Anti-Epidermal Growth Factor Receptor (EGFR) Antibodies Overcome Resistance of Ovarian Cancer Cells to Targeted Therapy and Natural Cytotoxicity

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
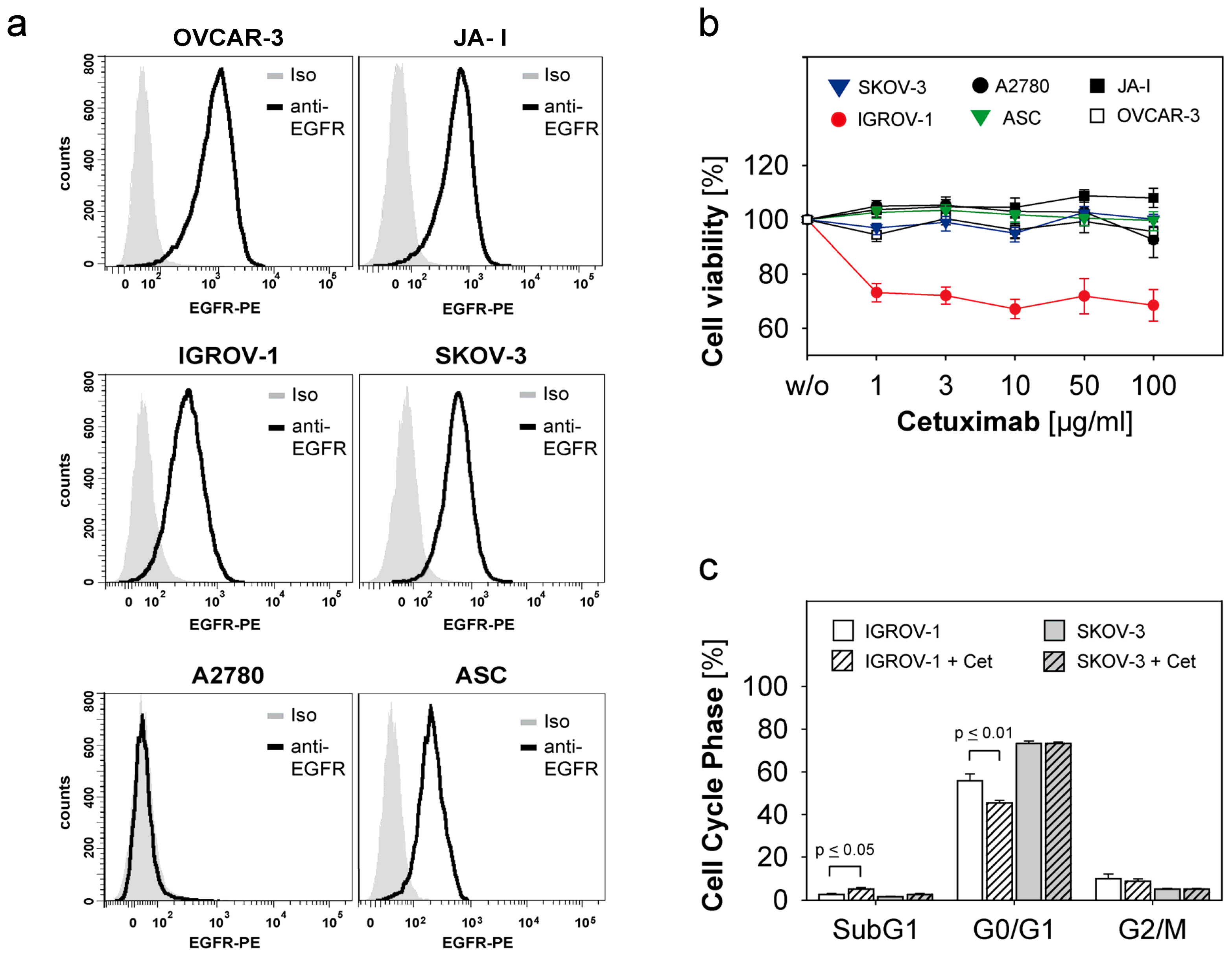
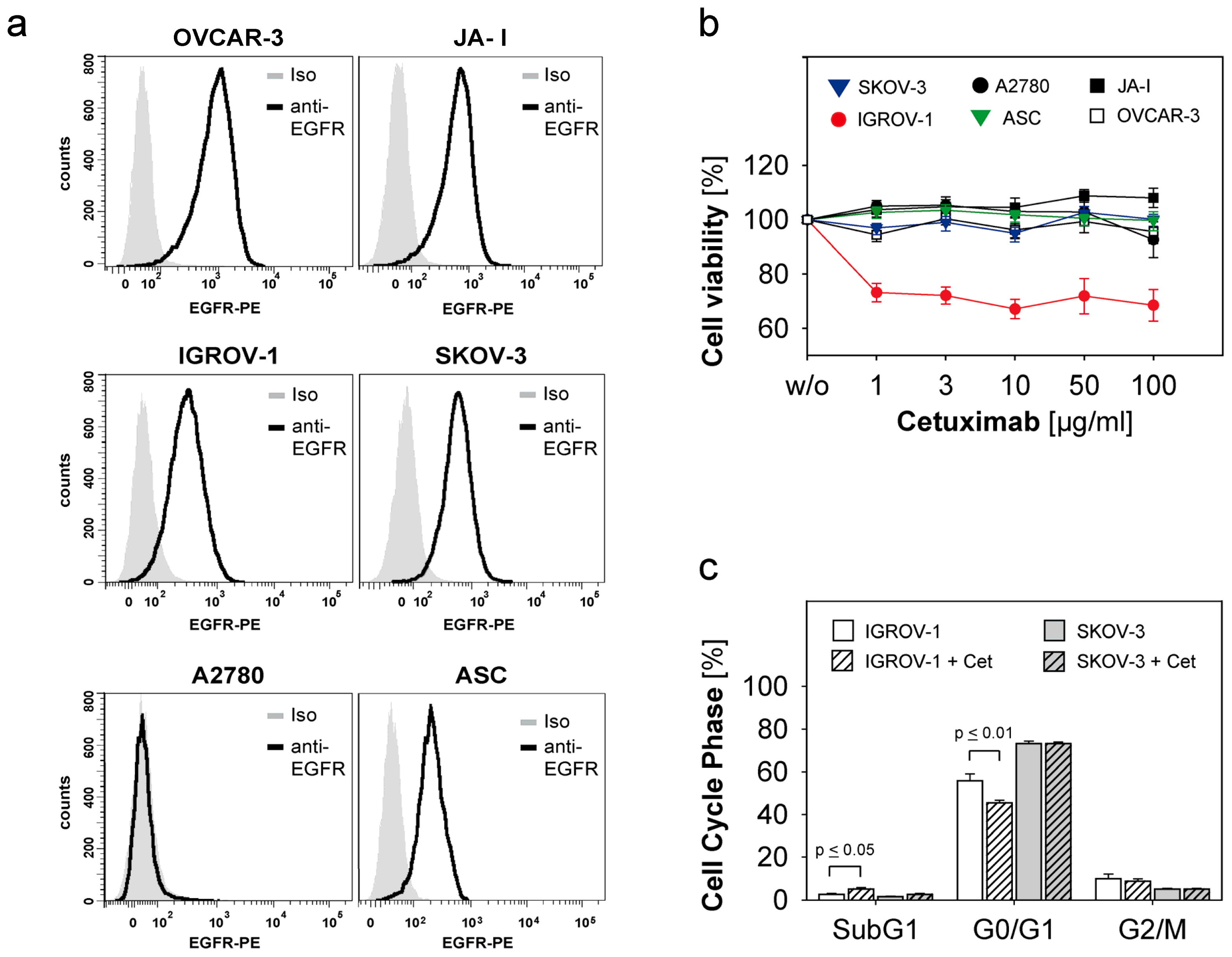
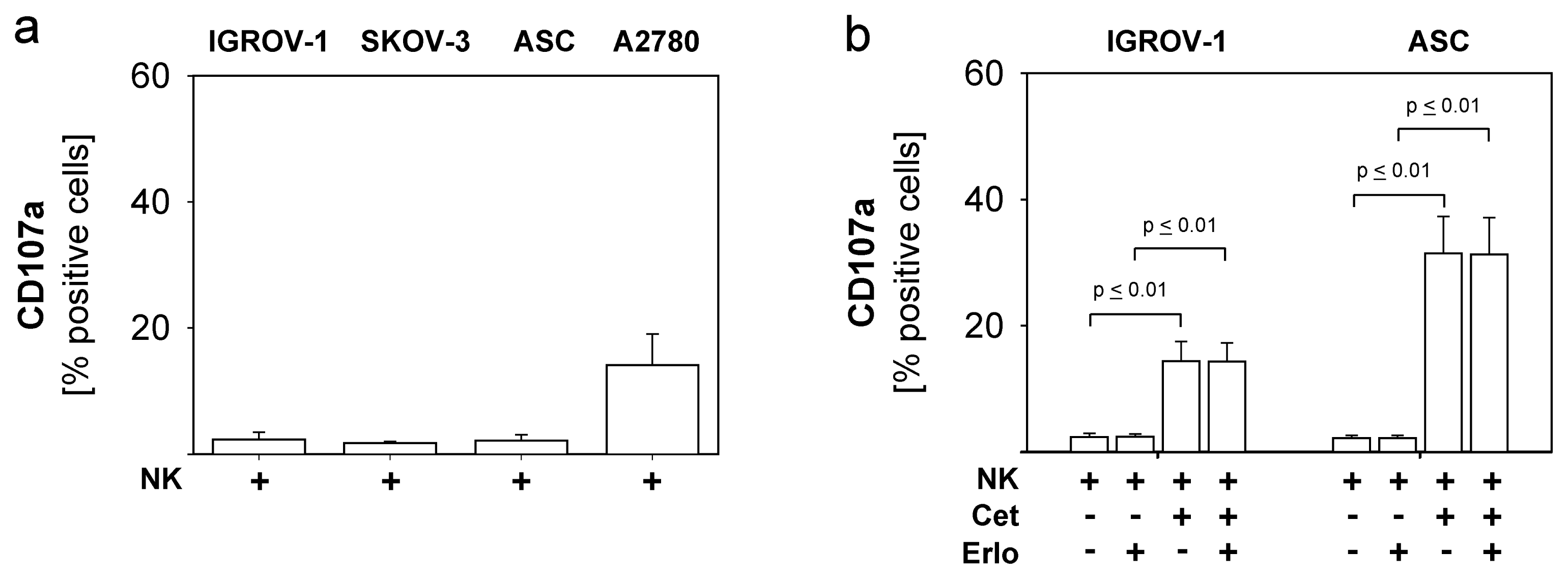
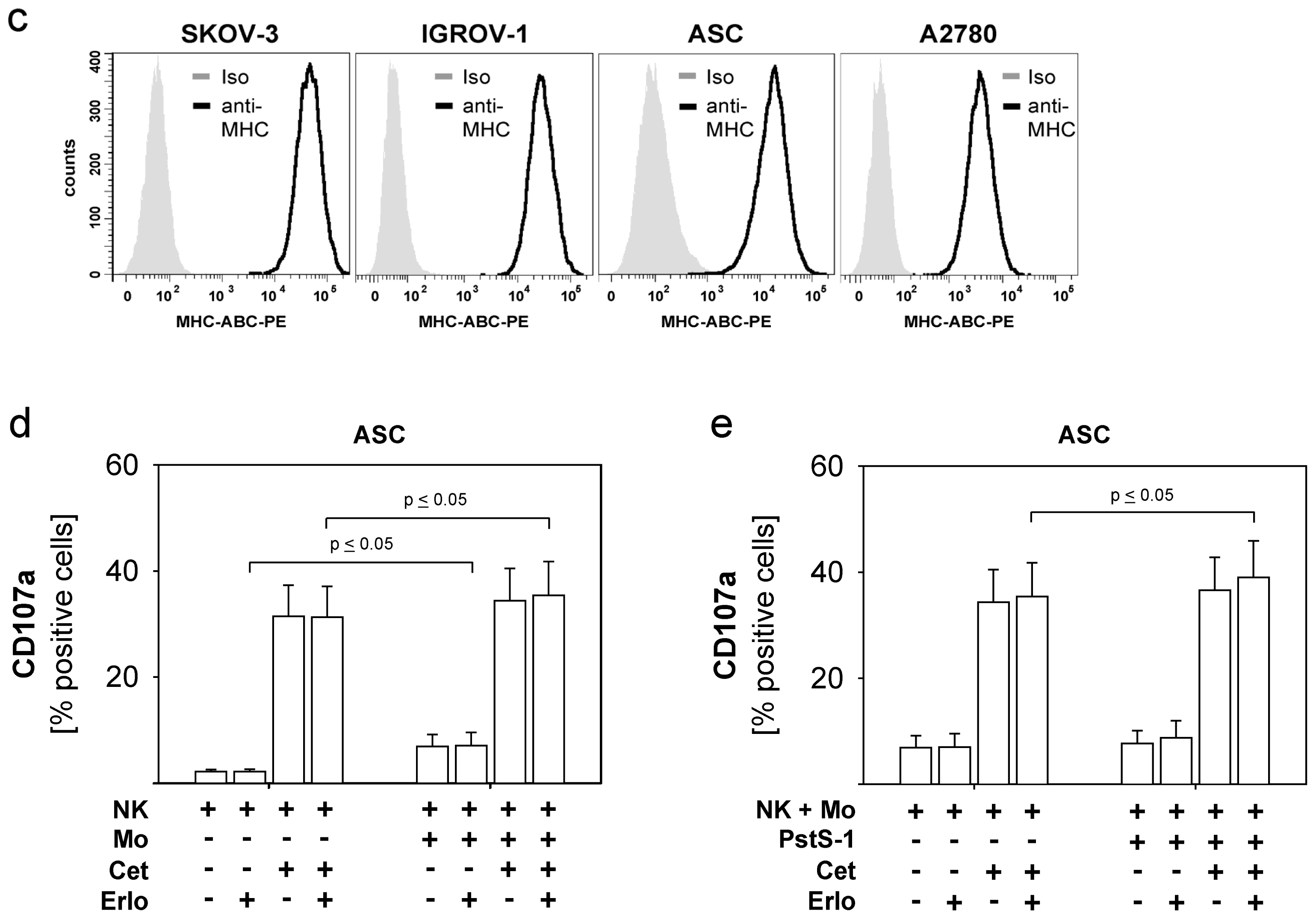
2.1. Susceptibility of Ovarian Cancer Cells to the Anti-EGFR-Antibody Cetuximab
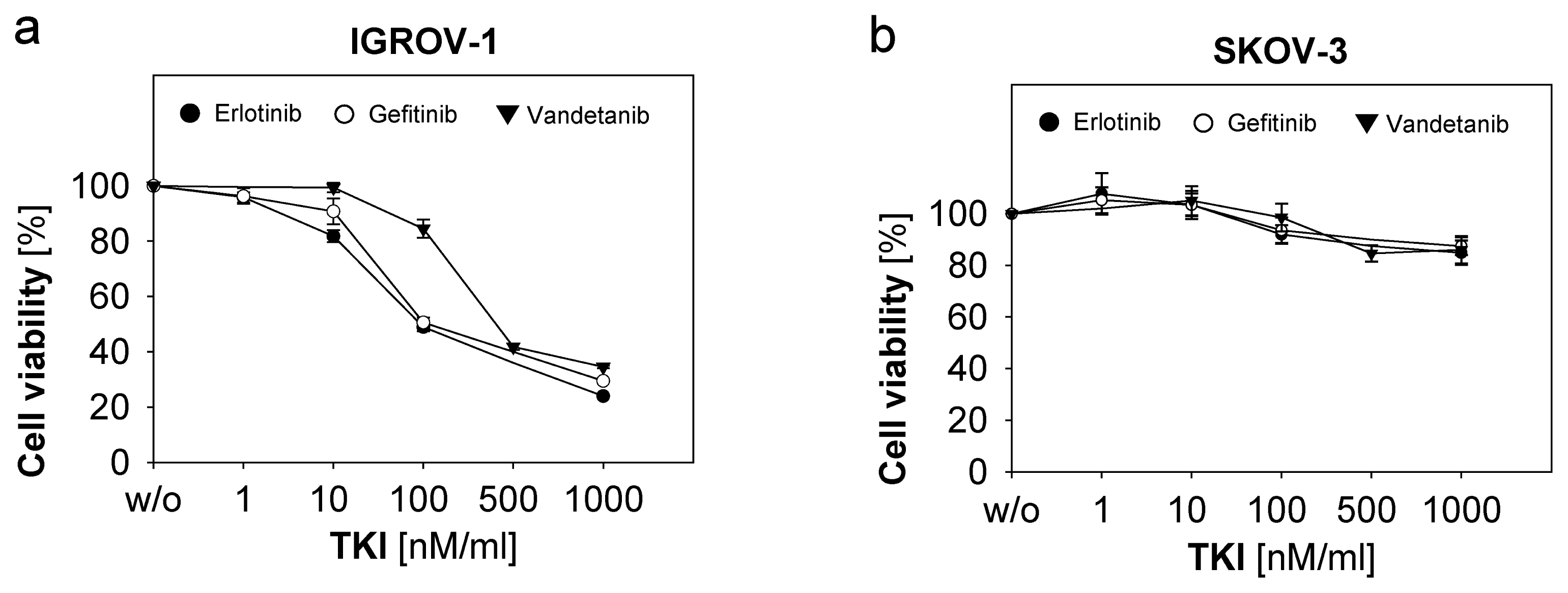
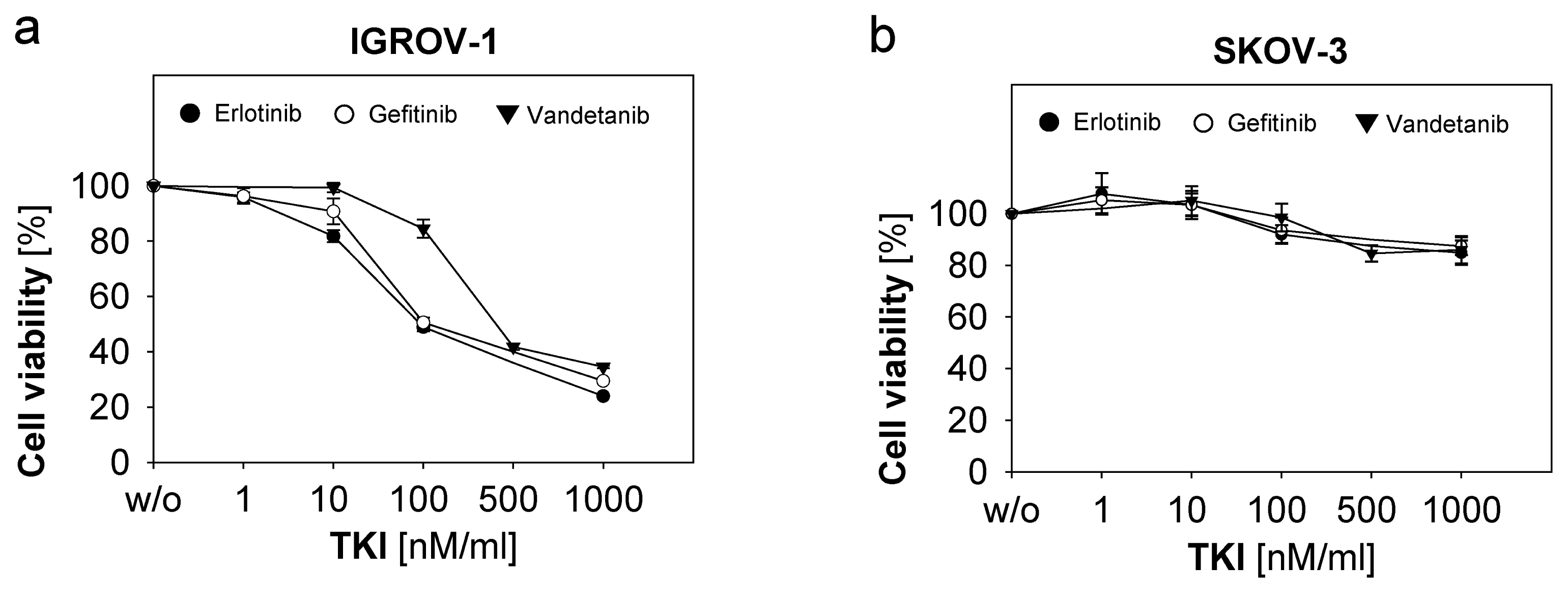
2.2. Response of Ovarian Cancer Cells to the Anti-EGFR-Tyrosine Kinase Inhibitors (TKI)
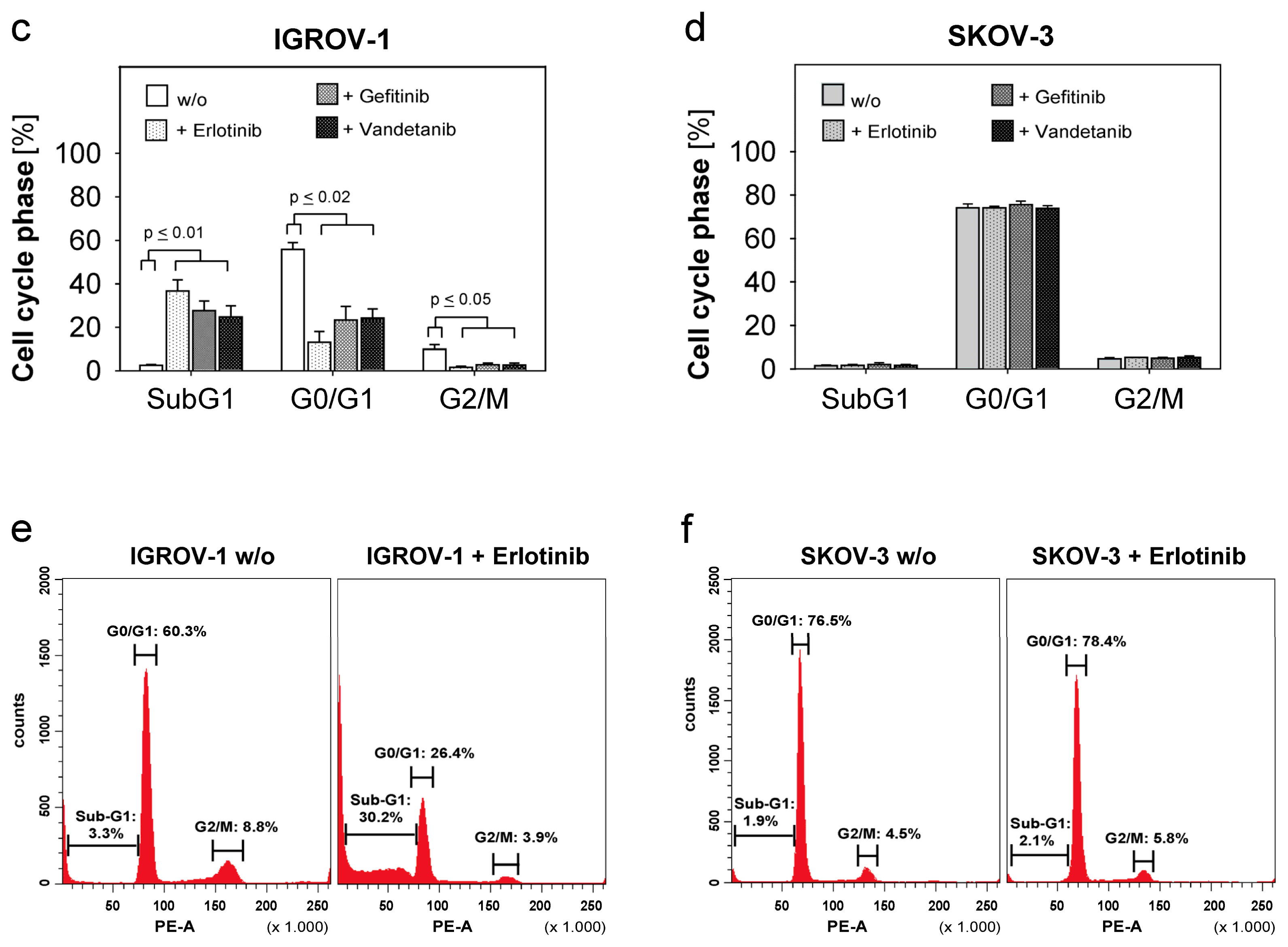
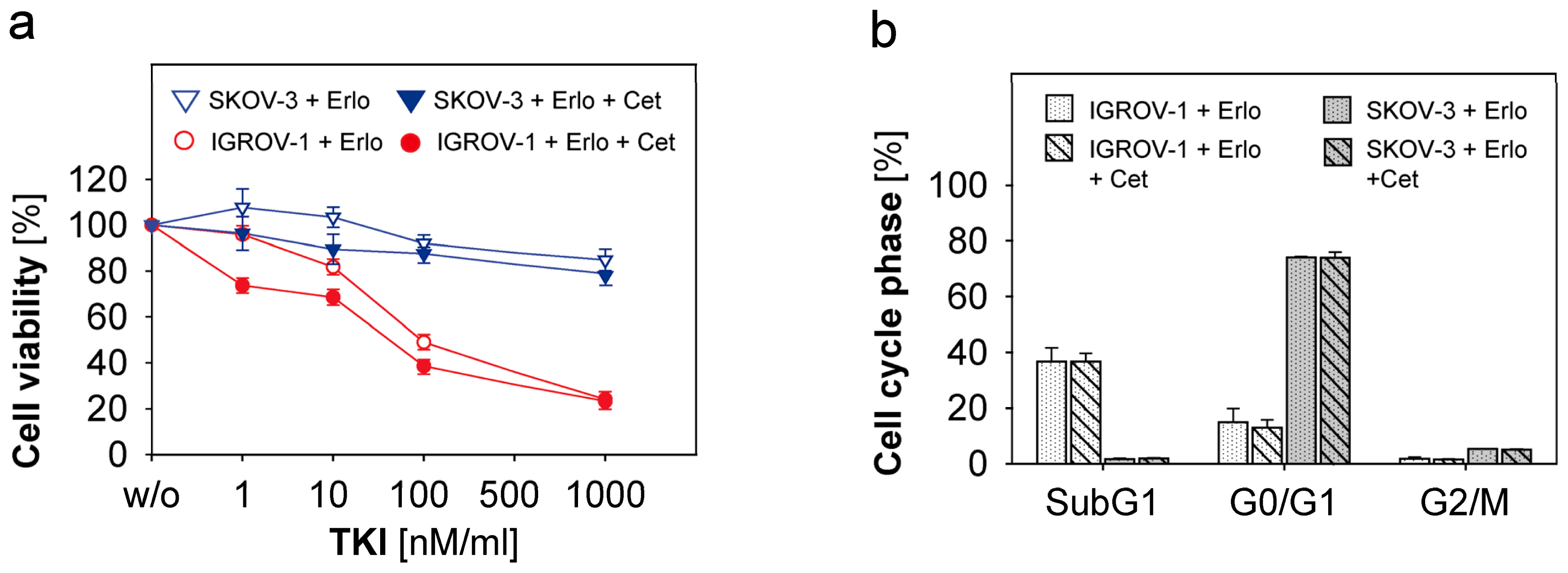
2.3. Combined Use of Anti-EGFR-Tyrosine Kinase Inhibitors (TKI) and Anti-EGFR-Antibody Cetuximab in Ovarian Cancer Cells
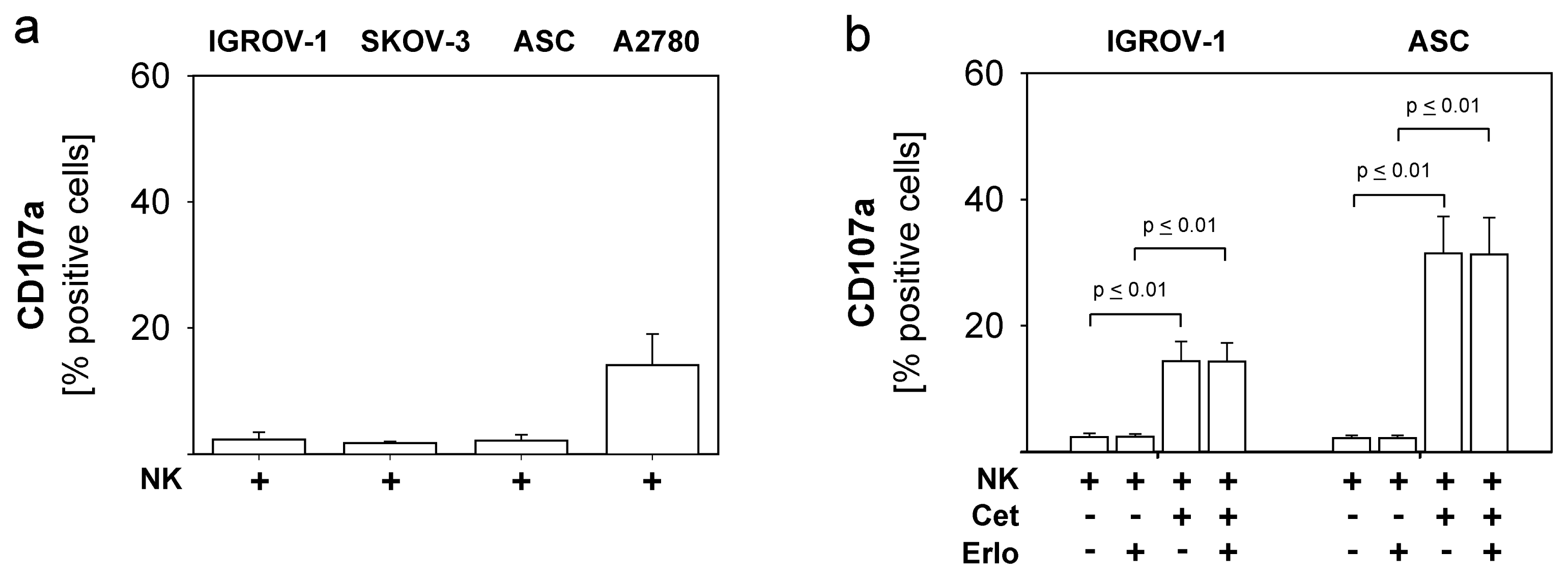
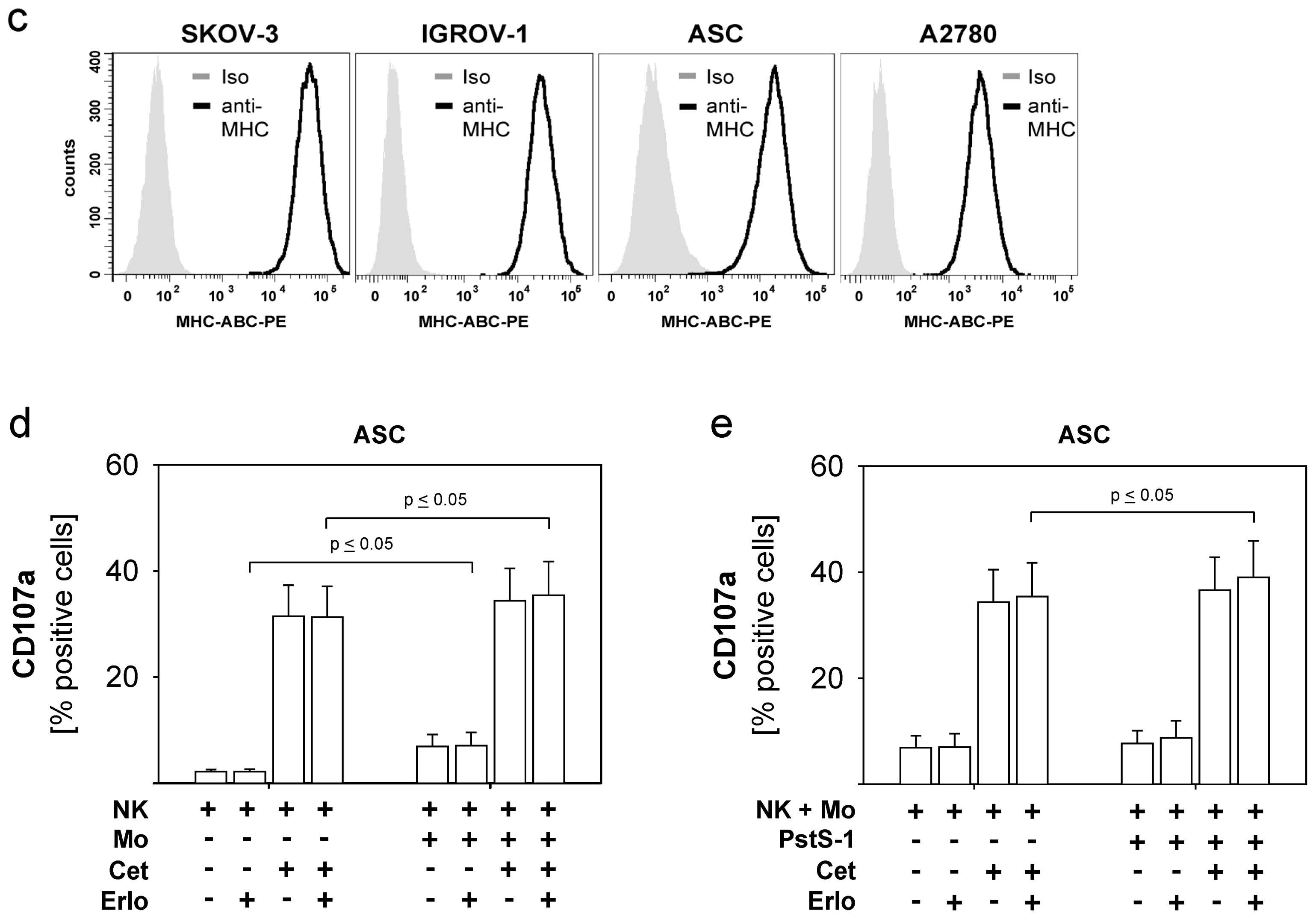
2.4. Combined Use of TKI, Cetuximab, Immune Effector Cells and Immunostimulatory Bacterial PstS-1
3. Experimental Section
3.1. Cell lines and Cell Culture
3.2. MTT-Proliferation-Assay
3.3. Isolation of NK Cells and Monocytes from PBMC’s of Healthy Donors
3.4. Stimulation of Purified NK Cells and Monocytes with PstS-1
3.5. Flow Cytometric Analysis (FACS)
3.6. CD107a Degranulation Assay and ADCC
3.7. Flow Cytometric DNA-Staining and Cell Cycle Analysis
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
- Conflict of InterestsThe authors declare no conflict of interest.
References
- Howlader, N.A.; Krapcho, M.; Neyman, N.; Aminou, R.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; Cho, H.; Mariotto, A.; et al. Seer cancer statistics review, 1975–2009 (vintage 2009 populations). Available online: http://seer.cancer.gov/csr/1975_2009_pops09 accessed on 11 September 2012.
- Bartlett, J.M.; Langdon, S.P.; Simpson, B.J.; Stewart, M.; Katsaros, D.; Sismondi, P.; Love, S.; Scott, W.N.; Williams, A.R.; Lessells, A.M.; et al. The prognostic value of epidermal growth factor receptor mRNA expression in primary ovarian cancer. Br. J. Cancer 1996, 73, 301–306. [Google Scholar]
- Fischer-Colbrie, J.; Witt, A.; Heinzl, H.; Speiser, P.; Czerwenka, K.; Sevelda, P.; Zeillinger, R. EGFR and steroid receptors in ovarian carcinoma: Comparison with prognostic parameters and outcome of patients. Anticancer Res 1997, 17, 613–619. [Google Scholar]
- Mendelsohn, J.; Baselga, J. Status of epidermal growth factor receptor antagonists in the biology and treatment of cancer. J. Clin. Oncol 2003, 21, 2787–2799. [Google Scholar]
- Bijman, M.N.; van Berkel, M.P.; Kok, M.; Janmaat, M.L.; Boven, E. Inhibition of functional HER family members increases the sensitivity to docetaxel in human ovarian cancer cell lines. AntiCancer Drugs 2009, 20, 450–460. [Google Scholar]
- Konner, J.; Schilder, R.J.; DeRosa, F.A.; Gerst, S.R.; Tew, W.P.; Sabbatini, P.J.; Hensley, M.L.; Spriggs, D.R.; Aghajanian, C.A. A phase II study of cetuximab/paclitaxel/carboplatin for the initial treatment of advanced-stage ovarian, primary peritoneal, or fallopian tube cancer. Gynecol. Oncol 2008, 110, 140–145. [Google Scholar]
- Secord, A.A.; Blessing, J.A.; Armstrong, D.K.; Rodgers, W.H.; Miner, Z.; Barnes, M.N.; Lewandowski, G.; Mannel, R.S. Gynecologic Oncology Group. Phase II trial of cetuximab and carboplatin in relapsed platinum-sensitive ovarian cancer and evaluation of epidermal growth factor receptor expression: A gynecologic oncology group study. Gynecol. Oncol 2008, 108, 493–499. [Google Scholar]
- Schilder, R.J.; Pathak, H.B.; Lokshin, A.E.; Holloway, R.W.; Alvarez, R.D.; Aghajanian, C.; Min, H.; Devarajan, K.; Ross, E.; Drescher, C.W.; et al. Phase II trial of single agent cetuximab in patients with persistent or recurrent epithelial ovarian or primary peritoneal carcinoma with the potential for dose escalation to rash. Gynecol. Oncol 2009, 113, 21–27. [Google Scholar]
- Gordon, A.N.; Finkler, N.; Edwards, R.P.; Garcia, A.A.; Crozier, M.; Irwin, D.H.; Barrett, E. Efficacy and safety of erlotinib HCl, an epidermal growth factor receptor (HER-1/EGFR) tyrosine kinase inhibitor, in patients with advanced ovarian carcinoma: Results from a phase II multicenter study. Int. J. Gynecol. Cancer 2005, 15, 785–792. [Google Scholar]
- Blank, S.V.; Christos, P.; Curtin, J.P.; Goldman, N.; Runowicz, C.D.; Sparano, J.A.; Liebes, L.; Chen, H.X.; Muggia, F.M. Erlotinib added to carboplatin and paclitaxel as first-line treatment of ovarian cancer: A phase II study based on surgical reassessment. Gynecol. Oncol 2010, 119, 451–456. [Google Scholar]
- Vasey, P.A.; Gore, M.; Wilson, R.; Rustin, G.; Gabra, H.; Guastalla, J.P.; Lauraine, E.P.; Paul, J.; Carty, K.; Kaye, S. Scottish Gynecological Cancer Trials Group. A phase Ib trial of docetaxel, carboplatin and erlotinib in ovarian, fallopian tube and primary peritoneal cancers. Br. J. Cancer 2008, 98, 1774–1780. [Google Scholar]
- Chambers, S.K.; Clouser, M.C.; Baker, A.F.; Roe, D.J.; Cui, H.; Brewer, M.A.; Hatch, K.D.; Gordon, M.S.; Janicek, M.F.; Isaacs, J.D.; et al. Overexpression of tumour vascular endothelial growth factor A may portend an increased likelihood of progression in a phase II trial of bevacizumab and erlotinib in resistant ovarian cancer. Clin. Cancer Res 2010, 16, 5320–5328. [Google Scholar]
- Schilder, R.J.; Sill, M.W.; Chen, X.; Darcy, K.M.; Decesare, S.L.; Lewandowski, G.; Lee, R.B.; Arciero, C.A.; Wu, H.; Godwin, A.K. Phase II study of gefitinib in patients with relapsed or persistent ovarian or primary peritoneal carcinoma and evaluation of epidermal growth factor receptor mutations and immunohistochemical expression: A Gynecologic Oncology Group study. Clin. Cancer Res 2005, 11, 5539–5548. [Google Scholar]
- Posadas, E.M.; Liel, M.S.; Kwitkowski, V.; Minasian, L.; Godwin, A.K.; Hussain, M.M.; Espina, V.; Wood, B.J.; Steinberg, S.M.; Kohn, E.C. A phase II and pharmacodynamic study of gefitinib in patients with refractory or recurrent epithelial ovarian cancer. Cancer 2007, 109, 1323–1330. [Google Scholar]
- Ciardiello, F.; Caputo, R.; Bianco, R.; Damiano, V.; Pomatico, G.; de Placido, S.; Bianco, A.R.; Tortora, G. Antitumour effect and potentiation of cytotoxic drugs activity in human cancer cells by ZD-1839 (Iressa), an epidermal growth factor receptor-selective tyrosine kinase inhibitor. Clin. Cancer Res 2000, 6, 2053–2063. [Google Scholar]
- Pautier, P.; Joly, F.; Kerbrat, P.; Bougnoux, P.; Fumoleau, P.; Petit, T.; Rixe, O.; Ringeisen, F.; Carrasco, A.T.; Lhomme, C. Phase II study of gefitinib in combination with paclitaxel (p) and carboplatin (c) as second-line therapy for ovarian, tubal or peritoneal adenocarcinoma (1839IL/0074). Gynecol. Oncol 2010, 116, 157–162. [Google Scholar]
- Annunziata, C.M.; Walker, A.J.; Minasian, L.; Yu, M.; Kotz, H.; Wood, B.J.; Calvo, K.; Choyke, P.; Kimm, D.; Steinberg, S.M.; et al. Vandetanib, designed to inhibit VEGFR2 and EGFR signaling, had no clinical activity as monotherapy for recurrent ovarian cancer and no detectable modulation of VEGFR2. Clin. Cancer Res 2010, 16, 664–672. [Google Scholar]
- Zhang, W.; Gordon, M.; Schultheis, A.M.; Yang, D.Y.; Nagashima, F.; Azuma, M.; Chang, H.M.; Borucka, E.; Lurje, G.; Sherrod, A.E.; et al. FCGR2a and FCGR3a polymorphisms associated with clinical outcome of epidermal growth factor receptor expressing metastatic colorectal cancer patients treated with single-agent cetuximab. J. Clin. Oncol 2007, 25, 3712–3718. [Google Scholar]
- Screpanti, V.; Wallin, R.P.; Grandien, A.; Ljunggren, H.G. Impact of FasL-induced apoptosis in the elimination of tumour cells by NK cells. Mol. Immunol 2005, 42, 495–499. [Google Scholar]
- Salih, J.; Hilpert, J.; Placke, T.; Grunebach, F.; Steinle, A.; Salih, H.R.; Krusch, M. The Bcr/abl-inhibitors imatinib, nilotinib and dasatinib differentially affect NK cell reactivity. Int. J. Cancer 2010, 127, 2119–2128. [Google Scholar]
- Lamm, D.L.; Blumenstein, B.A.; Crawford, E.D.; Montie, J.E.; Scardino, P.; Grossman, H.B.; Stanisic, T.H.; Smith, J.A., Jr; Sullivan, J.; Sarosdy, M.F.; et al. A randomized trial of intravesical doxorubicin and immunotherapy with bacille calmette-guerin for transitional-cell carcinoma of the bladder. N. Engl. J. Med. 1991, 325, 1205–1209. [Google Scholar]
- Brandau, S.; Riemensberger, J.; Jacobsen, M.; Kemp, D.; Zhao, W.; Zhao, X.; Jocham, D.; Ratliff, T.L.; Bohle, A. NK cells are essential for effective BCG immunotherapy. Int. J. Cancer 2001, 92, 697–702. [Google Scholar]
- Suttmann, H.; Jacobsen, M.; Reiss, K.; Jocham, D.; Bohle, A.; Brandau, S. Mechanisms of bacillus calmette-guerin mediated natural killer cell activation. J. Urol 2004, 172, 1490–1495. [Google Scholar]
- Singh, M.; Andersen, A.B.; McCarthy, J.E.; Rohde, M.; Schutte, H.; Sanders, E.; Timmis, K.N. The mycobacterium tuberculosis 38-kDa antigen: Overproduction in escherichia coli, purification and characterization. Gene 1992, 117, 53–60. [Google Scholar]
- Rodriguez, A.; Troye-Blomberg, M.; Lindroth, K.; Ivanyi, J.; Singh, M.; Fernandez, C. B- and T-cell responses to the mycobacterium surface antigen PstS-1 in the respiratory tract and adjacent tissues. Role of adjuvants and routes of immunization. Vaccine 2003, 21, 458–467. [Google Scholar]
- Sanger, C.; Busche, A.; Bentien, G.; Spallek, R.; Jonas, F.; Bohle, A.; Singh, M.; Brandau, S. Immunodominant PstS-1 antigen of mycobacterium tuberculosis is a potent biological response modifier for the treatment of bladder cancer. BMC Cancer 2004, 4, 86. [Google Scholar]
- Jung, S.B.; Yang, C.S.; Lee, J.S.; Shin, A.R.; Jung, S.S.; Son, J.W.; Harding, C.V.; Kim, H.J.; Park, J.K.; Paik, T.H.; et al. The mycobacterial 38-kilodalton glycolipoprotein antigen activates the mitogen-activated protein kinase pathway and release of proinflammatory cytokines through Toll-like receptors 2 and 4 in human monocytes. Infect. Immun 2006, 74, 2686–2696. [Google Scholar]
- Sanchez, A.; Espinosa, P.; Esparza, M.A.; Colon, M.; Bernal, G.; Mancilla, R. Mycobacterium tuberculosis 38-kda lipoprotein is apoptogenic for human monocyte-derived macrophages. Scand. J. Immunol 2009, 69, 20–28. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar]
- Riccardi, C.; Nicoletti, I. Analysis of apoptosis by propidium iodide staining and flow cytometry. Nature Protoc 2006, 1, 1458–1461. [Google Scholar]
- Prewett, M.; Rockwell, P.; Rose, C.; Goldstein, N. Anti-tumour and cell cycle responses in KB cells treated with a chimeric anti-EGFR monoclonal antibody in combination with cisplatin. Int. J. Oncol 1996, 9, 217–224. [Google Scholar]
- Karapetis, C.S.; Khambata-Ford, S.; Jonker, D.J.; O’Callaghan, C.J.; Tu, D.; Tebbutt, N.C.; Simes, R.J.; Chalchal, H.; Shapiro, J.D.; Robitaille, S.; et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N. Engl. J. Med 2008, 359, 1757–1765. [Google Scholar]
- Wheeler, D.L.; Huang, S.; Kruser, T.J.; Nechrebecki, M.M.; Armstrong, E.A.; Benavente, S.; Gondi, V.; Hsu, K.T.; Harari, P.M. Mechanisms of acquired resistance to cetuximab: Role of HER (ErbB) family members. Oncogene 2008, 27, 3944–3956. [Google Scholar]
- Grunt, T.W.; Wagner, R.; Grusch, M.; Berger, W.; Singer, C.F.; Marian, B.; Zielinski, C.C.; Lupu, R. Interaction between fatty acid synthase- and ErbB-systems in ovarian cancer cells. Biochem. Biophys. Res. Commun 2009, 385, 454–459. [Google Scholar]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med 2004, 350, 2129–2139. [Google Scholar]
- Bull Phelps, S.L.; Schorge, J.O.; Peyton, M.J.; Shigematsu, H.; Xiang, L.L.; Miller, D.S.; Lea, J.S. Implications of EGFR inhibition in ovarian cancer cell proliferation. Gynecol. Oncol 2008, 109, 411–417. [Google Scholar]
- Tanaka, Y.; Terai, Y.; Tanabe, A.; Sasaki, H.; Sekijima, T.; Fujiwara, S.; Yamashita, Y.; Kanemura, M.; Ueda, M.; Sugita, M.; et al. Prognostic effect of epidermal growth factor receptor gene mutations and the aberrant phosphorylation of Akt and ERK in ovarian cancer. Cancer Biol. Ther 2011, 11, 50–57. [Google Scholar]
- Huether, A.; Hopfner, M.; Baradari, V.; Schuppan, D.; Scherubl, H. EGFR blockade by cetuximab alone or as combination therapy for growth control of hepatocellular cancer. Biochem. Pharmacol 2005, 70, 1568–1578. [Google Scholar]
- Huang, S.; Armstrong, E.A.; Benavente, S.; Chinnaiyan, P.; Harari, P.M. Dual-agent molecular targeting of the epidermal growth factor receptor (EGFR): Combining anti-EGFR antibody with tyrosine kinase inhibitor. Cancer Res 2004, 64, 5355–5362. [Google Scholar]
- Janjigian, Y.Y.; Azzoli, C.G.; Krug, L.M.; Pereira, L.K.; Rizvi, N.A.; Pietanza, M.C.; Kris, M.G.; Ginsberg, M.S.; Pao, W.; Miller, V.A.; et al. Phase I/II trial of cetuximab and erlotinib in patients with lung adenocarcinoma and acquired resistance to erlotinib. Clin. Cancer Res 2011, 17, 2521–2527. [Google Scholar]
- Weickhardt, A.J.; Price, T.J.; Chong, G.; Gebski, V.; Pavlakis, N.; Johns, T.G.; Azad, A.; Skrinos, E.; Fluck, K.; Dobrovic, A.; et al. Dual targeting of the epidermal growth factor receptor using the combination of cetuximab and erlotinib: Preclinical evaluation and results of the phase II DUX study in chemotherapy-refractory, advanced colorectal cancer. J. Clin. Oncol 2012, 30, 1505–1512. [Google Scholar]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar]

© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gottschalk, N.; Kimmig, R.; Lang, S.; Singh, M.; Brandau, S. Anti-Epidermal Growth Factor Receptor (EGFR) Antibodies Overcome Resistance of Ovarian Cancer Cells to Targeted Therapy and Natural Cytotoxicity. Int. J. Mol. Sci. 2012, 13, 12000-12016. https://doi.org/10.3390/ijms130912000
Gottschalk N, Kimmig R, Lang S, Singh M, Brandau S. Anti-Epidermal Growth Factor Receptor (EGFR) Antibodies Overcome Resistance of Ovarian Cancer Cells to Targeted Therapy and Natural Cytotoxicity. International Journal of Molecular Sciences. 2012; 13(9):12000-12016. https://doi.org/10.3390/ijms130912000
Chicago/Turabian StyleGottschalk, Nina, Rainer Kimmig, Stephan Lang, Mahavir Singh, and Sven Brandau. 2012. "Anti-Epidermal Growth Factor Receptor (EGFR) Antibodies Overcome Resistance of Ovarian Cancer Cells to Targeted Therapy and Natural Cytotoxicity" International Journal of Molecular Sciences 13, no. 9: 12000-12016. https://doi.org/10.3390/ijms130912000