The Potential of Antimicrobial Peptides as Biocides


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Antimicrobial Peptides
1.2. Cationic Antimicrobial Peptides
1.3. Anionic Antimicrobial Peptides
1.4. Amphibian Antimicrobial Peptides
1.5. Rational Design and Selection of an Antimicrobial Peptide Motif
1.6. Ultrashort Cationic Antimicrobial Peptides
1.7. Lipopeptides
1.8. Licensed and Commercially Available Lipopeptides
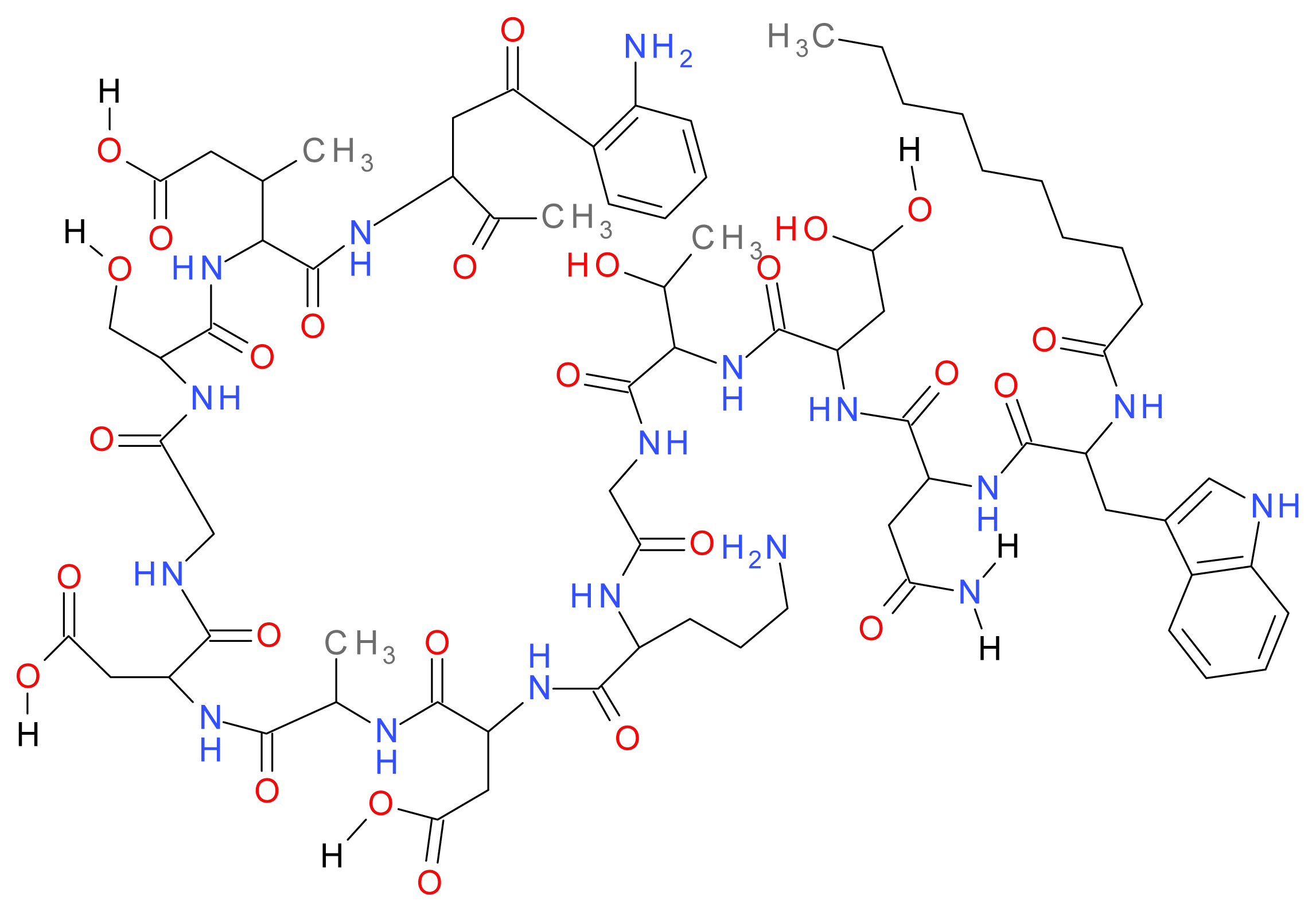
1.8.1. Daptomycin
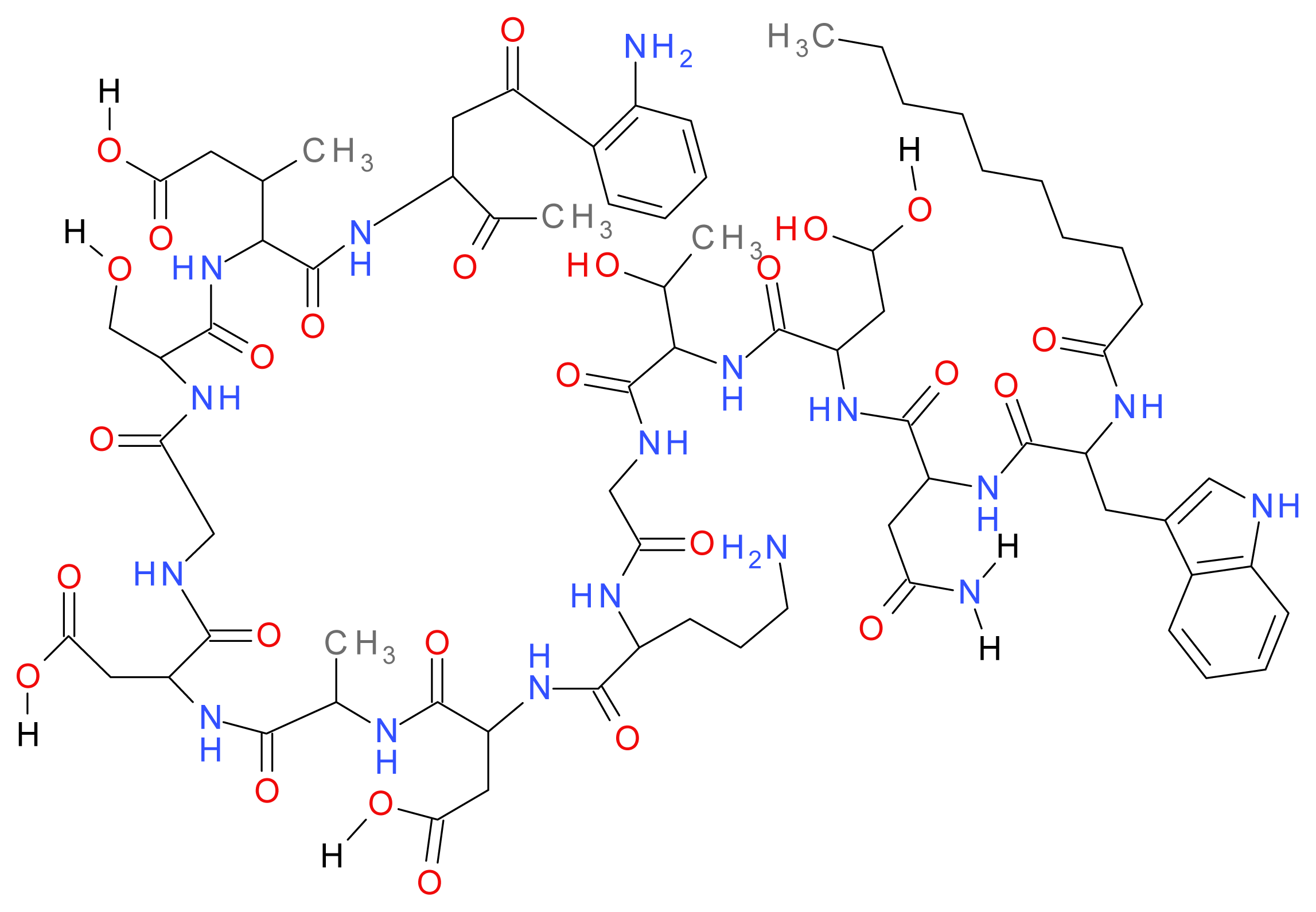
1.8.2. Polymyxins (B and E)
2. Mechanism of Action of Antimicrobial Peptides
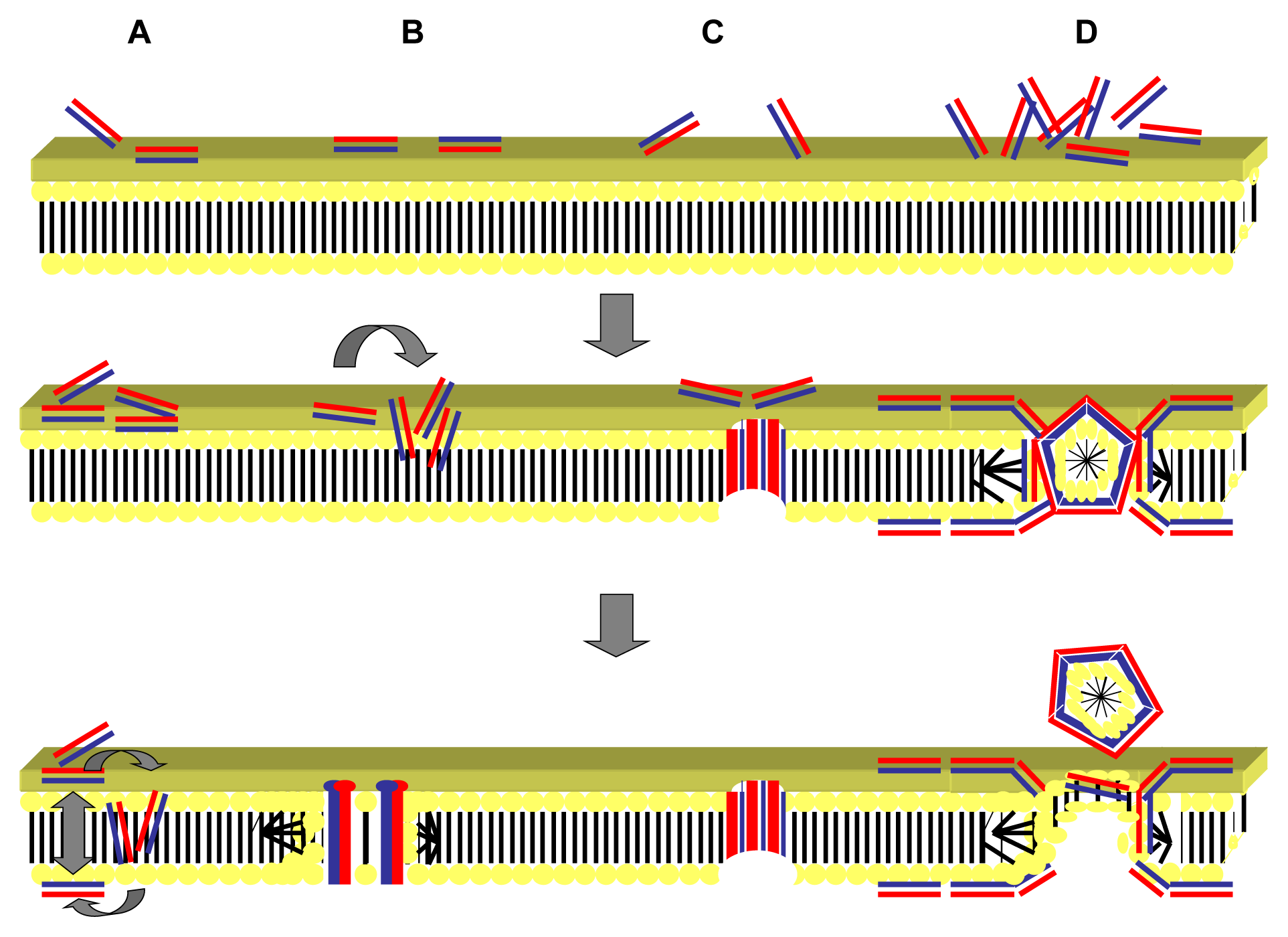
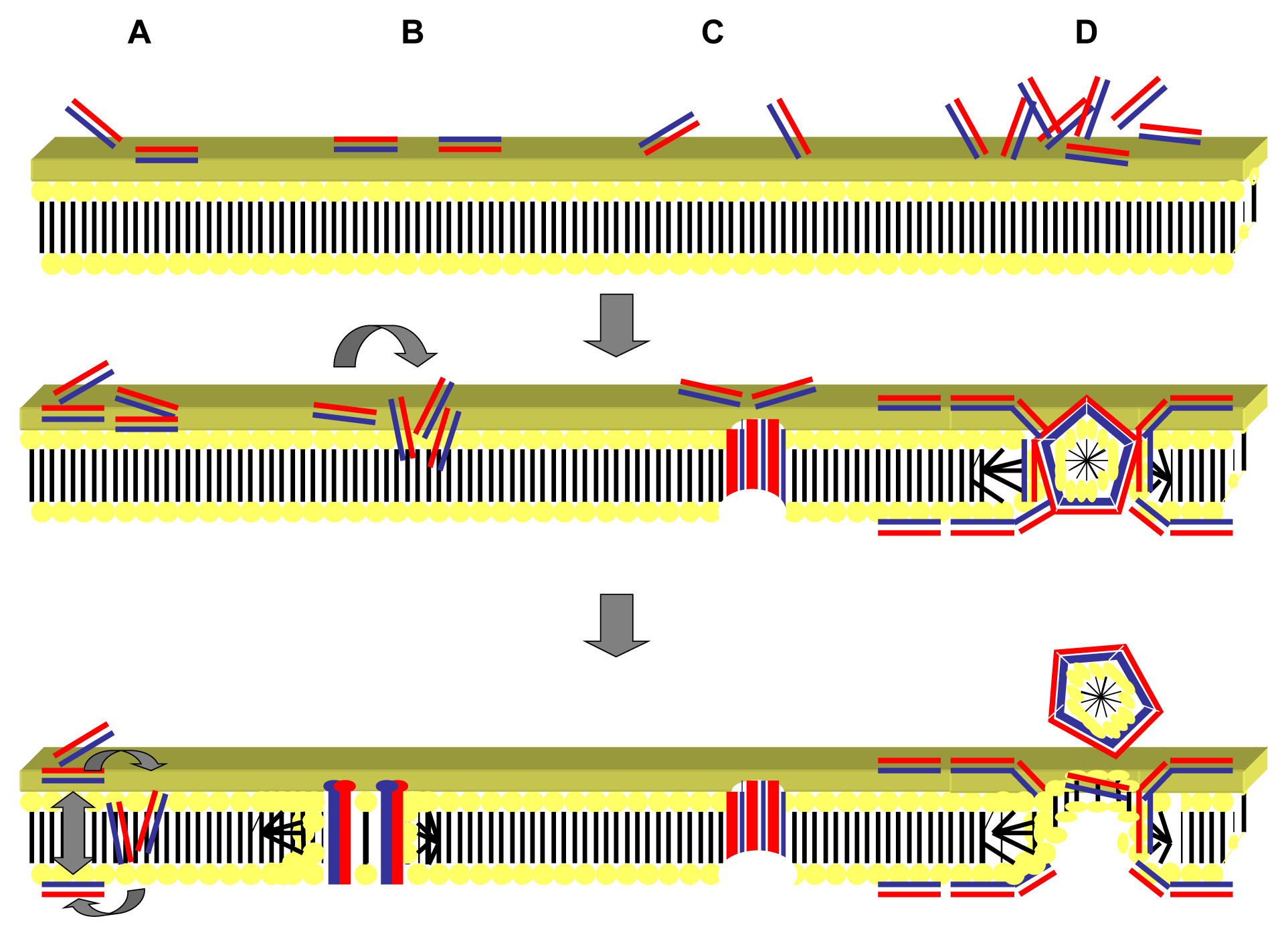
2.1. Targeting of the Microbial Cell Membrane
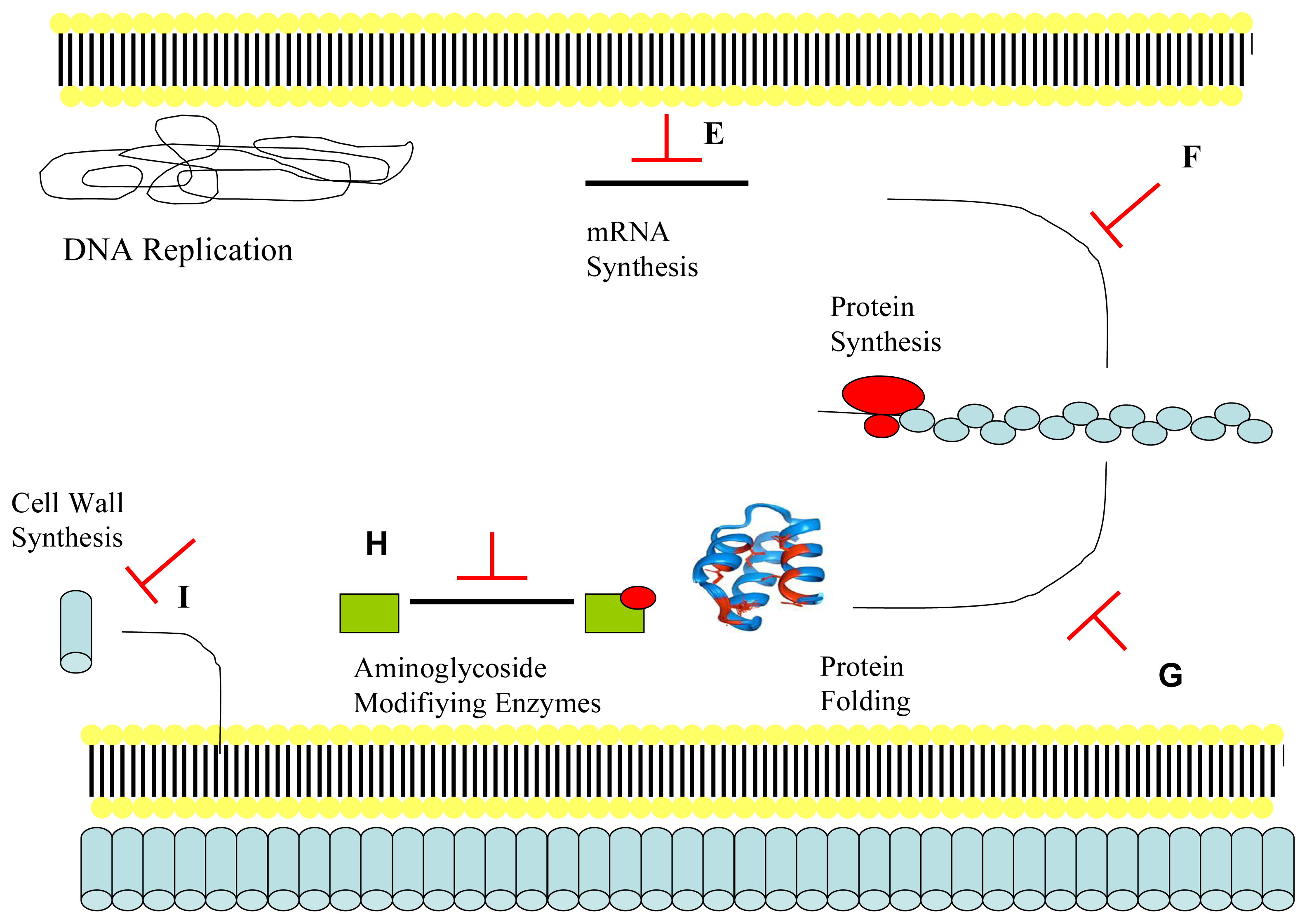
2.2. Mechanism of Action of Antimicrobial Peptides: Intracellular Targeting
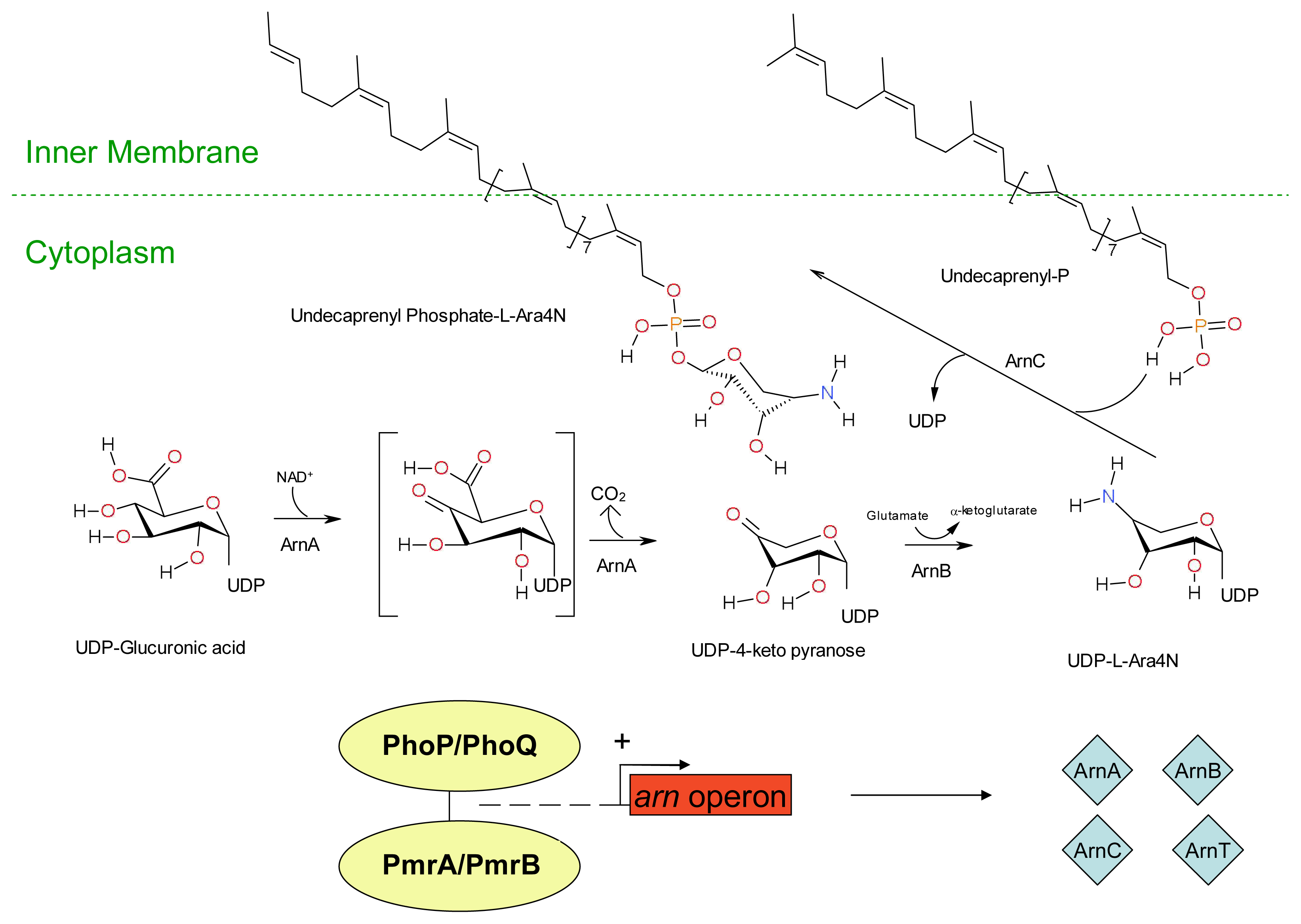
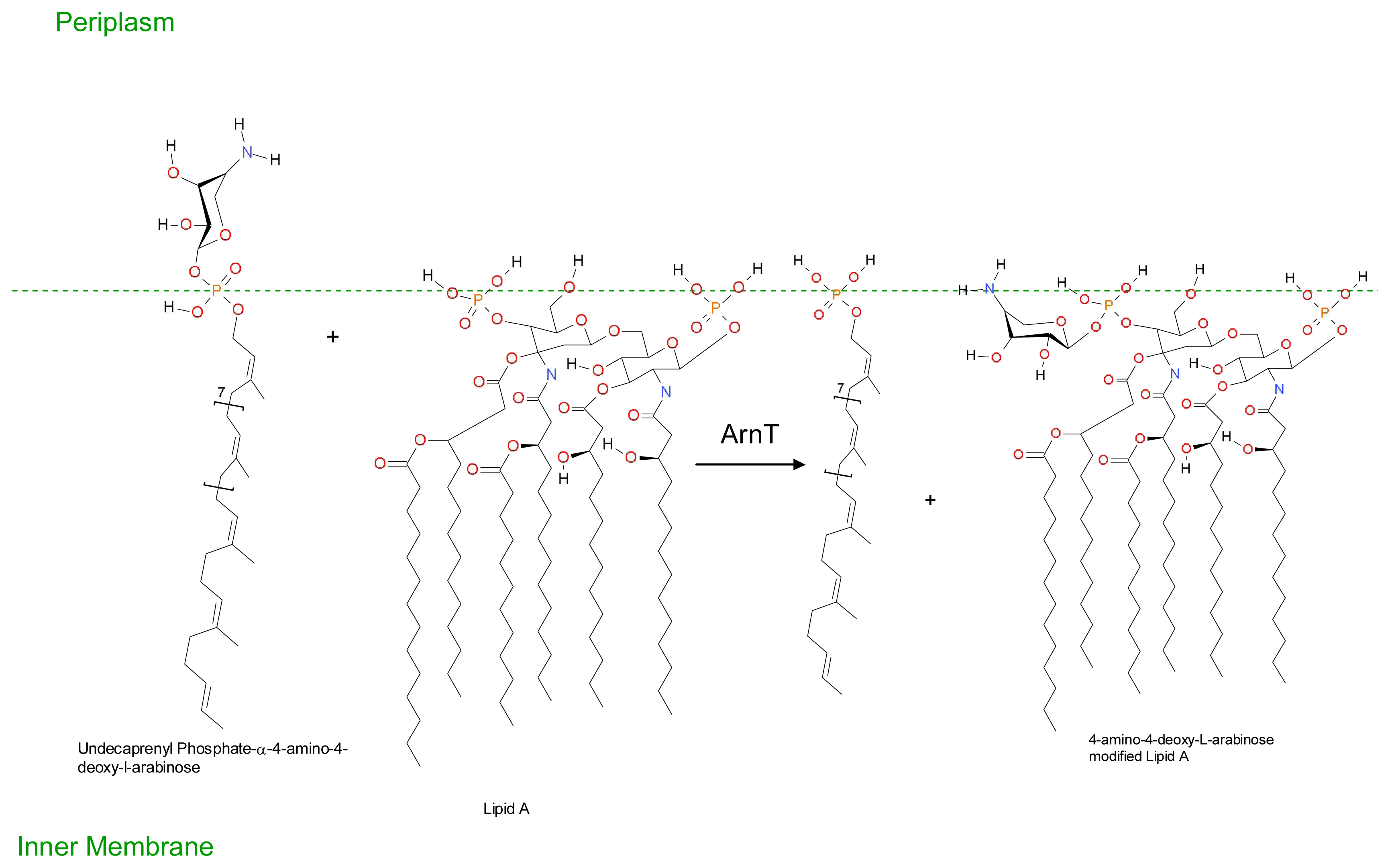
3. Development of Resistance to Antimicrobial Peptides
4. Future Perspectives
Acknowledgements
References
- Falagas, ME; Fragoulis, KN; Karydis, I. A comparative study on the cost of new antibiotics and drugs of other therapeutic categories. PLoS ONE 2006, 1, 1–4. [Google Scholar]
- Chambers, HF; Deleo, FR. Waves of resistance: Staphylococcus aureus in the antibiotic era. Nat. Rev. Microbiol 2009, 7, 629–641. [Google Scholar]
- Wenzel, RP. The antibiotic pipeline-challenges, costs and values. N Engl J Med 2001, 523–526. [Google Scholar]
- Boucher, HW; Talbot, GH; Bradley, JS; Edwards, JE; Gilbert, D; Rice, LB; Scheld, M; Spellberg, B; Bartlett, J. Bad bugs no drugs: No ESKAPE! An update from the infectious diseases society of America. Clin. Infect. Dis 2009, 48, 1–12. [Google Scholar]
- Rice, LB. Do we really need new anti-infective drugs? Curr. Opin. Pharmacol 2003, 3, 459–463. [Google Scholar]
- Andreu, D; Rivas, L. Animal antimicrobial peptides: An overview. Biopolymers 1998, 47, 415–433. [Google Scholar]
- Joerger, RD. Alternatives to antibiotics: Bacteriocins, antimicrobial peptides and bacteriophages. Poult. Sci 2003, 82, 640–647. [Google Scholar]
- Brown, KL; Hancock, RE. Cationic host defense (antimicrobial) peptides. Curr. Opin. Immunol 2006, 18, 24–30. [Google Scholar]
- Bowdish, DM; Davidson, DJ; Lau, YE; Lee, K; Scott, MG; Hancock, RE. Impact of LL-37 on anti-infective immunity. J. Leukoc. Biol 2005, 77, 451–459. [Google Scholar]
- Rosenfeld, Y; Sahl, HG; Shai, Y. Parameters involved in antimicrobial and endotoxin detoxification activities of antimicrobial peptides. Biochemistry 2008, 47, 6468–6478. [Google Scholar]
- Elsbach, P. What is the real role of antimicrobial polypeptides that can mediate several other inflammatory responses? J. Clin. Invest 2003, 111, 1643–1645. [Google Scholar]
- Yeaman, MR; Yount, NY. Mechanisms of antimicrobial peptide action and resistance. Pharmacol. Rev 2003, 55, 27–55. [Google Scholar]
- Brogden, KA. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol 2005, 3, 238–250. [Google Scholar]
- Ganz, T; Lehrer, RI. Antibiotic peptides from higher eukaryotes: Biology and applications. Mol. Med. Today 1999, 5, 292–297. [Google Scholar]
- Van’t Hof, W; Veerman, EC; Helmerhorst, EJ; Amerongen, AV. Antimicrobial peptides: Properties and applicability. Biol. Chem 2001, 382, 597–619. [Google Scholar]
- Hancock, RE; Diamond, G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol 2000, 8, 402–410. [Google Scholar]
- Brahmachary, M; Krishnan, SP; Koh, JL; Khan, AM; Seah, SH; Tan, TW; Brusic, V; Bajic, VB. ANTIMIC: A database of antimicrobial sequences. Nucleic Acids Res 2004, 32, D586–D589. [Google Scholar]
- Hwang, PM; Vogel, HJ. Structure-function relationships of antimicrobial peptides. Biochem. Cell Biol 1998, 76, 235–246. [Google Scholar]
- Powers, JP; Hancock, RE. The relationship between peptide structure and antibacterial activity. Peptides 2003, 24, 1681–1691. [Google Scholar]
- Hilpert, K; Elliott, MR; Volkmer-Engert, R; Henklein, P; Donini, O; Zhou, Q; Winkler, DF; Hancock, RE. Sequence requirements and an optimization strategy for short antimicrobial peptides. Chem. Biol 2006, 13, 1101–1107. [Google Scholar]
- Bisht, GS; Rawat, DS; Kumar, A; Kumar, R; Pasha, S. Antimicrobial activity of rationally designed amino terminal modified peptides. Bioorg. Med. Chem. Lett 2007, 17, 4343–4346. [Google Scholar]
- Hicks, RP; Bhonsle, JB; Venugopal, D; Koser, BW; Magill, AJ. De novo design of selective antibiotic peptides by incorporation of unnatural amino acids. J. Med. Chem 2007, 50, 3026–3036. [Google Scholar]
- Laverty, G; McLaughlin, M; Shaw, C; Gorman, SP; Gilmore, BF. Antimicrobial activity of short, synthetic cationic lipopeptides. Chem. Biol. Drug Des 2010, 75, 563–569. [Google Scholar]
- Marshall, SH; Arenas, G. Antimicrobial Peptides: A natural alternative to chemical antibiotics and a potential for applied biotechnology. Electron. J. Biomed 2003, 6, 272–284. [Google Scholar]
- Hancock, RE; Brown, KL; Mookherjee, N. Host defence peptides from invertebrates—Emerging antimicrobial strategies. Immunobiology 2006, 211, 315–322. [Google Scholar]
- McPhee, JB; Hancock, RE. Function and therapeutic potential of host defence peptides. J. Pept. Sci 2005, 11, 677–687. [Google Scholar]
- Guskey, MT; Tsuji, BT. A comparative review of the lipoglycopeptides: Oritavancin, dalbavancin, and telavancin. Pharmacotherapy 2010, 30, 80–94. [Google Scholar]
- Zelezetsky, I; Pag, U; Sahl, HG; Tossi, A. Tuning the biological properties of amphipathic alpha-helical antimicrobial peptides: Rational use of minimal amino acid substitutions. Peptides 2005, 26, 2368–2376. [Google Scholar]
- Thomas, S; Karnik, S; Barai, RS; Jayaraman, VK; Idicula-Thomas, S. CAMP: A useful resource for research on antimicrobial peptides. Nucleic Acids Res 2010, 38, D774–D780. [Google Scholar]
- Maloy, WL; Kari, UP. Structure-activity studies on magainins and other host defense peptides. Biopolymers 1995, 37, 105–122. [Google Scholar]
- Saberwal, G; Nagaraj, R. Cell-lytic and antibacterial peptides that act by perturbing the barrier function of membranes: Facets of their conformational features, structure-function correlations and membrane-perturbing abilities. Biochim. Biophys. Acta 1994, 1197, 109–131. [Google Scholar]
- Ganz, T; Lehrer, RI. Antimicrobial peptides of vertebrates. Curr. Opin. Immunol 1998, 10, 41–44. [Google Scholar]
- Hancock, RE; Chapple, DS. Peptide antibiotics. Antimicrob. Agents Chemother 1999, 43, 1317–1323. [Google Scholar]
- Shai, Y. From innate immunity to de-novo designed antimicrobial peptides. Curr. Pharm. Des 2002, 8, 715–725. [Google Scholar]
- Bhunia, A; Domadia, PN; Bhattacharjya, S. Structural and thermodynamic analyses of the interaction between melittin and lipopolysaccharide. Biochim. Biophys. Acta 2007, 1768, 3282–3291. [Google Scholar]
- Zasloff, M. Magainins, a class of antimicrobial peptides from xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar]
- Bulet, P; Hetru, C; Dimarcq, JL; Hoffmann, D. Antimicrobial peptides in insects; Structure and function. Dev. Comp. Immunol 1999, 23, 329–344. [Google Scholar]
- Powers, JP; Rozek, A; Hancock, RE. Structure-activity relationships for the beta-hairpin cationic antimicrobial peptide polyphemusin I. Biochim. Biophys. Acta 2004, 1698, 239–250. [Google Scholar]
- Boman, HG. Peptide antibiotics and their role in innate immunity. Annu. Rev. Immunol 1995, 13, 61–92. [Google Scholar]
- Yang, ST; Yub Shin, SY; Kim, YC; Kim, Y; Hahm, KS; Kim, JI. Conformation-dependent antibiotic activity of tritrpticin, a cathelicidin-derived antimicrobial peptide. Biochem. Biophys. Res. Commun 2002, 296, 1044–1050. [Google Scholar]
- Selsted, ME; Novotny, MJ; Morris, WL; Tang, YQ; Smith, W; Cullor, JS. Indolicidin, a novel bactericidal tridecapeptide amide from neutrophils. J. Biol. Chem 1992, 267, 4292–4295. [Google Scholar]
- Kim, SM; Kim, JM; Joshi, BP; Cho, H; Lee, KH. Indolicidin-derived antimicrobial peptide analogs with greater bacterial selectivity and requirements for antibacterial and hemolytic activities. Biochim. Biophys. Acta 2009, 1794, 185–192. [Google Scholar]
- Halevy, R; Rozek, A; Kolusheva, S; Hancock, RE; Jelinek, R. Membrane binding and permeation by indolicidin analogs studied by a biomimetic lipid/polydiacetylene vesicle assay. Peptides 2003, 24, 1753–1761. [Google Scholar]
- Foster, JW; Woodruff, HB. Antibiotic substances produced by bacteria. Ann. N. Y. Acad. Sci 2010, 1213, 125–136. [Google Scholar]
- Greenhalgh, DG. Topical antimicrobial agents for burn wounds. Clin. Plast. Surg 2009, 36, 597–606. [Google Scholar]
- Laforce, FM; Boose, DS. Sublethal damage of Escherichia coli by lung lavage. Am. Rev. Respir. Dis 1981, 124, 733–737. [Google Scholar]
- LaForce, FM; Boose, DS. Effect of zinc and phosphate on an antibacterial peptide isolated from lung lavage. Infect. Immun 1984, 45, 692–696. [Google Scholar]
- Harris, F; Dennison, SR; Phoenix, DA. Anionic antimicrobial peptides from eukaryotic organisms. Curr. Protein Pept. Sci 2009, 10, 585–606. [Google Scholar]
- Lai, R; Liu, H; Hui Lee, W; Zhang, Y. An anionic antimicrobial peptide from toad. Bombina Maxima. Biochem. Biophys. Res. Commun 2002, 295, 796–799. [Google Scholar]
- Steffen, H; Rieg, S; Wiedemann, I; Kalbacher, H; Deeg, M; Sahl, HG; Peschel, A; Gotz, F; Garbe, C; Schittek, B. Naturally processed dermcidin-derived peptides do not permeabilize bacterial membranes and kill microorganisms irrespective of their charge. Antimicrob. Agents Chemother 2006, 50, 2608–2620. [Google Scholar]
- Goumon, Y; Lugardon, K; Gadroy, P; Strub, JM; Welters, ID; Stefano, GB; Aunis, D; Metz-Boutigue, MH. Processing of proenkephalin-a in bovine chromaffin cells. Identification of natural derived fragments by N-terminal sequencing and matrix-assisted laser desorption ionization-time of flight mass spectrometry. J. Biol. Chem 2000, 275, 38355–38362. [Google Scholar]
- Diego-Garcia, E; Batista, CV; Garcia-Gomez, BI; Lucas, S; Candido, DM; Gomez-Lagunas, F; Possani, LD. The Brazilian scorpion Tityus costatus karsch: Genes, peptides and function. Toxicon 2005, 45, 273–283. [Google Scholar]
- Bruhn, H; Winkelmann, J; Andersen, C; Andra, J; Leippe, M. Dissection of the mechanisms of cytolytic and antibacterial activity of lysenin, a defence protein of the annelid Eisenia Fetida. Dev. Comp. Immunol 2006, 30, 597–606. [Google Scholar]
- Prochazkova, P; Silerova, M; Felsberg, J; Joskova, R; Beschin, A; de Baetselier, P; Bilej, M. Relationship between hemolytic molecules in Eisenia Fetida earthworms. Dev. Comp. Immunol 2006, 30, 381–392. [Google Scholar]
- Brogden, KA; de Lucca, AJ; Bland, J; Elliott, S. Isolation of an ovine pulmonary surfactant-associated anionic peptide bactericidal for Pasteurella haemolytica. Proc. Natl. Acad. Sci. USA 1996, 93, 412–416. [Google Scholar]
- Grubor, B; Meyerholz, DK; Ackermann, MR. Collectins and cationic antimicrobial peptides of the respiratory epithelia. Vet. Pathol 2006, 43, 595–612. [Google Scholar]
- Schutte, BC; McCray, PB, Jr. [Beta]-defensins in lung host defense. Annu. Rev. Physiol 2002, 64, 709–748. [Google Scholar]
- Brogden, KA; Ackermann, M; McCray, PB, Jr; Tack, BF. Antimicrobial peptides in animals and their role in host defences. Int. J. Antimicrob Agents 2003, 22, 465–478. [Google Scholar]
- Vandendriessche, L. Inhibitors of ribonuclease activity. Arch. Biochem. Biophys 1956, 65, 347–353. [Google Scholar]
- Sela, M. Inhibition of ribonuclease by copolymers of glutamic acid and aromatic amino acids. J. Biol. Chem 1962, 237, 418–421. [Google Scholar]
- Conlon, JM; Al-Ghaferi, N; Abraham, B; Leprince, J. Strategies for transformation of naturally-occurring amphibian antimicrobial peptides into therapeutically valuable anti-infective agents. Methods 2007, 42, 349–357. [Google Scholar]
- Conlon, JM; Raza, H; Coquet, L; Jouenne, T; Leprince, J; Vaudry, H; King, JD. Purification of peptides with differential cytolytic activities from the skin secretions of the central American frog, Lithobates Vaillanti (Ranidae). Comp. Biochem. Physiol. C. Toxicol. Pharmacol 2009, 150, 150–154. [Google Scholar]
- VanCompernolle, SE; Taylor, RJ; Oswald-Richter, K; Jiang, J; Youree, BE; Bowie, JH; Tyler, MJ; Conlon, JM; Wade, D; Aiken, C; et al. Antimicrobial peptides from amphibian skin potently inhibit human immunodeficiency virus infection and transfer of virus from dendritic cells to T cells. J. Virol 2005, 79, 11598–11606. [Google Scholar]
- Nicolas, P; Mor, A. Peptides as weapons against microorganisms in the chemical defense system of vertebrates. Annu. Rev. Microbiol 1995, 49, 277–304. [Google Scholar]
- Simmaco, M; Mignogna, G; Barra, D. Antimicrobial peptides from amphibian skin: What do they tell us? Biopolymers 1998, 47, 435–450. [Google Scholar]
- Tyler, MJ; Stone, DJ; Bowie, JH. A novel method for the release and collection of dermal, glandular secretions from the skin of frogs. J. Pharmacol. Toxicol. Methods 1992, 28, 199–200. [Google Scholar]
- Nutkins, JC; Williams, DH. Identification of highly acidic peptides from processing of the skin prepropeptides of Xenopus laevis. Eur. J. Biochem 1989, 181, 97–102. [Google Scholar]
- Mor, A; Hani, K; Nicolas, P. The vertebrate peptide antibiotics dermaseptins have overlapping structural features but target specific microorganisms. J. Biol. Chem 1994, 269, 31635–31641. [Google Scholar]
- Conlon, JM; Kolodziejek, J; Nowotny, N. Antimicrobial peptides from ranid frogs: Taxonomic and phylogenetic markers and a potential source of new therapeutic agents. Biochim. Biophys. Acta 2004, 1696, 1–14. [Google Scholar]
- Tossi, A; Sandri, L; Giangaspero, A. Amphipathic, alpha-helical antimicrobial peptides. Biopolymers 2000, 55, 4–30. [Google Scholar]
- Mignogna, G; Simmaco, M; Kreil, G; Barra, D. Antibacterial and haemolytic peptides containing d-alloisoleucine from the skin of Bombina variegata. EMBO J 1993, 12, 4829–4832. [Google Scholar]
- Clark, DP; Durell, S; Maloy, WL; Zasloff, M. Ranalexin. A novel antimicrobial peptide from bullfrog (Rana Catesbeiana) skin, structurally related to the bacterial antibiotic, polymyxin. J. Biol. Chem 1994, 269, 10849–10855. [Google Scholar]
- Mor, A; Nguyen, VH; Delfour, A; Migliore-Samour, D; Nicolas, P. Isolation, amino acid sequence, and synthesis of dermaseptin, a novel antimicrobial peptide of amphibian skin. Biochemistry 1991, 30, 8824–8830. [Google Scholar]
- Raftery, MJ; Waugh, RJ; Bowie, JH; Wallace, JC; Tyler, MJ. The structures of the frenatin peptides from the skin secretion of the giant tree frog Litoria infrafrenata. J. Pept. Sci 1996, 2, 117–124. [Google Scholar]
- Lee, WH; Li, Y; Lai, R; Li, S; Zhang, Y; Wang, W. Variety of antimicrobial peptides in the bombina maxima toad and evidence of their rapid diversification. Eur. J. Immunol 2005, 35, 1220–1229. [Google Scholar]
- Lai, R; Zheng, YT; Shen, JH; Liu, GJ; Liu, H; Lee, WH; Tang, SZ; Zhang, Y. Antimicrobial peptides from skin secretions of Chinese red belly toad Bombina maxima. Peptides 2002, 23, 427–435. [Google Scholar]
- Hernandez, C; Mor, A; Dagger, F; Nicolas, P; Hernandez, A; Benedetti, EL; Dunia, I. Functional and structural damage in Leishmania mexicana exposed to the cationic peptide dermaseptin. Eur. J. Cell Biol 1992, 59, 414–424. [Google Scholar]
- Strom, MB; Haug, BE; Skar, ML; Stensen, W; Stiberg, T; Svendsen, JS. The pharmacophore of short cationic antibacterial peptides. J. Med. Chem 2003, 46, 1567–1570. [Google Scholar]
- Haug, BE; Stensen, W; Stiberg, T; Svendsen, JS. Bulky nonproteinogenic amino acids permit the design of very small and effective cationic antibacterial peptides. J. Med. Chem 2004, 47, 4159–4162. [Google Scholar]
- Sieber, P. Modification of tryptophan residues during acidolysis of 4-methoxy-2,3,6-trimethylbenzenesulfonyl groups. Effects of scavengers. Tetrahedron Lett 1987, 28, 1637–1640. [Google Scholar]
- Choi, H; Aldrich, JV. Comparison of methods for the fmoc solid-phase synthesis and cleavage of a peptide containing both tryptophan and arginine. Int. J. Pept. Protein Res 1993, 42, 58–63. [Google Scholar]
- Makovitzki, A; Baram, J; Shai, Y. Antimicrobial lipopolypeptides composed of palmitoyl di- and tricationic peptides: In vitro and in vivo activities, self-assembly to nanostructures, and a plausible mode of action. Biochemistry 2008, 47, 10630–10636. [Google Scholar]
- Vallon-Eberhard, A; Makovitzki, A; Beauvais, A; Latge, JP; Jung, S; Shai, Y. Efficient clearance of aspergillus fumigatus in murine lungs by an ultrashort antimicrobial lipopeptide, palmitoyl-Lys-Ala-DAla-Lys. Antimicrob. Agents Chemother 2008, 52, 3118–3126. [Google Scholar]
- Makovitzki, A; Avrahami, D; Shai, Y. Ultrashort antibacterial and antifungal lipopeptides. Proc. Natl. Acad. Sci. USA 2006, 103, 15997–16002. [Google Scholar]
- Hancock, RE; Sahl, HG. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol 2006, 24, 1551–1557. [Google Scholar]
- Schmidtchen, A; Frick, IM; Andersson, E; Tapper, H; Bjorck, L. Proteinases of common pathogenic bacteria degrade and inactivate the antibacterial peptide LL-37. Mol. Microbiol 2002, 46, 157–168. [Google Scholar]
- Chen, Y; Vasil, AI; Rehaume, L; Mant, CT; Burns, JL; Vasil, ML; Hancock, RE; Hodges, RS. Comparison of biophysical and biologic properties of alpha-helical enantiomeric antimicrobial peptides. Chem. Biol. Drug Des 2006, 67, 162–173. [Google Scholar]
- Papo, N; Oren, Z; Pag, U; Sahl, HG; Shai, Y. The consequence of sequence alteration of an amphipathic alpha-helical antimicrobial peptide and its diastereomers. J. Biol. Chem 2002, 277, 33913–33921. [Google Scholar]
- Thennarasu, S; Lee, DK; Tan, A; Prasad Kari, U; Ramamoorthy, A. Antimicrobial activity and membrane selective interactions of a synthetic lipopeptide MSI-843. Biochim. Biophys. Acta 2005, 1711, 49–58. [Google Scholar]
- Zhang, L; Falla, TJ. Antimicrobial peptides: Therapeutic potential. Expert Opin. Pharmacother 2006, 7, 653–663. [Google Scholar]
- Hilpert, K; Volkmer-Engert, R; Walter, T; Hancock, RE. High-throughput generation of small antibacterial peptides with improved activity. Nat. Biotechnol 2005, 23, 1008–1012. [Google Scholar]
- Haug, BE; Svendsen, JS. The role of tryptophan in the antibacterial activity of a 15-residue bovine lactoferricin peptide. J. Pept. Sci 2001, 7, 190–196. [Google Scholar]
- Lejon, T; Svendsen, JS; Haug, BE. Simple parameterization of non-proteinogenic amino acids for QSAR of antibacterial peptides. J. Pept. Sci 2002, 8, 302–306. [Google Scholar]
- Strom, MB; Rekdal, O; Svendsen, JS. Antibacterial activity of 15-residue lactoferricin derivatives. J. Pept. Res 2000, 56, 265–274. [Google Scholar]
- Strom, MB; Haug, BE; Rekdal, O; Skar, ML; Stensen, W; Svendsen, JS. Important structural features of 15-residue lactoferricin derivatives and methods for improvement of antimicrobial activity. Biochem. Cell Biol 2002, 80, 65–74. [Google Scholar]
- Strom, MB; Rekdal, O; Svendsen, JS. The effects of charge and lipophilicity on the antibacterial activity of undecapeptides derived from bovine lactoferricin. J. Pept. Sci 2002, 8, 36–43. [Google Scholar]
- Strom, MB; Rekdal, O; Svendsen, JS. Antimicrobial activity of short arginine- and tryptophan-rich peptides. J. Pept. Sci 2002, 8, 431–437. [Google Scholar]
- Japelj, B; Zorko, M; Majerle, A; Pristovsek, P; Sanchez-Gomez, S; Martinez de Tejada, G; Moriyon, I; Blondelle, SE; Brandenburg, K; Andra, J; et al. The acyl group as the central element of the structural organization of antimicrobial lipopeptide. J. Am. Chem. Soc 2007, 129, 1022–1023. [Google Scholar]
- Jerala, R. Synthetic lipopeptides: A novel class of anti-infectives. Expert Opin. Investig. Drugs 2007, 16, 1159–1169. [Google Scholar]
- Avrahami, D; Shai, Y. A new group of antifungal and antibacterial lipopeptides derived from non-membrane active peptides conjugated to palmitic acid. J. Biol. Chem 2004, 279, 12277–12285. [Google Scholar]
- Kabara, JJ; Swieczkowski, DM; Conley, AJ; Truant, JP. Fatty acids and derivatives as antimicrobial agents. Antimicrob. Agents Chemother 1972, 2, 23–28. [Google Scholar]
- Kitahara, T; Koyama, N; Matsuda, J; Aoyama, Y; Hirakata, Y; Kamihira, S; Kohno, S; Nakashima, M; Sasaki, H. Antimicrobial activity of saturated fatty acids and fatty amines against methicillin-resistant Staphylococcus aureus. Biol. Pharm. Bull 2004, 27, 1321–1326. [Google Scholar]
- Shalev, DE; Rotem, S; Fish, A; Mor, A. Consequences of N-acylation on structure and membrane binding properties of dermaseptin derivative K4-S4-(1–13). J. Biol. Chem 2006, 281, 9432–9438. [Google Scholar]
- Tsubery, H; Ofek, I; Cohen, S; Fridkin, M. N-terminal modifications of polymyxin B nonapeptide and their effect on antibacterial activity. Peptides 2001, 22, 1675–1681. [Google Scholar]
- Malina, A; Shai, Y. Conjugation of fatty acids with different lengths modulates the antibacterial and antifungal activity of a cationic biologically inactive peptide. Biochem. J 2005, 390, 695–702. [Google Scholar]
- Arbeit, RD; Maki, D; Tally, FP; Campanaro, E; Eisenstein, BI. Daptomycin 98–01 and 99–01 Investigators. The safety and efficacy of daptomycin for the treatment of complicated skin and skin-structure infections. Clin. Infect. Dis 2004, 38, 1673–1681. [Google Scholar]
- Livermore, DM. Future directions with daptomycin. J Antimicrob Chemother 2008, 62, iii41–iii49. [Google Scholar]
- Mohr, JF; Friedrich, LV; Yankelev, S; Lamp, KC. Daptomycin for the treatment of Enterococcal bacteraemia: Results from the Cubicin Outcomes Registry and Experience (CORE). Int. J. Antimicrob. Agents 2009, 33, 543–548. [Google Scholar]
- Andrew, JH; Wale, MC; Wale, LJ; Greenwood, D. The effect of cultural conditions on the activity of LY146032 against Staphylococci and Streptococci. J. Antimicrob. Chemother 1987, 20, 213–221. [Google Scholar]
- Chow, AW; Cheng, N. In vitro activities of daptomycin (LY146032) and paldimycin (U-70,138F) against anaerobic gram-positive bacteria. Antimicrob. Agents Chemother 1988, 32, 788–790. [Google Scholar]
- Alder, JD. Daptomycin: A new drug class for the treatment of gram-positive infections. Drugs Today (Barc. ) 2005, 41, 81–90. [Google Scholar]
- Silverman, JA; Mortin, LI; Vanpraagh, AD; Li, T; Alder, J. Inhibition of daptomycin by pulmonary surfactant: In vitro modeling and clinical impact. J. Infect. Dis 2005, 191, 2149–2152. [Google Scholar]
- Steenbergen, JN; Alder, J; Thorne, GM; Tally, FP. Daptomycin: A lipopeptide antibiotic for the treatment of serious gram-positive infections. J. Antimicrob. Chemother 2005, 55, 283–288. [Google Scholar]
- Jung, D; Rozek, A; Okon, M; Hancock, RE. Structural transitions as determinants of the action of the calcium-dependent antibiotic daptomycin. Chem. Biol 2004, 11, 949–957. [Google Scholar]
- Silverman, JA; Perlmutter, NG; Shapiro, HM. Correlation of daptomycin bactericidal activity and membrane depolarization in Staphylococcus aureus. Antimicrob. Agents Chemother 2003, 47, 2538–2544. [Google Scholar]
- Shoemaker, DM; Simou, J; Roland, WE. A review of daptomycin for injection (cubicin) in the treatment of complicated skin and skin structure infections. Ther. Clin. Risk Manag 2006, 2, 169–174. [Google Scholar]
- Ainsworth, GC; Brown, AM; Brownlee, G. Aerosporin, an antibiotic produced by Bacillus aerosporus Greer. Nature 1947, 160, 263. [Google Scholar]
- Stansly, PG; Shepherd, RG; White, HL. Polymyxin: A new chemotherapeutic agent. Johns. Hopk. Hosp. Bull 1947, 81, 43–54. [Google Scholar]
- Benedict, RG; Langlykke, AF. Antibiotic activity of Bacillus polymyxa. J. Bacteriol 1947, 54, 24–25. [Google Scholar]
- Koyama, Y; Kurosasa, A; Tsuchiya, A; Takakuta, K. A new antibiotic ‘colistin’ produced by spore-forming soil bacteria. J. Antibiot 1950, 3, 457–458. [Google Scholar]
- Kanazawa, K; Sato, Y; Ohki, K; Okimura, K; Uchida, Y; Shindo, M; Sakura, N. Contribution of each amino acid residue in polymyxin B(3) to antimicrobial and lipopolysaccharide binding activity. Chem. Pharm. Bull. (Tokyo) 2009, 57, 240–244. [Google Scholar]
- Kwa, A; Kasiakou, SK; Tam, VH; Falagas, ME. Polymyxin B: Similarities to and differences from colistin (polymyxin E). Expert Rev. Anti Infect. Ther 2007, 5, 811–821. [Google Scholar]
- Falagas, ME; Kasiakou, SK. Toxicity of polymyxins: A systematic review of the evidence from old and recent studies. Crit Care 2006, 10. [Google Scholar] [CrossRef]
- Falagas, ME; Kasiakou, SK. Colistin: The revival of polymyxins for the management of multidrug-resistant gram-negative bacterial infections. Clin. Infect. Dis 2005, 40, 1333–1341. [Google Scholar]
- British Medical Association; Royal Pharmaceutical Society of Great Britain, British National Formulary; Pharmaceutical Press: London, UK, 2009.
- Li, J; Nation, RL; Milne, RW; Turnidge, JD; Coulthard, K. Evaluation of colistin as an agent against multi-resistant gram-negative bacteria. Int. J. Antimicrob. Agents 2005, 25, 11–25. [Google Scholar]
- Catchpole, CR; Andrews, JM; Brenwald, N; Wise, R. A reassessment of the in-vitro activity of colistin sulphomethate sodium. J. Antimicrob. Chemother 1997, 39, 255–260. [Google Scholar]
- Vaara, M. Agents that increase the permeability of the outer membrane. Microbiol. Rev 1992, 56, 395–411. [Google Scholar]
- Vaara, M; Porro, M. Group of peptides that act synergistically with hydrophobic antibiotics against gram-negative enteric bacteria. Antimicrob. Agents Chemother 1996, 40, 1801–1805. [Google Scholar]
- Clausell, A; Garcia-Subirats, M; Pujol, M; Busquets, MA; Rabanal, F; Cajal, Y. Gram-negative outer and inner membrane models: Insertion of cyclic cationic lipopeptides. J. Phys. Chem. B 2007, 111, 551–563. [Google Scholar]
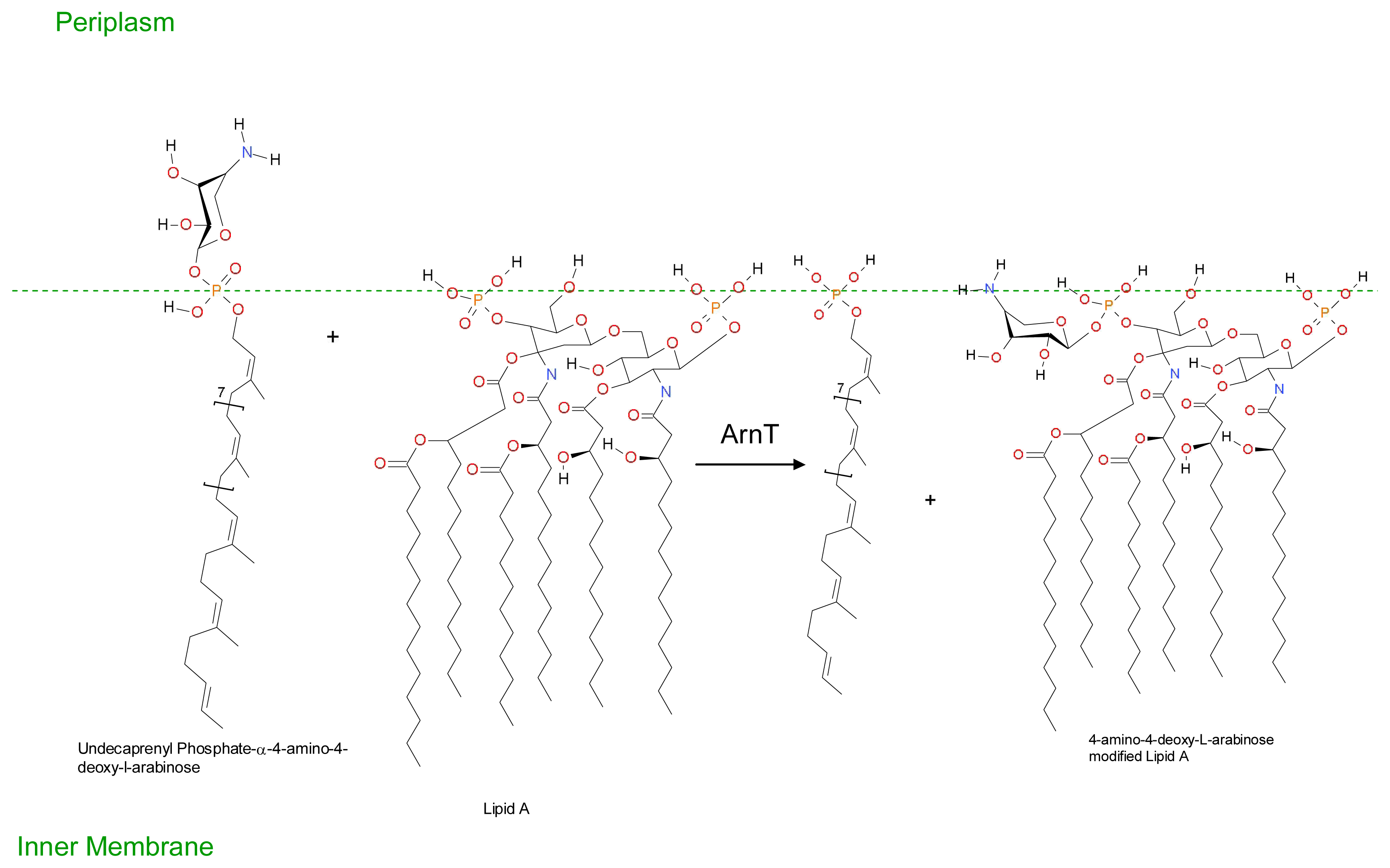
- Groisman, EA; Kayser, J; Soncini, FC. Regulation of polymyxin resistance and adaptation to low-Mg2+ environments. J. Bacteriol 1997, 179, 7040–7045. [Google Scholar]
- Conrad, RS; Galanos, C. Fatty acid alterations and polymyxin B binding by lipopolysaccharides from Pseudomonas aeruginosa adapted to polymyxin B resistance. Antimicrob. Agents Chemother 1989, 33, 1724–1728. [Google Scholar]
- Gilleland, HE, Jr; Lyle, RD. Chemical alterations in cell envelopes of polymyxin-resistant Pseudomonas aeruginosa isolates. J. Bacteriol 1979, 138, 839–845. [Google Scholar]
- Shand, GH; Anwar, H; Brown, MR. Outer membrane proteins of polymyxin resistant Pseudomonas aeruginosa: Effect of magnesium depletion. J. Antimicrob. Chemother 1988, 22, 811–821. [Google Scholar]
- Warren, HS; Kania, SA; Siber, GR. Binding and neutralization of bacterial lipopolysaccharide by colistin nonapeptide. Antimicrob. Agents Chemother 1985, 28, 107–112. [Google Scholar]
- Cardoso, LS; Araujo, MI; Goes, AM; Pacifico, LG; Oliveira, RR; Oliveira, SC. Polymyxin B as inhibitor of LPS contamination of Schistosoma mansoni recombinant proteins in human cytokine analysis. Microb. Cell. Fact 2007, 6, 1–6. [Google Scholar]
- Koch-Weser, J; Sidel, VW; Federman, EB; Kanarek, P; Finer, DC; Eaton, AE. Adverse effects of sodium colistimethate. manifestations and specific reaction rates during 317 courses of therapy. Ann. Intern. Med 1970, 72, 857–868. [Google Scholar]
- Brown, JM; Dorman, DC; Roy, LP. Acute renal failure due to overdosage of colistin. Med. J. Aust 1970, 2, 923–924. [Google Scholar]
- Falagas, ME; Rizos, M; Bliziotis, IA; Rellos, K; Kasiakou, SK; Michalopoulos, A. Toxicity after prolonged (more than four weeks) administration of intravenous colistin. BMC Infect Dis 2005, 5. [Google Scholar] [CrossRef] [Green Version]
- Bergen, PJ; Li, J; Rayner, CR; Nation, RL. Colistin methanesulfonate is an inactive prodrug of colistin against Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2006, 50, 1953–1958. [Google Scholar]
- Matsuzaki, K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta 1999, 1462, 1–10. [Google Scholar]
- Neuhaus, FC; Baddiley, J. A continuum of anionic charge: Structures and functions of d-alanyl-teichoic acids in gram-positive bacteria. Microbiol. Mol. Biol. Rev 2003, 67, 686–723. [Google Scholar]
- Muhle, SA; Tam, JP. Design of gram-negative selective antimicrobial peptides. Biochemistry 2001, 40, 5777–5785. [Google Scholar]
- Avrahami, D; Shai, Y. Bestowing antifungal and antibacterial activities by lipophilic acid conjugation to d,l-amino acid-containing antimicrobial peptides: A plausible mode of action. Biochemistry 2003, 42, 14946–14956. [Google Scholar]
- Sung, WS; Lee, DG. Pleurocidin-derived antifungal peptides with selective membrane-disruption effect. Biochem. Biophys. Res. Commun 2008, 369, 858–861. [Google Scholar]
- Lopez-Garcia, B; Marcos, JF; Abad, C; Perez-Paya, E. Stabilisation of mixed peptide/lipid complexes in selective antifungal hexapeptides. Biochim. Biophys. Acta 2004, 1660, 131–137. [Google Scholar]
- Jenssen, H; Hamill, P; Hancock, RE. Peptide antimicrobial agents. Clin. Microbiol. Rev 2006, 19, 491–511. [Google Scholar]
- Hallock, KJ; Lee, DK; Ramamoorthy, A. MSI-78, an analogue of the magainin antimicrobial peptides, disrupts lipid bilayer structure via positive curvature strain. Biophys. J 2003, 84, 3052–3060. [Google Scholar]
- Cirac, AD; Moiset, G; Mika, JT; Kocer, A; Salvador, P; Poolman, B; Marrink, SJ; Sengupta, D. The molecular basis for antimicrobial activity of pore-forming cyclic peptides. Biophys. J 2011, 100, 2422–2431. [Google Scholar]
- Matsuzaki, K. Magainins as paradigm for the mode of action of pore forming polypeptides. Biochim. Biophys. Acta 1998, 1376, 391–400. [Google Scholar]
- Powers, JP; Tan, A; Ramamoorthy, A; Hancock, RE. Solution structure and interaction of the antimicrobial polyphemusins with lipid membranes. Biochemistry 2005, 44, 15504–15513. [Google Scholar]
- Ehrenstein, G; Lecar, H. Electrically gated ionic channels in lipid bilayers. Q. Rev. Biophys 1977, 10, 1–34. [Google Scholar]
- Shai, Y; Oren, Z. From “carpet” mechanism to de-novo designed diastereomeric cell-selective antimicrobial peptides. Peptides 2001, 22, 1629–1641. [Google Scholar]
- He, K; Ludtke, SJ; Worcester, DL; Huang, HW. Neutron scattering in the plane of membranes: Structure of alamethicin pores. Biophys. J 1996, 70, 2659–2666. [Google Scholar]
- Oren, Z; Shai, Y. Mode of action of linear amphipathic alpha-helical antimicrobial peptides. Biopolymers 1998, 47, 451–463. [Google Scholar]
- Bechinger, B; Lohner, K. Detergent-like actions of linear amphipathic cationic antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1529–1539. [Google Scholar]
- Sato, H; Feix, JB. Peptide-membrane interactions and mechanisms of membrane destruction by amphipathic alpha-helical antimicrobial peptides. Biochim. Biophys. Acta 2006, 1758, 1245–1256. [Google Scholar]
- Sawyer, JG; Martin, NL; Hancock, RE. Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect. Immun 1988, 56, 693–698. [Google Scholar]
- Black, CB; Huang, H-W; Cowan, JA. Biological Coordination chemistry of magnesium, sodium, and potassium ions. Protein and nucleotide binding sites. Coord Chem Rev 1994, 135–136, 165–202. [Google Scholar]
- Chen, CZ; Cooper, SL. Interactions between dendrimer biocides and bacterial membranes. Biomaterials 2002, 23, 3359–3368. [Google Scholar]
- Giacometti, A; Cirioni, O; Barchiesi, F; Fortuna, M; Scalise, G. In-vitro activity of cationic peptides alone and in combination with clinically used antimicrobial agents against Pseudomonas aeruginosa. J. Antimicrob. Chemother 1999, 44, 641–645. [Google Scholar]
- Giacometti, A; Cirioni, O; Del Prete, MS; Paggi, AM; D’Errico, MM; Scalise, G. Combination studies between polycationic peptides and clinically used antibiotics against gram-positive and gram-negative bacteria. Peptides 2000, 21, 1155–1160. [Google Scholar]
- Kumar, M; Chaturvedi, AK; Kavishwar, A; Shukla, PK; Kesarwani, AP; Kundu, B. Identification of a novel antifungal nonapeptide generated by combinatorial approach. Int. J. Antimicrob. Agents 2005, 25, 313–320. [Google Scholar]
- Lehrer, RI; Szklarek, D; Ganz, T; Selsted, ME. Correlation of binding of rabbit granulocyte peptides to Candida albicans with candidacidal activity. Infect. Immun 1985, 49, 207–211. [Google Scholar]
- Park, Y; Lee, DG; Hahm, KS. HP(2–9)-Magainin 2(1–12), a synthetic hybrid peptide, exerts its antifungal effect on candida albicans by damaging the plasma membrane. J. Pept. Sci 2004, 10, 204–209. [Google Scholar]
- Bellamy, W; Wakabayashi, H; Takase, M; Kawase, K; Shimamura, S; Tomita, M. Killing of Candida albicans by lactoferricin B, a potent antimicrobial peptide derived from the N-terminal region of bovine lactoferrin. Med. Microbiol. Immunol 1993, 182, 97–105. [Google Scholar]
- Ajesh, K; Sreejith, K. Peptide antibiotics: An alternative and effective antimicrobial strategy to circumvent fungal infections. Peptides 2009, 30, 999–1006. [Google Scholar]
- Maget-Dana, R; Peypoux, F. Iturins, a special class of pore-forming lipopeptides: Biological and physicochemical properties. Toxicology 1994, 87, 151–174. [Google Scholar]
- Vylkova, S; Sun, JN; Edgerton, M. The role of released ATP in killing Candida albicans and other extracellular microbial pathogens by cationic peptides. Purinergic Signal 2007, 3, 91–97. [Google Scholar]
- Park, CB; Kim, HS; Kim, SC. Mechanism of action of the antimicrobial peptide buforin II: Buforin II kills microorganisms by penetrating the cell membrane and inhibiting cellular functions. Biochem. Biophys. Res. Commun 1998, 244, 253–257. [Google Scholar]
- Boehr, DD; Draker, KA; Koteva, K; Bains, M; Hancock, RE; Wright, GD. Broad-spectrum peptide inhibitors of aminoglycoside antibiotic resistance enzymes. Chem. Biol 2003, 10, 189–196. [Google Scholar]
- Falla, TJ; Karunaratne, DN; Hancock, RE. Mode of action of the antimicrobial peptide indolicidin. J. Biol. Chem 1996, 271, 19298–19303. [Google Scholar]
- Lee, DG; Kim, HK; Kim, SA; Park, Y; Park, SC; Jang, SH; Hahm, KS. Fungicidal effect of indolicidin and its interaction with phospholipid membranes. Biochem. Biophys. Res. Commun 2003, 305, 305–310. [Google Scholar]
- Ginsburg, I; Koren, E. Are cationic antimicrobial peptides also ‘double-edged swords’? Expert Rev. Anti Infect. Ther 2008, 6, 453–462. [Google Scholar]
- Ginsburg, I. Bactericidal cationic peptides can also function as bacteriolysis-inducing agents mimicking beta-lactam antibiotics? It is enigmatic why this concept is consistently disregarded. Med. Hypotheses 2004, 62, 367–374. [Google Scholar]
- Boman, HG; Agerberth, B; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun 1993, 61, 2978–2984. [Google Scholar]
- Holzl, MA; Hofer, J; Steinberger, P; Pfistershammer, K; Zlabinger, GJ. Host antimicrobial proteins as endogenous immunomodulators. Immunol. Lett 2008, 119, 4–11. [Google Scholar]
- Xiong, YQ; Yeaman, MR; Bayer, AS. In vitro antibacterial activities of platelet microbicidal protein and neutrophil defensin against Staphylococcus aureus are influenced by antibiotics differing in mechanism of action. Antimicrob. Agents Chemother 1999, 43, 1111–1117. [Google Scholar]
- Ulvatne, H; Samuelsen, O; Haukland, HH; Kramer, M; Vorland, LH. Lactoferricin B inhibits bacterial macromolecular synthesis in Escherichia coli and Bacillus subtilis. FEMS Microbiol. Lett 2004, 237, 377–384. [Google Scholar]
- Subbalakshmi, C; Sitaram, N. Mechanism of antimicrobial action of indolicidin. FEMS Microbiol. Lett 1998, 160, 91–96. [Google Scholar]
- Healy, DP; Gardner, JC; Puthoff, BK; Kagan, RJ; Neely, AN. Antibiotic-mediated bacterial filamentation: A potentially important laboratory phenomenon. Clin. Microbiol. Newsl 2007, 29, 22–24. [Google Scholar]
- Hsu, CH; Chen, C; Jou, ML; Lee, AY; Lin, YC; Yu, YP; Huang, WT; Wu, SH. Structural and DNA-binding studies on the bovine antimicrobial peptide, indolicidin: Evidence for multiple conformations involved in binding to membranes and DNA. Nucleic Acids Res 2005, 33, 4053–4064. [Google Scholar]
- Iimura, M; Gallo, RL; Hase, K; Miyamoto, Y; Eckmann, L; Kagnoff, MF. Cathelicidin mediates innate intestinal defense against colonization with epithelial adherent bacterial pathogens. J. Immunol 2005, 174, 4901–4907. [Google Scholar]
- Eckmann, L. Defence molecules in intestinal innate immunity against bacterial infections. Curr. Opin. Gastroenterol 2005, 21, 147–151. [Google Scholar]
- Agerberth, B; Charo, J; Werr, J; Olsson, B; Idali, F; Lindbom, L; Kiessling, R; Jornvall, H; Wigzell, H; Gudmundsson, GH. The human antimicrobial and chemotactic peptides LL-37 and alpha-defensins are expressed by specific lymphocyte and monocyte populations. Blood 2000, 96, 3086–3093. [Google Scholar]
- Zanetti, M. Cathelicidins, multifunctional peptides of the innate immunity. J. Leukoc. Biol 2004, 75, 39–48. [Google Scholar]
- Li, J; Post, M; Volk, R; Gao, Y; Li, M; Metais, C; Sato, K; Tsai, J; Aird, W; Rosenberg, RD; et al. PR39, a peptide regulator of angiogenesis. Nat. Med 2000, 6, 49–55. [Google Scholar]
- Rotem, S; Mor, A. Antimicrobial peptide mimics for improved therapeutic properties. Biochim. Biophys. Acta 2009, 1788, 1582–1592. [Google Scholar]
- Patrzykat, A; Friedrich, CL; Zhang, L; Mendoza, V; Hancock, RE. Sublethal concentrations of pleurocidin-derived antimicrobial peptides inhibit macromolecular synthesis in Escherichia coli. Antimicrob. Agents Chemother 2002, 46, 605–614. [Google Scholar]
- Park, CB; Kim, MS; Kim, SC. A novel antimicrobial peptide from Bufo Bufo gargarizans. Biochem. Biophys. Res. Commun 1996, 218, 408–413. [Google Scholar]
- Piddock, LJ. Antibacterials-mechanisms of action. Curr. Opin. Microbiol 1998, 1, 502–508. [Google Scholar]
- Hale, JD; Hancock, RE. Alternative mechanisms of action of cationic antimicrobial peptides on bacteria. Expert Rev. Anti Infect. Ther 2007, 5, 951–959. [Google Scholar]
- Otvos, L, Jr; Rogers, ME; Consolvo, PJ; Condie, BA; Lovas, S; Bulet, P; Blaszczyk-Thurin, M. Interaction between heat shock proteins and antimicrobial peptides. Biochemistry 2000, 39, 14150–14159. [Google Scholar]
- Kragol, G; Lovas, S; Varadi, G; Condie, BA; Hoffmann, R; Otvos, L, Jr. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry 2001, 40, 3016–3026. [Google Scholar]
- Otvos, L, Jr; Snyder, C; Condie, B; Bulet, P; Wade, JD. Chimeric antimicrobial peptides exhibit multiple modes of action. Int. J. Pept. Res. Ther 2005, 11, 29–42. [Google Scholar]
- Brotz, H; Bierbaum, G; Leopold, K; Reynolds, PE; Sahl, HG. The lantibiotic mersacidin inhibits peptidoglycan synthesis by targeting lipid II. Antimicrob. Agents Chemother 1998, 42, 154–160. [Google Scholar]
- Brotz, H; Sahl, HG. New insights into the mechanism of action of lantibiotics—Diverse biological effects by binding to the same molecular target. J. Antimicrob. Chemother 2000, 46, 1–6. [Google Scholar]
- Marr, AK; Gooderham, WJ; Hancock, RE. Antibacterial peptides for therapeutic use: Obstacles and realistic outlook. Curr. Opin. Pharmacol 2006, 6, 468–472. [Google Scholar]
- Helmerhorst, EJ; Breeuwer, P; van’t Hof, W; Walgreen-Weterings, E; Oomen, LC; Veerman, EC; Amerongen, AV; Abee, T. The cellular target of histatin 5 on Candida albicans is the energized mitochondrion. J. Biol. Chem 1999, 274, 7286–7291. [Google Scholar]
- Kavanagh, K; Dowd, S. Histatins: Antimicrobial peptides with therapeutic potential. J. Pharm. Pharmacol 2004, 56, 285–289. [Google Scholar]
- Hodges, RL; Hodges, DW; Goggans, K; Xuei, X; Skatrud, P; McGilvray, D. Genetic modification of an Echinocandin B-producing strain of Aspergillus nidulans to produce mutants blocked in sterigmatocystin biosynthesis. J. Ind. Microbiol 1994, 13, 372–381. [Google Scholar]
- Lorand, T; Kocsis, B. Recent advances in antifungal agents. Mini Rev. Med. Chem 2007, 7, 900–911. [Google Scholar]
- Gonzalez, GM; Tijerina, R; Najvar, LK; Bocanegra, R; Luther, M; Rinaldi, MG; Graybill, JR. Correlation between antifungal susceptibilities of Coccidioides immitis in vitro and antifungal treatment with caspofungin in a mouse model. Antimicrob. Agents Chemother 2001, 45, 1854–1859. [Google Scholar]
- Douglas, CM. Fungal beta(1,3)-d-glucan synthesis. Med. Mycol 2001, 39, 55–66. [Google Scholar]
- Giuliani, A; Pirri, G; Nicoletto, SF. Antimicrobial peptides: An overview of a promising class of therapeutics. Eur. J. Biol 2007, 2, 1–33. [Google Scholar]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother 2001, 45, 999–1007. [Google Scholar]
- Donlan, RM. Role of biofilms in antimicrobial resistance. ASAIO J 2000, 46, S47–S52. [Google Scholar]
- Mulcahy, H; Charron-Mazenod, L; Lewenza, S. Extracellular DNA chelates cations and induces antibiotic resistance in Pseudomonas aeruginosa biofilms. PLoS Pathog 2008, 4, 1–9. [Google Scholar]
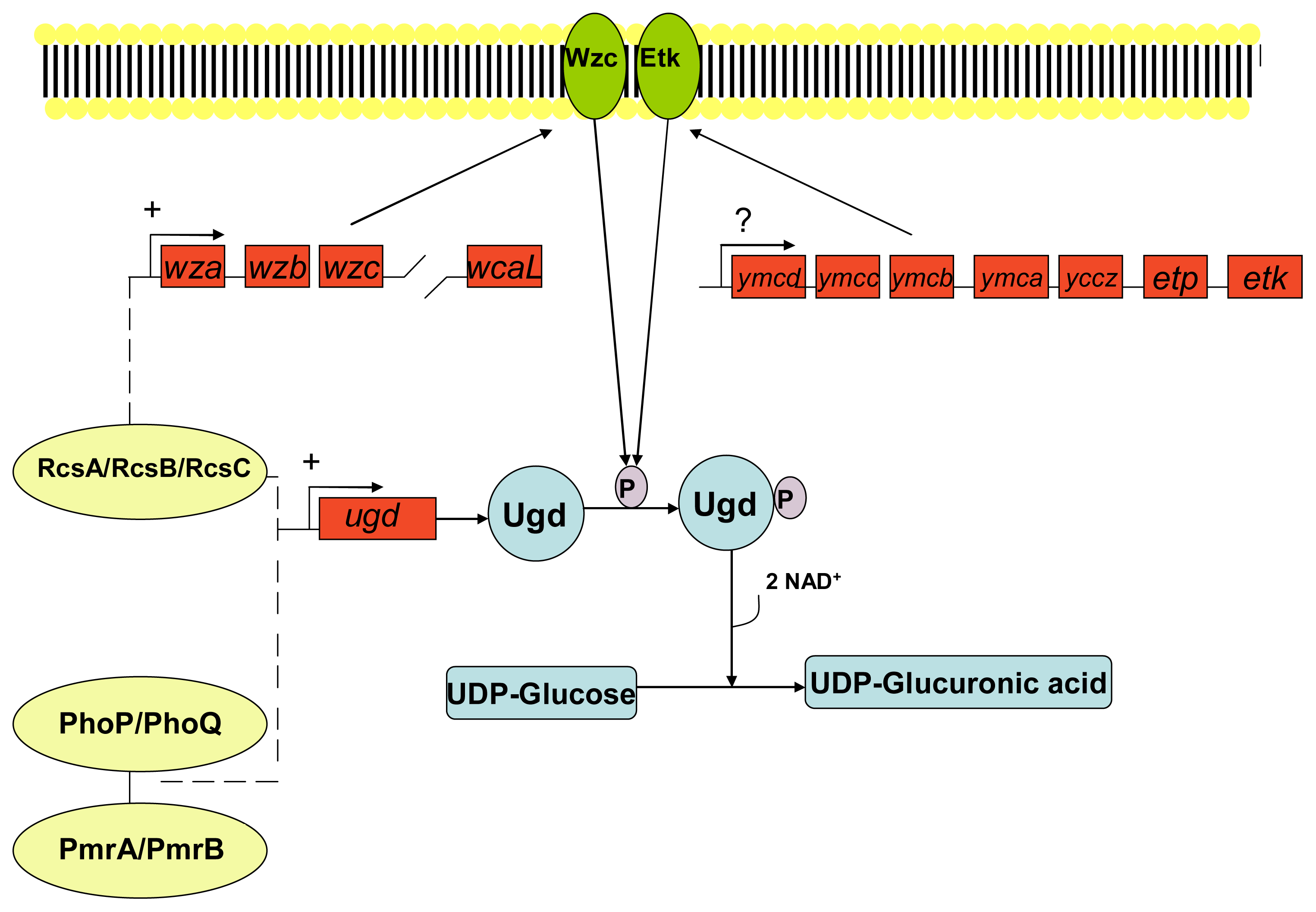
- Grangeasse, C; Cozzone, AJ; Deutscher, J; Mijakovic, I. Tyrosine phosphorylation: An emerging regulatory device of bacterial physiology. Trends Biochem. Sci 2007, 32, 86–94. [Google Scholar]
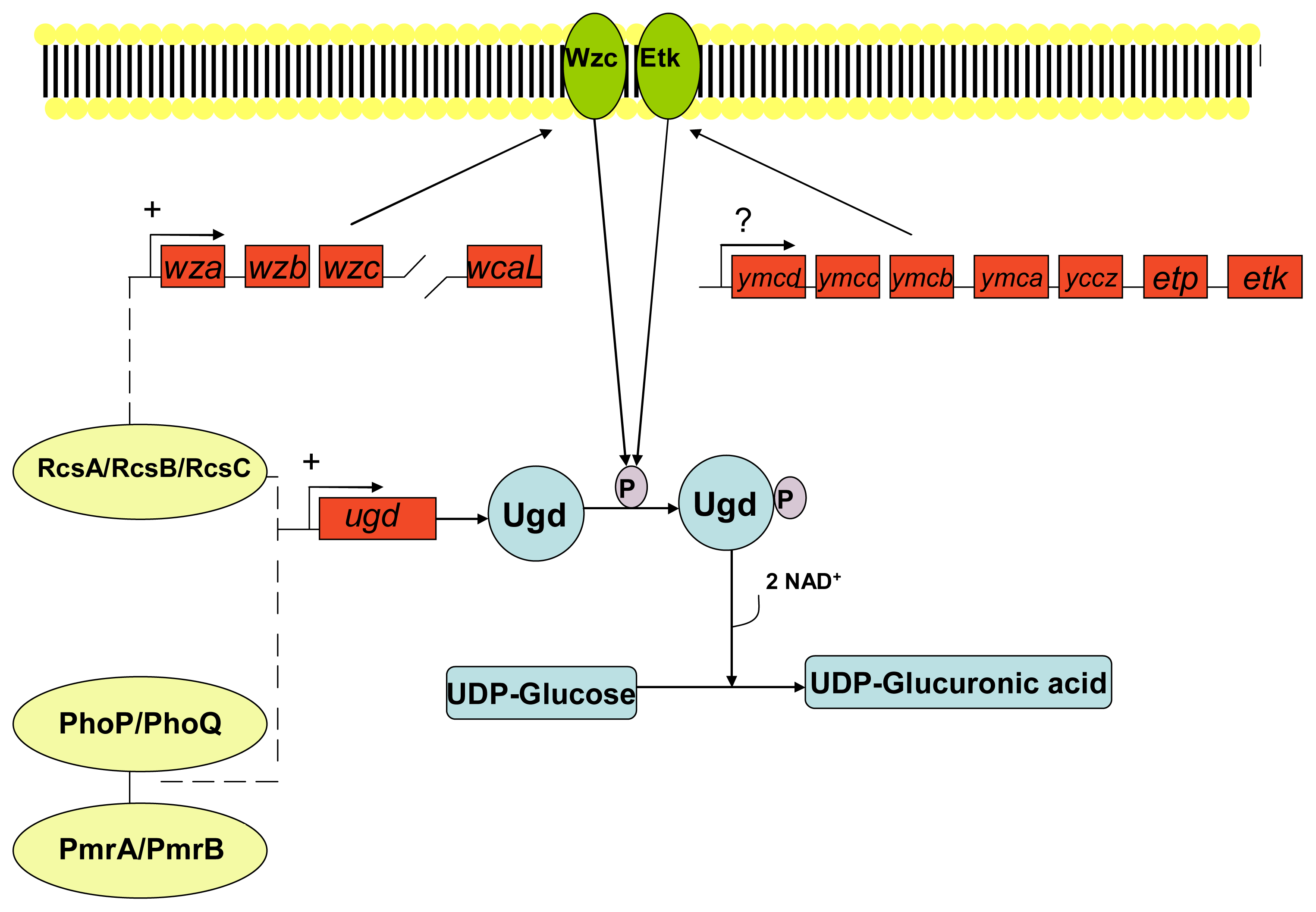
- Vincent, C; Doublet, P; Grangeasse, C; Vaganay, E; Cozzone, AJ; Duclos, B. Cells of Escherichia coli contain a protein-tyrosine kinase, wzc, and a phosphotyrosine-protein phosphatase, wzb. J. Bacteriol 1999, 181, 3472–3477. [Google Scholar]
- Peleg, A; Shifrin, Y; Ilan, O; Nadler-Yona, C; Nov, S; Koby, S; Baruch, K; Altuvia, S; Elgrably-Weiss, M; Abe, CM; et al. Identification of an Escherichia coli operon required for formation of the O-antigen capsule. J. Bacteriol 2005, 187, 5259–5266. [Google Scholar]
- Lacour, S; Bechet, E; Cozzone, AJ; Mijakovic, I; Grangeasse, C. Tyrosine phosphorylation of the UDP-glucose dehydrogenase of Escherichia coli is at the crossroads of colanic acid synthesis and polymyxin resistance. PLoS One 2008, 3, 1–9. [Google Scholar]
- Pirri, G; Giuliani, A; Nicoletto, SF; Pizzuto, L; Rinaldi, AC. Lipopeptides as anti-infectives: A practical perspective. Cent. Eur. J. Biol 2009, 4, 258–273. [Google Scholar]
- Jensen, T; Pedersen, SS; Garne, S; Heilmann, C; Hoiby, N; Koch, C. Colistin inhalation therapy in cystic fibrosis patients with chronic Pseudomonas aeruginosa lung infection. J. Antimicrob. Chemother 1987, 19, 831–838. [Google Scholar]
- Kun, P; Landau, LI; Phelan, PD. Nebulized gentamicin in children and adolescents with cystic fibrosis. Aust. Paediatr. J 1984, 20, 43–45. [Google Scholar]
- Hoskin, DW; Ramamoorthy, A. Studies on anticancer activities of antimicrobial peptides. Biochim. Biophys. Acta 2008, 1778, 357–375. [Google Scholar]
- Sambri, V; Marangoni, A; Giacani, L; Gennaro, R; Murgia, R; Cevenini, R; Cinco, M. Comparative in vitro activity of five cathelicidin-derived synthetic peptides against Leptospira, Borrelia and Treponema Pallidum. J. Antimicrob. Chemother 2002, 50, 895–902. [Google Scholar]
- Robinson, WE, Jr; McDougall, B; Tran, D; Selsted, ME. Anti-HIV-1 activity of indolicidin, an antimicrobial peptide from neutrophils. J. Leukoc. Biol 1998, 63, 94–100. [Google Scholar]
- Reddy, KV; Shahani, SK; Meherji, PK. Spermicidal activity of magainins: In vitro and in vivo studies. Contraception 1996, 53, 205–210. [Google Scholar]
- Yedery, RD; Reddy, KV. Antimicrobial peptides as microbicidal contraceptives: Prophecies for prophylactics—A mini review. Eur. J. Contracept. Reprod. Health Care 2005, 10, 32–42. [Google Scholar]
- Henriques, ST; Melo, MN; Castanho, MA. Cell-penetrating peptides and antimicrobial peptides: How different are they. Biochem J 2006, 399, 1–7. [Google Scholar]
- Reddy, KV; Yedery, RD; Aranha, C. Antimicrobial peptides: Premises and promises. Int. J. Antimicrob. Agents 2004, 24, 536–547. [Google Scholar]
- Carpenter, CF; Chambers, HF. Daptomycin: Another novel agent for treating infections due to drug-resistant gram-positive pathogens. Clin. Infect. Dis 2004, 38, 994–1000. [Google Scholar]
- Oleson, FB, Jr; Berman, CL; Kirkpatrick, JB; Regan, KS; Lai, JJ; Tally, FP. Once-daily dosing in dogs optimizes daptomycin safety. Antimicrob. Agents Chemother 2000, 44, 2948–2953. [Google Scholar]


© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laverty, G.; Gorman, S.P.; Gilmore, B.F. The Potential of Antimicrobial Peptides as Biocides. Int. J. Mol. Sci. 2011, 12, 6566-6596. https://doi.org/10.3390/ijms12106566
Laverty G, Gorman SP, Gilmore BF. The Potential of Antimicrobial Peptides as Biocides. International Journal of Molecular Sciences. 2011; 12(10):6566-6596. https://doi.org/10.3390/ijms12106566
Chicago/Turabian StyleLaverty, Garry, Sean P. Gorman, and Brendan F. Gilmore. 2011. "The Potential of Antimicrobial Peptides as Biocides" International Journal of Molecular Sciences 12, no. 10: 6566-6596. https://doi.org/10.3390/ijms12106566