The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Use of AMPs in Preventing Multidrug-Resistant Bacteria
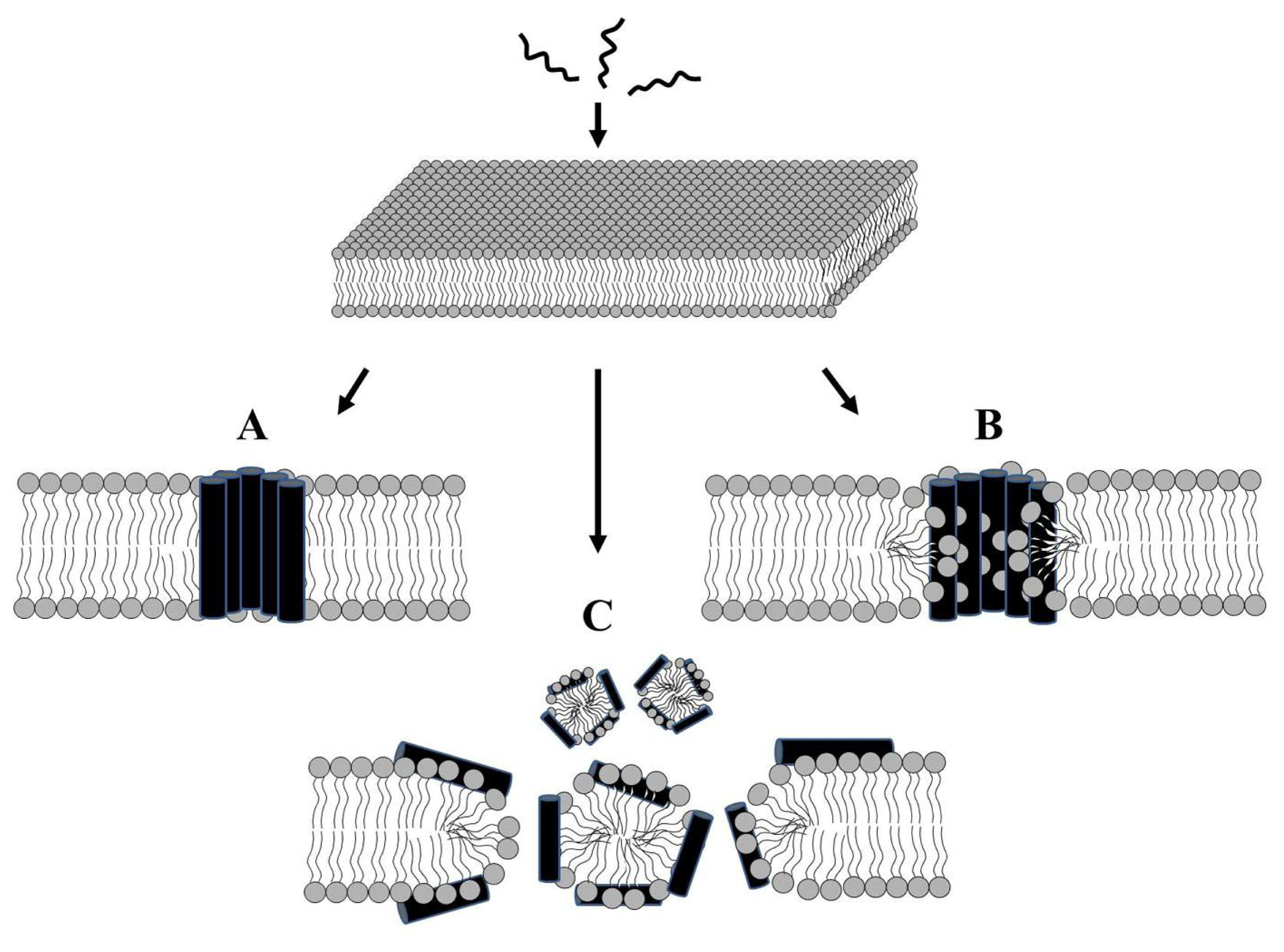
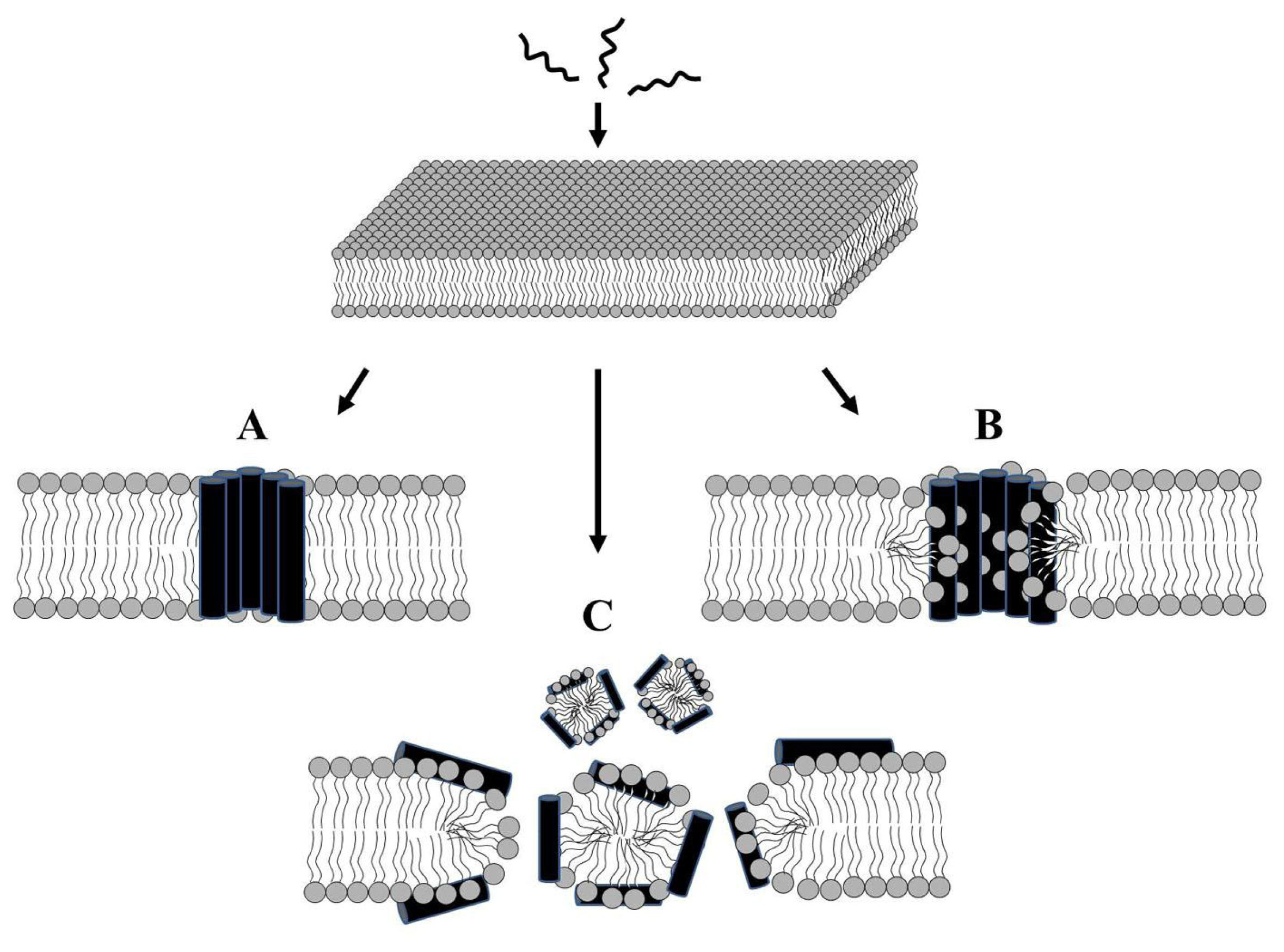
2.1. Modes of Antibacterial Action
2.1.1. Lipopolysaccharide (LPS) Neutralization or Disaggregation by Antimicrobial Peptides
2.2. Cell Wall-Lipid II
2.3. Alteration of Membrane Potential or Induction of Membrane Permeabilization
2.4. Inhibition of Cytoplasmic Proteins Related to Cell Division or Survival
2.5. Inhibition of Macromolecular Synthesis through Interaction with Nucleic Acids
3. Synergetic Effects between Antimicrobial Peptides and Clinically used Antibiotics
4. In Vivo Application of Antimicrobial Peptides
5. Clinical Development of Antimicrobial Peptides
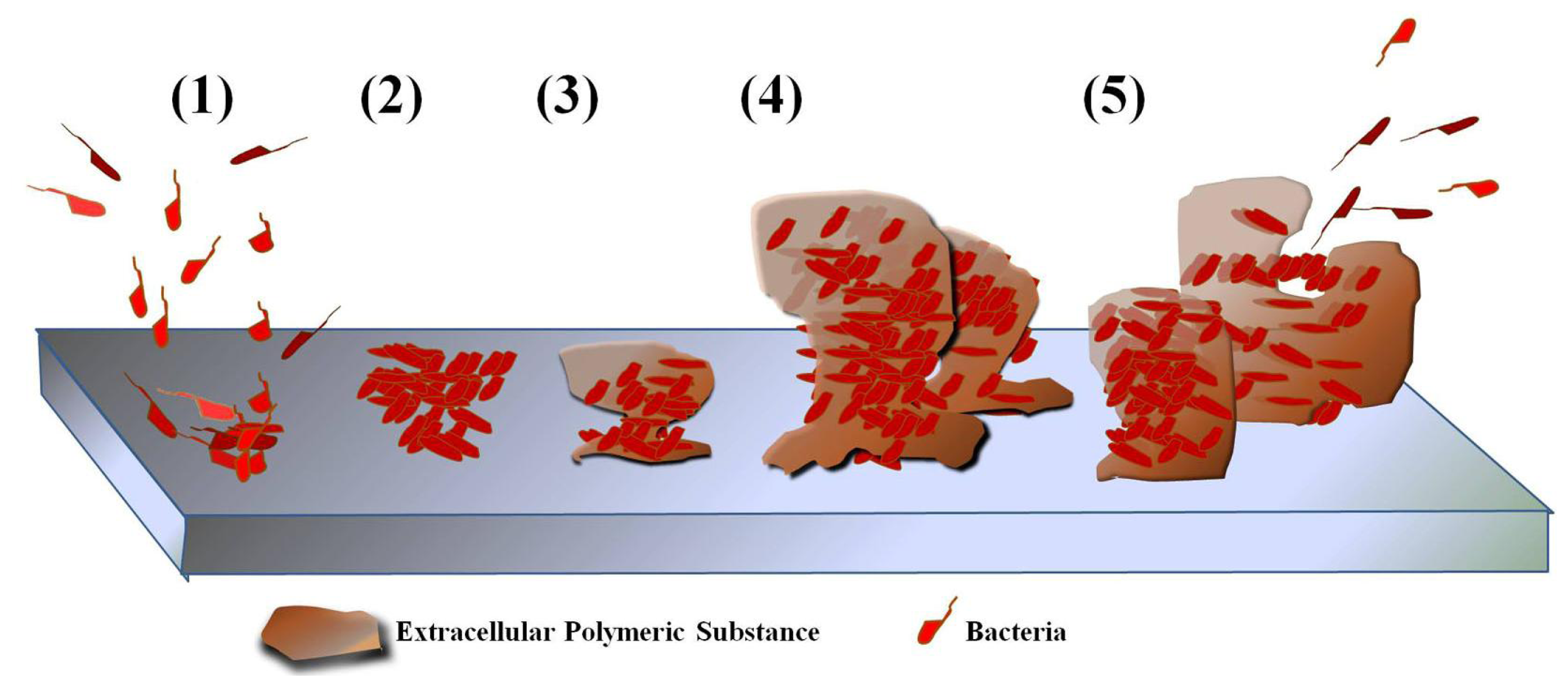
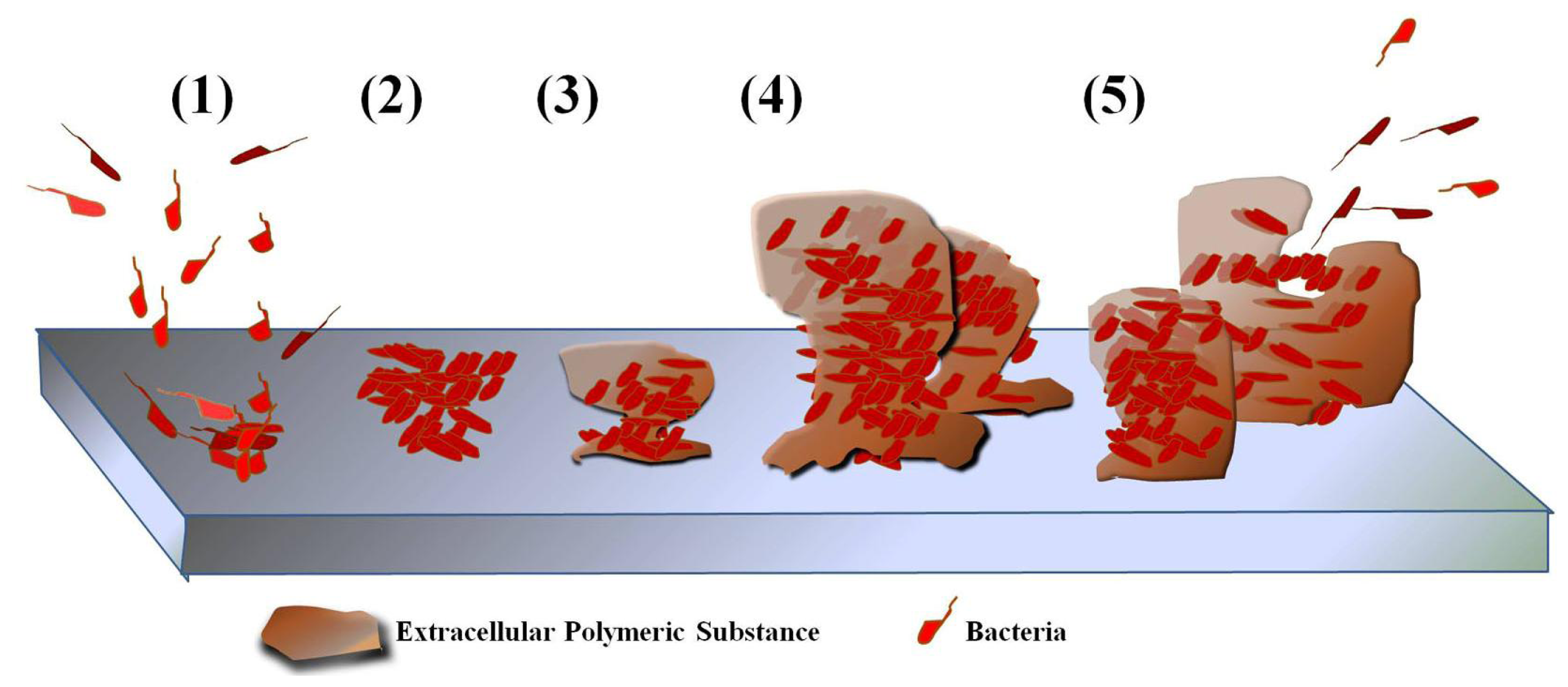
6. Use of AMPs in Preventing Biofilm
6.1. Biofilm Formation
6.2. Applications to Prevent or Remove Biofilms
6.2.1. In Vitro Anti-Biofilm Activity of Antimicrobial Peptides against Biofilm of MDR Bacteria
6.2.2. Anti-Biofilm Activity in Medical Devices
6.2.3. Anti-Biofilm Activity against Oral Plaque
6.2.4. Others
8. Conclusions
Acknowledgements
References
- Klein, E; Smith, DL; Laxminarayan, R. Hospitalizations and deaths caused by methicillin-resistant Staphylococcus aureus, United States, 1999–2005. Emerg Infect Dis 2007, 13, 1840–1846. [Google Scholar]
- Klevens, RM; Edwards, JR; Richards, CL, Jr; Horan, TC; Gaynes, RP; Pollock, DA; Cardo, DM. Estimating health care-associated infections and deaths in U.S. hospitals, 2002. Public Health Rep 2007, 122, 160–166. [Google Scholar]
- Johnson, AP; Pearson, A; Duckworth, G. Surveillance and epidemiology of MRSA bacteraemia in the UK. J Antimicrob Chemother 2005, 56, 455–462. [Google Scholar]
- Monroe, D. Looking for chinks in the armor of bacterial biofilms. PLoS Biol 2007, 5, e307. [Google Scholar]
- Hall-Stoodley, L; Costerton, JW; Stoodley, P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2004, 2, 95–108. [Google Scholar]
- Stewart, PS; Costerton, JW. Antibiotic resistance of bacteria in biofilms. Lancet 2001, 358, 135–138. [Google Scholar]
- Stamm, WE. Catheter-associated urinary tract infections: Epidemiology, pathogenesis, and prevention. Am J Med 1991, 91, 65S–71S. [Google Scholar]
- Nickel, JC; Costerton, JW; McLean, RJ; Olson, M. Bacterial biofilms: influence on the pathogenesis, diagnosis and treatment of urinary tract infections. J Antimicrob Chemother 1994, 33, 31–41. [Google Scholar]
- Hojo, K; Nagaoka, S; Ohshima, T; Maeda, N. Bacterial interactions in dental biofilm development. J Dent Res 2009, 88, 982–990. [Google Scholar]
- O’Grady, NP; Aexander, M; Dellinger, EP; Gerberding, JL; Heard, SO; Maki, DG; Masur, H; McCormick, RD; Mermel, LA; Pearson, ML; et al. Guidelines for the prevention of intravascular catheter-related infections. Centers for disease control and prevention. MMWR Recomm Rep 2002, 51, 1–29. [Google Scholar]
- Zasloff, M. Antimicrobial peptides of multicellular organisms. Nature 2002, 415, 389–395. [Google Scholar]
- Chan, DI; Prenner, EJ; Vogel, HJ. Tryptophan- and arginine-rich antimicrobial peptides: structures and mechanisms of action. Biochim Biophys Acta 2006, 1758, 1184–1202. [Google Scholar]
- Oppenheim, JJ; Biragyn, A; Kwak, LW; Yang, D. Roles of antimicrobial peptides such as defensins in innate and adaptive immunity. Ann Rheum Dis 2003, 62, ii17–21. [Google Scholar]
- Steiner, H; Hultmark, D; Engström, A; Bennich, H; Boman, HG. Sequence and specificity of two antibacterial proteins involved in insect immunity. Nature 1981, 292, 246–248. [Google Scholar]
- Gibson, BW; Poulter, L; Williams, DH. A mass spectrometric assay for novel peptides: Application to Xenopus laevis skin secretions. Peptides 1985, 6, 23–27. [Google Scholar]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc Natl Acad Sci USA 1987, 84, 5449–5453. [Google Scholar]
- Yeaman, MR; Yount, NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol Rev 2003, 55, 27–55. [Google Scholar]
- Brogden, KA. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 2005, 3, 238–250. [Google Scholar]
- Pieters, RJ; Arnusch, CJ; Breukink, E. Membrane permeabilization by multivalent anti-microbial peptides. Protein Pept Lett 2009, 16, 736–742. [Google Scholar]
- Nicias, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J 2009, 276, 6483–6496. [Google Scholar]
- Friedrich, C; Scott, MG; Karunaratne, N; Yan, H; Hancock, RE. Salt-resistant alpha-helical cationic antimicrobial peptides. Antimicrob Agents Chemother 1999, 43, 1542–1548. [Google Scholar]
- Mukhopadhyay, K; Whitmire, W; Xiong, YQ; Molden, J; Jones, T; Peschel, A; Staubitz, P; Adler-Moore, J; McNamara, PJ; Proctor, RA; Yeaman, MR; Bayer, AS. In vitro susceptibility of Staphylococcus aureus to thrombin-induced platelet microbicidal protein-1 (tPMP-1) is influenced by cell membrane phospholipid composition and asymmetry. Microbiology 2007, 153, 1187–1197. [Google Scholar]
- Thwaite, JE; Hibbs, S; Titball, RW; Atkins, TP. Proteolytic degradation of human antimicrobial peptide LL-37 by Bacillus anthracis may contribute to virulence. Antimicrob Agents Chemother 2006, 50, 2316–2322. [Google Scholar]
- McGillivray, SM; Ebrahimi, CM; Fisher, N; Sabet, M; Zhang, DX; Chen, Y; Haste, NM; Aroian, RV; Gallo, RL; Guiney, DG; Friedlander, AM; Koehler, TM; Nizet, V. ClpX contributes to innate defense peptide resistance and virulence phenotypes of Bacillus anthracis. J Innate Immun 2009, 1, 494–506. [Google Scholar]
- Giacometti, A; Cirioni, O; Kamysz, W; D’Amato, G; Silvestri, C; Simona Del Prete, M; Lukasiak, J; Scalise, G. In vitro activity and killing effect of the synthetic hybrid cecropin A-melittin peptide CA(1-7)M(2-9)NH(2) on methicillin-resistant nosocomial isolates of Staphylococcus aureus and interactions with clinically used antibiotics. Diagn Microbiol Infect Dis 2004, 49, 197–200. [Google Scholar]
- Park, Y; Park, SN; Park, SC; Shin, SO; Kim, JY; Kang, SJ; Kim, MH; Jeong, CY; Hahm, KS. Synergism of Leu-Lys rich antimicrobial peptides and chloramphenicol against bacterial cells. Biochim Biophys Acta 2006, 1764, 24–32. [Google Scholar]
- Park, Y; Kim, HJ; Hahm, KS. Antibacterial synergism of novel antibiotic peptides with chloramphenicol. Biochem Biophys Res Commun 2004, 321, 109–115. [Google Scholar]
- Gutsmann, T; Hagge, SO; David, A; Roes, S; Böhling, A; Hammer, MU; Seydel, U. Lipid-mediated resistance of Gram-negative bacteria against various pore-forming antimicrobial peptides. J Endotoxin Res 2005, 11, 167–173. [Google Scholar]
- Gee, K; Kozlowski, M; Kumar, A. Tumor necrosis factor-alpha induces functionally active hyaluronan-adhesive CD44 by activating sialidase through p38 mitogen-activated protein kinase in lipopolysaccharide-stimulated human monocytic cells. J Biol Chem 2003, 278, 37275–37287. [Google Scholar]
- Blanqué, R; Meakin, C; Millet, S; Gardner, CR. Hypothermia as an indicator of the acute effects of lipopolysaccharides: Comparison with serum levels of IL1 beta, IL6 and TNF alpha. Gen Pharmacol 1996, 27, 973–977. [Google Scholar]
- Freudenberg, MA; Tchaptchet, S; Keck, S; Fejer, G; Huber, M; Schütze, N; Beutler, B; Galanos, C. Lipopolysaccharide sensing an important factor in the innate immune response to Gram-negative bacterial infections: Benefits and hazards of LPS hypersensitivity. Immunobiology 2008, 213, 193–203. [Google Scholar]
- Li, P; Wohland, T; Ho, B; Ding, JL. Perturbation of Lipopolysaccharide (LPS) Micelles by Sushi 3 (S3) antimicrobial peptide. The importance of an intermolecular disulfide bond in S3 dimer for binding, disruption, and neutralization of LPS. J Biol Chem 2004, 279, 50150–50156. [Google Scholar]
- Bhattacharjya, S; Domadia, PN; Bhunia, A; Malladi, S; David, SA. High-resolution solution structure of a designed peptide bound to lipopolysaccharide: transferred nuclear overhauser effects, micelle selectivity, and anti-endotoxic activity. Biochemistry 2007, 46, 5864–5874. [Google Scholar]
- Storm, DR; Rosental, KS; Swanson, PE. Polymyxin and related peptide antibiotics. Annu Rev Biochem 1977, 46, 723–763. [Google Scholar]
- Kwa, A; Kasiakou, SK; Tam, VH; Falagas, ME. Polymyxin B: Similarities to and differences from colistin (polymyxin E). Expert Rev Anti-Infect Ther 2007, 5, 811–821. [Google Scholar]
- Mogi, T; Kita, K. Gramicidin S and polymyxins: The revival of cationic cyclic peptide antibiotics. Cell Mol Life Sci 2007, 66, 3821–3826. [Google Scholar]
- Ding, JL; Li, P; Ho, B. The Sushi peptides: structural characterization and mode of action against Gram-negative bacteria. Cell Mol Life Sci 2008, 65, 1202–1219. [Google Scholar]
- Kim, JY; Park, SC; Yoon, MY; Hahm, KS; Park, Y. C-terminal amidation of PMAP-23: Translocation to the inner membrane of Gram-negative bacteria. Amino Acids 2011, 40, 183–195. [Google Scholar]
- Gough, M; Hancock, RE; Kelly, NM. Antiendotoxin activity of cationic peptide antimicrobial agents. Infect Immun 1996, 64, 4922–4927. [Google Scholar]
- Scott, MG; Yan, H; Hancock, RE. Biological properties of structurally related alpha-helical cationic antimicrobial peptides. Infect Immun 1999, 67, 2005–2009. [Google Scholar]
- Giacometti, A; Cirioni, O; Ghiselli, R; Mocchegiani, F; Del Prete, MS; Viticchi, C; Kamysz, W; Lempicka, E; Saba, V; Scalise, G. Potential therapeutic role of cationic peptides in three experimental models of septic shock. Antimicrob Agents Chemother 2002, 46, 2132–2136. [Google Scholar]
- Guo, L; Lim, KB; Poduje, CM; Daniel, M; Gunn, JS; Hackett, M; Miller, SI. Lipid A acylation and bacterial resistance against vertebrate antimicrobial peptides. Cell 1998, 95, 189–198. [Google Scholar]
- Zhou, Z; Lin, S; Cotter, RJ; Raetz, CR. Lipid A modifications characteristic of Salmonella typhimurium are induced by NH4VO3 in Escherichia coli K12. Detection of 4-amino-4-deoxy-l-arabinose, phosphoethanolamine and palmitate. J Biol Chem 1999, 274, 18503–18514. [Google Scholar]
- Ernst, RK; Guina, T; Miller, SI. How intracellular bacteria survive: surface modifications that promote resistance to host innate immune responses. J Infect Dis 1999, 179, S326–S330. [Google Scholar]
- Glaser, L. Bacterial cell surface polysaccharides. Annu Rev Biochem 1973, 42, 91–112. [Google Scholar]
- Di Guilmi, AM; Dessen, A; Dideberg, O; Vernet, T. Bifunctional penicillin-binding proteins: Focus on the glycosyltransferase domain and its specific inhibitor moenomycin. Curr Pharm Biotechnol 2002, 3, 63–75. [Google Scholar]
- Wilke, MS; Lovering, AL; Strynadka, NC. Beta-lactam antibiotic resistance: a current structural perspective. Curr Opin Microbiol 2005, 8, 525–533. [Google Scholar]
- Brakstad, OG; Maeland, JA. Mechanisms of methicillin resistance in staphylococci. APMIS 1997, 105, 264–276. [Google Scholar]
- Berger-Bächi, B; Rohrer, S. Factors influencing methicillin resistance in staphylococci. Arch Microbiol 2002, 178, 165–171. [Google Scholar]
- Gin, AS; Zhanel, GG. Vancomycin-resistant enterococci. Ann Pharmacother 1996, 30, 615–624. [Google Scholar]
- Bierbaum, G; Sahl, HG. Lantibiotics: Mode of action, biosynthesis and bioengineering. Curr Pharm Biotechnol 2009, 10, 2–18. [Google Scholar]
- Mattick, AT; Hirsch, A. Further observations on an inhibitory substance (nisin) from lactic streptococci. Lancet 1947, 2, 5–8. [Google Scholar]
- Jansen, EF; Hirschmann, DJ. Binding of simple carbohydrates and some N-acetyllactosamine-containing oligosaccharides to Erythrina cristagalli agglutinin as followed with a fluorescent indicator ligand. Arch Biochem 1994, 4, 297–304. [Google Scholar]
- Allgaier, H; Jung, G; Werner, RG; Schneider, U; Zähner, H. Epidermin: sequencing of a heterodetic tetracyclic 21-peptide amide antibiotic. Eur J Biochem 1986, 160, 9–22. [Google Scholar]
- Sahl, HG; Grossgarten, M; Widger, WR; Cramer, WA; Brandis, H. Structural similarities of the staphylococcin-like peptide Pep-5 to the peptide antibiotic nisin. Antimicrob Agents Chemother 1985, 27, 836–840. [Google Scholar]
- Chatterjee, S; Chatterjee, S; Lad, SJ; Phansalkar, MS; Rupp, RH; Ganguli, BN; Fehlhaber, HW; Kogler, H. Mersacidin, a new antibiotic from Bacillus. Fermentation, isolation, purification and chemical characterization. J Antibiot 1992, 45, 832–838. [Google Scholar]
- Fredenhagen, A; Fendrich, G; Märki, F; Märki, W; Gruner, J; Raschdorf, F; Peter, HH. Duramycins B and C, two new lanthionine containing antibiotics as inhibitors of phospholipase A2. Structural revision of duramycin and cinnamycin. J Antibiot 1990, 43, 1403–1412. [Google Scholar]
- Hansen, JN. Nisin as a model food preservative. Crit Rev Food Sci Nutr 1994, 34, 69–93. [Google Scholar]
- Hasper, HE; Kramer, NE; Smith, JL; Hillman, JD; Zachariah, C; Kuipers, OP; de Kruijff, B; Breukink, E. An alternative bactericidal mechanism of action for lantibiotic peptides that target lipid II. Science 2006, 313, 1636–1637. [Google Scholar]
- Bierbaum, G; Sahl, HG. Lantibiotics: Mode of action, biosynthesis and bioengineering. Curr Pharm Biotechnol 2009, 10, 2–18. [Google Scholar]
- Hsu, ST; Breukink, E; Tischenko, E; Lutters, MA; de Kruijff, B; Kaptein, R; Bonvin, AM; van Nuland, NA. The nisin-lipid II complex reveals a pyrophosphate cage that provides a blueprint for novel antibiotics. Nat Struct Mol Biol 2004, 11, 963–967. [Google Scholar]
- Parisot, J; Carey, S; Breukink, E; Chan, WC; Narbad, A; Bonev, B. Molecular mechanism of target recognition by subtilin, a class I lanthionine antibiotic. Antimicrob Agents Chemother 2008, 52, 612–618. [Google Scholar]
- Sahl, HG; Jack, RW; Bierbaum, G. Biosynthesis and biological activities of lantibiotics with unique post-translational modifications. Eur J Biochem 1995, 230, 827–853. [Google Scholar]
- Brötz, H; Bierbaum, G; Leopold, K; Reynolds, PE; Sahl, HG. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob Agents Chemother 1998, 42, 154–160. [Google Scholar]
- Alibert-Franco, S; Pradines, B; Mahamoud, A; Davin-Regli, A; Pagès, JM. Efflux mechanism, an attractive target to combat multidrug resistant Plasmodium falciparum and Pseudomonas aeruginosa. Curr Med Chem 2009, 16, 301–317. [Google Scholar]
- Langton, KP; Henderson, PJ; Herbert, RB. Antibiotic resistance: Multidrug efflux proteins, a common transport mechanism? Nat Prod Rep 2005, 22, 439–451. [Google Scholar]
- Zgurskaya, HI; Krishnamoorthy, G; Tikhonova, EB; Lau, SY; Stratton, KL. Mechanism of antibiotic efflux in Gram-negative bacteria. Front Biosci 2003, 8, s862–s873. [Google Scholar]
- Pérez, A; Canle, D; Latasa, C; Poza, M; Beceiro, A; Tomás Mdel, M; Fernández, A; Mallo, S; Pérez, S; Molina, F; et al. Cloning, nucleotide sequencing, and analysis of the AcrAB-TolC efflux pump of Enterobacter cloacae and determination of its involvement in antibiotic resistance in a clinical isolate. Antimicrob Agents Chemother 2007, 51, 3247–3253. [Google Scholar]
- Nicolas, P. Multifunctional host defense peptides: Intracellular-targeting antimicrobial peptides. FEBS J 2009, 276, 6483–6496. [Google Scholar]
- Oren, Z; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar]
- Mihajlovic, M; Lazaridis, T. Antimicrobial peptides in toroidal and cylindrical pores. Biochim Biophys Acta 2010, 1798, 1485–1493. [Google Scholar]
- Gottler, LM; Ramamoorthy, A. Structure, membrane orientation, mechanism, and function of pexiganan--a highly potent antimicrobial peptide designed from magainin. Biochim Biophys Acta 2009, 1788, 1680–1686. [Google Scholar]
- Shai, Y; Oren, Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides 2001, 22, 1629–1641. [Google Scholar]
- Ramamoorthy, A; Lee, DK; Narasimhaswamy, T; Nanga, RP. Cholesterol reduces pardaxin’s dynamics-a barrel-stave mechanism of membrane disruption investigated by solid-state NMR. Biochim Biophys Acta 2010, 1798, 223–227. [Google Scholar]
- Cantor, RS. Size distribution of barrel-stave aggregates of membrane peptides: Influence of the bilayer lateral pressure profile. Biophys J 2002, 82, 2520–2525. [Google Scholar]
- Bessin, Y; Saint, N; Marri, L; Marchini, D; Molle, G. Antibacterial activity and pore-forming properties of ceratotoxins: A mechanism of action based on the barrel stave model. Biochim Biophys Acta 2004, 1667, 148–156. [Google Scholar]
- Gazit, E; Miller, IR; Biggin, PC; Sansom, MS; Shai, Y. Structure and orientation of the mammalian antibacterial peptide cecropin P1 within phospholipid membranes. J Mol Biol 1996, 258, 860–870. [Google Scholar]
- Wong, H; Bowie, JH; Carver, JA. The solution structure and activity of caerin 1.1, an antimicrobial peptide from the Australian green tree frog, Litoria splendida. Eur J Biochem 1997, 247, 545–557. [Google Scholar]
- Ludtke, SJ; He, K; Heller, WT; Harroun, TA; Yang, L; Huang, HW. Membrane pores induced by magainin. Biochemistry 1996, 35, 13723–13728. [Google Scholar]
- Tamba, Y; Yamazaki, M. Magainin 2-induced pore formation in the lipid membranes depends on its concentration in the membrane interface. J Phys Chem B 2009, 113, 4846–4852. [Google Scholar]
- Basanez, G; Shinnar, AE; Zimmerberg, J. Interaction of hagfish cathelicidin antimicrobial peptides with model lipid membranes. FEBS Lett 2002, 532, 115–120. [Google Scholar]
- Henzler Wildman, KA; Lee, DK; Ramamoorthy, A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003, 42, 6545–6558. [Google Scholar]
- Park, SC; Kim, MH; Hossain, MA; Shin, SY; Kim, Y; Stella, L; Wade, JD; Park, Y; Hahm, KS. Amphipathic alpha-helical peptide, HP (2-20), and its analogues derived from Helicobacter pylori: Pore formation mechanism in various lipid compositions. Biochim Biophys Acta 2008, 1778, 229–241. [Google Scholar]
- Lee, TH; Hall, KN; Swann, MJ; Popplewell, JF; Unabia, S; Park, Y; Hahm, KS; Aguilar, MI. The membrane insertion of helical antimicrobial peptides from the N-terminus of Helicobacter pylori ribosomal protein L1. Biochim Biophys Acta 2010, 1798, 544–557. [Google Scholar]
- Park, SC; Kim, JY; Shin, SO; Jeong, CY; Kim, MH; Shin, SY; Cheong, GW; Park, Y; Hahm, KS. Investigation of toroidal pore and oligomerization by melittin using transmission electron microscopy. Biochem Biophys Res Commun 2006, 343, 222–228. [Google Scholar]
- Han, M; Mei, Y; Khant, H; Ludtke, SJ. Characterization of antibiotic peptide pores using cryo-EM and comparison to neutron scattering. Biophys J 2009, 97, 164–172. [Google Scholar]
- Yang, L; Harroun, TA; Weiss, TM; Ding, L; Huang, HW. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys J 2001, 81, 1475–1485. [Google Scholar]
- Allende, D; Simon, SA; McIntosh, TJ. Melittin-induced bilayer leakage depends on lipid material properties: evidence for toroidal pores. Biophys J 2005, 88, 1828–1837. [Google Scholar]
- Kragol, G; Hoffmann, R; Chattergoon, MA; Lovas, S; Cudic, M; Bulet, P; Condie, BA; Rosengren, KJ; Montaner, LJ; Otvos, L, Jr. Identification of crucial residues for the antibacterial activity of the proline-rich peptide, pyrrhocoricin. Eur J Biochem 2002, 269, 4226–4237. [Google Scholar]
- Kragol, G; Lovas, S; Varadi, G; Condie, BA; Hoffmann, R; Otvos, L, Jr. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar]
- Otvos, L, Jr; Insug, O; Rogers, ME; Consolvo, PJ; Condie, BA; Lovas, S; Bulet, P; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar]
- Vizan, JL; Hernández-Chico, C; del Castillo, I; Moreno, F. The peptide antibiotic microcin B17 induces double-strand cleavage of DNA mediated by E. coli DNA gyrase. EMBO J 1991, 10, 467–476. [Google Scholar]
- Park, CB; Kim, HS; Kim, SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem Biophys Res Commun 1998, 244, 253–257. [Google Scholar]
- Boman, HG; Agerberth, B; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect Immun 1993, 61, 2978–2984. [Google Scholar]
- Subbalakshmi, C; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol Lett 1998, 160, 91–96. [Google Scholar]
- Xiong, YQ; Bayer, AS; Yeaman, MR. Inhibition of intracellular macromolecular synthesis in Staphylococcus aureus by thrombin-induced platelet microbicidal proteins. J Infect Dis 2002, 185, 348–356. [Google Scholar]
- Cho, JH; Sung, BH; Kim, SC. Buforins: histone H2A-derived antimicrobial peptides from toad stomach. Biochim Biophys Acta 2009, 1788, 1564–1569. [Google Scholar]
- Hsu, CH; Chen, C; Jou, ML; Lee, AY; Lin, YC; Yu, YP; Huang, WT; Wu, SH. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res 2005, 33, 4053–4064. [Google Scholar]
- Barriere, SL. Bacterial resistance to beta-lactams, and its prevention with combination antimicrobial therapy. Pharmacotherapy 1992, 12, 397–402. [Google Scholar]
- Wu, YL; Scott, EM; Po, AL; Tariq, VN. Ability of azlocillin and tobramycin in combination to delay or prevent resistance development in Pseudomonas aeruginosa. J Antimicrob Chemother 1999, 44, 389–392. [Google Scholar]
- Steenbergen, JN; Mohr, JF; Thorne, GM. Effects of daptomycin in combination with other antimicrobial agents: A review of in vitro and animal model studies. J Antimicrob Chemother 2009, 64, 1130–1138. [Google Scholar]
- Anantharaman, A; Rizvi, MS; Sahal, D. Synergy with rifampin and kanamycin enhances potency, kill kinetics, and selectivity of de novo-designed antimicrobial peptides. Antimicrob Agents Chemother 2010, 54, 1693–1699. [Google Scholar]
- Cirioni, O; Silvestri, C; Ghiselli, R; Orlando, F; Riva, A; Mocchegiani, F; Chiodi, L; Castelletti, S; Gabrielli, E; Saba, V; et al. Protective effects of the combination of alpha-helical antimicrobial peptides and rifampicin in three rat models of Pseudomonas aeruginosa infection. J Antimicrob Chemother 2008, 62, 1332–1338. [Google Scholar]
- Cirioni, O; Ghiselli, R; Silvestri, C; Kamysz, W; Orlando, F; Mocchegiani, F; Di Matteo, F; Riva, A; Lukasiak, J; Scalise, G; et al. Efficacy of tachyplesin III, colistin, and imipenem against a multiresistant Pseudomonas aeruginosa strain. Antimicrob Agents Chemother 2007, 51, 2005–2010. [Google Scholar]
- Jeong, N; Kim, JY; Park, SC; Lee, JK; Gopal, R; Yoo, S; Son, BK; Hahm, JS; Park, Y; Hahm, KS. Antibiotic and synergistic effect of Leu-Lys rich peptide against antibiotic resistant microorganisms isolated from patients with cholelithiasis. Biochem Biophys Res Commun 2010, 399, 581–586. [Google Scholar]
- McGregor, DP. Discovering and improving novel peptide therapeutics. Curr Opin Pharmacol 2008, 8, 616–619. [Google Scholar]
- Svenson, J; Stensen, W; Brandsdal, BO; Haug, BE; Monrad, J; Svendsen, JS. Antimicrobial peptides with stability toward tryptic degradation. Biochemistry 2008, 47, 3777–3788. [Google Scholar]
- Park, KH; Nan, YH; Park, Y; Kim, JI; Park, IS; Hahm, KS; Shin, SY. Cell specificity, anti-inflammatory activity, and plausible bactericidal mechanism of designed Trp-rich model antimicrobial peptides. Biochim Biophys Acta 2009, 1788, 1193–1203. [Google Scholar]
- Whitcomb, DC; Lowe, ME. Human pancreatic digestive enzymes. Dig Dis Sci 2007, 52, 1–17. [Google Scholar]
- Gray, GM; Cooper, HL. Protein digestion and absorption. Gastroenterology 1971, 61, 535–544. [Google Scholar]
- Park, Y; Park, SC; Kim, JY; Park, JO; Seo, CH; Nah, JW; Hahm, KS. In vitro efficacy of a synthetic all-d antimicrobial peptide against clinically isolated drug-resistant strains. Int J Antimicrob Agents 2010, 35, 208–209. [Google Scholar]
- Wang, P; Nan, YH; Yang, ST; Kang, SW; Kim, Y; Park, IS; Hahm, KS; Shin, SY. Cell selectivity and anti-inflammatory activity of a Leu/Lys-rich alpha-helical model antimicrobial peptide and its diastereomeric peptides. Peptides 2010, 31, 1251–1261. [Google Scholar]
- Dartois, V; Sanchez-Quesada, J; Cabezas, E; Chi, E; Dubbelde, C; Dunn, C; Granja, J; Gritzen, C; Weinberger, D; Ghadiri, MR; et al. Systemic antibacterial activity of novel synthetic cyclic peptides. Antimicrob Agents Chemother 2005, 49, 3302–3310. [Google Scholar]
- Rozek, A; Powers, JP; Friedrich, CL; Hancock, RE. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003, 42, 14130–14138. [Google Scholar]
- Avrahami, D; Shai, Y. Conjugation of a magainin analogue with lipophilic acids controls hydrophobicity, solution assembly, and cell selectivity. Biochemistry 2002, 41, 2254–2263. [Google Scholar]
- Chongsiriwatana, NP; Patch, JA; Czyzewski, AM; Dohm, MT; Ivankin, A; Gidalevitz, D; Zuckermann, RN; Barron, AE. Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci USA 2008, 105, 2794–2799. [Google Scholar]
- Wang, P; Bang, JK; Kim, HJ; Kim, JK; Kim, Y; Shin, SY. Antimicrobial specificity and mechanism of action of disulfide-removed linear analogs of the plant-derived Cys-rich antimicrobial peptide Ib-AMP1. Peptides 2009, 30, 2144–2149. [Google Scholar]
- Meng, H; Kumar, K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J Am Chem Soc 2007, 129, 15615–15622. [Google Scholar]
- Porter, EA; Weisblum, B; Gellman, SH. Mimicry of host-defense peptides by unnatural oligomers: antimicrobial beta-peptides. J Am Chem Soc 2002, 124, 7324–7330. [Google Scholar]
- Radzishevsky, IS; Rotem, S; Bourdetsky, D; Navon-Venezia, S; Carmeli, Y; Mor, A. Improved antimicrobial peptides based on acyl-lysine oligomers. Nat Biotechnol 2007, 25, 657–659. [Google Scholar]
- Kruszewska, D; Sahl, HG; Bierbaum, G; Pag, U; Hynes, SO; Ljungh, A. Mersacidin eradicates methicillin-resistant Staphylococcus aureus (MRSA) in a mouse rhinitis model. J Antimicrob Chemother 2004, 54, 648–653. [Google Scholar]
- Chatterjee, S; Chatterjee, DK; Jani, RH; Blumbach, J; Ganguli, BN; Klesel, N; Limbert, M; Seibert, G. Mersacidin, a new antibiotic from Bacillus. In vitro and in vivo antibacterial activity. J Antibiot (Tokyo) 1992, 45, 839–845. [Google Scholar]
- Jevsevar, S; Kunstelj, M; Porekar, VG. PEGylation of therapeutic proteins. Biotechnol J 2010, 5, 113–128. [Google Scholar]
- Pasut, G; Veronese, FM. PEG conjugates in clinical development or use as anticancer agents: An overview. Adv Drug Deliv Rev 2009, 61, 1177–1188. [Google Scholar]
- Veronese, FM; Mero, A. The impact of PEGylation on biological therapies. BioDrugs 2008, 22, 315–329. [Google Scholar]
- Imura, Y; Nishida, M; Ogawa, Y; Takakura, Y; Matsuzaki, K. Action mechanism of tachyplesin I and effects of PEGylation. Biochim Biophys Acta 2007, 1768, 1160–1169. [Google Scholar]
- Imura, Y; Nishida, M; Matsuzaki, K. Action mechanism of PEGylated magainin 2 analogue peptide. Biochim Biophys Acta 2007, 1768, 2578–2585. [Google Scholar]
- Zhang, G; Han, B; Lin, X; Wu, X; Yan, H. Modification of antimicrobial peptide with low molar mass poly(ethylene glycol). J Biochem 2008, 144, 781–788. [Google Scholar]
- Isaacson, RE. MBI-226. Micrologix/Fujisawa. Curr Opin Investig Drugs 2003, 4, 999–1003. [Google Scholar]
- Rubinchik, E; Dugourd, D; Algara, T; Pasetka, C; Friedland, HD. Antimicrobial and antifungal activities of a novel cationic antimicrobial peptide, omiganan, in experimental skin colonisation models. Int J Antimicrob Agents 2009, 34, 457–461. [Google Scholar]
- Yeung, AT; Gellatly, SL; Hancock, RE. Multifunctional cationic host defence peptides and their clinical applications. Cell Mol Life Sci 2011, 68, 2161–2176. [Google Scholar]
- Hancock, REW; Sahl, HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 2006, 24, 1551–1557. [Google Scholar]
- Zaiou, M. Multifunctional antimicrobial peptides: Therapeutic targets in several human diseases. J Mol Med 2007, 85, 317–329. [Google Scholar]
- Zhang, L; Falla, TJ. Antimicrobial peptides: Therapeutic potential. Expert Opin Pharmacother 2006, 7, 653–663. [Google Scholar]
- Costerton, JW; Irvin, RT; Cheng, KJ. The bacterial glycocalyx in nature and disease. Ann Rev Microbiol 1981, 35, 299–324. [Google Scholar]
- Costerton, J. Introduction to biofilm. Int J Antimicrob Agents 1999, 11, 217–221. [Google Scholar]
- Hojo, K; Nagaoka, S; Ohshima, T; Maeda, N. Bacterial interactions in dental biofilm development. J Dent Res 2009, 88, 982–990. [Google Scholar]
- Paju, S; Scannapieco, FA. Oral biofilms, periodontitis, and pulmonary infections. Oral Dis 2007, 13, 508–512. [Google Scholar]
- Palmer, J. Bacterial biofilms in chronic rhinosinusitis. Ann Otol Rhinol Laryngol Suppl 2006, 196, 35–39. [Google Scholar]
- Wagner, VE; Iglewski, BH. P. aeruginosa Biofilms in CF Infection. Clin Rev Allergy Immunol 2008, 35, 124–134. [Google Scholar]
- Lau, GW; Hassett, DJ; Britigan, BE. Modulation of lung epithelial functions by Pseudomonas aeruginosa. Trends Microbiol 2005, 13, 389–397. [Google Scholar]
- Khardori, N; Yassien, M. Biofilms in device-related infections. J Ind Microbiol 1995, 15, 141–147. [Google Scholar]
- Habash, M; Reid, G. Microbial biofilms: their development and significance for medical device-related infections. J Clin Pharmacol 1999, 39, 887–898. [Google Scholar]
- Hoffman, LR; D’Argenio, DA; MacCoss, MJ; Zhang, Z; Jones, RA; Miller, SI. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature 2005, 436, 1171–1175. [Google Scholar]
- Costerton, JW; Stewart, PS; Greenberg, EP. Bacterial biofilms: a common cause of persistent infections. Science 1999, 284, 1318–1322. [Google Scholar]
- Hall-Stoodley, L; Stoodley, P. Evolving concepts in biofilm infections. Cell Microbiol 2009, 11, 1034–1043. [Google Scholar]
- Landini, P; Antoniani, D; Burgess, JG; Nijland, R. Molecular mechanisms of compounds affecting bacterial biofilm formation and dispersal. Appl Microbiol Biotechnol 2010, 86, 813–823. [Google Scholar]
- Horswill, AR; Stoodley, P; Stewart, PS; Parsek, MR. The effect of the chemical, biological, and physical environment on quorum sensing in structured microbial communities. Anal Bioanal Chem 2007, 387, 371–380. [Google Scholar]
- Spoering, AL; Gilmore, MS. Quorum sensing and DNA release in bacterial biofilms. Curr Opin Microbiol 2006, 9, 133–137. [Google Scholar]
- Suntharalingam, P; Cvitkovitch, DG. Quorum sensing in streptococcal biofilm formation. Trends Microbiol 2005, 13, 3–6. [Google Scholar]
- Chen, G; Swem, LR; Swem, DL; Stauff, DL; O’Loughlin, CT; Jeffrey, PD; Bassler, BL; Hughson, FM. A strategy for antagonizing quorum sensing. Mol Cell 2011, 42, 199–209. [Google Scholar]
- Waters, CM; Bassler, BL. Quorum sensing: Cell-to-cell communication in bacteria. Annu Rev Cell Dev Biol 2005, 21, 319–346. [Google Scholar]
- Sutherland, I. Biofilm exopolysaccharides: A strong and sticky framework. Microbiology 2001, 147, 3–9. [Google Scholar]
- Moreau-Marquis, S; Stanton, BA; O’Toole, GA. Pseudomonas aeruginosa biofilm formation in the cystic fibrosis airway. Pulm Pharmacol Ther 2008, 21, 595–599. [Google Scholar]
- Overhage, J; Campisano, A; Bains, M; Torfs, EC; Rehm, BH; Hancock, RE. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun 2008, 76, 4176–4182. [Google Scholar]
- Hell, E; Giske, CG; Nelson, A; Römling, U; Marchini, G. Human cathelicidin peptide LL37 inhibits both attachment capability and biofilm formation of Staphylococcus epidermidis. Lett Appl Microbiol 2010, 50, 211–215. [Google Scholar]
- Amer, LS; Bishop, BM; van Hoek, ML. Antimicrobial and antibiofilm activity of cathelicidins and short, synthetic peptides against Francisella. Biochem Biophys Res Commun 2010, 396, 246–251. [Google Scholar]
- Porat, Y; Marynka, K; Tam, A; Steinberg, D; Mor, A. Acyl-substituted dermaseptin S4 derivatives with improved bactericidal properties, including on oral microflora. Antimicrob Agents Chemother 2005, 50, 4153–4160. [Google Scholar]
- Laverty, G; McLaughlin, M; Shaw, C; Gorman, SP; Gilmore, BF. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem Biol Drug Des 2010, 75, 563–569. [Google Scholar]
- Roveta, S; Marchese, A; Schito, GC. Activity of daptomycin on biofilms produced on a plastic support by Staphylococcus spp. Int J Antimicrob Agents 2008, 31, 321–328. [Google Scholar]
- H⊘iby, N; Bjarnsholt, T; Givskov, M; Molin, S; Ciofu, O. Antibiotic resistance of bacterial biofilms. Int J Antimicrob Agents 2010, 35, 322–332. [Google Scholar]
- Yoshinari, M; Kato, T; Matsuzaka, K; Hayakawa, T; Shiba, K. Prevention of biofilm formation on titanium surfaces modified with conjugated molecules comprised of antimicrobial and titanium-binding peptides. Biofouling 2010, 26, 103–110. [Google Scholar]
- Willcox, MD; Hume, EB; Aliwarga, Y; Kumar, N; Cole, N. A novel cationic-peptide coating for the prevention of microbial colonization on contact lenses. J Appl Microbiol 2008, 105, 1817–1825. [Google Scholar]
- Cole, N; Hume, EB; Vijay, AK; Sankaridurg, P; Kumar, N; Willcox, MD. In vivo performance of melimine as an antimicrobial coating for contact lenses in models of CLARE and CLPU. Invest Ophthalmol Vis Sci 2010, 51, 390–395. [Google Scholar]
- Cirioni, O; Giacometti, A; Ghiselli, R; Kamysz, W; Orlando, F; Mocchegiani, F; Silvestri, C; Licci, A; Chiodi, L; Lukasiak, J; Saba, V; Scalise, G. Citropin 1.1-treated central venous catheters improve the efficacy of hydrophobic antibiotics in the treatment of experimental staphylococcal catheter-related infection. Peptides 2006, 27, 1210–1216. [Google Scholar]
- Marsh, PD. Dental plaque: Biological significance of a biofilm and community life-style. J Clin Periodontol 2005, 32, 7–15. [Google Scholar]
- Rudney, JD. Saliva and dental plaque. Adv Dent Res 2000, 14, 29–39. [Google Scholar]
- Marsh, PD; Bradshaw, DJ. Dental plaque as a biofilm. J Ind Microbiol 1995, 15, 169–175. [Google Scholar]
- Sissons, CH; Anderson, SA; Wong, L; Coleman, MJ; White, DC. Microbiota of plaque microcosm biofilms: Effect of three times daily sucrose pulses in different simulated oral environments. Caries Res 2007, 41, 413–422. [Google Scholar]
- Filoche, S; Wong, L; Sissons, CH. Oral biofilms: Emerging concepts in microbial ecology. J Dent Res 2010, 89, 8–18. [Google Scholar]
- Leung, KP; Abercrombie, JJ; Campbell, TM; Gilmore, KD; Bell, CA; Faraj, JA; DeLuca, PP. Antimicrobial peptides for plaque control. Adv Dent Res 2009, 21, 57–62. [Google Scholar]
- Eberhard, J; Pietschmann, R; Falk, W; Jepsen, S; Dommisch, H. The immune response of oral epithelial cells induced by single-species and complex naturally formed biofilms. Oral Microbiol Immunol 2009, 24, 325–330. [Google Scholar]
- Lobos, O; Padilla, A; Padilla, C. In vitro antimicrobial effect of bacteriocin PsVP-10 in combination with chlorhexidine and triclosan against Streptococcus mutans and Streptococcus sobrinus strains. Arch Oral Biol 2009, 54, 230–234. [Google Scholar]
- Wakabayashi, H; Yamauchi, K; Kobayashi, T; Yaeshima, T; Iwatsuki, K; Yoshie, H. Inhibitory effects of lactoferrin on growth and biofilm formation of Porphyromonas gingivalis and Prevotella intermedia. Antimicrob Agents Chemother 2009, 53, 3308–3316. [Google Scholar]
- Arslan, SY; Leung, KP; Wu, CD. The effect of lactoferrin on oral bacterial attachment. Oral Microbiol Immunol 2009, 24, 411–416. [Google Scholar]
- Wakabayashi, H; Kondo, I; Kobayashi, T; Yamauchi, K; Toida, T; Iwatsuki, K; Yoshie, H. Periodontitis, periodontopathic bacteria and lactoferrin. Biometals 2010, 23, 419–424. [Google Scholar]
- Darouiche, RO; Mansouri, MD; Gawande, PV; Madhyastha, S. Antimicrobial and antibiofilm efficacy of triclosan and DispersinB combination. J Antimicrob Chemother 2009, 64, 88–93. [Google Scholar]
- Kaplan, JB. Therapeutic potential of biofilm-dispersing enzymes. Int J Artif Organs 2009, 32, 545–554. [Google Scholar]
- Izano, EA; Amarante, MA; Kher, WB; Kaplan, JB. Differential roles of poly-N-acetylglucosamine surface polysaccharide and extracellular DNA in Staphylococcus aureus and Staphylococcus epidermidis biofilms. Appl Environ Microbiol 2008, 74, 470–476. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Park, S.-C.; Park, Y.; Hahm, K.-S. The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation. Int. J. Mol. Sci. 2011, 12, 5971-5992. https://doi.org/10.3390/ijms12095971
Park S-C, Park Y, Hahm K-S. The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation. International Journal of Molecular Sciences. 2011; 12(9):5971-5992. https://doi.org/10.3390/ijms12095971
Chicago/Turabian StylePark, Seong-Cheol, Yoonkyung Park, and Kyung-Soo Hahm. 2011. "The Role of Antimicrobial Peptides in Preventing Multidrug-Resistant Bacterial Infections and Biofilm Formation" International Journal of Molecular Sciences 12, no. 9: 5971-5992. https://doi.org/10.3390/ijms12095971