Hereditary and Sporadic Forms of Aβ-Cerebrovascular Amyloidosis and Relevant Transgenic Mouse Models

Abstract
:1. Introduction
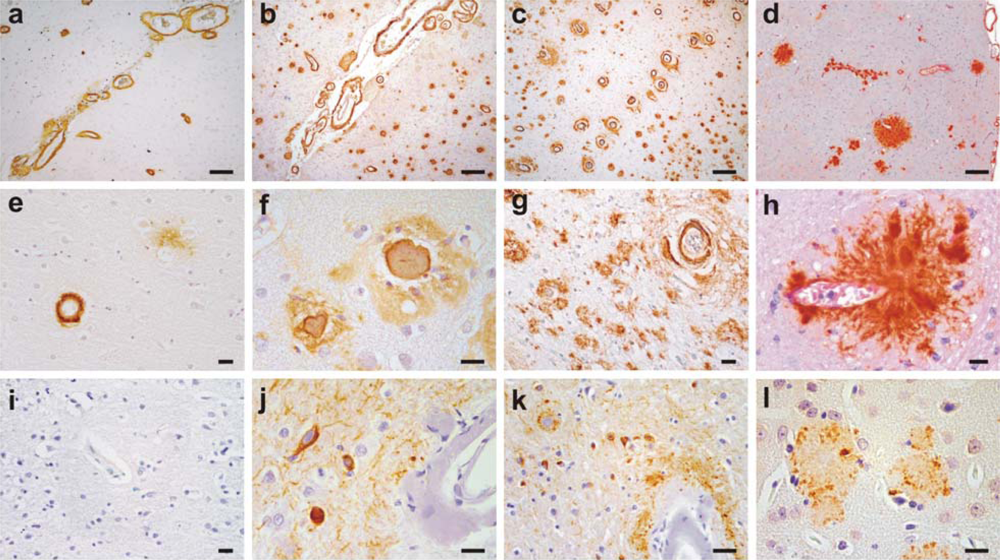
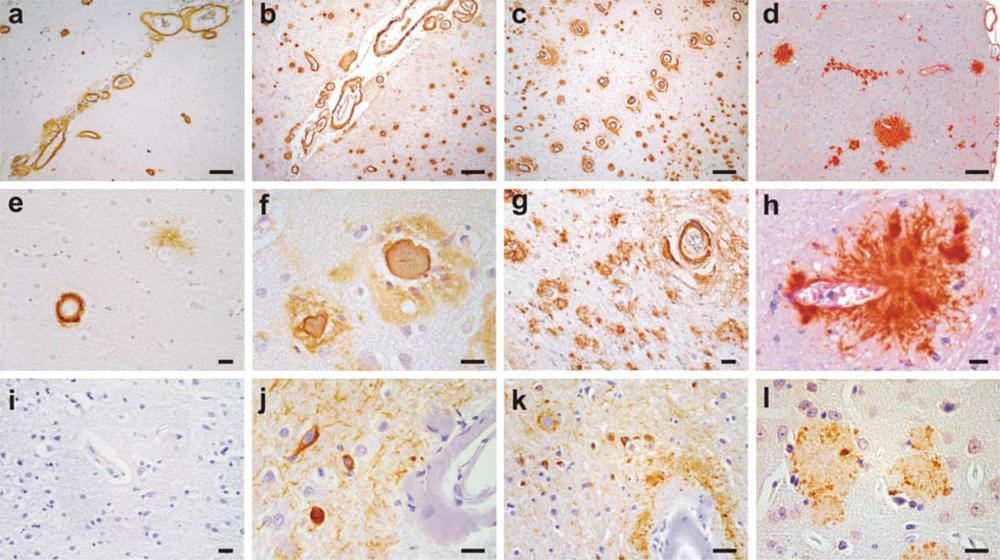
2. Clinical and Pathological Consequences of CAA
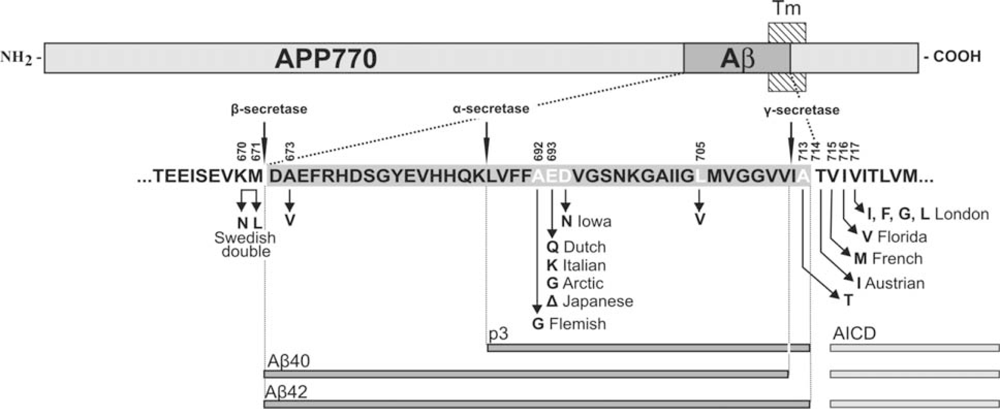
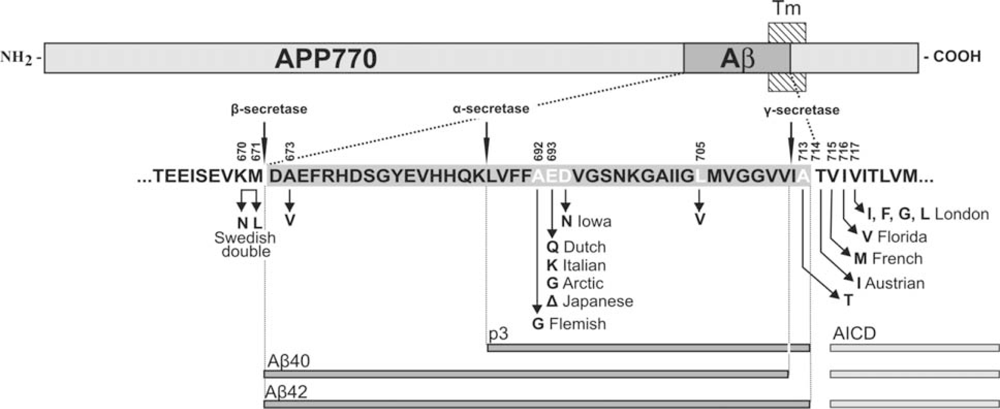
3. APP Mutations within the Aβ Sequence Associated with Cerebrovascular Amyloidosis
4. Other Genetic Risk Factors for CAA
5. Mouse Models of Cerebrovascular Amyloidosis
- Mouse models have given a definitive view that Aβ42 is essential for Aβ deposition not only in parenchyma but also in vessels. When APP/Sw mice (producing both Aβ40 and Aβ42) are bred with mice expressing mutant PS1 (that increases brain Aβ42/Aβ40 ratio), the crossbred mice have drastically increased amyloid deposition in parenchyma and vessels compared to single transgenic controls [97,98]. These APP/PS1 mice also have a higher number but smaller sizes of amyloid deposits most likely due to the extra “seeds” provided by Aβ42 [99]. A similar crossbreeding of APP Dutch mice with mice expressing AD-related PS1 G384A mutation also increases amyloid depositions and shifts the pathology from vascular to the parenchymal compartment [91]. The premise that Aβ42 is essential for Aβ deposition is also neatly answered by BRI-Aβ40 and BRI-Aβ42 mice that produce only Aβ40 or Aβ42 [100]. In these mice, a fusion construct is utilized wherein the carboxyl terminus sequence of BRI protein, involved in amyloid deposition in familial British and Danish dementia, is replaced by a sequence encoding either Aβ40 or Aβ42. A proteolytic cleavage of this fusion protein at a furin cleavage site immediately preceding Aβ results in high-levels of Aβ40 or Aβ42 secretion. While BRI-Aβ40 mice expressing high levels of Aβ40 do not develop overt amyloid pathology, the BRI-Aβ42 mice line expressing lower levels of Aβ42 develop all types of brain amyloid deposits including CAA [100]. Crossbreeding of BRI-Aβ42 mice with Tg2576 mice again leads to a massive increase in amyloid deposition. These data establish that Aβ42 is essential for amyloid deposition in the parenchyma and also in vessels.
- Mouse models have also supported a “protective” role of Aβ40 in plaque deposition and therefore in AD pathology, especially when the levels of Aβ40 exceed a critical level. The initial data to support a protective role of Aβ40 came from in vitro studies where Aβ40 was shown to directly interfere with Aβ42 aggregation by delaying the Aβ42-mediated nucleation step at an early stage in the fibrillogenesis process [101]. More recently, γ-secretase site APP mutations, like the Austrian (T714I) and French (V715M) mutations, have also been shown to cause a drastic decrease in Aβ40 production [102,103]. For instance, Austrian APP reduces Aβ40 by ≈ 80% and because Aβ40 is the major physiologically produced peptide (≈ 9 times more than Aβ42), a sharp reduction in total Aβ also occurs [102,104]. A similar decrease in absolute Aβ40 levels has also been shown for a number of clinical PS mutants [80,81] and interestingly, age-of-onset of PS1-linked AD not only correlates inversely with Aβ42 but also directly with Aβ40 levels [80,105]. Further studies on mouse models have also provided compelling data to support the premise that Aβ40 is anti-amyloidotic. First, results from transgenic mice expressing wild-type and various mutant forms of APP suggest that increased Aβ40 levels reduce amyloid deposition [106]. Secondly, at least two independent studies utilizing knockin PS familial AD mutations crossbred with Tg2576 mice show a greatly accelerated plaque pathology accompanied by decreased production of Aβ40 without an increase in secreted brain Aβ42 levels [107,108]. Similarly, BRI-Aβ40 mice crossbred with either BRI-Aβ42 or Tg2576 mice show greatly reduced brain amyloid deposition compared to singly transgenic BRI-Aβ42 or Tg2576 mice [100]. These data all suggest that in the absence of Aβ42, the clearance of Aβ40 is very high, and depending upon the critical levels of Aβ42, Aβ40 might even be anti-amyloidotic [72].
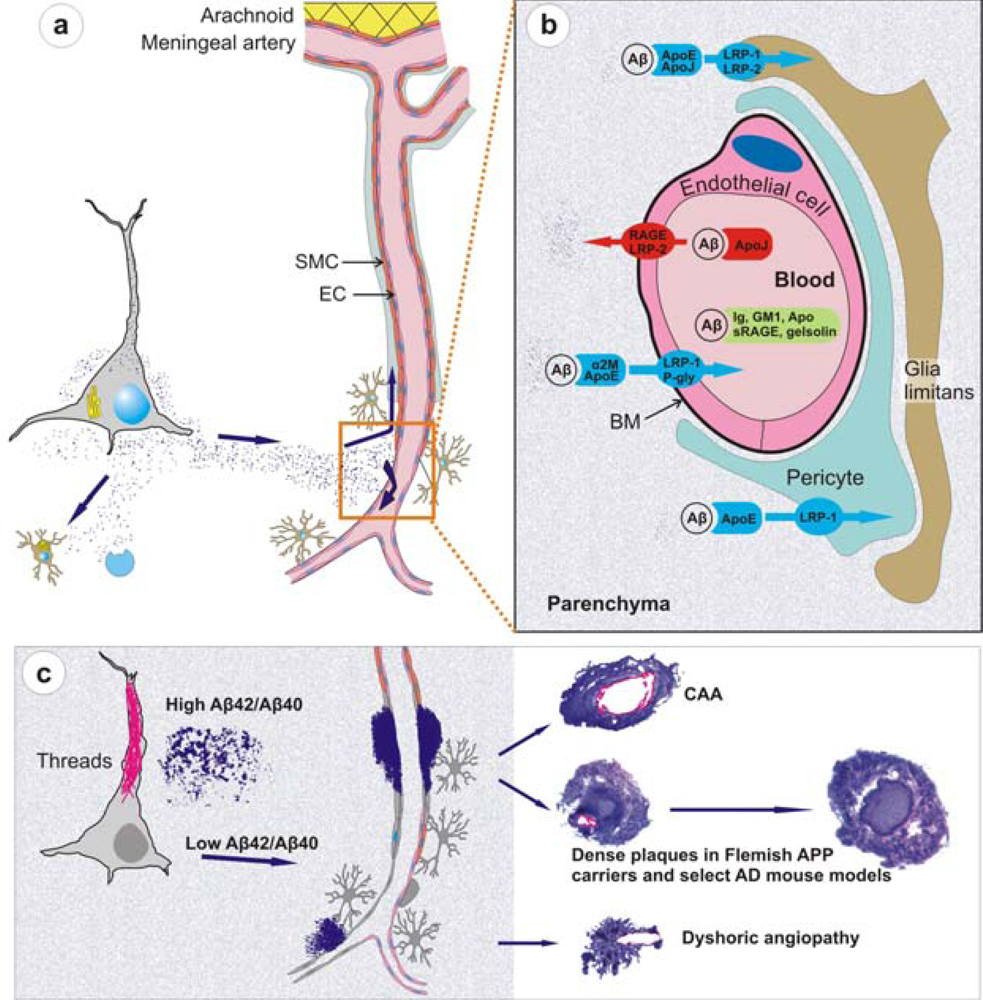
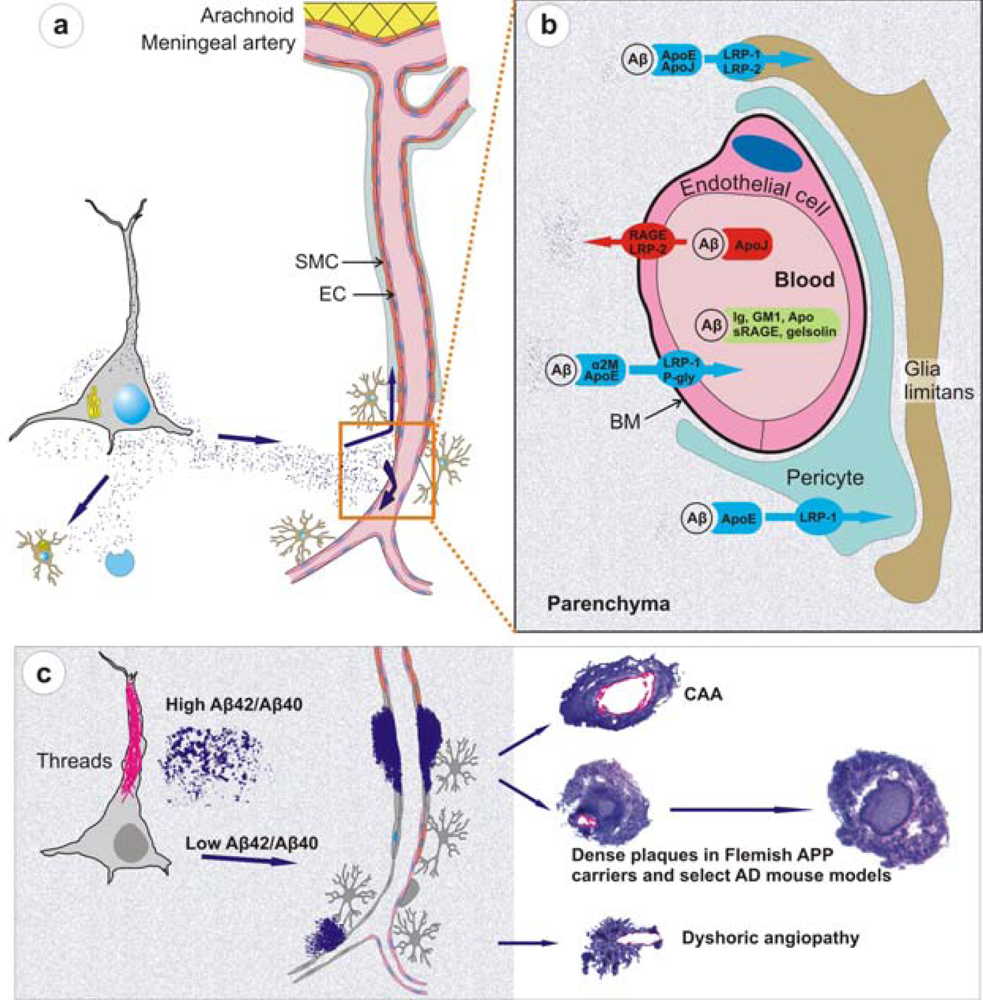
- Recent work on knockout and transgenic mice has provided evidence that Aβ is cleared from brain by mechanisms involving microglial uptake, degradation by cellular or extracellular proteases, as well as by clearance through vascular route. For instance, in vivo imaging of live mice has shown that Aβ is not only taken up by microglia but astrocytes also play a direct role in degradation of Aβ [109]. Secondly, knockout mouse models have supported data of Aβ degradation by brain proteases such as neprilysin, insulin-degrading enzyme (IDE), endothelin-converting enzymes-1 and -2, and matrix metalloproteinase-2 and -9 [110]. Consider, for example, neprilysin homozygous knockout mice expressing mutant APP and showing an expectedly higher burden of brain amyloid including CAA [111]. Thirdly, studies on mouse models have demonstrated that Aβ that cannot be locally degraded has the potential to diffuse away from the site of production. Non-transgenic brain tissue grafted in APP23 hosts develop both diffuse and congophilic amyloid plaques [112]. And finally, experimental studies in mice have shown that diffusible Aβ is transported directly across the BBB into the blood, mediated by low density lipoprotein receptor-related protein-1 (LRP-1)/α2-macroglobulin and ApoE [113,57]. Mouse models of mutant Aβ have also shown that some of these mutant Aβ such as the Dutch/Iowa mutants could be deficient in its clearance across the BBB explaining the robust CAA seen in these mutation carriers (Deane et al., 2004; Davis et al., 2006). Studies on rodents also support a second vessel-related Aβ clearance route along the periarterial spaces [56,113,114]. Exogenous tracer studies in rats show that tracers injected into the parenchyma of grey matter in the rat brain drain along perivascular spaces around leptomeningeal arteries to the base of the brain, pass through the cribriform plate and, via nasal lymphatics, to deep cervical lymph nodes [115].
- Studies on mouse models have indicated that local production of Aβ by vascular elements is not absolutely essential for the development of CAA. This is important as, for instance, hypoxia followed by reoxygenation in microvascular smooth vessel cells has been shown to upregulate APP and proposed to be an initiating event in the pathogenesis of amyloid angiopathy [116]. However, mice that solely produce APP/Aβ from neuronal cells in an endogenous APP knockout background drive Aβ pathology in both parenchyma and vessels [114]. Furthermore, these studies also lend support to the hypothesis that failure of Aβ vascular drainage and entrapment of Aβ in the periarterial space leads to development of CAA [56]. Similarly, pathological studies on some of the mouse models have suggested that in certain situations, dense plaques in mouse models are also associated with vascular walls as has been proposed earlier for sporadic AD [117] and shown for Flemish APP pathology [33]. Recent data from Tg2576 and PSAPP mice show that up to 90% of these dense plaques, but not diffuse plaques, are centred on vessel walls or reside in the immediate perivascular regions [99]. Similar observations have also been made on Tg-SwDI mice where all dense plaques were observed to be associated with vessel walls [93]. These data suggest that the mechanisms involved in the formation of “neuritic” dense (core) plaques in select AD such as Flemish APP pathology or in select transgenic mice such as Tg2576 and PSAPP mice are similar to CAA formation, but distinct from those involved in the formation of non-neuritic, diffuse plaques (Figure 3).
- Mouse models demonstrate that similar to large vessel-CAA in humans [119], amyloid associated with vessel walls is predominantly of the Aβ40 type. A similar biochemical profile is also observed for the compact dense-core plaques, which is in contrast to diffuse plaques that are predominantly composed of Aβ42 [120]. Conversely, situations that lead to a drastic reduction of Aβ40 also drastically reduce the prevalence of CAA and dense-core plaques as observed in Austrian APP pathology [102]. Tg2576 and PSAPP mouse models preferentially producing Aβ40 also deposit Aβ40-enriched cerebrovascular amyloid [121,99]. Dutch APP mice also deposit more mutant Aβ40 than mutant Aβ42 in vessels [91]. Furthermore, experimental work on rodents has demonstrated that Aβ40 is more prone to compact as dense deposits in contrast to the faster aggregating Aβ42 that preferentially deposits as diffuse plaques. When soluble Aβ40 and Aβ42 are injected in rat brain, soluble Aβ40 forms congophilic, fibrillar dense deposits while Aβ1-42 forms only diffuse deposits [122]. Lastly, BRI-Aβ42 mice that solely secrete Aβ42 also have Aβ42 peptide trafficked and deposited in vessels [100]. Thus, mouse models support the viewpoint that although “Aβ40” plays an important role in development of vessel-associated compact plaques, most likely due to a more efficient vascular clearance and its high abundance, Aβ42 on its own also has the potential to migrate and deposit in association with vessels; and without a critical relative level of Aβ42, Aβ40 clearance is too efficient to allow deposition.
- Mouse models have partly elucidated the role of ApoE which is otherwise poorly understood. ApoE has been shown to bind to Aβ [123], and studies on transgenic mice suggest that ApoE, especially ApoE-ε4, has a role in Aβ fibrillization [124]. When APP London mice are crossbred with ApoE knockout mice, Aβ chiefly deposits as diffuse, nonfibrillar plaques. However, when these mice were crossbred with mice transgenically expressing human ApoE-ε4, they develop far more fibrillogenic, dense-core plaques and CAA than when crossbred with mice expressing the ApoE-ε3 isoform [124]. Because dense-core plaques and CAA are rich in Aβ40, these data suggest that ApoE-ε4 has a role in Aβ40 fibrillization as also suggested by studies on AD [125].
- Transgenic mouse models serve as a useful model to study CAA-associated pathological changes. Vascular Aβ deposits in mouse models are shown to cause degeneration of vascular smooth muscle cells and of other vascular components typically identified in AD and hereditary cerebral amyloidosis [126,91,11]. Some mouse models also show ultrastructural microvascular abnormalities in non-amyloidotic vessels such as endothelial cell loss, basement membrane thickening, and degeneration of smooth muscle cells and pericytes as shown for AD [99,11]. Additionally, mouse models have shown that basement membrane abnormalities could also contribute to development of CAA as capillary basement membrane thickening precedes the development of CAA in TGF-β transgenic mice [127]. And lastly, transgenic mouse models such as APP23 and Tg2576 have shown that vascular amyloidosis is indeed the cause of spontaneous haemorrhages as both cerebral microhaemorrhages and fatal lobar haemorrhages occur in these mice [126,99].
- Finally, mouse models of amyloidosis have proved to be essential in testing therapeutic amyloid targeting from vessels. A number of active and passive immunotherapeutic approaches such as peripheral sequestering utilizing non-immune mechanisms have been successfully tried in these mouse models [128,129,130,131,132]. In an active immunization trial on an AD mouse model, behavioural and cognitive abnormalities were shown to be reversed coinciding with ≈ 50% reduction in dense-core plaques [129]. Similarly, a passive immunization approach in Tg2576 mouse model has been shown to revert some of the BBB abnormalities observed in these mice [133]. Furthermore, mouse models also reproduce some of the side effects of anti-Aβ vaccinations. For instance, similar to one of the encephalitic patients from the Aβ active immunization trial revealing presence of multiple cortical haemorrhages in association with Tcell inflammatory infiltrates [134], APP23 mice receiving passive anti-Aβ immunization were also shown to have infrequent but severe CAA-associated microhaemorrhages [135]. Furthermore, similar to breakdown of BBB seen in Tg2576 [99], autopsy of one of the encephalitic patients from the active immunization trial revealed that antibody titers in cerebrospinal fluid equalled those in plasma, again indicating a severe breakdown of the BBB [136]. These data indicate that mouse models of amyloidosis could be instrumental in understanding some of the ill effects of anti-amyloid drug targeting. However, data from mouse models should always be viewed with caution as mouse models also have serious limitations as reviewed recently [72]. As an example, vaccination trials targeting Aβ N-terminus in humanized mouse models could easily miss the adverse effects caused by sequestration of physiological Aβ because human and murine Aβ have a different N-terminus. Despite these limitations, mouse models would continue to provide important clues in the understanding of the processes involved in vascular amyloidosis and in causing dementia.
6. Conclusions
Acknowledgments
References
- Vinters, HV; Wang, ZZ; Secor, DL. Brain parenchymal and microvascular amyloid in Alzheimer's disease. Brain Pathol 1996, 6, 179–195. [Google Scholar]
- Jellinger, KA. Alzheimer disease and cerebrovascular pathology: an update. J. Neural Transm 2002, 109, 813–836. [Google Scholar]
- Revesz, T; Holton, JL; Lashley, T; Plant, G; Frangione, B; Rostagno, A; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol 2009. [Google Scholar]
- Hardy, J; Selkoe, DJ. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 2002, 297, 353–356. [Google Scholar]
- Scholtz, W. Studien zur Pathologie der Hirngefässe. II. Die drüsige Entartung der Hirnarterien und -capillären. Z.Gesamte.Neurol.Psychiat 1938, 162, 694–715. [Google Scholar]
- Mandybur, TI. The incidence of cerebral amyloid angiopathy in Alzheimer's disease. Neurology 1975, 25, 120–126. [Google Scholar]
- Thal, DR; Ghebremedhin, E; Rub, U; Yamaguchi, H; Del Tredici, K; Braak, H. Two types of sporadic cerebral amyloid angiopathy. J. Neuropathol. Exp. Neurol 2002, 61, 282–293. [Google Scholar]
- Surbeck, E. L'angiopathie dyshorique (Morel) d l'ecorce cerebrale Etude anatomoclinique et statistique: Aspect genetique; Thesis. University of Geneva: Geneva, Switzerland, 1961. [Google Scholar]
- Vonsattel, JP; Myers, RH; Hedley-Whyte, ET; Ropper, AH; Bird, ED; Richardson, EP, Jr. Cerebral amyloid angiopathy without and with cerebral hemorrhages: a comparative histological study. Ann. Neurol 1991, 30, 637–649. [Google Scholar]
- Kalaria, RN; Ballard, C. Overlap between pathology of Alzheimer disease and vascular dementia. Alzheimer Dis Assoc Disord 1999, 13(Suppl 3). [Google Scholar]
- Farkas, E; Luiten, PGM. Cerebral microvascular pathology in aging and Alzheimer's disease. Prog. Neurobiol 2001, 64, 575–611. [Google Scholar]
- Okazaki, H; Reagan, TJ; Campbell, RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin.Proc 1979, 54, 22–31. [Google Scholar]
- Mandybur, TI. Cerebral amyloid angiopathy: the vascular pathology and complications. J. Neuropathol. Exp. Neurol 1986, 45, 79–90. [Google Scholar]
- Vinters, HV. Cerebral amyloid angiopathy. A critical review. Stroke 1987, 18, 311–324. [Google Scholar]
- O'Donnell, HC; Rosand, J; Knudsen, KA; Furie, KL; Segal, AZ; Chiu, RI; Ikeda, D; Greenberg, SM. Apolipoprotein E genotype and the risk of recurrent lobar intracerebral hemorrhage. N. Engl. J. Med 2000, 342, 240–245. [Google Scholar]
- Jellinger, KA; Lauda, F; Attems, J. Sporadic cerebral amyloid angiopathy is not a frequent cause of spontaneous brain hemorrhage. Eur. J. Neurol 2007, 14, 923–928. [Google Scholar]
- Snowdon, DA; Greiner, LH; Mortimer, JA; Riley, KP; Greiner, PA; Markesbery, WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study [see comments]. JAMA 1997, 277, 813–817. [Google Scholar]
- Cadavid, D; Mena, H; Koeller, K; Frommelt, RA. Cerebral beta amyloid angiopathy is a risk factor for cerebral ischemic infarction. A case control study in human brain biopsies. J. Neuropathol. Exp. Neurol 2000, 59, 768–773. [Google Scholar]
- Suter, OC; Sunthorn, T; Kraftsik, R; Straubel, J; Darekar, P; Khalili, K; Miklossy, J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke 2002, 33, 1986–1992. [Google Scholar]
- Gray, F; Dubas, F; Roullet, E; Escourolle, R. Leukoencephalopathy in diffuse hemorrhagic cerebral amyloid angiopathy. Ann. Neurol 1985, 18, 54–59. [Google Scholar]
- Greenberg, SM; Vonsattel, JP; Stakes, JW; Gruber, M; Finklestein, SP. The clinical spectrum of cerebral amyloid angiopathy: presentations without lobar hemorrhage. Neurology 1993, 43, 2073–2079. [Google Scholar]
- Attems, J; Quass, M; Jellinger, KA; Lintner, F. Topographical distribution of cerebral amyloid angiopathy and its effect on cognitive decline are influenced by Alzheimer disease pathology. J. Neurol. Sci 2007, 257, 49–55. [Google Scholar]
- Vidal, R; Calero, M; Piccardo, P; Farlow, MR; Unverzagt, FW; Mendez, E; Jimenez-Huete, A; Beavis, R; Gallo, G; Gomez-Tortosa, E; Ghiso, J; Hyman, BT; Frangione, B; Ghetti, B. Senile dementia associated with amyloid beta protein angiopathy and tau perivascular pathology but not neuritic plaques in patients homozygous for the APOE-epsilon4 allele. Acta Neuropathol. (Berl) 2000, 100, 1–12. [Google Scholar]
- Attems, J; Jellinger, KA. Only cerebral capillary amyloid angiopathy correlates with Alzheimer pathology—a pilot study. Acta Neuropathol. (Berl) 2004, 107, 83–90. [Google Scholar]
- Glenner, GG; Wong, CW. Alzheimer's disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun 1984, 122, 885–890. [Google Scholar]
- van Duinen, SG; Castaño, EM; Prelli, F; Bots, GTAB; Luyendijk, W; Frangione, B. Hereditary cerebral hemorrhage with amyloidosis in patients of Dutch origin is related to Alzheimer disease. Proc. Natl. Acad. Sci. USA 1987, 84, 5991–5994. [Google Scholar]
- Haan, J; Hardy, JA; Roos, RAC. Hereditary cerebral hemorrhage with amyloidosis-Dutch type: its importance for Alzheimer research. TINS 1991, 14, 231–234. [Google Scholar]
- Maat-Schieman, ML; Yamaguchi, H; van Duinen, SG; Natte, R; Roos, RA. Age-related plaque morphology and C-terminal heterogeneity of amyloid beta in Dutch-type hereditary cerebral hemorrhage with amyloidosis. Acta Neuropathol. (Berl) 2000, 99, 409–419. [Google Scholar]
- Levy, E; Carman, MD; Fernandez-Madrid, IJ; Power, MD; Lieberburg, I; van Duinen, SG; Bots, GT; Luyendijk, W; Frangione, B. Mutation of the Alzheimer's disease amyloid gene in hereditary cerebral hemorrhage, Dutch type. Science 1990, 248, 1124–1126. [Google Scholar]
- Van Broeckhoven, C; Haan, J; Bakker, E; Hardy, JA; Van Hul, W; Wehnert, A; Vegter-Van, dV; Roos, RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 1990, 248, 1120–1122. [Google Scholar]
- Hendriks, L; van Duijn, CM; Cras, P; Cruts, M; Van Hul, W; van Harskamp, F; Warren, A; McInnis, MG; Antonarakis, SE; Martin, J-J; Hofman, A; Van Broeckhoven, C. Presenile dementia and cerebral haemorrhage linked to a mutation at condon 692 of the β-amyloid precursor protein gene. Nat. Genet 1992, 1, 218–221. [Google Scholar]
- Roks, G; van Harskamp, F; De, KI; Cruts, M; De Jonghe, C; Kumar-Singh, S; Tibben, A; Tanghe, H; Niermeijer, MF; Hofman, A; van Swieten, JC; Van Broeckhoven, C; van Duijn, CM. Presentation of amyloidosis in carriers of the codon 692 mutation in the amyloid precursor protein gene (APP692). Brain 2000, 123, 2130–2140. [Google Scholar]
- Kumar-Singh, S; Cras, P; Wang, R; Kros, JM; van Swieten, J; Lubke, U; Ceuterick, C; Serneels, S; Vennekens, K; Timmermans, J-P; Van Marck, E; Martin, J-J; van Duijn, C; Van Broeckhoven, C. Dense-core senile plaques in the Flemish variant of Alzheimer's disease are vasocentric. Am J Pathol 2002, 161, 507–520. [Google Scholar]
- Cras, P; van Harskamp, F; Hendriks, L; Ceuterick, C; van Duijn, CM; Stefanko, SZ; Hofman, A; Kros, JM; Van Broeckhoven, C; Martin, JJ. Presenile Alzheimer dementia characterized by amyloid angiopathy and large amyloid core type senile plaques in the APP 692Ala–>Gly mutation. Acta Neuropathol 1998, 96, 253–260. [Google Scholar]
- Brooks, WS; Kwok, JB; Halliday, GM; Godbolt, AK; Rossor, MN; Creasey, H; Jones, AO; Schofield, PR. Hemorrhage is uncommon in new Alzheimer family with Flemish amyloid precursor protein mutation. Neurology 2004, 63, 1613–1617. [Google Scholar]
- Miravalle, L; Tokuda, T; Chiarle, R; Giaccone, G; Bugiani, O; Tagliavini, F; Frangione, B; Ghiso, J. Substitutions at codon 22 of Alzheimer's A{beta} peptide induce conformational changes and diverse apoptotic effects in human cerebral endothelial cells. J. Biol. Chem 2000, 275, 27110–27116. [Google Scholar]
- Obici, L; Demarchi, A; de Rosa, G; Bellotti, V; Marciano, S; Donadei, S; Arbustini, E; Palladini, G; Diegoli, M; Genovese, E; Ferrari, G; Coverlizza, S; Merlini, G. A novel AbetaPP mutation exclusively associated with cerebral amyloid angiopathy. Ann. Neurol 2005, 58, 639–644. [Google Scholar]
- Nilsberth, C; Westlind-Danielsson, A; Eckman, C; Condron, MM; Axelman, K; Forsell, C; Stenh, C; Luthman, H; Teplow, DB; Younkin, SG; Naslund, J; Lannfelt, L. The ‘Arctic’ APP mutation (E693G) causes Alzheimer's disease by enhanced Abeta protofibril formation. Nat. Neurosci 2001, 4, 887–893. [Google Scholar]
- Basun, H; Bogdanovic, N; Ingelsson, M; Almkvist, O; Naslund, J; Axelman, K; Bird, TD; Nochlin, D; Schellenberg, GD; Wahlund, LO; Lannfelt, L. Clinical and neuropathological features of the arctic APP gene mutation causing early-onset Alzheimer disease. Arch. Neurol 2008, 65, 499–505. [Google Scholar]
- Grabowski, TJ; Cho, HS; Vonsattel, JP; Rebeck, GW; Greenberg, SM. Novel Amyloid Precursor Protein Mutation in an Iowa Family with Dementia and Severe Cerebral Amyloid Angiopathy. Ann. Neurol 2001, 49, 697–705. [Google Scholar]
- Greenberg, SM; Shin, Y; Grabowski, TJ; Cooper, GE; Rebeck, GW; Iglesias, S; Chapon, F; Tournier-Lasserve, E; Baron, JC. Hemorrhagic stroke associated with the Iowa amyloid precursor protein mutation. Neurology 2003, 60, 1020–1022. [Google Scholar]
- Rossi, G; Giaccone, G; Maletta, R; Morbin, M; Capobianco, R; Mangieri, M; Giovagnoli, AR; Bizzi, A; Tomaino, C; Perri, M; Di, NM; Tagliavini, F; Bugiani, O; Bruni, AC. A family with Alzheimer disease and strokes associated with A713T mutation of the APP gene. Neurology 2004, 63, 910–912. [Google Scholar]
- Di Fede, G; Catania, M; Morbin, M; Rossi, G; Suardi, S; Mazzoleni, G; Merlin, M; Giovagnoli, AR; Prioni, S; Erbetta, A; Falcone, C; Gobbi, M; Colombo, L; Bastone, A; Beeg, M; Manzoni, C; Francescucci, B; Spagnoli, A; Cantu, L; Del, FE; Levy, E; Salmona, M; Tagliavini, F. A recessive mutation in the APP gene with dominant-negative effect on amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar]
- Van Broeckhoven, C; Haan, J; Bakker, E; Hardy, JA; Van Hul, W; Wehnert, A; Vegter-Van, dV; Roos, RA. Amyloid beta protein precursor gene and hereditary cerebral hemorrhage with amyloidosis (Dutch). Science 1990, 248, 1120–1122. [Google Scholar]
- Tomiyama, T; Nagata, T; Shimada, H; Teraoka, R; Fukushima, A; Kanemitsu, H; Takuma, H; Kuwano, R; Imagawa, M; Ataka, S; Wada, Y; Yoshioka, E; Nishizaki, T; Watanabe, Y; Mori, H. A new amyloid beta variant favoring oligomerization in Alzheimer's-type dementia. Ann. Neurol 2008, 63, 377–387. [Google Scholar]
- Kumar-Singh, S; De Jonghe, C; Cruts, M; Kleinert, R; Wang, R; Mercken, M; De Strooper, B; Vanderstichele, H; Lofgren, A; Vanderhoeven, I; Backhovens, H; Vanmechelen, E; Kroisel, PM; Van Broeckhoven, C. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated abeta(42) in Alzheimer's disease. Hum. Mol. Genet 2000, 9, 2589–2598. [Google Scholar]
- Jarrett, JT; Berger, EP; Lansbury, PT, Jr. The carboxy terminus of the -amyloid protein is critical for the seeding of amyloid formation: Implications for the pathogenesis of Alzheimer's disease. Biochemistry 1993, 32, 4693–4697. [Google Scholar]
- Wisniewski, T; Ghiso, J; Frangione, B. Peptides homologous to the amyloid protein of Alzheimer's disease containing a glutamine for glutamic acid substitution have accelerated amyloid fibril formation. Biochem. Biophys. Res. Commun 1991, 179, 1247–1254. [Google Scholar]
- Fraser, PE; Nguyen, JT; Inouye, H; Surewicz, WK; Selkoe, DJ; Podlisny, MB; Kirschner, DA. Fibril formation by primate, rodent and Dutch-hemorrhagic analogues of Alzheimer amyloid β-protein. Biochemistry 1992, 31, 10716–10723. [Google Scholar]
- Walsh, DM; Hartley, DM; Condron, MM; Selkoe, DJ; Teplow, DB. In vitro studies of amyloid beta-protein fibril assembly and toxicity provide clues to the aetiology of Flemish variant (Ala692–>Gly) Alzheimer's disease. Biochem. J 2001, 355, 869–877. [Google Scholar]
- Kumar-Singh, S; Julliams, A; Nuyens, D; Labeur, C; Vennekens, K; Serneels, S; Van Osta, P; Geerts, H; De Strooper, B; Van Broeckhoven, C. In vitro studies of Flemish, Dutch, and wild type Amyloid ß (Aß) provide evidence for a two-stage Aß neurotoxicity. Neurobiol Dis 2002, 11, 300–310. [Google Scholar]
- Walsh, DM; Klyubin, I; Fadeeva, JV; Cullen, WK; Anwyl, R; Wolfe, MS; Rowan, MJ; Selkoe, DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 2002, 416, 535–539. [Google Scholar]
- Davis-Salinas, J; Van Nostrand, WE. Amyloid beta-protein aggregation nullifies its pathologic properties in cultured cerebrovascular smooth muscle cells. J. Biol. Chem 1995, 270, 20887–20890. [Google Scholar]
- Maat-Schieman, ML; van Duinen, SG; Bornebroek, M; Haan, J; Roos, RA. Hereditary cerebral hemorrhage with amyloidosis-Dutch type (HCHWA-D): II–A review of histopathological aspects. Brain Pathol 1996, 6, 115–120. [Google Scholar]
- Farzan, M; Schnitzler, CE; Vasilieva, N; Leung, D; Choe, H. BACE2, a beta -secretase homolog, cleaves at the beta site and within the amyloid-beta region of the amyloid-beta precursor protein. Proc Natl Acad Sci USA 2000, 97, 9712–9717. [Google Scholar]
- Weller, RO; Massey, A; Newman, TA; Hutchings, M; Kuo, YM; Roher, AE. Cerebral amyloid angiopathy: amyloid beta accumulates in putative interstitial fluid drainage pathways in Alzheimer's disease. Am. J. Pathol 1998, 153, 725–733. [Google Scholar]
- Zlokovic, BV. Neurovascular mechanisms of Alzheimer's neurodegeneration. Trends Neurosci 2005, 28, 202–208. [Google Scholar]
- Cullen, KM; Kocsi, Z; Stone, J. Microvascular pathology in the aging human brain: Evidence that senile plaques are sites of microhaemorrhages. Neurobiol Aging 2006, 27, 1786–1796. [Google Scholar]
- Melchor, JP; McVoy, L; Van Nostrand, WE. Charge alterations of E22 enhance the pathogenic properties of the amyloid beta-protein. J.Neurochem 2000, 74, 2209–2212. [Google Scholar]
- Murakami, K; Irie, K; Morimoto, A; Ohigashi, H; Shindo, M; Nagao, M; Shimizu, T; Shirasawa, T. Synthesis, aggregation, neurotoxicity, and secondary structure of various A beta 1- 42 mutants of familial Alzheimer's disease at positions 21–23. Biochem. Biophys. Res. Commun 2002, 294, 5–10. [Google Scholar]
- Tsubuki, S; Takaki, Y; Saido, TC. Dutch, Flemish, Italian, and Arctic mutations of APP and resistance of Abeta to physiologically relevant proteolytic degradation. Lancet 2003, 361, 1957–1958. [Google Scholar]
- Betts, V; Leissring, MA; Dolios, G; Wang, R; Selkoe, DJ; Walsh, DM. Aggregation and catabolism of disease-associated intra-Abeta mutations: reduced proteolysis of AbetaA21G by neprilysin. Neurobiol. Dis 2008, 31, 442–450. [Google Scholar]
- Yamamoto, N; Van Nostrand, WE; Yanagisawa, K. Further evidence of local ganglioside-dependent amyloid beta-protein assembly in brain. Neuroreport 2006, 17, 1735–1737. [Google Scholar]
- Deane, R; Wu, ZH; Sagare, A; Davis, J; Yan, SD; Hamm, K; Xu, F; Parisi, M; Larue, B; Hu, HW; Spijkers, P; Guo, H; Song, XM; Lenting, PJ; Van Nostrand, WE; Zlokovic, BV. LRP/amyloid beta-peptide interaction mediates differential brain efflux of A beta isoforms. Neuron 2004, 43, 333–344. [Google Scholar]
- Davis, J; Xu, F; Miao, J; Previti, ML; Romanov, G; Ziegler, K; Van Nostrand, WE. Deficient cerebral clearance of vasculotropic mutant Dutch/Iowa Double A beta in human A betaPP transgenic mice. Neurobiol. Aging 2006, 27, 946–954. [Google Scholar]
- Lemere, CA; Blusztajn, JK; Yamaguchi, H; Wisniewski, T; Saido, TC; Selkoe, DJ. Sequence of deposition of heterogeneous amyloid beta-peptides and APOE in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol. Dis 1996, 3, 16–32. [Google Scholar]
- Belza, MG; Urich, H. Cerebral amyloid angiopathy in Down's syndrome. Clin.Neuropathol 1986, 5, 257–260. [Google Scholar]
- Donahue, JE; Khurana, JS; Adelman, LS. Intracerebral hemorrhage in two patients with Down's syndrome and cerebral amyloid angiopathy. Acta Neuropathol. (Berl) 1998, 95, 213–216. [Google Scholar]
- McCarron, MO; Nicoll, JA; Graham, DI. A quartet of Down's syndrome, Alzheimer's disease, cerebral amyloid angiopathy, and cerebral haemorrhage: interacting genetic risk factors. J. Neurol. Neurosurg. Psychiatry 1998, 65, 405–406. [Google Scholar]
- Rovelet-Lecrux, A; Hannequin, D; Raux, G; Meur, NL; Laquerriere, A; Vital, A; Dumanchin, C; Feuillette, S; Brice, A; Vercelletto, M; Dubas, F; Frebourg, T; Campion, D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet 2006, 38, 24–26. [Google Scholar]
- Sleegers, K; Brouwers, N; Gijselinck, I; Theuns, J; Goossens, D; Wauters, J; Del Favero, J; Cruts, M; van Duijn, CM; Van Broeckhoven, C. APP duplication is sufficient to cause early onset Alzheimer's dementia with cerebral amyloid angiopathy. Brain 2006, 129, 2977–2983. [Google Scholar]
- Kumar-Singh, S. Cerebral amyloid angiopathy: pathogenetic mechanisms and link to dense amyloid plaques. Genes Brain Behav 2008, 7, 67–82. [Google Scholar]
- Ikeda, M; Sharma, V; Sumi, SM; Rogaeva, EA; Poorkaj, P; Sherrington, R; Nee, L; Tsuda, T; Oda, N; Watanabe, M; Aoki, M; Shoji, M; Abe, K; Itoyama, Y; Hirai, S; Schellenberg, GD; Bird, TD; George-Hyslop, PH. The clinical phenotype of two missense mutations in the presenilin I gene in Japanese patients. Ann. Neurol 1996, 40, 912–917. [Google Scholar]
- Yasuda, M; Maeda, K; Ikejiri, Y; Kawamata, T; Kuroda, S; Tanaka, C. A novel missense mutation in the presenilin-1 gene in a familial Alzheimer's disease pedigree with abundant amyloid angiopathy. Neurosci. Lett 1997, 232, 29–32. [Google Scholar]
- Wegiel, J; Wisniewski, HM; Kuchna, I; Tarnawski, M; Badmajew, E; Popovitch, E; Kulczycki, J; Dowjat, WK; Wisniewski, T. Cell-type-specific enhancement of amyloid-beta deposition in a novel presenilin-1 mutation (P117L). J. Neuropathol. Exp. Neurol 1998, 57, 831–838. [Google Scholar]
- Singleton, AB; Hall, R; Ballard, CG; Perry, RH; Xuereb, JH; Rubinsztein, DC; Tysoe, C; Matthews, P; Cordell, B; Kumar-Singh, S; De Jonghe, C; Cruts, M; Van Broeckhoven, C; Morris, CM. Pathology of early-onset Alzheimer's disease cases bearing the Thr113- 114ins presenilin-1 mutation. Brain 2000, 123, 2467–2474. [Google Scholar]
- Dermaut, B; Kumar-Singh, S; De Jonghe, C; Cruts, M; Lofgren, A; Lubke, U; Cras, P; Dom, R; De Deyn, PP; Martin, JJ; Van Broeckhoven, C. Cerebral amyloid angiopathy is a pathogenic lesion in Alzheimer's disease due to a novel presenilin 1 mutation. Brain 2001, 124, 2383–2392. [Google Scholar]
- Nochlin, D; Bird, TD; Nemens, EJ; Ball, MJ; Sumi, SM. Amyloid angiopathy in a Volga German family with Alzheimer's disease and a presenilin-2 mutation (N141I). Ann. Neurol 1998, 43, 131–135. [Google Scholar]
- Mann, DM; Pickering-Brown, SM; Takeuchi, A; Iwatsubo, T. Amyloid Angiopathy and Variability in Amyloid beta Deposition Is Determined by Mutation Position in Presenilin-1- Linked Alzheimer's Disease. Am. J. Pathol 2001, 158, 2165–2175. [Google Scholar]
- Kumar-Singh, S; Theuns, J; Van Broeck, B; Pirici, D; Vennekens, K; Corsmit, E; Cruts, M; Dermaut, B; Wang, R; Van Broeckhoven, C. Mean age-of-onset of familial alzheimer disease caused by presenilin mutations correlates with both increased Abeta42 and decreased Abeta40. Hum. Mutat 2006, 27, 686–695. [Google Scholar]
- Bentahir, M; Nyabi, O; Verhamme, J; Tolia, A; Horre, K; Wiltfang, J; Esselmann, H; De Strooper, B. Presenilin clinical mutations can affect gamma-secretase activity by different mechanisms. J. Neurochem 2006, 96, 732–742. [Google Scholar]
- Greenberg, SM; Rebeck, GW; Vonsattel, JP; Gomez-Isla, T; Hyman, BT. Apolipoprotein E epsilon 4 and cerebral hemorrhage associated with amyloid angiopathy. Ann. Neurol 1995, 38, 254–259. [Google Scholar]
- Premkumar, DR; Cohen, DL; Hedera, P; Friedland, RP; Kalaria, RN. Apolipoprotein Eepsilon4 alleles in cerebral amyloid angiopathy and cerebrovascular pathology associated with Alzheimer's disease. Am. J. Pathol 1996, 148, 2083–2095. [Google Scholar]
- Attems, J; Lauda, F; Jellinger, KA. Unexpectedly low prevalence of intracerebral hemorrhages in sporadic cerebral amyloid angiopathy: an autopsy study. J. Neurol 2008, 255, 70–76. [Google Scholar]
- Nicoll, JA; Burnett, C; Love, S; Graham, DI; Dewar, D; Ironside, JW; Stewart, J; Vinters, HV. High frequency of apolipoprotein E epsilon 2 allele in hemorrhage due to cerebral amyloid angiopathy. Ann. Neurol 1997, 41, 716–721. [Google Scholar]
- McCarron, MO; Nicoll, JA; Stewart, J; Ironside, JW; Mann, DM; Love, S; Graham, DI; Dewar, D. The apolipoprotein E epsilon2 allele and the pathological features in cerebral amyloid angiopathy-related hemorrhage. J. Neuropathol. Exp. Neurol 1999, 58, 711–718. [Google Scholar]
- Bertram, L; Tanzi, RE. Thirty years of Alzheimer's disease genetics: the implications of systematic meta-analyses. Nat. Rev. Neurosci 2008, 9, 768–778. [Google Scholar]
- Yamada, M; Sodeyama, N; Itoh, Y; Takahashi, A; Otomo, E; Matsushita, M; Mizusawa, H. Association of neprilysin polymorphism with cerebral amyloid angiopathy. J. Neurol. Neurosurg. Psychiatry 2003, 74, 749–751. [Google Scholar]
- Howland, DS; Savage, MJ; Huntress, FA; Wallace, RE; Schwartz, DA; Loh, T; Melloni, RHJ; DeGennaro, LJ; Greenberg, BD; Siman, R. Mutant and native human beta-amyloid precursor proteins in transgenic mouse brain. Neurobiol. Aging 1995, 16, 685–699. [Google Scholar]
- Kumar-Singh, S; Dewachter, I; Moechars, D; Lubke, U; De Jonghe, C; Ceuterick, C; Checler, F; Naidu, A; Cordell, B; Cras, P; Van Broeckhoven, C; Van Leuven, F. Behavioral disturbances without amyloid deposits in mice overexpressing human amyloid precursor protein with Flemish (A692G) or Dutch (E693Q) mutation. Neurobiol Dis 2000, 7, 9–22. [Google Scholar]
- Herzig, MC; Winkler, DT; Burgermeister, P; Pfeifer, M; Kohler, E; Schmidt, SD; Danner, S; Abramowski, D; Sturchler-Pierrat, C; Burki, K; van Duinen, SG; Maat-Schieman, MLC; Staufenbiel, M; Mathews, PM; Jucker, M. A beta is targeted to the vasculature in a mouse model of hereditary cerebral hemorrhage with amyloidosis. Nat. Neurosci 2004, 7, 954–960. [Google Scholar]
- Davis, J; Xu, F; Deane, R; Romanov, G; Previti, ML; Zeigler, K; Zlokovic, BV; Van Nostrand, WE. Early-onset and robust cerebral microvascular accumulation of amyloid betaprotein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem 2004, 279, 20296–20306. [Google Scholar]
- Miao, J; Xu, F; Davis, J; Otte-Holler, I; Verbeek, MM; Van Nostrand, WE. Cerebral microvascular Aß protein deposition induces vascular degeneration and neuroinflammation in transgenic mice expressing human vasculotropic mutant AßPP. Am. J. Pathol 2005, 167, 505–515. [Google Scholar]
- Hsiao, K; Chapman, P; Nilsen, S; Eckman, C; Harigaya, Y; Younkin, S; Yang, F; Cole, G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science 1996, 274, 99–102. [Google Scholar]
- Sturchler-Pierrat, C; Abramowski, D; Duke, M; Wiederhold, KH; Mistl, C; Rothacher, S; Ledermann, B; Burki, K; Frey, P; Paganetti, PA; Waridel, C; Calhoun, ME; Jucker, M; Probst, A; Staufenbiel, M; Sommer, B. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA 1997, 94, 13287–13292. [Google Scholar]
- Domnitz, SB; Robbins, EM; Hoang, AW; Garcia-Alloza, M; Hyman, BT; Rebeck, GW; Greenberg, SM; Bacskai, BJ; Frosch, MP. Progression of cerebral amyloid angiopathy in transgenic mouse models of Alzheimer disease. J. Neuropathol. Exp. Neurol 2005, 64, 588–594. [Google Scholar]
- Borchelt, DR; Ratovitski, T; vanLare, J; Lee, MK; Gonzales, V; Jenkins, NA; Copeland, NG; Price, DL; Sisodia, SS. Accelerated amyloid deposition in the brains of transgenic mice coexpressing mutant presenilin 1 and amyloid precursor proteins. Neuron 1997, 19, 939–945. [Google Scholar]
- Holcomb, L; Gordon, MN; McGowan, E; Yu, X; Benkovic, S; Jantzen, P; Wright, K; Saad, I; Mueller, R; Morgan, D; Sanders, S; Zehr, C; O'Campo, K; Hardy, J; Prada, CM; Eckman, C; Younkin, S; Hsiao, K; Duff, K. Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med 1998, 4, 97–100. [Google Scholar]
- Kumar-Singh, S; Pirici, D; McGowan, E; Serneels, S; Ceuterick, C; Hardy, J; Duff, K; Dickson, D; Van Broeckhoven, C. Dense core plaques in Tg2576 and PSAPP mouse models of Alzheimer's disease are centered on vessel walls. Am J Pathol 2005, 167, 527–543. [Google Scholar]
- McGowan, E; Pickford, F; Kim, J; Onstead, L; Eriksen, J; Yu, C; Skipper, L; Murphy, MP; Beard, J; Das, P; Jansen, K; Delucia, M; Lin, WL; Dolios, G; Wang, R; Eckman, CB; Dickson, DW; Hutton, M; Hardy, J; Golde, T. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron 2005, 47, 191–199. [Google Scholar]
- Snyder, SW; Ladror, US; Wade, WS; Wang, GT; Barrett, LW; Matayoshi, ED; Huffaker, HJ; Krafft, GA; Holzman, TF. Amyloid-Beta Aggregation - Selective-Inhibition of Aggregation in Mixtures of Amyloid with Different Chain Lengths. Biophys. J 1994, 67, 1216–1228. [Google Scholar]
- Kumar-Singh, S; De Jonghe, C; Cruts, M; Kleinert, R; Wang, R; Mercken, M; De Strooper, B; Vanderstichele, H; Lofgren, A; Vanderhoeven, I; Backhovens, H; Vanmechelen, E; Kroisel, PM; Van Broeckhoven, C. Nonfibrillar diffuse amyloid deposition due to a gamma(42)-secretase site mutation points to an essential role for N-truncated abeta(42) in Alzheimer's disease. Hum. Mol. Genet 2000, 9, 2589–2598. [Google Scholar]
- Ancolio, K; Dumanchin, C; Barelli, H; Warter, JM; Brice, A; Campion, D; Frebourg, T; Checler, F. Unusual phenotypic alteration of beta amyloid precursor protein (APP) maturation by a new Val-715 -> Met APP-770 mutation responsible for probable early-onset Alzheimer's disease. Proc. Natl.Acad. Sci. U.S.A 1999, 96, 4119–4124. [Google Scholar]
- De Jonghe, C; Esselens, C; Kumar-Singh, S; Craessaerts, K; Serneels, S; Checler, F; Annaert, W; Van Broeckhoven, C; De Strooper, B. Pathogenic APP mutations near the gammasecretase cleavage site differentially affect Abeta secretion and APP C-terminal fragment stability. Hum. Mol. Genet 2001, 10, 1665–1671. [Google Scholar]
- Strobel, G; Davies, P; Robakis, NK; Iwatsubo, T; Lee, HG; Nuno-Mura, A; Perry, G; Smith, MA; Zheng, H; De Strooper, B; Shen, J; Saura, CA; Golde, T; Hass, M; Yankner, B; Marambaud, P; Checler, F; Kopan, R; Tanzi, R; Neve, R; Davies, P; Marchesi, V; De Strooper, B; Tanzi, R; Van Leuven, F; Pimplikar, SW; Tanzi, R; Kumar-Singh, S. Alzheimer research forum discussion: Gain or loss of function - Time to shake up assumptions on gammasecretase in Alzheimer disease? J. Alzheimers Dis 2007, 11, 399–416. [Google Scholar]
- Mucke, L; Masliah, E; Yu, GQ; Mallory, M; Rockenstein, EM; Tatsuno, G; Hu, K; Kholodenko, D; Johnson-Wood, K; McConlogue, L. High-level neuronal expression of abeta 1- 42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci 2000, 20, 4050–4058. [Google Scholar]
- Wang, R; Wang, B; He, W; Zheng, H. Wild-type presenilin 1 protects against Alzheimer's disease mutation-induced amyloid pathology. J Biol. Chem 2006, 281, 15330–15336. [Google Scholar]
- Deng, Y; Tarassishin, L; Kallhoff, V; Peethumnongsin, E; Wu, L; Li, YM; Zheng, H. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired gamma-secretase activity and exacerbated amyloid pathology. J. Neurosci 2006, 26, 3845–3854. [Google Scholar]
- Wyss-Coray, T; Loike, JD; Brionne, TC; Lu, E; Anankov, R; Yan, F; Silverstein, SC; Husemann, J. Adult mouse astrocytes degrade amyloid-beta in vitro and in situ. Nat. Med 2003, 9, 453–457. [Google Scholar]
- Saido, TC; Iwata, N. Metabolism of amyloid beta peptide and pathogenesis of Alzheimer's disease. Towards presymptomatic diagnosis, prevention and therapy. Neurosci. Res 2006, 54, 235–253. [Google Scholar]
- Farris, W; Schutz, SG; Cirrito, JR; Shankar, GM; Sun, X; George, A; Leissring, MA; Walsh, DM; Qiu, WQ; Holtzman, DM; Selkoe, DJ. Loss of neprilysin function promotes amyloid plaque formation and causes cerebral amyloid angiopathy. Am. J. Pathol 2007, 171, 241–251. [Google Scholar]
- Meyer-Luehmann, M; Stalder, M; Herzig, MC; Kaeser, SA; Kohler, E; Pfeifer, M; Boncristiano, S; Mathews, PM; Mercken, M; Abramowski, D; Staufenbiel, M; Jucker, M. Extracellular amyloid formation and associated pathology in neural grafts. Nat. Neurosci 2003, 6, 370–377. [Google Scholar]
- Shibata, M; Yamada, S; Kumar, SR; Calero, M; Bading, J; Frangione, B; Holtzman, DM; Miller, CA; Strickland, DK; Ghiso, J; Zlokovic, BV. Clearance of Alzheimer's amyloid-ss(1- 40) peptide from brain by LDL receptor-related protein-1 at the blood-brain barrier. J. Clin. Invest 2000, 106, 1489–1499. [Google Scholar]
- Calhoun, ME; Burgermeister, P; Phinney, AL; Stalder, M; Tolnay, M; Wiederhold, KH; Abramowski, D; Sturchler-Pierrat, C; Sommer, B; Staufenbiel, M; Jucker, M. Neuronal overexpression of mutant amyloid precursor protein results in prominent deposition of cerebrovascular amyloid. Proc Natl Acad Sci USA 1999, 96, 14088–14093. [Google Scholar]
- Kida, S; Pantazis, A; Weller, RO. CSF drains directly from the subarachnoid space into nasal lymphatics in the rat. Anatomy, histology and immunological significance. Neuropathol.Appl. Neurobiol 1993, 19, 480–488. [Google Scholar]
- Wang, Z; Wu, D; Vinters, HV. Hypoxia and reoxygenation of brain microvascular smooth muscle cells in vitro: cellular responses and expression of cerebral amyloid angiopathyassociated proteins. APMIS 2002, 110, 423–434. [Google Scholar]
- Miyakawa, T; Shimoji, A; Kuramoto, R; Higuchi, Y. The relationship between senile plaques and cerebral blood vessels in Alzheimer's disease and senile dementia. Morphological mechanism of senile plaque production. Virchows Arch. B. Cell Pathol. Incl. Mol. Pathol 1982, 40, 121–129. [Google Scholar]
- Pirici, D; Van Broeckhoven, C; Kumar-Singh, S. Animal models of dementia; De Deyn, PP, Van Dam, D, Eds.; Neuromethods. Humana Press Inc: Totowa NJ, 2009. [Google Scholar]
- Gravina, SA; Ho, L; Eckman, CB; Long, KE; Otvos, L; Younkin, LH; Suzuki, N; Younkin, SG. Amyloid beta protein (A beta) in Alzheimer's disease brain. Biochemical and immunocytochemical analysis with antibodies specific for forms ending at A beta 40 or A beta 42(43). J. Biol. Chem 1995, 270, 7013–7016. [Google Scholar]
- Iwatsubo, T; Saido, TC; Mann, DM; Lee, VM; Trojanowski, JQ. Full-length amyloid-beta (1–42(43)) and amino-terminally modified and truncated amyloid-beta 42(43) deposit in diffuse plaques. Am. J. Pathol 1996, 149, 1823–1830. [Google Scholar]
- McGowan, E; Sanders, S; Iwatsubo, T; Takeuchi, A; Saido, T; Zehr, C; Yu, X; Uljon, S; Wang, R; Mann, D; Dickson, D; Duff, K. Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol. Dis 1999, 6, 231–244. [Google Scholar]
- Shin, RW; Ogino, K; Kondo, A; Saido, TC; Trojanowski, JQ; Kitamoto, T; Tateishi, J. Amyloid beta-protein (Abeta) 1–40 but not Abeta1-42 contributes to the experimental formation of Alzheimer disease amyloid fibrils in rat brain. J. Neurosci 1997, 17, 8187–8193. [Google Scholar]
- Strittmatter, WJ; Weisgraber, KH; Huang, D; Dong, L-M; Salvesen, GS; Pericak-Vance, M; Schmechel, D; Saunders, AM; Goldgaber, D; Roses, AD. Binding of human apolipoprotein E to synthetic amymloid β peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar]
- Holtzman, DM; Bales, KR; Tenkova, T; Fagan, AM; Parsadanian, M; Sartorius, LJ; Mackey, B; Olney, J; McKeel, D; Wozniak, D; Paul, SM. Apolipoprotein E isoformdependent amyloid deposition and neuritic degeneration in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. USA 2000, 97, 2892–2897. [Google Scholar]
- Mann, DM; Iwatsubo, T; Pickering-Brown, SM; Owen, F; Saido, TC; Perry, RH. Preferential deposition of amyloid beta protein (Abeta) in the form Abeta40 in Alzheimer's disease is associated with a gene dosage effect of the apolipoprotein E E4 allele. Neurosci. Lett 1997, 221, 81–84. [Google Scholar]
- Winkler, DT; Bondolfi, L; Herzig, MC; Jann, L; Calhoun, ME; Wiederhold, KH; Tolnay, M; Staufenbiel, M; Jucker, M. Spontaneous hemorrhagic stroke in a mouse model of cerebral amyloid angiopathy. J. Neurosci 2001, 21, 1619–1627. [Google Scholar]
- Wyss-Coray, T; Lin, C; Sanan, DA; Mucke, L; Masliah, E. Chronic overproduction of transforming growth factor-beta1 by astrocytes promotes Alzheimer's disease-like microvascular degeneration in transgenic mice. Am. J. Pathol 2000, 156, 139–150. [Google Scholar]
- Schenk, D; Barbour, R; Dunn, W; Gordon, G; Grajeda, H; Guido, T; Hu, K; Huang, J; Johnson-Wood, K; Khan, K; Kholodenko, D; Lee, M; Liao, Z; Lieberburg, I; Motter, R; Mutter, L; Soriano, F; Shopp, G; Vasquez, N; Vandevert, C; Walker, S; Wogulis, M; Yednock, T; Games, D; Seubert, P. Immunization with amyloid-beta attenuates Alzheimerdisease- like pathology in the PDAPP mouse [see comments]. Nature 1999, 400, 173–177. [Google Scholar]
- Janus, C; Pearson, J; McLaurin, J; Mathews, PM; Jiang, Y; Schmidt, SD; Chishti, MA; Horne, P; Heslin, D; French, J; Mount, HT; Nixon, RA; Mercken, M; Bergeron, C; Fraser, PE; George-Hyslop, P; Westaway, D. A beta peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer's disease. Nature 2000, 408, 979–982. [Google Scholar]
- DeMattos, RB; Bales, KR; Cummins, DJ; Paul, SM; Holtzman, DM. Brain to plasma amyloid-beta efflux: a measure of brain amyloid burden in a mouse model of Alzheimer's disease. Science 2002, 295, 2264–2267. [Google Scholar]
- Matsuoka, Y; Saito, M; LaFrancois, J; Saito, M; Gaynor, K; Olm, V; Wang, LL; Casey, E; Lu, YF; Shiratori, C; Lemere, C; Duff, K. Novel therapeutic approach for the treatment of Alzheimer's disease by peripheral administration of agents with an affinity to beta-amyloid. J. Neurosci 2003, 23, 29–33. [Google Scholar]
- Deane, R; Du, YS; Submamaryan, RK; Larue, B; Jovanovic, S; Hogg, E; Welch, D; Manness, L; Lin, C; Yu, J; Zhu, H; Ghiso, J; Frangione, B; Stern, A; Schmidt, AM; Armstrong, DL; Arnold, B; Liliensiek, B; Nawroth, P; Hofman, F; Kindy, M; Stern, D; Zlokovic, B. RAGE mediates amyloid-beta peptide transport across the blood-brain barrier and accumulation in brain. Nat. Med 2003, 9, 907–913. [Google Scholar]
- Dickstein, DL; Biron, KE; Ujiie, M; Pfeifer, CG; Jeffries, AR; Jefferies, WA. Abeta peptide immunization restores blood-brain barrier integrity in Alzheimer disease. FASEB J 2006, 20, 426–433. [Google Scholar]
- Ferrer, I; Boada, RM; Sanchez Guerra, ML; Rey, MJ; Costa-Jussa, F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol 2004, 14, 11–20. [Google Scholar]
- Pfeifer, M; Boncristiano, S; Bondolfi, L; Stalder, A; Deller, T; Staufenbiel, M; Mathews, PM; Jucker, M. Cerebral hemorrhage after passive anti-Abeta immunotherapy. Science 2002, 298, 1379. [Google Scholar]
- Hock, C; Konietzko, U; Streffer, JR; Tracy, J; Signorell, A; Muller-Tillmanns, B; Lemke, U; Henke, K; Moritz, E; Garcia, E; Wollmer, MA; Umbricht, D; de Quervain, DJ; Hofmann, M; Maddalena, A; Papassotiropoulos, A; Nitsch, RM. Antibodies against betaamyloid slow cognitive decline in Alzheimer's disease. Neuron 2003, 38, 547–554. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
| CAA | Cerebral amyloid angiopathy; pathological changes occurring in cerebral blood vessels caused by deposition of amyloid protein of different origins, but here due to Aβ. |
| Dense-core plaques | Also referred to as “neuritic”, “senile”, “classic”, or “mature” plaques. The central compact dense-core is surrounded by a corona of diffuse plaque. The corona contains predominantly Aβ42 and is ThS-negative. The core is ThS-positive. Almost always associated with proximate phospho-tau pathology. |
| Dense plaques | ThS-positive compact amyloid without the corona of diffuse plaques. Major compact plaque type in transgenic mouse models. Biochemically resembles cores of the dense-core plaques. In this review, also sometimes used to include dense-core plaques. |
| Diffuse plaques | Loosely arranged fibrils that are usually ThS-negative. Best recognized with immunohistochemistry and are constituted predominantly of Aβ42. Are not commonly associated with proximate phospho-tau pathology. |
| APP Codon* | Substitution | Position within Aβ | Proximity to secretases | Nickname | Disease | References |
|---|---|---|---|---|---|---|
| 673 | Ala→Val | A2V | β-secretase | [43] | ||
| 692 | Ala→Gly | A21G | α-secretase | Flemish | AD/ Cerebral haemorrhage | [31,33] |
| 693 | Glu→Gln | E22Q | merge with α-secretase | Dutch | Cerebral haemorrhage | [29,44] |
| Glu→Lys | E22K | Italian | Cerebral haemorrhage | [36] | ||
| 693 (merge the cell with above) | Glu→Gly | E22G | also α- secretase (merge the cell with above) | Arctic | AD/ severe CAA but no strokes | [38,39] |
| Glu–>Del | E22Δ | |||||
| Japanese | AD; pathology unavailable | [45] | ||||
| 694 | Asp→Asn | D23N | ||||
| Iowa | AD/ severe CAA but no strokes | [40,41] | ||||
| 705 | Leu→Val | L34V | ||||
| - | Severe CAA | [37] | ||||
| 713 | Ala→Thr | A42T | γ-secretase | - | AD/ Cerebral haemorrhage | [42] |
| 714 | Thr→Iso | T43I | also γ- secretase(merge with above) | Austrian | AD | [46] |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kumar-Singh, S. Hereditary and Sporadic Forms of Aβ-Cerebrovascular Amyloidosis and Relevant Transgenic Mouse Models. Int. J. Mol. Sci. 2009, 10, 1872-1895. https://doi.org/10.3390/ijms10041872
Kumar-Singh S. Hereditary and Sporadic Forms of Aβ-Cerebrovascular Amyloidosis and Relevant Transgenic Mouse Models. International Journal of Molecular Sciences. 2009; 10(4):1872-1895. https://doi.org/10.3390/ijms10041872
Chicago/Turabian StyleKumar-Singh, Samir. 2009. "Hereditary and Sporadic Forms of Aβ-Cerebrovascular Amyloidosis and Relevant Transgenic Mouse Models" International Journal of Molecular Sciences 10, no. 4: 1872-1895. https://doi.org/10.3390/ijms10041872