Retro-MoRFs: Identifying Protein Binding Sites by Normal and Reverse Alignment and Intrinsic Disorder Prediction



Abstract
:1. Introduction
2. Results and Discussion
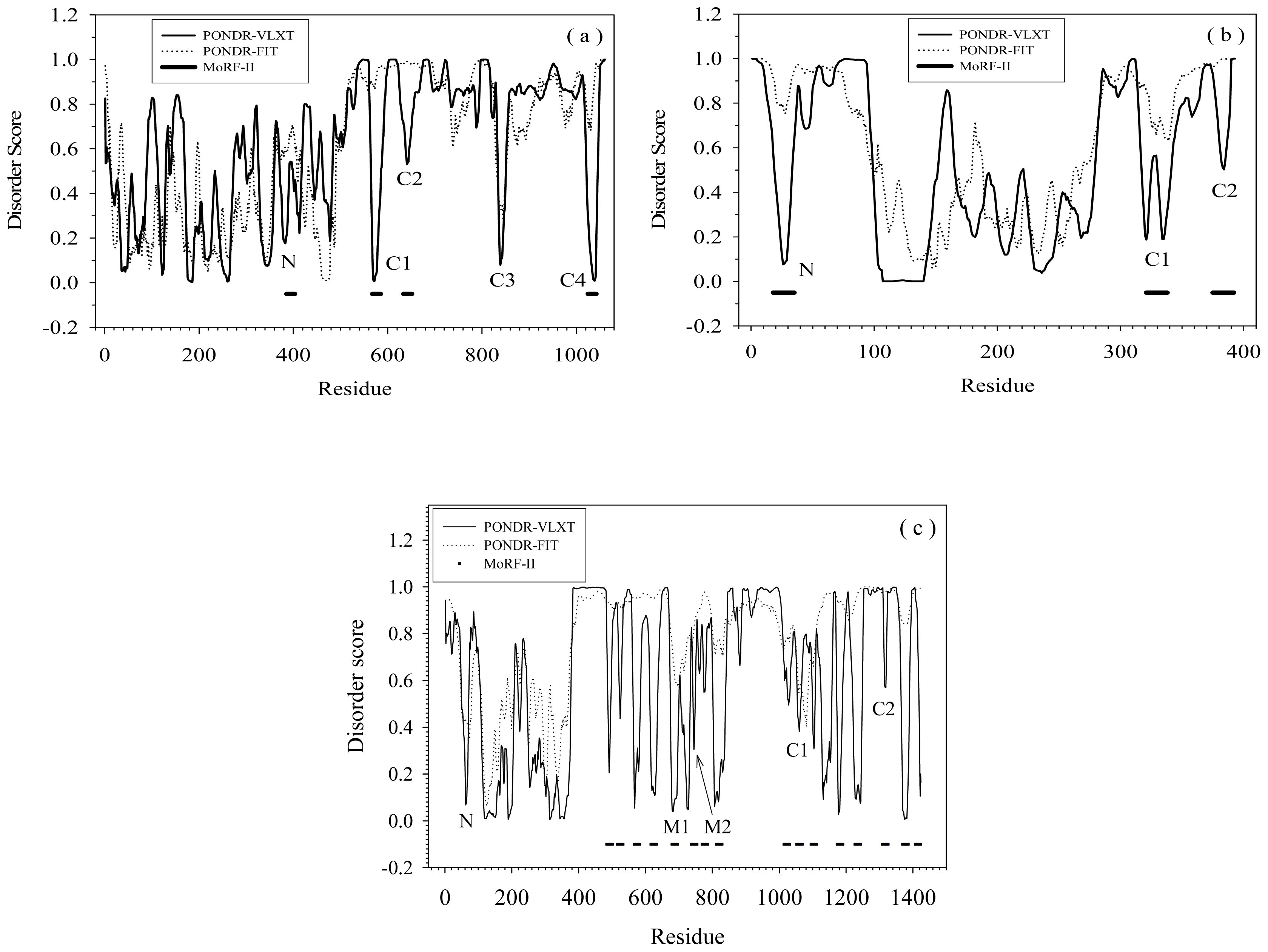
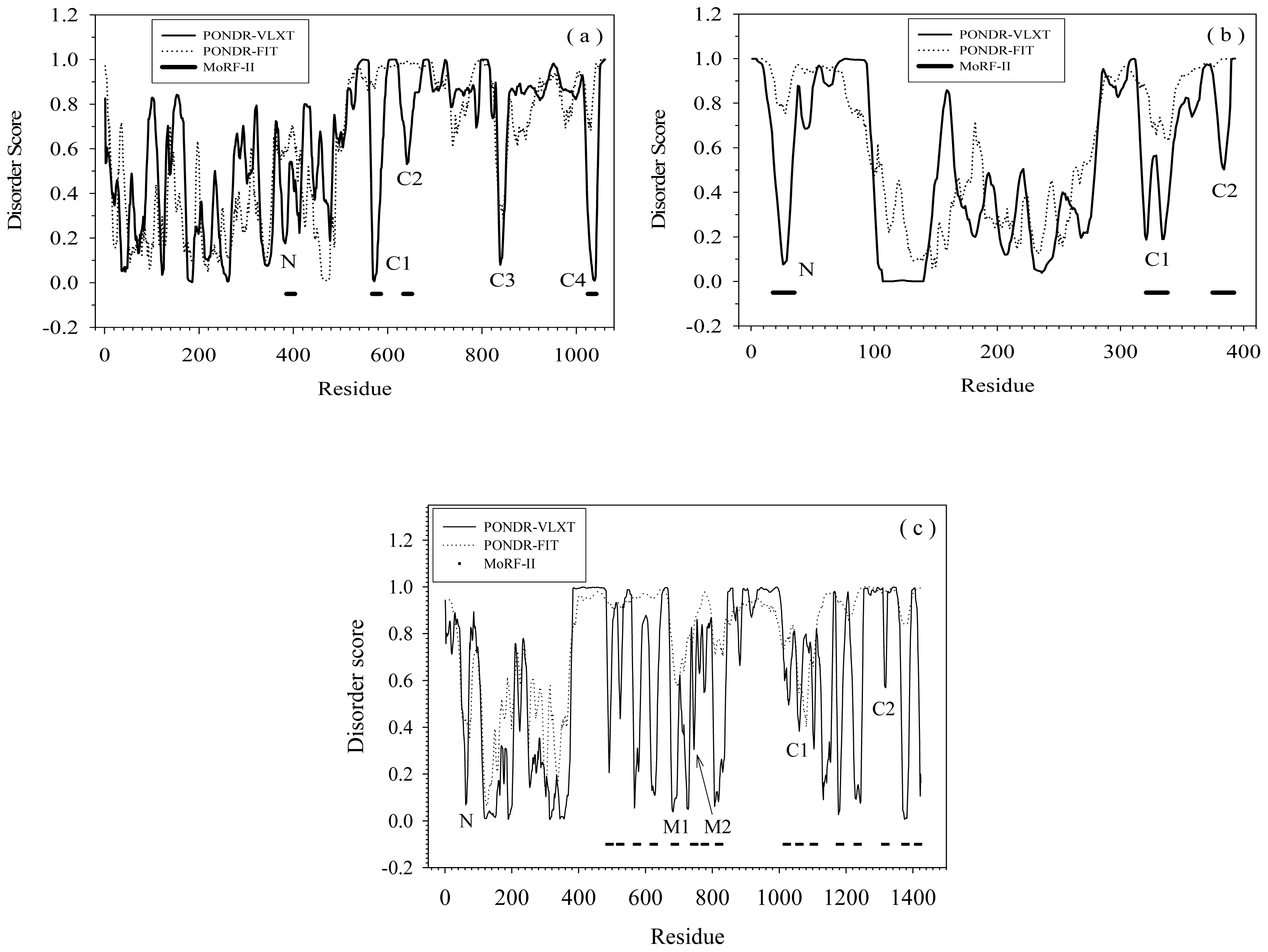
2.1. Functional Roles of MoRF Regions in Three Proteins
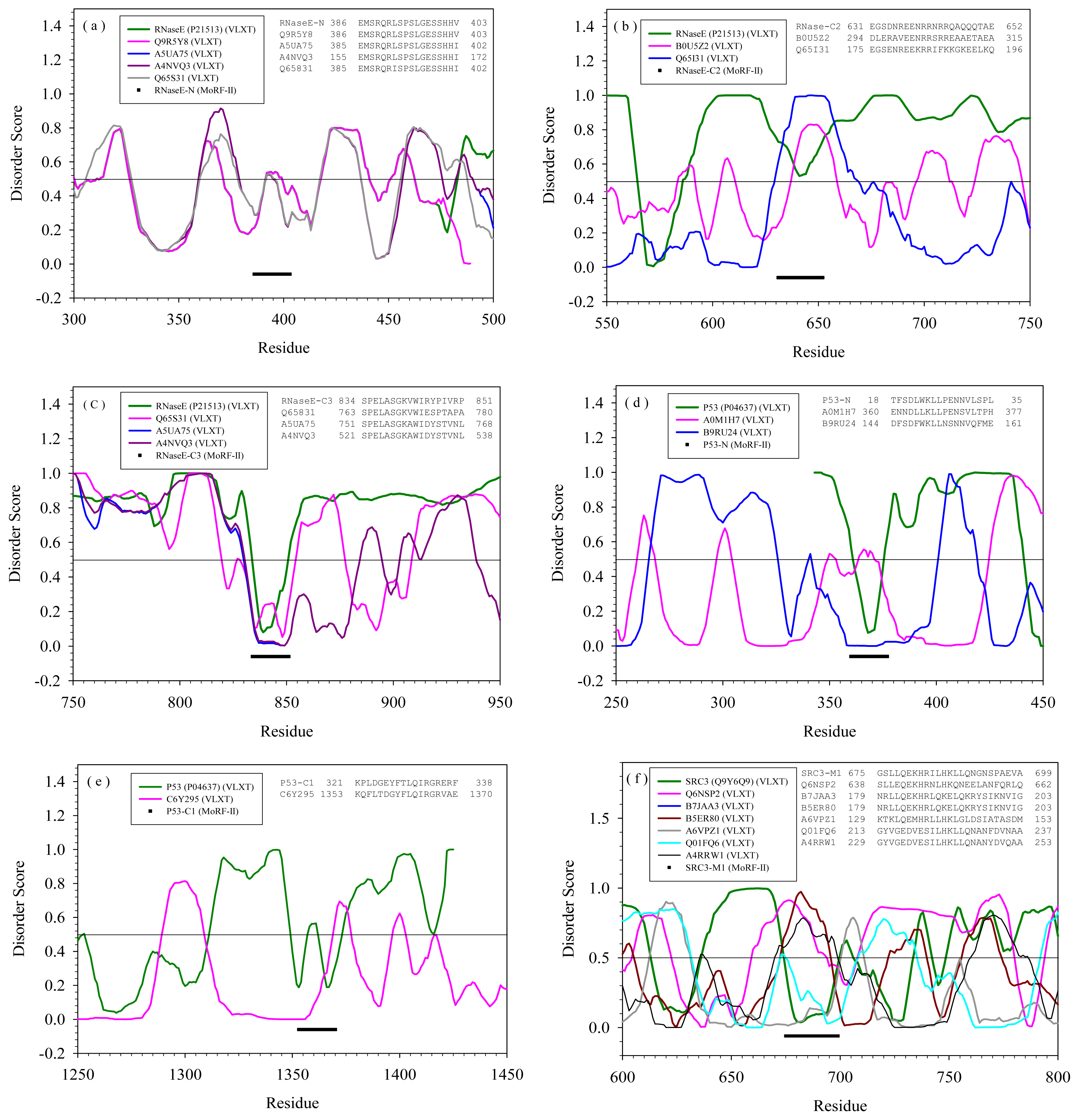
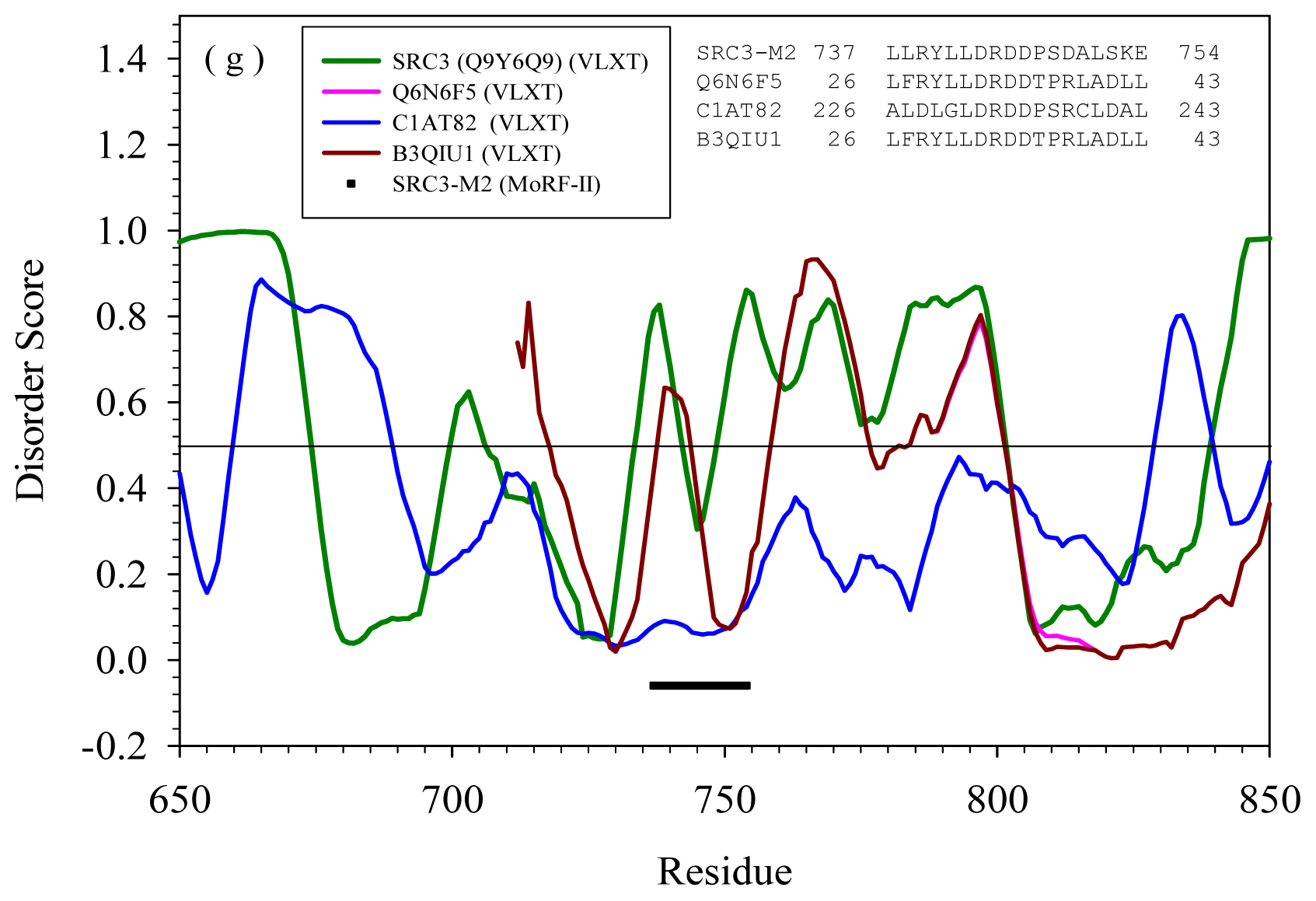
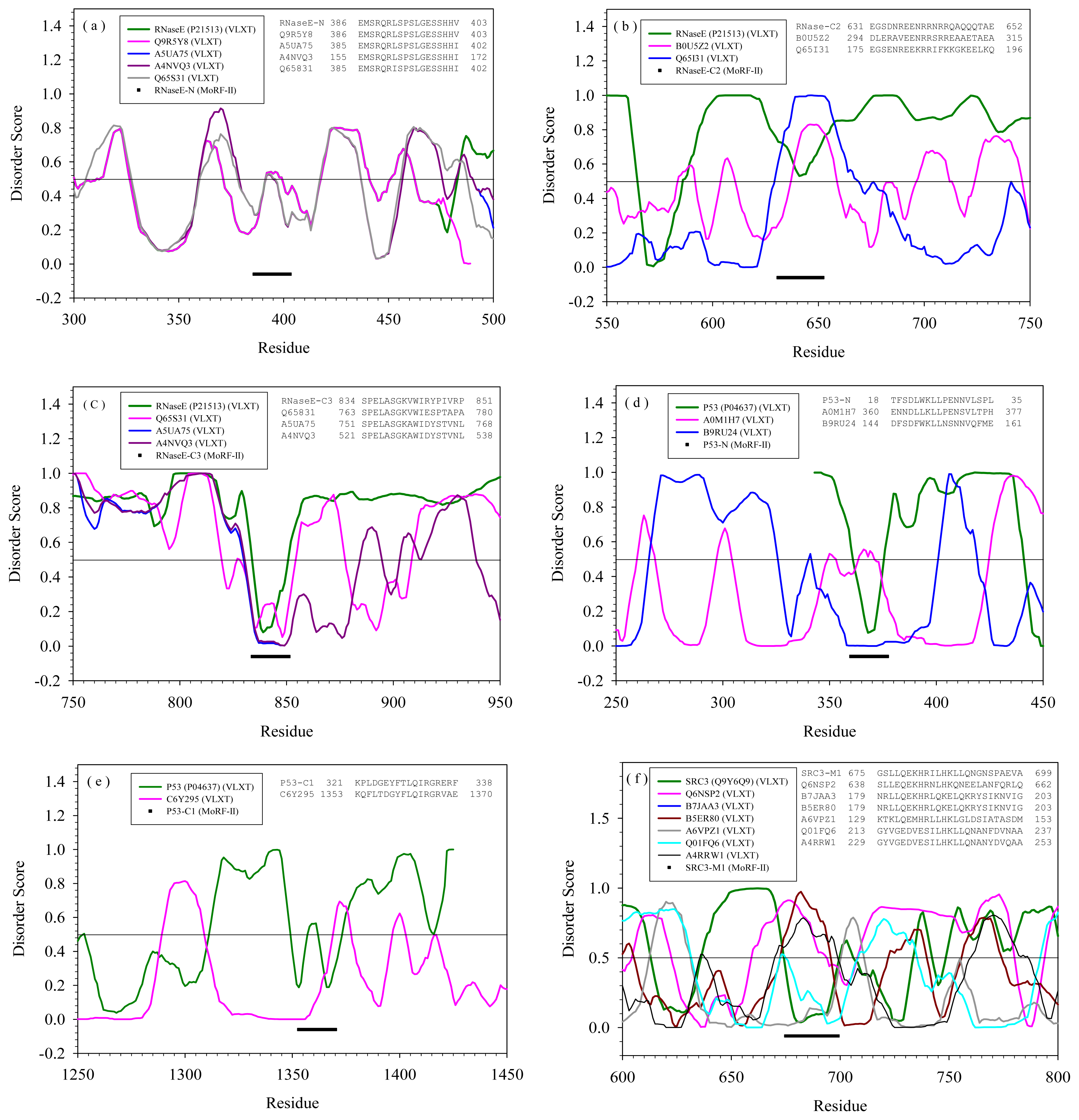
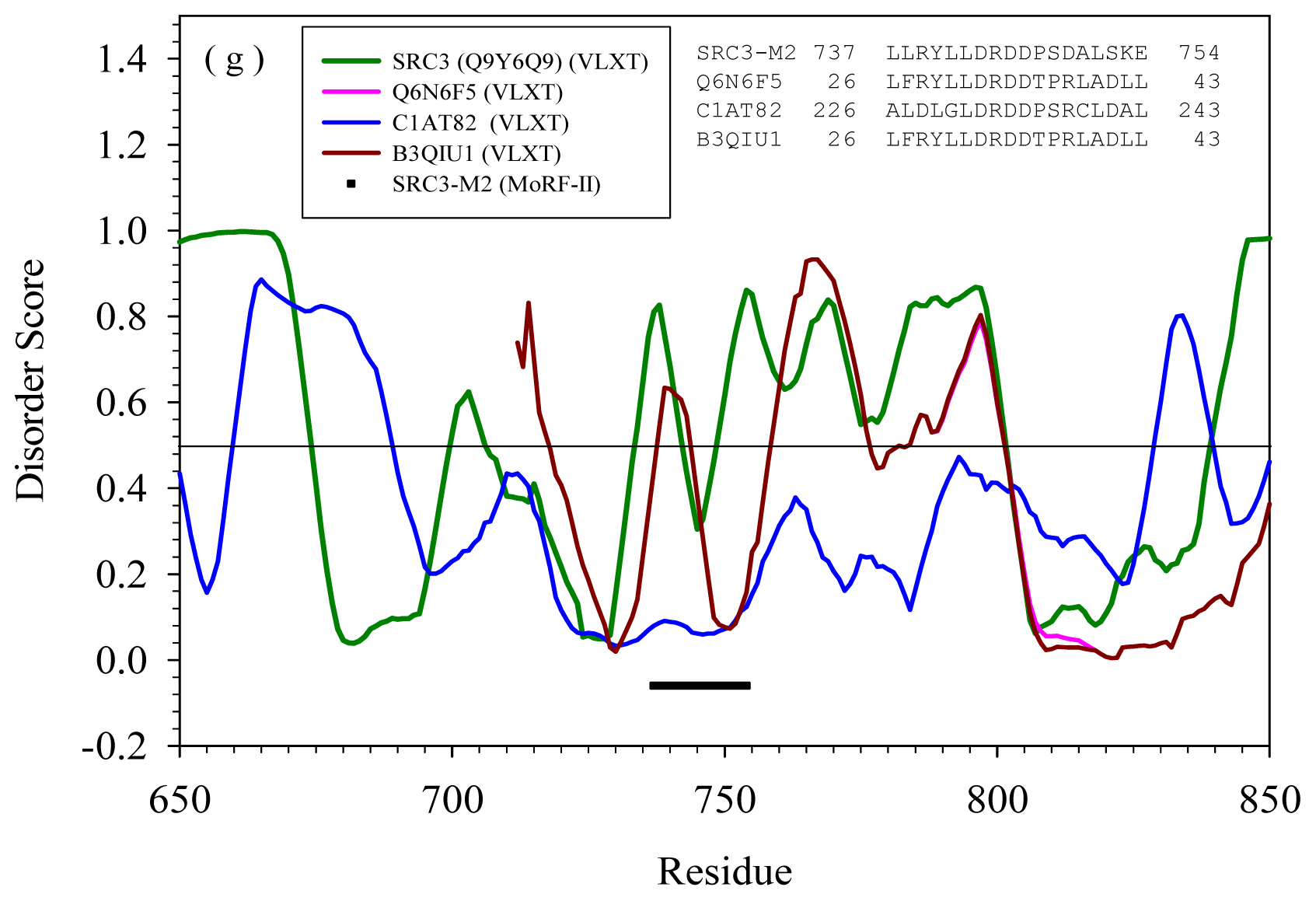
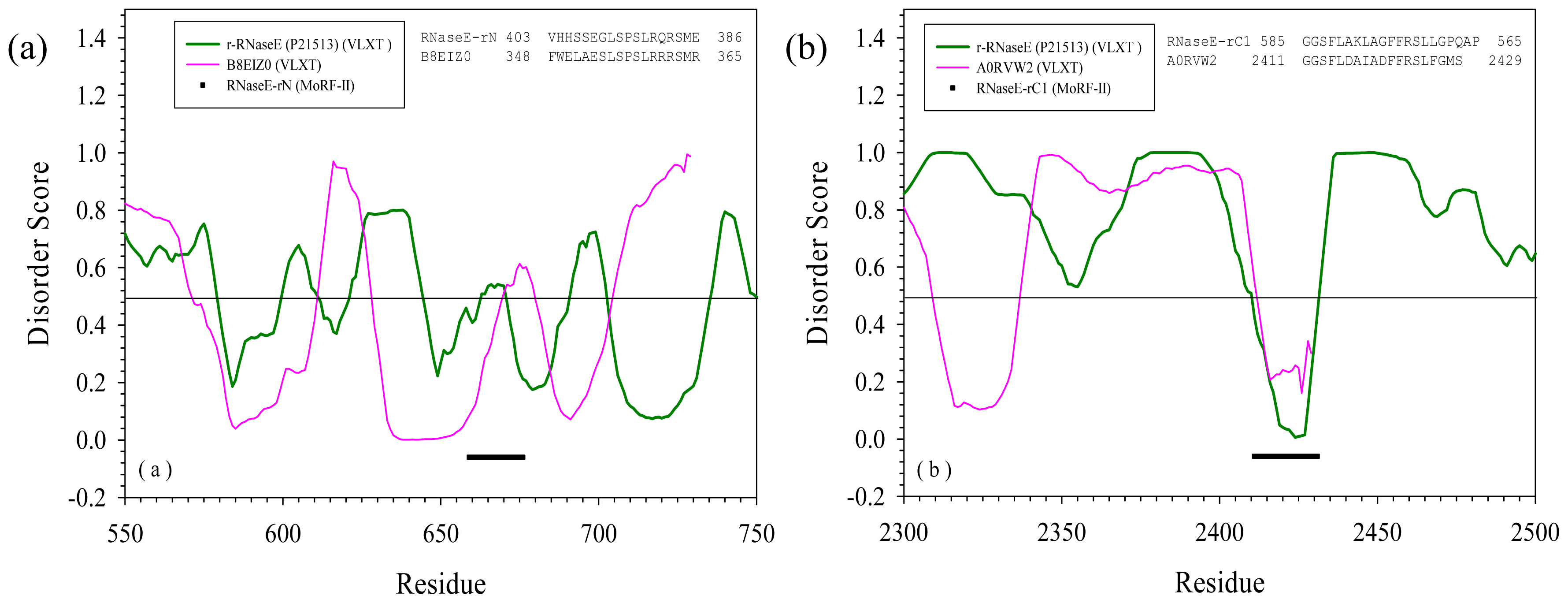
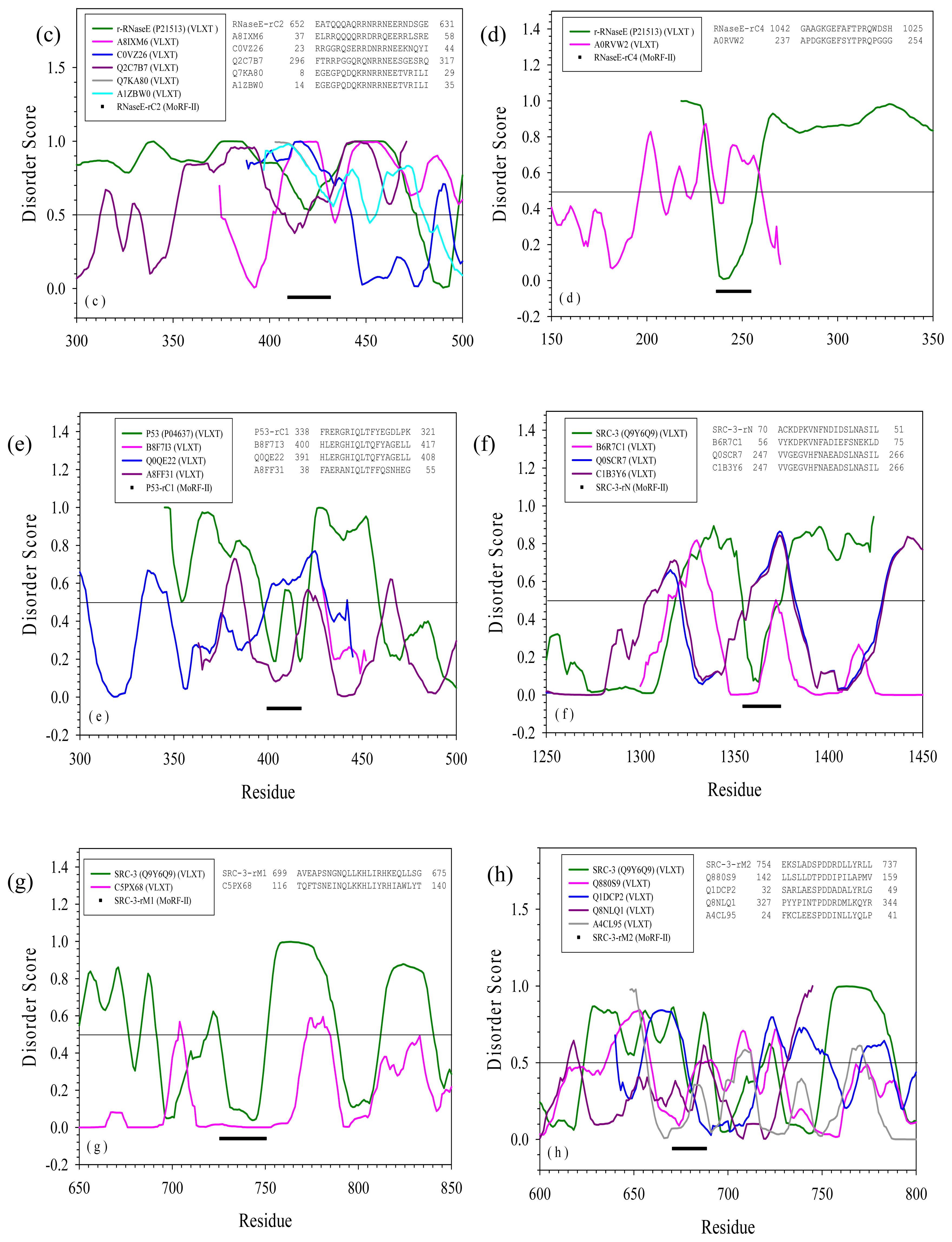
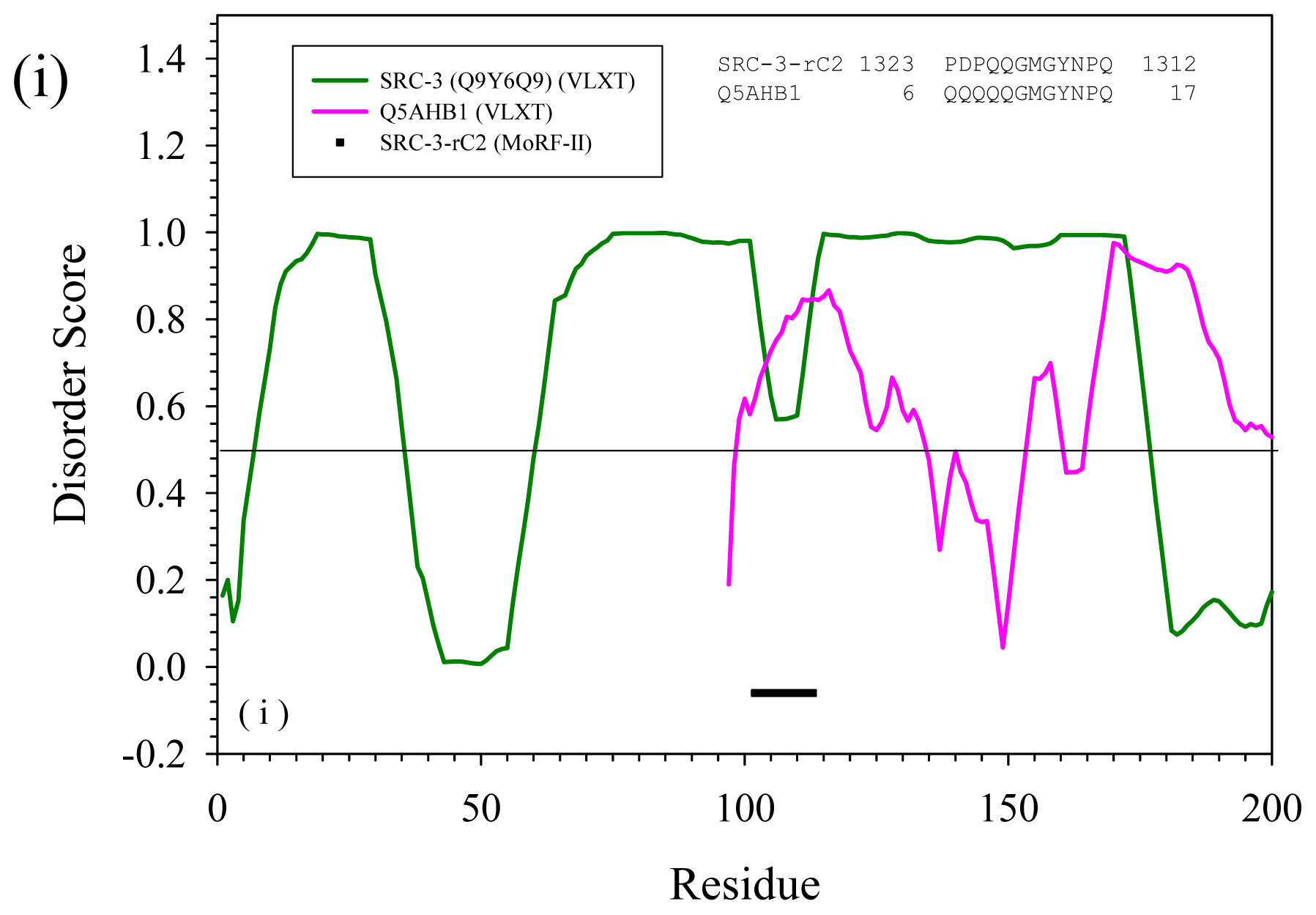
2.2. Alignment of MoRFs by Normal Sequential Order
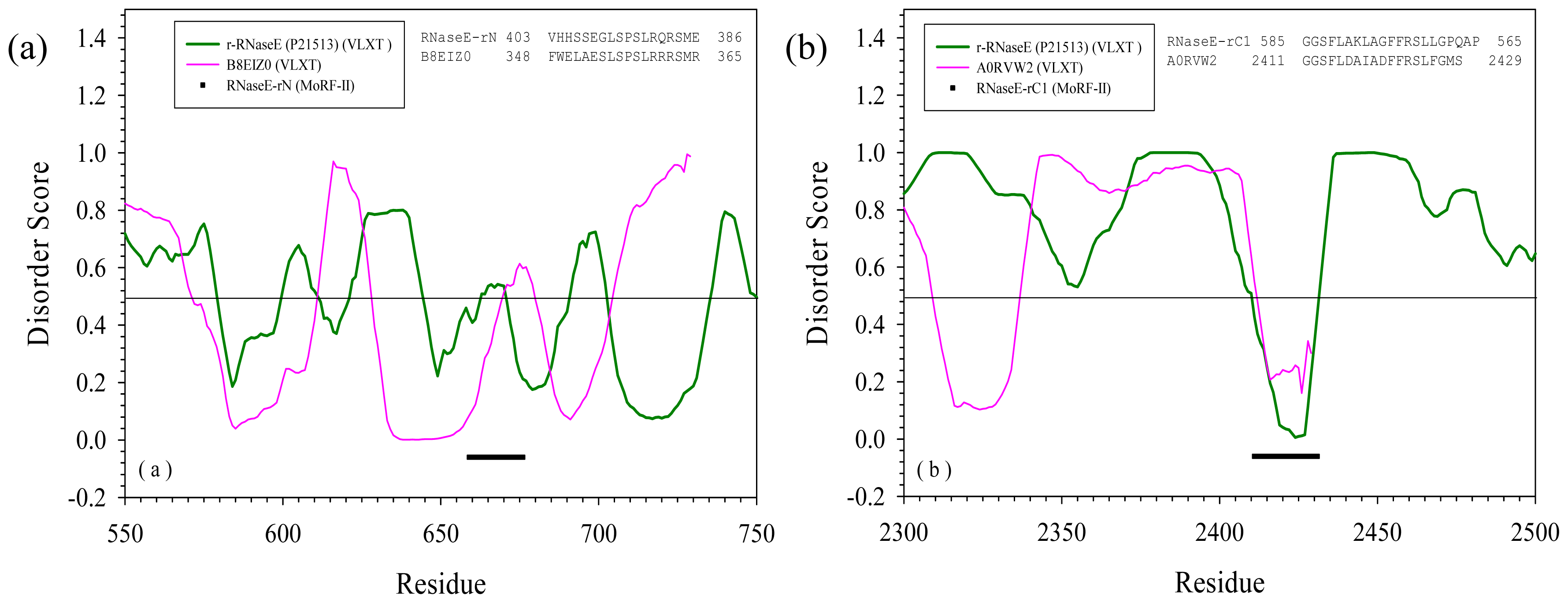
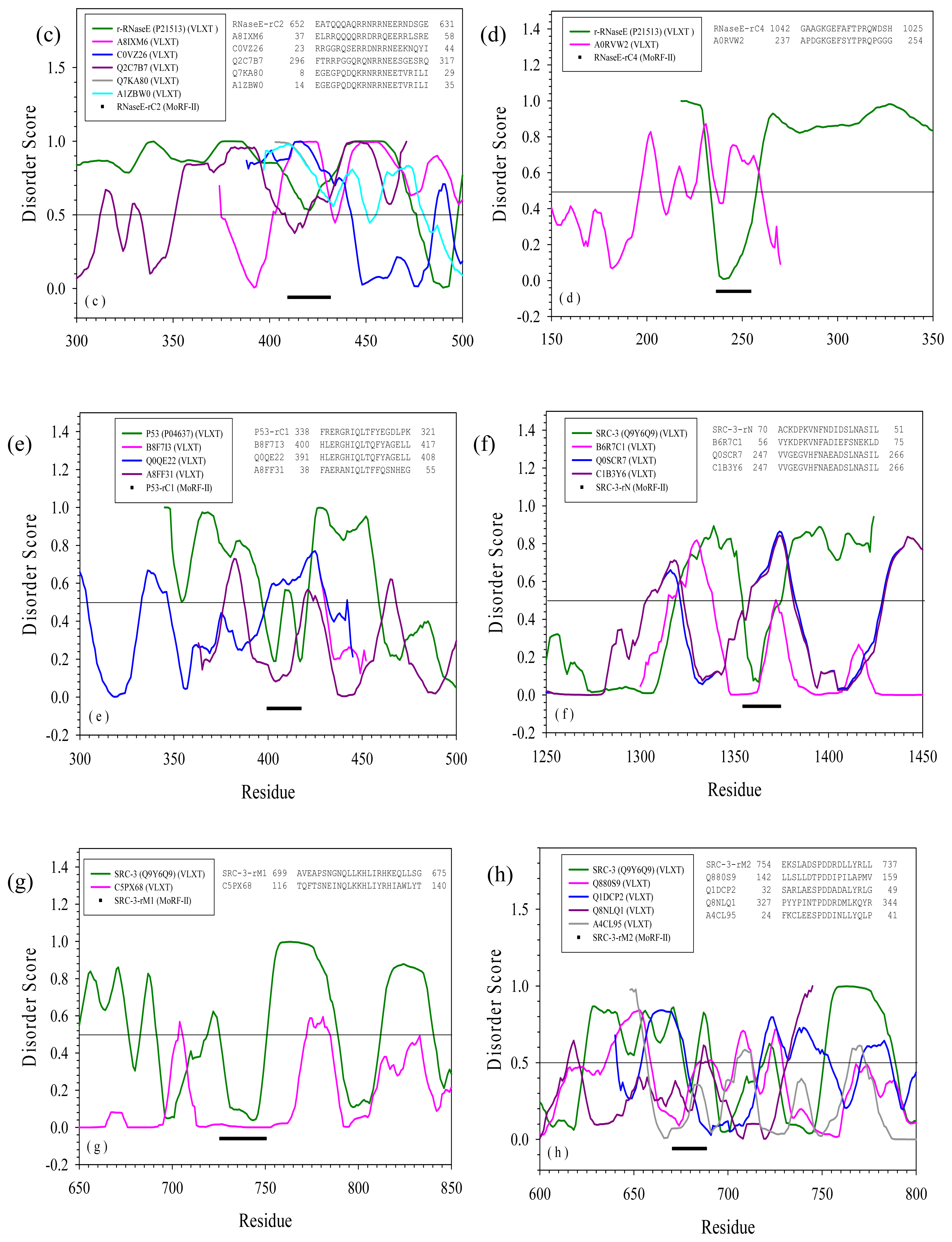
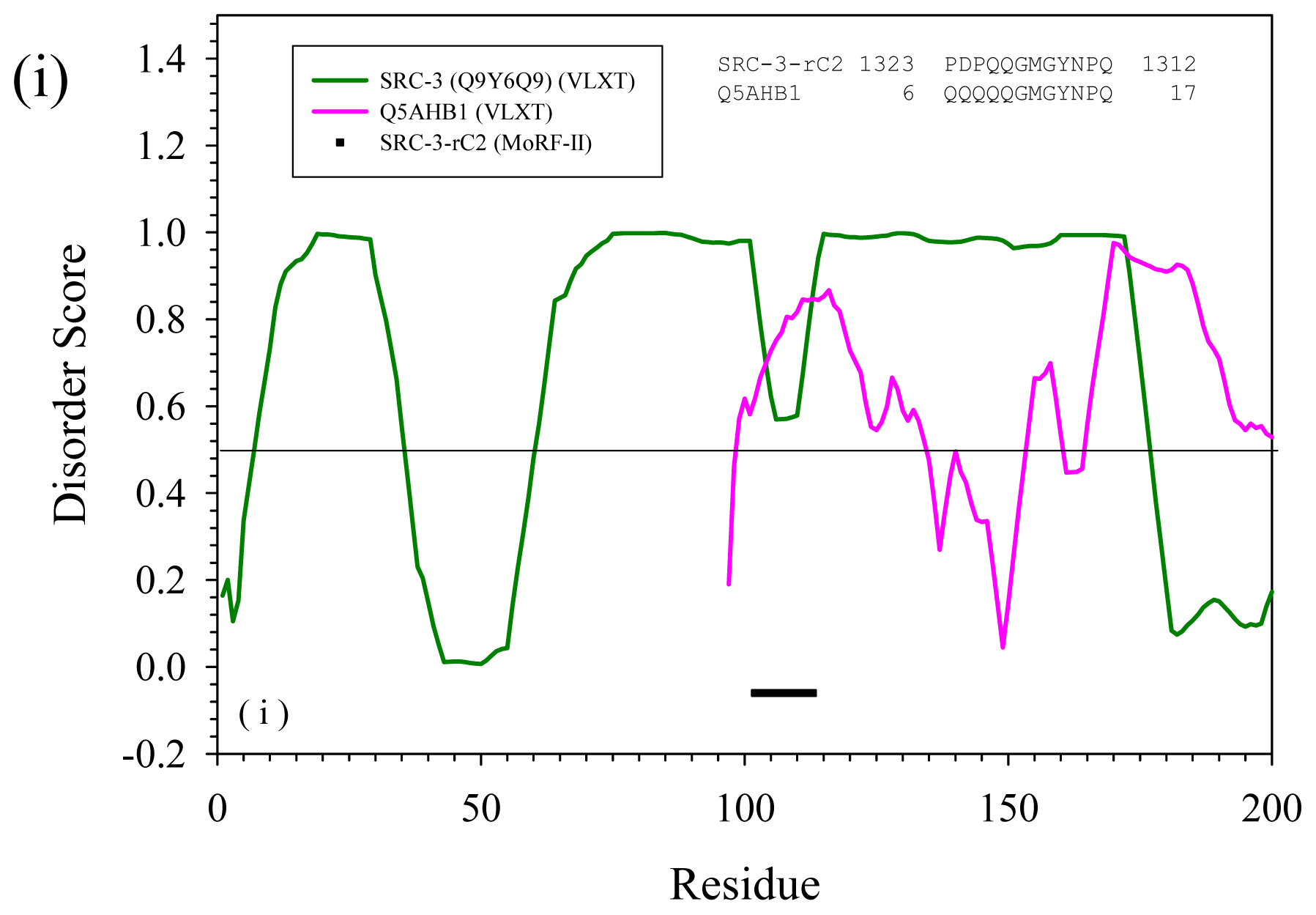
2.3. Reversely Identified Potential Binding Sites
3. Method Section
3.1. PONDR-RIBS (Reversely Identified Binding Sites)
3.2. Disorder Prediction
3.3. Proteins Studied by PONDR-RIBS
3.4. Alignment against Various Protein Databases
4. Conclusions
Acknowledgements
References
- Schoniger, M; Waterman, MS. A local algorithm for DNA sequence alignment with inversions. Bull. Math. Biol 1992, 54, 521–536. [Google Scholar]
- Olszewski, KA; Kolinski, A; Skolnick, J. Does a backwardly read protein sequence have a unique native state? Protein Eng 1996, 9, 5–14. [Google Scholar]
- Lacroix, E; Viguera, AR; Serrano, L. Reading protein sequences backwards. Fold Des 1998, 3, 79–85. [Google Scholar]
- Rai, J. Retroinverso mimetics of S peptide. Chem. Biol. Drug. Des 2007, 70, 552–526. [Google Scholar]
- Pal-Bhowmick, I; Pandey, RP; Jarori, GK; Kar, S; Sahal, D. Structural and functional studies on Ribonuclease S, retro S and retro-inverso S peptides. Biochem. Biophys. Res. Commun 2007, 364, 608–613. [Google Scholar]
- Preissner, R; Goede, A; Michalski, E; Frommel, C. Inverse sequence similarity in proteins and its relation to the three-dimensional fold. FEBS Lett 1997, 414, 425–429. [Google Scholar]
- Lorenzen, S; Gille, C; Preissner, R; Frommel, C. Inverse sequence similarity of proteins does not imply structural similarity. FEBS Lett 2003, 545, 105–109. [Google Scholar]
- Mittl, PR; Deillon, C; Sargent, D; Liu, N; Klauser, S; Thomas, RM; Gutte, B; Grutter, MG. The retro-GCN4 leucine zipper sequence forms a stable three-dimensional structure. Proc. Natl. Acad. Sci. USA 2000, 97, 2562–6256. [Google Scholar]
- Pan, PK; Zheng, ZF; Lyu, PC; Huang, PC. Why reversing the sequence of the alpha domain of human metallothionein-2 does not change its metal-binding and folding characteristics. Eur. J. Biochem 1999, 266, 33–39. [Google Scholar]
- Cheley, S; Braha, O; Lu, X; Conlan, S; Bayley, H. A functional protein pore with a “retro” transmembrane domain. Protein Sci 1999, 8, 1257–1267. [Google Scholar]
- Shukla, A; Raje, M; Guptasarma, P. A backbone-reversed form of an all-beta alpha-crystallin domain from a small heat-shock protein (retro-HSP12.6) folds and assembles into structured multimers. J. Biol. Chem 2003, 278, 26505–26510. [Google Scholar]
- Ahmed, S; Shukla, A; Guptasarma, P. Folding behavior of a backbone-reversed protein: Reversible polyproline type II to beta-sheet thermal transitions in retro-GroES multimers with GroES-like features. Biochim. Biophys. Acta 2008, 1784, 916–923. [Google Scholar]
- Wright, PE; Dyson, HJ. Intrinsically unstructured proteins: Re-assessing the protein structure-function paradigm. J. Mol. Biol 1999, 293, 321–331. [Google Scholar]
- Dunker, AK; Brown, CJ; Obradovic, Z. Identification and functions of usefully disordered proteins. Adv. Protein Chem 2002, 62, 25–49. [Google Scholar]
- Dunker, AK; Brown, CJ; Lawson, JD; Iakoucheva, LM; Obradovic, Z. Intrinsic disorder and protein function. Biochemistry 2002, 41, 6573–6582. [Google Scholar]
- Minezaki, Y; Homma, K; Kinjo, AR; Nishikawa, K. Human transcription factors contain a high fraction of intrinsically disordered regions essential for transcriptional regulation. J. Mol. Biol 2006, 359, 1137–1149. [Google Scholar]
- Gan, HH; Perlow, RA; Roy, S; Ko, J; Wu, M; Huang, J; Yan, S; Nicoletta, A; Vafai, J; Sun, D; Wang, L; Noah, JE; Pasquali, S; Schlick, T. Analysis of protein sequence/structure similarity relationships. Biophys. J 2002, 83, 2781–2791. [Google Scholar]
- Dunker, AK; Lawson, JD; Brown, CJ; Williams, RM; Romero, P; Oh, JS; Oldfield, CJ; Campen, AM; Ratliff, CM; Hipps, KW; Ausio, J; Nissen, MS; Reeves, R; Kang, C; Kissinger, CR; Bailey, RW; Griswold, MD; Chiu, W; Garner, EC; Obradovic, Z. Intrinsically disordered protein. J. Mol. Graph. Model 2001, 19, 26–59. [Google Scholar]
- Romero, P; Obradovic, Z; Kissinger, CR; Villafranca, JE; Dunker, AK. Identifying Disordered regions in proteins from amino acid sequences. IEEE Int. Conf. Neural Networks 1997, 1, 90–95. [Google Scholar]
- Romero, P; Obradovic, Z; Li, X; Garner, EC; Brown, CJ; Dunker, AK. Sequence complexity of disordered protein. Proteins 2001, 42, 38–48. [Google Scholar]
- Oldfield, CJ; Cheng, Y; Cortese, MS; Brown, CJ; Uversky, VN; Dunker, AK. Comparing and combining predictors of mostly disordered proteins. Biochemistry 2005, 44, 1989–2000. [Google Scholar]
- Oldfield, CJ; Cheng, Y; Cortese, MS; Romero, P; Uversky, VN; Dunker, AK. Coupled folding and binding with alpha-helix-forming molecular recognition elements. Biochemistry 2005, 44, 12454–12470. [Google Scholar]
- Obradovic, Z; Peng, K; Vucetic, S; Radivojac, P; Brown, CJ; Dunker, AK. Predicting intrinsic disorder from amino acid sequence. Proteins 2003, 53, 566–572. [Google Scholar]
- Le Gall, T; Romero, PR; Cortese, MS; Uversky, VN; Dunker, AK. Intrinsic disorder in the Protein Data Bank. J. Biomol. Struct. Dyn 2007, 24, 325–342. [Google Scholar]
- Xie, H; Vucetic, S; Iakoucheva, LM; Oldfield, CJ; Dunker, AK; Uversky, VN; Obradovic, Z. Functional anthology of intrinsic disorder. 1. Biological processes and functions of proteins with long disordered regions. J. Proteome Res 2007, 6, 1882–1898. [Google Scholar]
- Cheng, Y; Oldfield, CJ; Meng, J; Romero, P; Uversky, VN; Dunker, AK. Mining alpha-helix-forming molecular recognition features with cross species sequence alignments. Biochemistry 2007, 46, 13468–13477. [Google Scholar]
- Koslover, DJ; Callaghan, AJ; Marcaida, MJ; Garman, EF; Martick, M; Scott, WG; Luisi, BF. The crystal structure of the Escherichia coli RNase E apoprotein and a mechanism for RNA degradation. Structure 2008, 16, 1238–1244. [Google Scholar]
- Callaghan, AJ; Marcaida, MJ; Stead, JA; McDowall, KJ; Scott, WG; Luisi, BF. Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 2005, 437, 1187–1191. [Google Scholar]
- Callaghan, AJ; Aurikko, JP; Ilag, LL; Gunter Grossmann, J; Chandran, V; Kuhnel, K; Poljak, L; Carpousis, AJ; Robinson, CV; Symmons, MF; Luisi, BF. Studies of the RNA degradosome-organizing domain of the Escherichia coli ribonuclease RNase E. J. Mol. Biol 2004, 340, 965–979. [Google Scholar]
- Uversky, VN; Oldfield, CJ; Midic, U; Xie, H; Xue, B; Vucetic, S; Iakoucheva, LM; Obradovic, Z; Dunker, AK. Unfoldomics of human diseases: Linking protein intrinsic disorder with diseases. BMC Genomics 2009, 10, S7. [Google Scholar]
- Kussie, PH; Gorina, S; Marechal, V; Elenbaas, B; Moreau, J; Levine, AJ; Pavletich, NP. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science 1996, 274, 948–953. [Google Scholar]
- Di Lello, P; Jenkins, LM; Jones, TN; Nguyen, BD; Hara, T; Yamaguchi, H; Dikeakos, JD; Appella, E; Legault, P; Omichinski, JG. Structure of the Tfb1/p53 complex: Insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell 2006, 22, 731–740. [Google Scholar]
- Poux, AN; Marmorstein, R. Molecular basis for Gcn5/PCAF histone acetyltransferase selectivity for histone and nonhistone substrates. Biochemistry 2003, 42, 14366–14374. [Google Scholar]
- Kuszewski, J; Gronenborn, AM; Clore, GM. Improving the packing and accuracy of NMR structures with a pseudopotential for the radius of gyration. J. Am. Chem. Soc 1999, 121, 2337–2338. [Google Scholar]
- Lowe, ED; Tews, I; Cheng, KY; Brown, NR; Gul, S; Noble, ME; Gamblin, SJ; Johnson, LN. Specificity determinants of recruitment peptides bound to phospho-CDK2/cyclin A. Biochemistry 2002, 41, 15625–15634. [Google Scholar]
- Avalos, JL; Celic, I; Muhammad, S; Cosgrove, MS; Boeke, JD; Wolberger, C. Structure of a Sir2 enzyme bound to an acetylated p53 peptide. Mol. Cell 2002, 10, 523–535. [Google Scholar]
- Mujtaba, S; He, Y; Zeng, L; Yan, S; Plotnikova, O; Sachchidanand; Sanchez, R; Zeleznik-Le, NJ; Ronai, Z; Zhou, MM. Structural mechanism of the bromodomain of the coactivator CBP in p53 transcriptional activation. Mol. Cell 2004, 13, 251–263. [Google Scholar]
- Rustandi, RR; Baldisseri, DM; Weber, DJ. Structure of the negative regulatory domain of p53 bound to S100B(betabeta). Nat. Struct. Biol 2000, 7, 570–574. [Google Scholar]
- Chen, SL; Dowhan, DH; Hosking, BM; Muscat, GE. The steroid receptor coactivator, GRIP-1, is necessary for MEF-2C-dependent gene expression and skeletal muscle differentiation. Genes Dev 2000, 14, 1209–1228. [Google Scholar]
- Belandia, B; Parker, MG. Functional interaction between the p160 coactivator proteins and the transcriptional enhancer factor family of transcription factors. J. Biol. Chem 2000, 275, 30801–30805. [Google Scholar]
- Segrest, JP; De Loof, H; Dohlman, JG; Brouillette, CG; Anantharamaiah, GM. Amphipathic helix motif: Classes and properties. Proteins 1990, 8, 103–117. [Google Scholar]
- Jones, MK; Anantharamaiah, GM; Segrest, JP. Computer programs to identify and classify amphipathic alpha helical domains. J. Lipid Res 1992, 33, 287–296. [Google Scholar]
- Torchia, J; Rose, DW; Inostroza, J; Kamei, Y; Westin, S; Glass, CK; Rosenfeld, MG. The transcriptional co-activator p/CIP binds CBP and mediates nuclear-receptor function. Nature 1997, 387, 677–684. [Google Scholar]
- Chen, H; Lin, RJ; Schiltz, RL; Chakravarti, D; Nash, A; Nagy, L; Privalsky, ML; Nakatani, Y; Evans, RM. Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with P/CAF and CBP/p300. Cell 1997, 90, 569–580. [Google Scholar]
- Heery, DM; Kalkhoven, E; Hoare, S; Parker, MG. A signature motif in transcriptional coactivators mediates binding to nuclear receptors. Nature 1997, 387, 733–736. [Google Scholar]
- Voegel, JJ; Heine, MJ; Tini, M; Vivat, V; Chambon, P; Gronemeyer, H. The coactivator TIF2 contains three nuclear receptor-binding motifs and mediates transactivation through CBP binding-dependent and -independent pathways. EMBO J 1998, 17, 507–519. [Google Scholar]
- Ding, XF; Anderson, CM; Ma, H; Hong, H; Uht, RM; Kushner, PJ; Stallcup, MR. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1): Multiple motifs with different binding specificities. Mol. Endocrinol 1998, 12, 302–313. [Google Scholar]
- Xu, J; Wu, RC; O’Malley, BW. Normal and cancer-related functions of the p160 steroid receptor co-activator (SRC) family. Nat. Rev. Cancer 2009, 9, 615–630. [Google Scholar]
- Guptasarma, P. Reversal of peptide backbone direction may result in the mirroring of protein structure. FEBS Lett 1992, 310, 205–210. [Google Scholar]
- Rath, A; Davidson, AR; Deber, CM. The structure of “unstructured” regions in peptides and proteins: Role of the polyproline II helix in protein folding and recognition. Biopolymers 2005, 80, 179–185. [Google Scholar]
- Creamer, TP; Campbell, MN. Determinants of the polyproline II helix from modeling studies. Adv. Protein Chem 2002, 62, 263–282. [Google Scholar]
- Kay, BK; Williamson, MP; Sudol, M. The importance of being proline: The interaction of proline-rich motifs in signaling proteins with their cognate domains. FASEB J 2000, 14, 231–241. [Google Scholar]
- Cesareni, G; Panni, S; Nardelli, G; Castagnoli, L. Can we infer peptide recognition specificity mediated by SH3 domains? FEBS Lett 2002, 513, 38–44. [Google Scholar]
- Dalgarno, DC; Botfield, MC; Rickles, RJ. SH3 domains and drug design: Ligands, structure, and biological function. Biopolymers 1997, 43, 383–400. [Google Scholar]
- Feng, S; Chen, JK; Yu, H; Simon, JA; Schreiber, SL. Two binding orientations for peptides to the Src SH3 domain: Development of a general model for SH3-ligand interactions. Science 1994, 266, 1241–1247. [Google Scholar]
- Thompson, JD; Higgins, DG; Gibson, TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Xue, B; Dunbrack, RL; Williams, RW; Dunker, AK; Uversky, VN. PONDR-FIT: a meta-predictor of intrinsically disordered amino acids. Biochim. Biophys. Acta 2010, 1804, 996–1010. [Google Scholar]
- Jensen, LJ; Kuhn, M; Stark, M; Chaffron, S; Creevey, C; Muller, J; Doerks, T; Julien, P; Roth, A; Simonovic, M; Bork, P; von Mering, C. STRING 8--a global view on proteins and their functional interactions in 630 organisms. Nucleic Acids Res 2009, 37, D412–416. [Google Scholar]
- Peng, K; Vucetic, S; Radivojac, P; Brown, CJ; Dunker, AK; Obradovic, Z. Optimizing long intrinsic disorder predictors with protein evolutionary information. J. Bioinform. Comput. Biol 2005, 3, 35–60. [Google Scholar]
- Peng, K; Radivojac, P; Vucetic, S; Dunker, AK; Obradovic, Z. Length-dependent prediction of protein intrinsic disorder. BMC Bioinformatics 2006, 7, 208. [Google Scholar]
- Prilusky, J; Felder, CE; Zeev-Ben-Mordehai, T; Rydberg, EH; Man, O; Beckmann, JS; Silman, I; Sussman, JL. FoldIndex: A simple tool to predict whether a given protein sequence is intrinsically unfolded. Bioinformatics 2005, 21, 3435–3438. [Google Scholar]
- Dosztanyi, Z; Csizmok, V; Tompa, P; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol 2005, 347, 827–839. [Google Scholar]
- Campen, A; Williams, RM; Brown, CJ; Meng, J; Uversky, VN; Dunker, AK. TOP-IDP-scale: A new amino acid scale measuring propensity for intrinsic disorder. Protein Pept. Lett 2008, 15, 956–963. [Google Scholar]
- Mohan, A; Oldfield, CJ; Radivojac, P; Vacic, V; Cortese, MS; Dunker, AK; Uversky, VN. Analysis of molecular recognition features (MoRFs). J. Mol. Biol 2006, 362, 1043–1059. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | MoRF | Proteins in PDB containing similar MoRF (a) | Proteins in SwissProt containing similar MoRF (a) | |||
|---|---|---|---|---|---|---|
| SwissProt id | Species | Name | Within IDR | |||
| RNase E | N | --- | Q9R5Y8 | E. Coli | Cell shape determining protein | Yes |
| A5UA75 | Haemophilus influenzae | Hydroxyethylthiazole kinase | Yes | |||
| A4NVQ3 | Haemophilus influenzae | rRNA pseudouridylate synthase C | Yes | |||
| Q65S31 | Mannheimia succiniciproducens | CafA protein | Yes | |||
| C1 | --- | --- | --- | --- | ||
| C2 | --- | B0U5Z2 | Xylella fastidiosa | Glutamyl-tRNA reductase | Yes | |
| Q65I31 | Bacillus licheniformis | Anthranilate synthaseTrpE | Yes | |||
| C3 | --- | A5UA75 | Haemophilus influenzae | Hydroxyethylthiazole kinase | Yes | |
| A4NVQ3 | Haemophilus influenzae | rRNA pseudouridylate synthase C | No | |||
| Q65S31 | Mannheimia succiniciproducens | CafA protein | No | |||
| C4 | --- | --- | --- | --- | ||
| p53 | N | --- | A0M1H7 | Gramella forsetii | Carbohydrate kinase | Yes |
| --- | B9RU24 | Ricinus communis | Mitochondrial respiratory chain complexes assembly protein, putative | |||
| C1 | --- | C6Y295 | Pedobacter heparinus | DNA polymerase III, α subunit | No/Yes | |
| C2 | --- | --- | --- | --- | ||
| SRC-3 | N | --- | --- | --- | --- | |
| M1 | --- | Q6NSP2 | Zebrafish | Rho/rac guanine nucleotide exchange factor (GEF) | Yes | |
| B7JAA3 | Acidithiobacillus ferrooxidans | Nif-specific regulatory protein | Yes | |||
| B5ER80 | Acidithiobacillus ferrooxidans | Transcriptional regulator, NifA, Fis Family | Yes | |||
| A6VPZ1 | Actinobacillus succinogenes | Sulfite reductase [NADPH] hemoprotein beta-component | No | |||
| Q01FQ6 | Ostreococcus tauri | CLP protease regulatory subunit CLPX (ISS) | Yes | |||
| A4RRW1 | Ostreococcus lucimarinus | Mitochondrial ClpX chaperone | Yes | |||
| M2 | --- | Q6N6F5 | Rhodopseudomo nas palustris | ATP-dependent DNA helicase | Yes | |
| C1AT82 | Rhodococcus opacus | Hypothetical membrane protein | No | |||
| B3QIU1 | Rhodopseudomonas palustris | DEAD/DEAH box helicase domain protein | Yes | |||
| C1 | --- | --- | --- | --- | ||
| C2 | --- | --- | --- | --- | ||
| Protein | MoRF(a) | Proteins in PDB containing similar MoRF (b) | Proteins in SwissProt containing similar MoRF (b) | |||
|---|---|---|---|---|---|---|
| SwissProt id | Species | Name | Within IDR | |||
| RNase E | rN | --- | B8EIZ0 | Methylocella silvestris | Glycosyl transferase family 2 | Yes |
| rC1 | --- | A0RVW2 | Cenarchaeum symbiosum | Putative uncharacterized protein | Yes | |
| rC2 | --- | A8IXM6 | Chlamydomonas reinhardtii | Dopamine beta-monooxygerase-like protein | Yes | |
| C0VZ26 | Actinomyces coleocanis | 30S ribosomal protein S5 | Yes | |||
| Q2C7B7 | Photobacterium | Pseudouridine synthase | Yes | |||
| Q7KA80 | Drosophila melanogaster | Heterogeneous nuclear ribonucleoprotein | Yes | |||
| A1ZBW0 | Drosophila melanogaster | Bancal isoform C | Yes | |||
| rC3 | --- | --- | --- | --- | ||
| rC4 | --- | B9LNU7 | Halorubrum lacusprofundi | Manganese containing catelase | Yes | |
| p53 | rN | --- | --- | --- | --- | |
| rC1 | --- | B8F7I3 | Haemophilus parasuis serovar 5 | tRNA modification GTPase TrmE | Yes | |
| Q0QE22 | Haemophilus parasuis | ThdF | Yes | |||
| A8FF31 | Bacillus pumilus | 3-dehydroquinate dehydratase | No | |||
| rC2 | --- | --- | --- | --- | ||
| SRC-3 | rN | --- | B6R7C1 | Pseudovibrio | Outer surface protein | No |
| Q0SCR7 | Rhodococcus | Aldehyde dehydrogenase | Yes | |||
| C1B3Y6 | Rhodococcus opacus | Phenylacetic acid degradation protein PaaN | Yes | |||
| rM1 | --- | C5PX68 | Sphingobacterium spiritivorum | Conserved hypothetical transmembrane protein | No | |
| rM2 | --- | Q880S9 | Pseudomonas syringae | AraC-family transcriptional regulator | No | |
| Q1DCP2 | Myxococcus xanthus | Tetratricopeptide repeat protein | No/Yes | |||
| Q8NLQ1 | Corynebacterium glutamicum | UDP-galactopyranose mutase | No | |||
| A4CL95 | Robiginitalea biformata | Type III restriction enzyme | No | |||
| rC1 | --- | --- | --- | --- | ||
| rC2 | --- | Q5AHB1 | Candida albicans | Actin cytoskeleton-regulatory complex protein PAN1 | Yes | |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Xue, B.; Dunker, A.K.; Uversky, V.N. Retro-MoRFs: Identifying Protein Binding Sites by Normal and Reverse Alignment and Intrinsic Disorder Prediction. Int. J. Mol. Sci. 2010, 11, 3725-3747. https://doi.org/10.3390/ijms11103725
Xue B, Dunker AK, Uversky VN. Retro-MoRFs: Identifying Protein Binding Sites by Normal and Reverse Alignment and Intrinsic Disorder Prediction. International Journal of Molecular Sciences. 2010; 11(10):3725-3747. https://doi.org/10.3390/ijms11103725
Chicago/Turabian StyleXue, Bin, A. Keith Dunker, and Vladimir N. Uversky. 2010. "Retro-MoRFs: Identifying Protein Binding Sites by Normal and Reverse Alignment and Intrinsic Disorder Prediction" International Journal of Molecular Sciences 11, no. 10: 3725-3747. https://doi.org/10.3390/ijms11103725