAGCAATAC | (T)13(G)10 | 90–98 | trnM |
| Satt126 | F:ATAAAACAAATTCGCTGATAT R:GCTTGGTAGCTGTAGGAA | (ATT)18 | 109–172 | B2 |
| Satt135 | F:TTCCAATACCTCCCAACTAAC R:CACGGATTTTAAATCATTATTACAT | (ATT)19 | 141–204 | D2 |
| Satt215 | F:GCGCCTTCTTCTGCTAAATCA R:CCCATTCAATTGAGATCCAAAATTAC | (ATT)11 | 114–221 | J |
| Satt216 | F:TACCCTTAATCACCGGACAA R:AGGGAACTAACACATTTAATCATCA | (ATT)20 | 137–251 | D1b |
| Satt221 | F:GCGGCAAACCATTATCTTCATT R:GCGATTGTACCACTAAAAACCATAG | (ATT)23 | 109–224 | D1a |
| Satt231 | F:GGCACGAATCAACATCAAAACTTC R:GCGTGTGCAAAATGTTCATCATCT | (ATT)32 | 160–328 | E |
| Satt233 | F:AAGCATACTCGTCGTAAC R:GCGGTGCAAAGATATTAGAAA | (ATT)16 | 169–238 | A2 |
| Satt270 | F:TGTGATGCCCCTTTTCT R:GCGCAGTGCATGGTTTTCTCA | (ATT)16 | 183–249 | I |
| satt277 | F:GGTGGTGGCGGGTTACTATTACT R:CCACGCTTCAGTTGATTCTTACA | (ATT)40 | 128–255 | C2 |
| satt288 | F:GCGGGGTGATTTAGTGTTTGACACCT R:GCGCTTATAATTAAGAGCAAAAGAAG | (ATT)17 | 195–273 | G |
| Satt294 | F:GCGCTCAGTGTGAAAGTTGTTTCTAT R:GCGGGTCAAATGCAAATTATTTTT | (ATT)23 | 237–303 | C1 |
| Satt373 | F:TCCGCGAGATAAATTCGTAAAAT R:GGCCAGATACCCAAGTTGTACTTGT | (TAT)21 | 210–279 | L |
| Satt423 | F:TTCGCTTGGGTTCAGTTACTT R:GTTGGGGAATTAAAAAAATG | (ATT)19 | 225–351 | F |
| Satt463 | F:CTGCAAATTTGATGCACATGTGTCTA R:TTGGATCTCATATTCAAACTTTCAAG | (ATT)19 | 100–214 | M |
| satt509 | F:GCGCAAGTGGCCAGCTCATCTATT R:GCGCTACCGTGTGGTGGTGTGCTACCT | (ATT)30 | 119–242 | B1 |
| Satt530 | F:CCAAGCGGGTGAAGAGGTTTTT R:CATGCATATTGACTTCATTATT | (ATT)12 | 201–279 | N |
| satt555 | F:GCGGTTGGCTTTGATGATGT R:TTACCGCATGTTCTTGGACTA | (ATT)13 | 234–312 | K |
| Satt568 | F:CGGACACCGGTCTACTAGGAAAGTAA R:GCGGAATAATCCAATTCAATTTA | (ATT)17 | 212–275 | H |
| satt572 | F:GCGGAGCATGTAAATCCAGCCTATTGA R:GCGGGCTAACTTATGTTACTAAACAAT | (ATT)14 | 130–241 | A1 |
| satt581 | F:CCAAAGCTGAGCAGCTGATAACT R:CCCTCACTCCTAGATTATTTGTTGT | (ATT)11 | 130–196 | O |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
He, S.; Wang, Y.; Volis, S.; Li, D.; Yi, T. Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine soja Sieb. et Zucc) Based on Nuclear and Chloroplast Microsatellite Variation. Int. J. Mol. Sci. 2012, 13, 12608-12628. https://doi.org/10.3390/ijms131012608
He S, Wang Y, Volis S, Li D, Yi T. Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine soja Sieb. et Zucc) Based on Nuclear and Chloroplast Microsatellite Variation. International Journal of Molecular Sciences. 2012; 13(10):12608-12628. https://doi.org/10.3390/ijms131012608
Chicago/Turabian StyleHe, Shuilian, Yunsheng Wang, Sergei Volis, Dezhu Li, and Tingshuang Yi. 2012. "Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine soja Sieb. et Zucc) Based on Nuclear and Chloroplast Microsatellite Variation" International Journal of Molecular Sciences 13, no. 10: 12608-12628. https://doi.org/10.3390/ijms131012608
APA StyleHe, S., Wang, Y., Volis, S., Li, D., & Yi, T. (2012). Genetic Diversity and Population Structure: Implications for Conservation of Wild Soybean (Glycine soja Sieb. et Zucc) Based on Nuclear and Chloroplast Microsatellite Variation. International Journal of Molecular Sciences, 13(10), 12608-12628. https://doi.org/10.3390/ijms131012608