Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer

Abstract
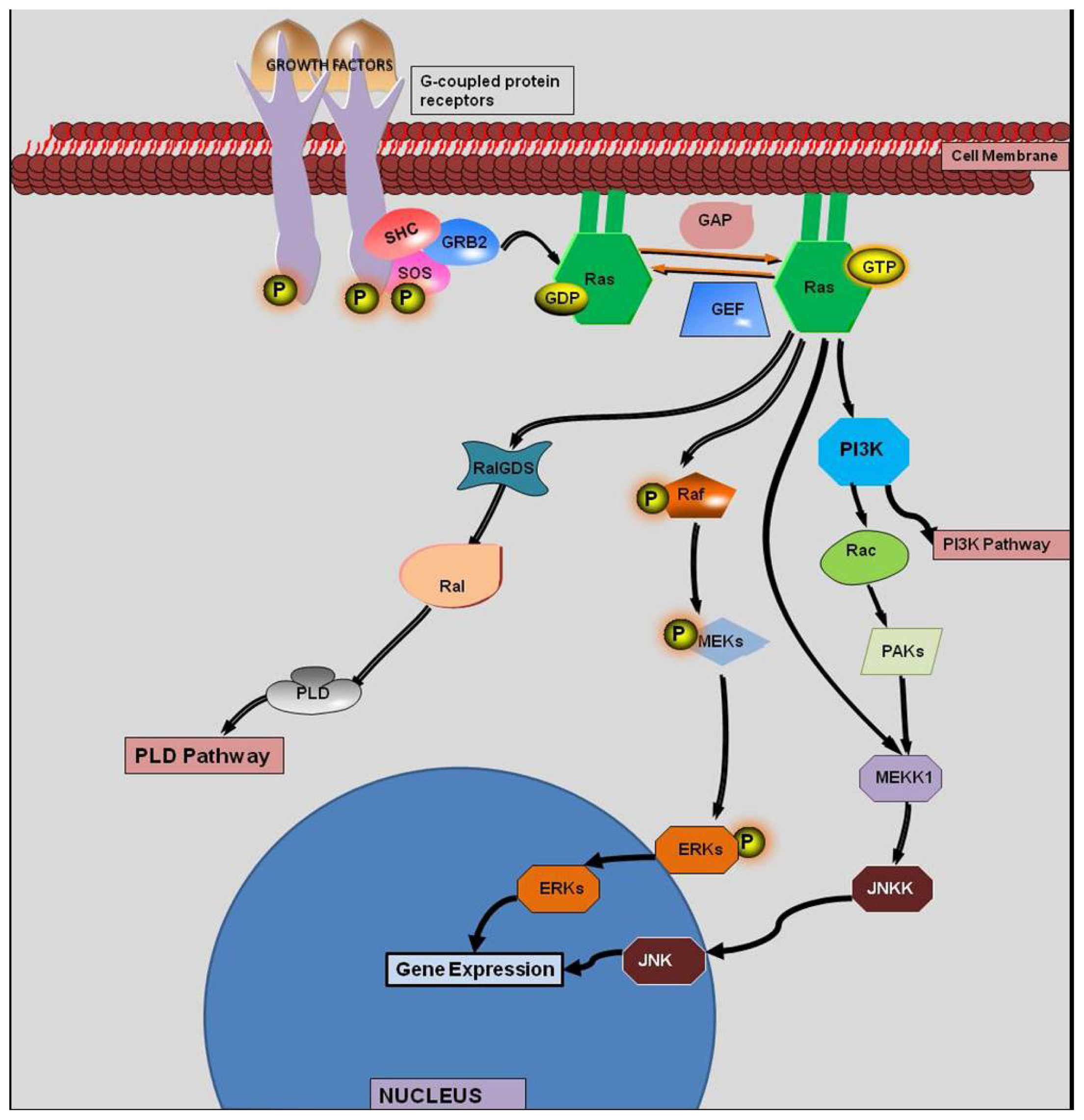
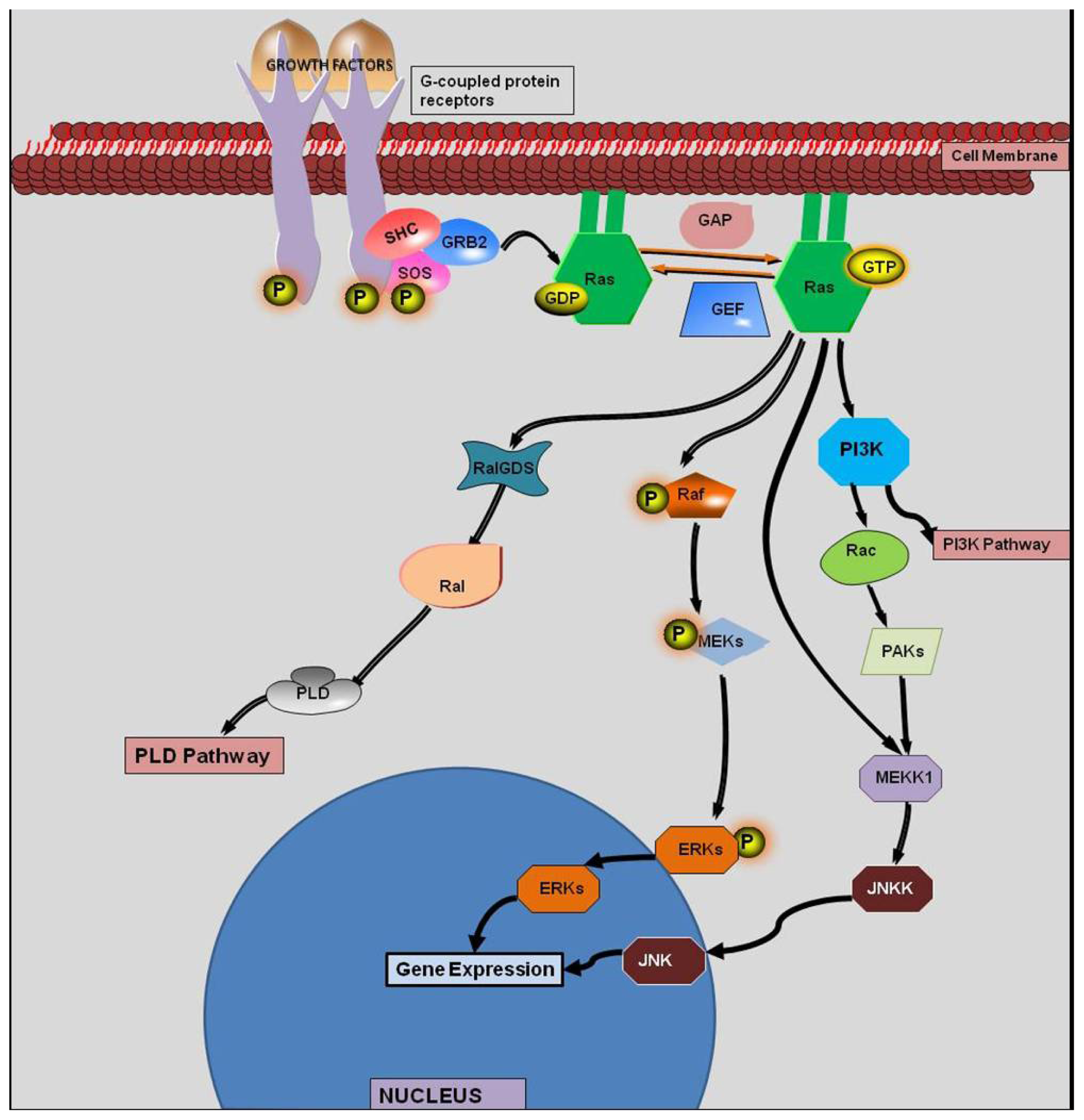
:1. Introduction
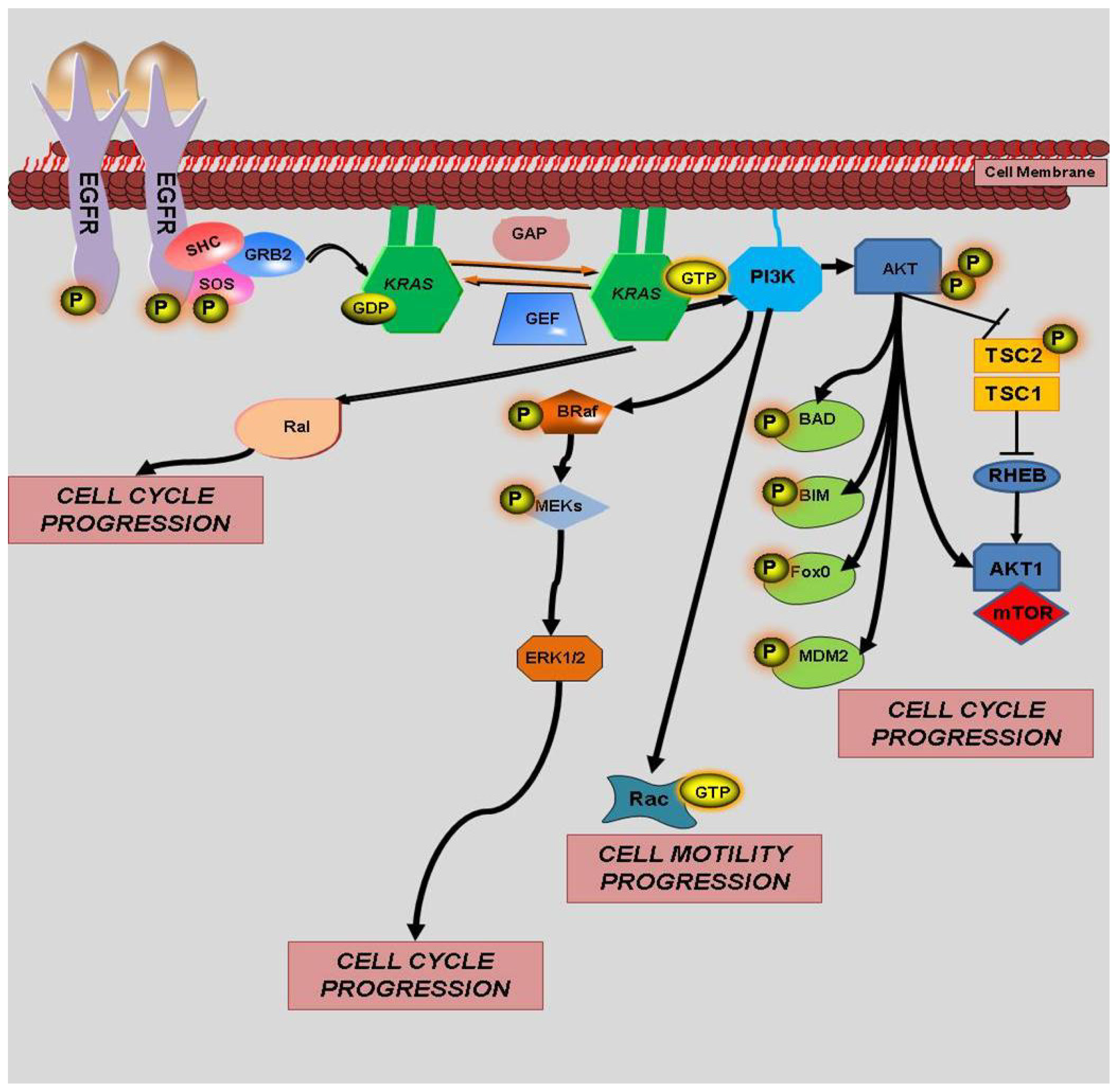
2. History of KRAS
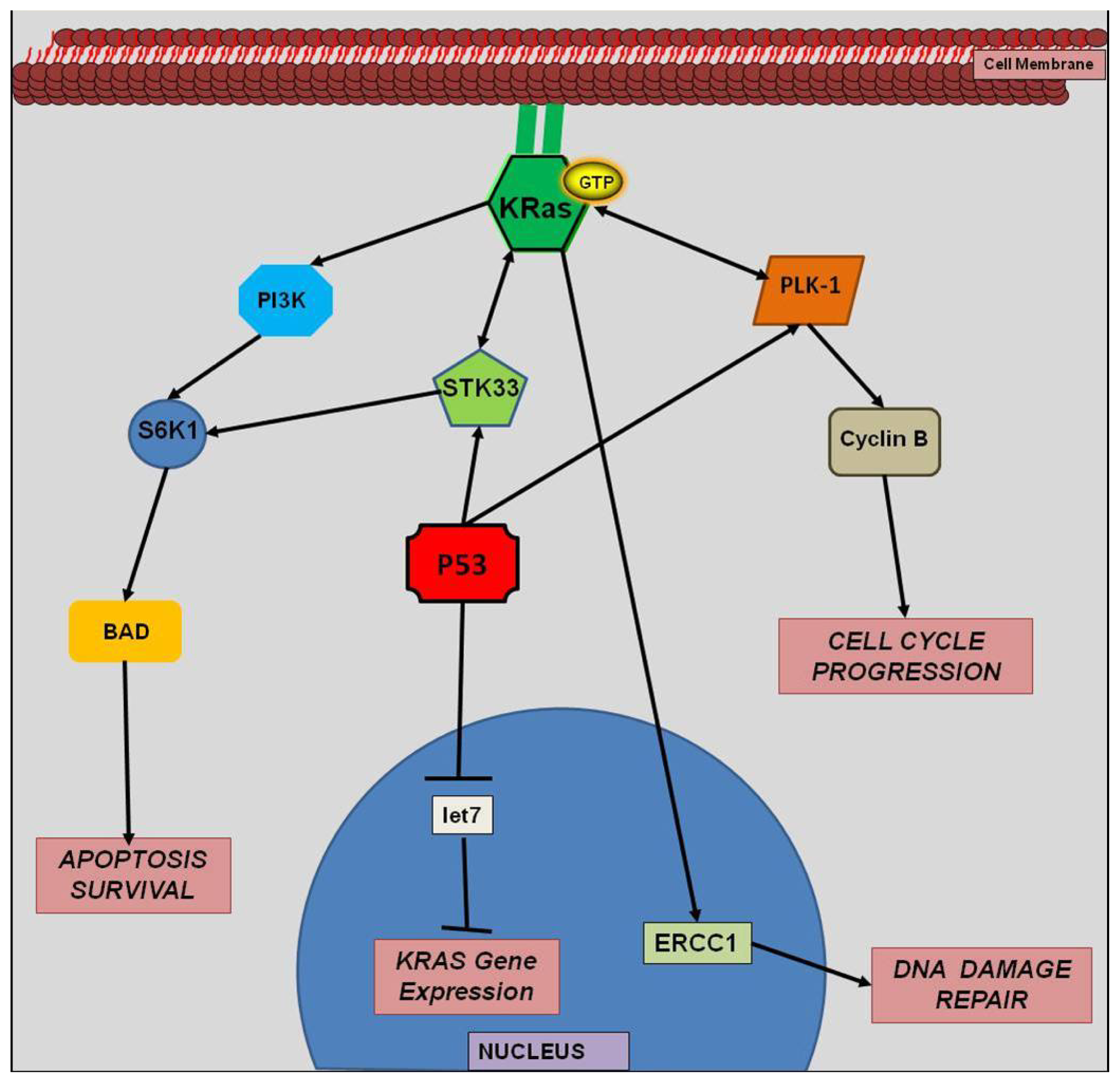
3. KRAS and Prognosis in Colorectal Cancer
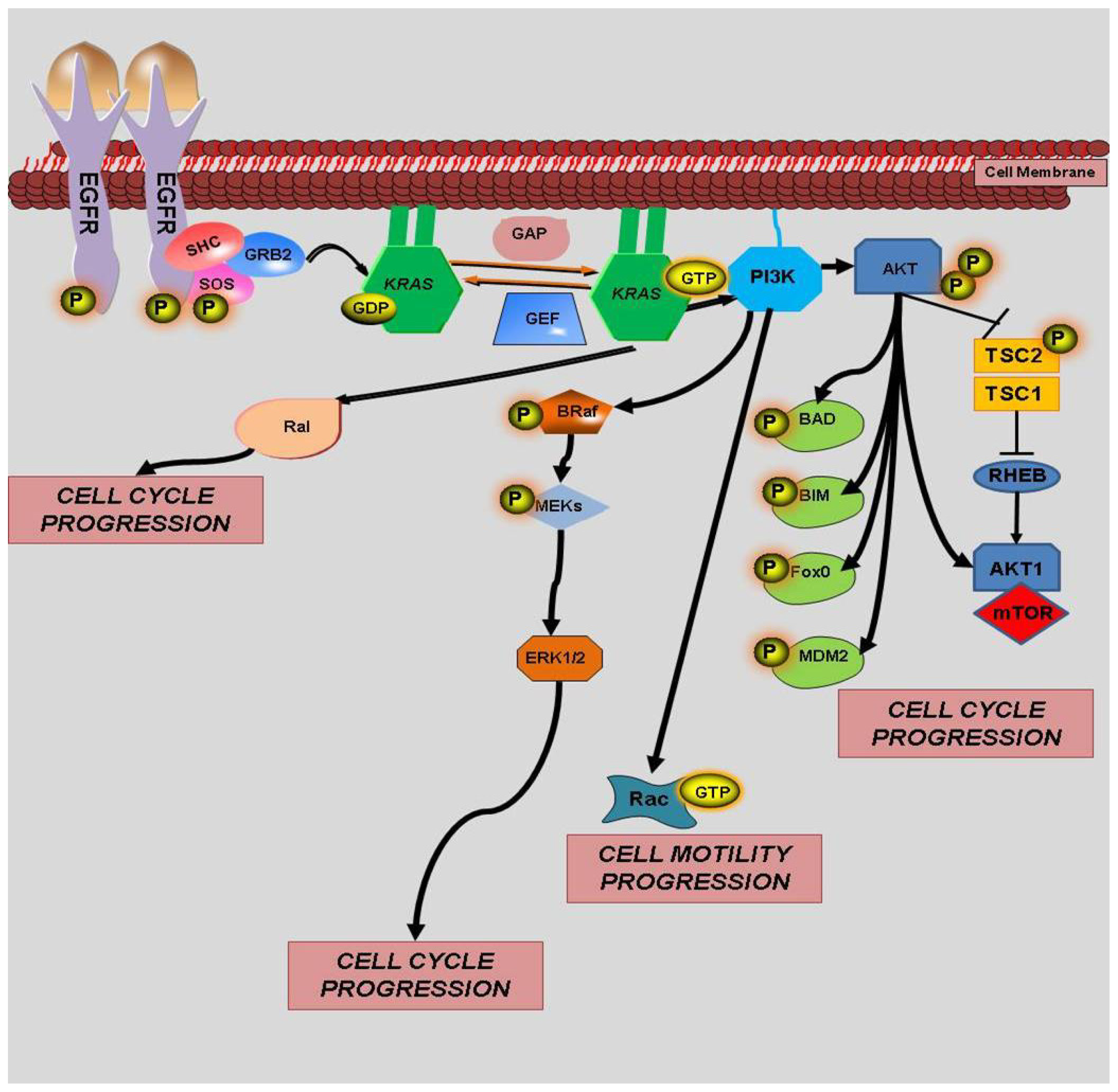
4. KRAS Mutations and Response to Therapy
4.1. KRAS and EGFR
4.2. KRAS and BRAF
4.3. KRAS Mutations in Locally Advanced Rectal Cancer
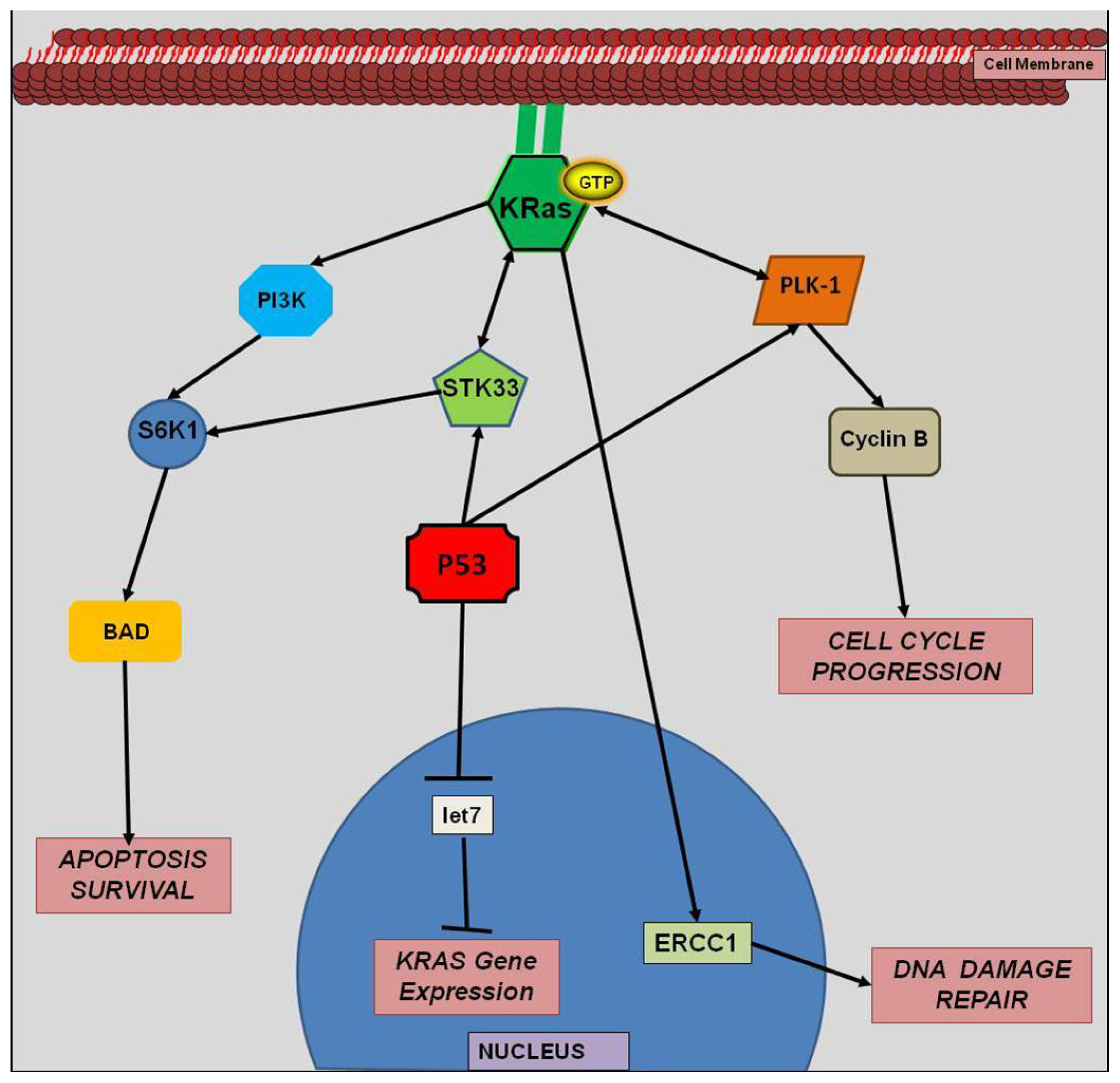
4.4. miRNA and KRAS Interactions
4.5. let-7 miRNA and KRAS
4.6. miR-143 and KRAS
5. Synthetic Lethality
6. Conclusion
References
- Almoguera, C.; Shibata, D.; Forrester, K.; Martin, J.; Arnheim, N.; Perucho, M. Most human carcinomas of the exocrine pancreas contain mutant C-K-Ras genes. Cell 1988, 53, 549–554. [Google Scholar]
- De Roock, W.; Piessevaux, H.; de Schutter, J.; Janssens, M.; de Hertogh, G.; Personeni, N.; Biesmans, B.; van Laethem, J.L.; Peeters, M.; Humblet, Y.; et al. KRAS wild-type state predicts survival and is associated to early radiological response in metastatic colorectal cancer treated with cetuximab. Ann. Oncol 2008, 19, 508–515. [Google Scholar]
- Bell, S.M.; Scott, N.; Cross, D.; Sagar, P.; Lewis, F.A.; Blair, G.E.; Taylor, G.R.; Dixon, M.F.; Quirke, P. Prognostic value of p53 overexpression and c-Ki-ras gene mutations in colorectal cancer. Gastroenterology 1993, 104, 57–64. [Google Scholar]
- Vogelstein, B.; Fearon, E.R.; Hamilton, S.R.; Kern, S.E.; Preisinger, A.C.; Leppert, M.; Nakamura, Y.; White, R.; Smits, A.M.; Bos, J.L. Genetic alterations during colorectal-tumor development. N. Engl. J. Med 1988, 319, 525–532. [Google Scholar]
- Kranenburg, O. The KRAS oncogene: Past, present, and future. Biochim. Biophys. Acta 2005, 1756, 81–82. [Google Scholar]
- Stratton, M.R.; Fisher, C.; Gusterson, B.A.; Cooper, C.S. Detection of point mutations in n-ras and k-ras genes of human embryonal rhabdomyosarcomas using oligonucleotide probes and the polymerase chain-reaction. Cancer Res 1989, 49, 6324–6327. [Google Scholar]
- Baines, A.T.; Xu, D.; Der, C.J. Inhibition of Ras for cancer treatment: The search continues. Future Med. Chem 2011, 3, 1787–1808. [Google Scholar]
- Bamford, S.; Dawson, E.; Forbes, S.; Clements, J.; Pettett, R.; Dogan, A.; Flanagan, A.; Teague, J.; Futreal, P.A.; Stratton, M.R.; et al. The COSMIC (Catalogue of Somatic Mutations in Cancer) database and website. Br. J. Cancer 2004, 91, 355–358. [Google Scholar]
- Rodenhuis, S.; Slebos, R.J.; Boot, A.J.; Evers, S.G.; Mooi, W.J.; Wagenaar, S.S.; van Bodegom, P.C.; Bos, J.L. Incidence and possible clinical significance of K-ras oncogene activation in adenocarcinoma of the human lung. Cancer Res 1988, 48, 5738–5741. [Google Scholar]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar]
- Shields, J.M.; Pruitt, K.; McFall, A.; Shaub, A.; Der, C.J. Understanding Ras: ‘it ain’t over ‘til it’s over’. Trends Cell Biol 2000, 10, 147–154. [Google Scholar]
- Bos, J.L.; Fearon, E.R.; Hamilton, S.R.; Verlaan-de Vries, M.; van Boom, J.H.; van der Eb, A.J.; Vogelstein, B. Prevalence of ras gene mutations in human colorectal cancers. Nature 1987, 327, 293–297. [Google Scholar]
- Forrester, K.; Almoguera, C.; Han, K.; Grizzle, W.E.; Perucho, M. Detection of high incidence of K-ras oncogenes during human colon tumorigenesis. Nature 1987, 327, 298–303. [Google Scholar]
- Capella, G.; Cronauer-Mitra, S.; Pienado, M.A.; Perucho, M. Frequency and spectrum of mutations at codons 12 and 13 of the c-K-ras gene in human tumors. Environ. Health Perspect 1991, 93, 125–131. [Google Scholar]
- Oudejans, J.J.; Slebos, R.J.C.; Zoetmulder, F.A.N.; Mooi, W.J.; Rodenhuis, S. Differential activation of ras genes by point mutation in human colon cancer with metastases to either lung or liver. Int. J. Cancer 1991, 49, 875–879. [Google Scholar]
- Burmer, G.C.; Loeb, L.A. Mutations in the KRAS2 oncogene during progressive stages of human colon carcinoma. Proc. Natl. Acad. Sci. USA 1989, 86, 2403–2407. [Google Scholar]
- Finkelstein, S.D.; Sayegh, R.; Christensen, S.; Swalsky, P.A. Genotypic classification of colorectal adenocarcinoma-biologic behavior correlates with K-Ras-2 mutation type. Cancer 1993, 71, 3827–3838. [Google Scholar]
- Andreyev, H.J.; Norman, A.R.; Cunningham, D.; Oates, J.R.; Clarke, P.A. Kirsten ras mutations in patients with colorectal cancer: The multicenter “RASCAL” study. J. Natl. Cancer Inst 1998, 90, 675–684. [Google Scholar]
- Seeburg, P.H.; Colby, W.W.; Capon, D.J.; Goeddel, D.V.; Levinson, A.D. Biological properties of human c-Ha-ras1 genes mutated at codon 12. Nature 1984, 312, 71–75. [Google Scholar]
- Chipperfield, R.G.; Jones, S.S.; Lo, K.M.; Weinberg, R.A. Activation of Ha-ras p21 by substitution, deletion, and insertion mutations. Mol. Cell Biol 1985, 5, 1809–1813. [Google Scholar]
- Andreyev, H.J.; Norman, A.R.; Cunningham, D.; Oates, J.; Dix, B.R.; Iacopetta, B.J.; Young, J.; Walsh, T.; Ward, R.; Hawkins, N.; et al. Kirsten ras mutations in patients with colorectal cancer: The “RASCAL II” study. Br. J. Cancer 2001, 85, 692–696. [Google Scholar]
- Suchy, B.; Zietz, C.; Rabes, H.M. K-ras point mutations in human colorectal carcinomas: Relation to aneuploidy and metastasis. Int. J. Cancer 1992, 52, 30–33. [Google Scholar]
- Tanaka, M.; Omura, K.; Watanabe, Y.; Oda, Y.; Nakanishi, I. Prognostic factors of colorectal-cancer - k-ras mutation, overexpression of the P53-protein, and cell proliferative activity. J. Surg. Oncol 1994, 57, 57–64. [Google Scholar]
- Pricolo, V.E.; Finkelstein, S.D.; Wu, T.T.; Keller, G.; Bakker, A.; Swalsky, P.A.; Bland, K.I. Prognostic value of TP53 and K-ras-2 mutational analysis in stage III carcinoma of the colon. Am. J. Surg 1996, 171, 41–46. [Google Scholar]
- Troungos, C.; Valavanis, C.; Kapranos, N.; Kittas, C. K-ras mutation in Greek patients with poorly and moderately differenciated tumours of the lower intestinal tract. Anticancer Res 1997, 17, 1399–1404. [Google Scholar]
- Rochlitz, C.F.; Heide, I.; Dekant, E.; Bohmer, R.; Peter, F.J.; Neuhaus, P.; Huhn, D.; Herrmann, R. Position specificity of ki-ras oncogene mutations during the progression of colorectal-carcinoma. Oncology 1993, 50, 70–76. [Google Scholar]
- Dix, B.R.; Robbins, P.; Soong, R.; Jenner, D.; House, A.K.; Iacopetta, B.J. The common molecular genetic alterations in Dukes’ B and C colorectal carcinomas are not short-term prognostic indicators of survival. Int. J. Cancer 1994, 59, 747–751. [Google Scholar]
- Morrin, M.; Kelly, M.; Barrett, N.; Delaney, P. Mutations of ki-ras and p53 genes in colorectal-cancer and their prognostic-significance. Gut 1994, 35, 1627–1631. [Google Scholar]
- Laurent-Puig, P.; Olschwang, S.; Delattre, O.; Remvikos, Y.; Asselain, B.; Melot, T.; Validire, P.; Muleris, M.; Girodet, J.; Salmon, R.J.; et al. Survival and acquired genetic alterations in colorectal cancer. Gastroenterology 1992, 102, 1136–1141. [Google Scholar]
- Inoue, Y.; Saigusa, S.; Iwata, T.; Okugawa, Y.; Toiyama, Y.; Tanaka, K.; Uchida, K.; Mohri, Y.; Kusunoki, M. The prognostic value of KRAS mutations in patients with colorectal cancer. Oncol. Rep 2012. [Google Scholar] [CrossRef]
- Reinacher-Schick, A.; Schulmann, K.; Modest, D.; Bruns, N.; Graeven, U.; Jaworska, M.; Greil, R.; Porschen, R.; Arnold, D.; Schmiegel, W.; et al. Effect of KRAS codon13 mutations in patients with advanced colorectal cancer (advanced CRC) under oxaliplatin containing chemotherapy. Results from a translational study of the AIO colorectal study group. BMC Cancer 2012, 12, 349. [Google Scholar]
- Zlobec, I.; Kovac, M.; Erzberger, P.; Molinari, F.; Bihl, M.P.; Rufle, A.; Foerster, A.; Frattini, M.; Terracciano, L.; Heinimann, K.; et al. Combined analysis of specific KRAS mutation, BRAF and microsatellite instability indentifies prognostic subgroups of sporadic and hereditary colorectal cancer. Int. J. Cancer 2010, 127, 2569–2575. [Google Scholar]
- Nash, G.M.; Gimbel, M.; Shia, J.; Nathanson, D.R.; Ndubuisi, M.I.; Zeng, Z.S.; Kemeny, N.; Paty, P.B. KRAS mutation correlates with accelerated metastatic progression in patients with colorectal liver metastases. Ann. Surg. Oncol 2010, 17, 572–578. [Google Scholar]
- Santini, D.; Loupakis, F.; Vincenzi, B.; Floriani, I.; Stasi, I.; Canestrari, E.; Rulli, E.; Maltese, P.E.; Andreoni, F.; Masi, G.; et al. High concordance of KRAS status between primary colorectal tumors and related metastatic sites: Implications for clinical practice. Oncologist 2008, 13, 1270–1275. [Google Scholar]
- Kim, M.J.; Lee, H.S.; Kim, J.H.; Kim, Y.J.; Kwon, J.H.; Lee, J.O.; Bang, S.M.; Park, K.U.; Kim, D.W.; Kang, S.B.; et al. Different metastatic pattern according to the KRAS mutational status and site-specific discordance of KRAS status in patients with colorectal cancer. BMC Cancer 2012, 12, 347. [Google Scholar]
- Louvet, C.; de Gramont, A.; Tournigand, C.; Artru, P.; Maindrault-Goebel, F.; Krulik, M. Correlation between progression free survival and response rate in patients with metastatic colorectal carcinoma. Cancer 2001, 91, 2033–2038. [Google Scholar]
- Lievre, A.; Bachet, J.; Le Corre, D.; Boige, V.; Landi, B.; Emile, J.; Cote, J.; Tomasic, G.; Penna, C.; Ducreux, M.; et al. KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 2006, 66, 3992–3995. [Google Scholar]
- Goldstein, N.S.; Armin, M. Epidermal growth factor receptor immunohistochemical reactivity in patients with American Joint Committee on Cancer Stage IV colon adenocarcinoma: Implications for a standardized scoring system. Cancer 2001, 92, 1331–1346. [Google Scholar]
- Salomon, D.S.; Brandt, R.; Ciardiello, F.; Normanno, N. Epidermal Growth Factor-Related Peptides and Their Receptors in Human Malignancies. Crit. Rev. Oncol./Hematol 1995, 19, 183–232. [Google Scholar]
- Hemming, A.W.; Davis, N.L.; Kluftinger, A.; Robinson, B.; Quenville, N.F.; Liseman, B.; LeRiche, J. Prognostic markers of colorectal cancer: An evaluation of DNA content, epidermal growth factor receptor, and Ki-67. J. Surg. Oncol 1992, 51, 147–152. [Google Scholar]
- Mayer, A.; Takimoto, M.; Fritz, E.; Schellander, G.; Kofler, K.; Ludwig, H. The prognostic significance of proliferating cell nuclear antigen, epidermal growth factor receptor, and mdr gene expression in colorectal cancer. Cancer 1993, 71, 2454–2460. [Google Scholar]
- Normanno, N.; Tejpar, S.; Morgillo, F.; de Luca, A.; van Cutsem, E.; Ciardiello, F. Implications for KRAS status and EGFR-targeted therapies in metastatic CRC. Nat. Rev. Clin. Oncol 2009, 6, 519–527. [Google Scholar]
- Bokemeyer, C.; Bondarenko, I.; Hartmann, J.T.; de Braud, F.; Schuch, G.; Zubel, A.; Celik, I.; Schlichting, M.; Koralewski, P. Efficacy according to biomarker status of cetuximab plus FOLFOX-4 as first-line treatment for metastatic colorectal cancer: The OPUS study. Ann. Oncol 2011, 22, 1535–1546. [Google Scholar]
- Van Cutsem, E.; Kohne, C.H.; Hitre, E.; Zaluski, J.; Chien, C.R.C.; Makhson, A.; D’Haens, G.; Pinter, T.; Lim, R.; Bodoky, G.; et al. Cetuximab and chemotherapy as initial treatment for metastatic colorectal cancer. N. Engl. J. Med 2009, 360, 1408–1417. [Google Scholar]
- Van Cutsem, E.; Kohne, C.H.; Lang, I.; Folprecht, G.; Nowacki, M.P.; Cascinu, S.; Shchepotin, I.; Maurel, J.; Cunningham, D.; Tejpar, S.; et al. Cetuximab plus irinotecan, fluorouracil, and leucovorin as first-line treatment for metastatic colorectal cancer: Updated analysis of overall survival according to tumor KRAS and BRAF mutation status. J. Clin. Oncol 2011, 29, 2011–2019. [Google Scholar]
- Bokemeyer, C.; Bondarenko, I.; Makhson, A.; Hartmann, J.T.; Aparicio, J.; de Braud, F.; Donea, S.; Ludwig, H.; et al. Fluorouracil, leucovorin, and oxaliplatin with and without cetuximab in the first-line treatment of metastatic colorectal cancer. J. Clin. Oncol 2009, 27, 663–671. [Google Scholar]
- Amado, R.G.; Wolf, M.; Peeters, M.; van Cutsem, E.; Siena, S.; Freeman, D.J.; Juan, T.; Sikorski, R.; Suggs, S.; Radinsky, R.; et al. Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J. Clin. Oncol 2008, 26, 1626–1634. [Google Scholar]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med 2008, 359, 1757–1765. [Google Scholar]
- De Roock, W.; Claes, B.; Bernasconi, D.; de Schutter, J.; Biesmans, B.; Fountzilas, G.; Kalogeras, K.T.; Kotoula, V.; Papamichael, D.; Laurent-Puig, P.; et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: A retrospective consortium analysis. Lancet Oncol 2010, 11, 753–762. [Google Scholar]
- De Roock, W.; Jonker, D.J.; di Nicolantonio, F.; Sartore-Bianchi, A.; Tu, D.; Siena, S.; Lamba, S.; Arena, S.; Frattini, M.; Piessevaux, H.; et al. Association of KRAS p.G13D mutation with outcome in patients with chemotherapy-refractory metastatic colorectal cancer treated with cetuximab. JAMA 2010, 304, 1812–1820. [Google Scholar]
- Tejpar, S.; Celik, I.; Schlichting, M.; Sartorius, U.; Bokemeyer, C.; van Cutsem, E. Association of KRAS G13D tumor mutations with outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. J. Clin. Oncol 2012. [Google Scholar] [CrossRef]
- Tejpar, S.; Bokemeyer, C.; Celik, I.; Schlichting, M.; Heeger, S.; van Cutsem, E. Kras mutations and outcome in patients with metastatic colorectal cancer treated with first-line chemotherapy with or without cetuximab. Ann. Oncol 2011, 22, v16–v17. [Google Scholar]
- Di Nicolantonio, F.; Martini, M.; Molinari, F.; Sartore-Bianchi, A.; Arena, S.; Saletti, P.; de Dosso, S.; Mazzucchelli, L.; Frattini, M.; Siena, S.; et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J. Clin. Oncol 2008, 26, 5705–5712. [Google Scholar]
- Siena, S.; Sartore-Bianchi, A.; di Nicolantonio, F.; Balfour, J.; Bardelli, A. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J. Natl. Cancer Inst 2009, 101, 1308–1324. [Google Scholar]
- Vaughn, C.P.; Zobell, S.D.; Furtado, L.V.; Baker, C.L.; Samowitz, W.S. Frequency of KRAS, BRAF, and NRAS mutations in colorectal cancer. Genes Chromosom. Cancer 2011, 50, 307–312. [Google Scholar]
- Bosset, J.F.; Collette, L.; Calais, G.; Mineur, L.; Maingon, P.; Radosevic-Jelic, L.; Daban, A.; Bardet, E.; Beny, A.; Ollier, J.C. Chemotherapy with preoperative radiotherapy in rectal cancer. N. Engl. J. Med 2006, 355, 1114–1123. [Google Scholar]
- Bosset, J.F.; Pavy, J.J.; Hamers, H.P.; Horiot, J.C.; Fabri, M.C.; Rougier, P.; Eschwege, F.; Schraub, S. Determination of the optimal dose of 5-fluorouracil when combined with low dose D,L-leucovorin and irradiation in rectal cancer: Results of three consecutive phase II studies. Eur. J. Cancer 1993, 29A, 1406–1410. [Google Scholar]
- Maas, M.; Nelemans, P.J.; Valentini, V.; Das, P.; Rodel, C.; Kuo, L.J.; Calvo, F.A.; Garcia-Aguilar, J.; Glynne-Jones, R.; Haustermans, K.; et al. Long-term outcome in patients with a pathological complete response after chemoradiation for rectal cancer: A pooled analysis of individual patient data. Lancet Oncol 2010, 11, 835–844. [Google Scholar]
- Bengala, C.; Bettelli, S.; Bertolini, F.; Sartori, G.; Fontana, A.; Malavasi, N.; Depenni, R.; Zironi, S.; Del Giovane, C.; Luppi, G.; et al. Prognostic role of EGFR gene copy number and KRAS mutation in patients with locally advanced rectal cancer treated with preoperative chemoradiotherapy. Br. J. Cancer 2010, 103, 1019–1024. [Google Scholar]
- Duldulao, M.P.; Lee, W.; Chen, Z.; Li, W.; Nelson, R.A.; Kim, J.; Garcia-Aguilar, J. Use of KRAS codon 13 mutations predict response to neoadjuvant chemoradiation therapy in patients with rectal adenocarcinoma. Ann. Surg. Oncol 2012, 19, S21–S22. [Google Scholar]
- Yu, S.N.; Lu, Z.H.; Liu, C.Z.; Meng, Y.X.; Ma, Y.H.; Zhao, W.G.; Liu, J.P.; Yu, J.; Chen, J. miRNA-96 suppresses KRAS and functions as a tumor suppressor gene in pancreatic cancer. Cancer Res 2010, 70, 6015–6025. [Google Scholar]
- Ambros, V. microRNAs: Tiny regulators with great potential. Cell 2001, 107, 823–826. [Google Scholar]
- Ambros, V. The function of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar]
- Slaby, O.; Svoboda, M.; Fabian, P.; Smerdova, T.; Knoflickova, D.; Bednarikova, M.; Nenutil, R.; Vyzula, R. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology 2007, 72, 397–402. [Google Scholar]
- Lea, M.A. Recently identified and potential targets for colon cancer treatment. Futur. Oncol 2010, 6, 993–1002. [Google Scholar]
- Wu, W.K.; Law, P.T.; Lee, C.W.; Cho, C.H.; Fan, D.; Wu, K.; Yu, J.; Sung, J.J. MicroRNA in colorectal cancer: From benchtop to bedside. Carcinogenesis 2011, 32, 247–253. [Google Scholar]
- Calin, G.A.; Sevignani, C.; Dan Dumitru, C.; Hyslop, T.; Noch, E.; Yendamuri, S.; Shimizu, M.; Rattan, S.; Bullrich, F.; Negrini, M.; et al. Human microRNA genes are frequently located at fragile sites and genomic regions involved in cancers. Proc. Natl. Acad. Sci. USA 2004, 101, 2999–3004. [Google Scholar]
- Earle, J.S.; Luthra, R.; Romans, A.; Abraham, R.; Ensor, J.; Yao, H.; Hamilton, S.R. Association of microRNA expression with microsatellite instability status in colorectal adenocarcinoma. J. Mol. Diagn 2010, 12, 433–440. [Google Scholar]
- Akao, Y.; Nakagawa, Y.; Hirata, I.; Iio, A.; Itoh, T.; Kojima, K.; Nakashima, R.; Kitade, Y.; Naoe, T. Role of anti-oncomirs miR-143 and -145 in human colorectal tumors. Cancer Gene Ther 2010, 17, 398–408. [Google Scholar]
- Johnson, S.M.; Grosshans, H.; Shingara, J.; Byrom, M.; Jarvis, R.; Cheng, A.; Labourier, E.; Reinert, K.L.; Brown, D.; Slack, F.J. RAS is regulated by the let-7 MicroRNA family. Cell 2005, 120, 635–647. [Google Scholar]
- Takamizawa, J.; Konishi, H.; Yanagisawa, K.; Tomida, S.; Osada, H.; Endoh, H.; Harano, T.; Yatabe, Y.; Nagino, M.; Nimura, Y.; et al. Reduced expression of the let-7 microRNAs in human lung cancers in association with shortened postoperative survival. Cancer Res 2004, 64, 3753–3756. [Google Scholar]
- Jeong, S.H.; Wu, H.G.; Park, W.Y. LIN28B confers radio-resistance through the posttranscriptional control of KRAS. Exp. Mol. Med 2009, 41, 912–918. [Google Scholar]
- Akao, Y.; Nakagawa, Y.; Naoe, T. let-7 microRNA functions as a potential growth suppressor in human colon cancer cells. Biol. Pharm. Bull 2006, 29, 903–906. [Google Scholar]
- King, C.E.; Cuatrecasas, M.; Castells, A.; Sepulveda, A.R.; Lee, J.S.; Rustgi, A.K. LIN28B promotes colon cancer progression and metastasis. Cancer Res 2011, 71, 4260–4268. [Google Scholar]
- King, C.E.; Wang, L.; Winograd, R.; Madison, B.B.; Mongroo, P.S.; Johnstone, C.N.; Rustgi, A.K. LIN28B fosters colon cancer migration, invasion and transformation through let-7-dependent and -independent mechanisms. Oncogene 2011, 30, 4185–4193. [Google Scholar]
- Graziano, F.; Canestrari, E.; Loupakis, F.; Ruzzo, A.; Galluccio, N.; Santini, D.; Rocchi, M.; Vincenzi, B.; Salvatore, L.; Cremolini, C.; et al. Genetic modulation of the Let-7 microRNA binding to KRAS 3′-untranslated region and survival of metastatic colorectal cancer patients treated with salvage cetuximab-irinotecan. Pharmacogn. J. 10 458–464.
- Chen, X.; Guo, X.; Zhang, H.; Xiang, Y.; Chen, J.; Yin, Y.; Cai, X.; Wang, K.; Wang, G.; Ba, Y.; et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene 2009, 28, 1385–1392. [Google Scholar]
- Pichler, M.; Winter, E.; Stotz, M.; Eberhard, K.; Samonigg, H.; Lax, S.; Hoefler, G. Down-regulation of KRAS-interacting miRNA-143 predicts poor prognosis but not response to EGFR-targeted agents in colorectal cancer. Br. J. Cancer 2012, 106, 1826–1832. [Google Scholar]
- Borralho, P.M.; Kren, B.T.; Castro, R.E.; da Silva, I.B.; Steer, C.J.; Rodrigues, C.M. MicroRNA-143 reduces viability and increases sensitivity to 5-fluorouracil in HCT116 human colorectal cancer cells. FEBS J 2009, 276, 6689–6700. [Google Scholar]
- Kaelin, W.G., Jr. The concept of synthetic lethality in the context of anticancer therapy. Nat. Rev. Cancer 2005, 5, 689–698. [Google Scholar]
- Luo, J.; Emanuele, M.J.; Li, D.; Creighton, C.J.; Schlabach, M.R.; Westbrook, T.F.; Wong, K.K.; Elledge, S.J. A genome-wide RNAi screen identifies multiple synthetic lethal interactions with the Ras oncogene. Cell 2009, 137, 835–848. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| KRAS mutations at codons 12 and 13 from RASCAL and RASCAL II studies | ||||
|---|---|---|---|---|
| Mutation | % of specific codon mutations | % of all codon 12/13 mutations | ||
| Codon 12 | RASCAL I [18] | RASCAL II [21] | RASCAL I [18] | RASCAL II [21] |
| G→A | ||||
| Glycine (GGT)→Serine (AGT) | 7.5% | 8.3% | 6.8% | 6.2% |
| Glycine (GGT)→Aspartate (GAT) | 31.5% | 39.3% | 28.5% | 39.6% |
| G→T | ||||
| Glycine (GGT)→Cysteine (TGT) | 8.9% | 10.2% | 8.0% | 7.7% |
| Glycine (GGT)→Valine (GGT) | 24.2% | 33.3% | 21.9% | 25.8% |
| G→C | ||||
| Glycine (GGT)→Arginine (CGT) | 3.8% | 3.5% | ||
| Glycine (GGT)→Alanine (GCT) | 6.2% | 8.8% | 5.6% | 6.6% |
| Codon 12, unknown Point Mutation | 17.8% | 16.0% | ||
| Codon 13 | ||||
| G→A | ||||
| Glycine (GGT)→Aspartate (GAC) | 83.9% | 100% | 14.5% | 24.8% |
| G→T | ||||
| Glycine (GGT)→Cysteine (TGC) | 6.8% | 1.2% | ||
| Glycine (GGT)→Valine (GTC) | 2.1% | 0.4% | ||
| G→C | ||||
| Glycine (GGT)→Arginine (CGC) | 0.7% | 0.1% | ||
| Glycine (GGT)→Alanine (GCC) | 2.1% | 0.4% | ||
| Codon 13, unknown Point Mutation | 5.5% | 1.0% | ||
| % of Colorectal Cancer Patients with KRAS mutation | 37.7% | 34.8% | ||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arrington, A.K.; Heinrich, E.L.; Lee, W.; Duldulao, M.; Patel, S.; Sanchez, J.; Garcia-Aguilar, J.; Kim, J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. Int. J. Mol. Sci. 2012, 13, 12153-12168. https://doi.org/10.3390/ijms131012153
Arrington AK, Heinrich EL, Lee W, Duldulao M, Patel S, Sanchez J, Garcia-Aguilar J, Kim J. Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer. International Journal of Molecular Sciences. 2012; 13(10):12153-12168. https://doi.org/10.3390/ijms131012153
Chicago/Turabian StyleArrington, Amanda K., Eileen L. Heinrich, Wendy Lee, Marjun Duldulao, Supriya Patel, Julian Sanchez, Julio Garcia-Aguilar, and Joseph Kim. 2012. "Prognostic and Predictive Roles of KRAS Mutation in Colorectal Cancer" International Journal of Molecular Sciences 13, no. 10: 12153-12168. https://doi.org/10.3390/ijms131012153