Neuronal Aneuploidy in Health and Disease:A Cytomic Approach to Understand the Molecular Individuality of Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Aneuploidy in the Normal and Diseased Brain
1.1. The overall rate of aneuploidy for the full complement of chromosomes of any vertebrate brain remains to be determined
1.2. It is unclear what degree of aneuploidy a single cell can support and still survive in the adult brain
1.3. Caution for stem cell therapy
1.4. The functional significance of a brain composed of an intermixed population of aneuploid and euploid neurons is currently unknown
1.5. Chromosomal mosaicism confined to the brain might involve different chromosomes relevant to aging and common brain disorders
2. Selective Vulnerability of Cortical Neurons Reflect the Modular Cortical Structure
3. Ontogenetic and Phylogenetic Aspects of Brain Pathology
4. A Cytomic Approach to Study Molecular Brain Architecture
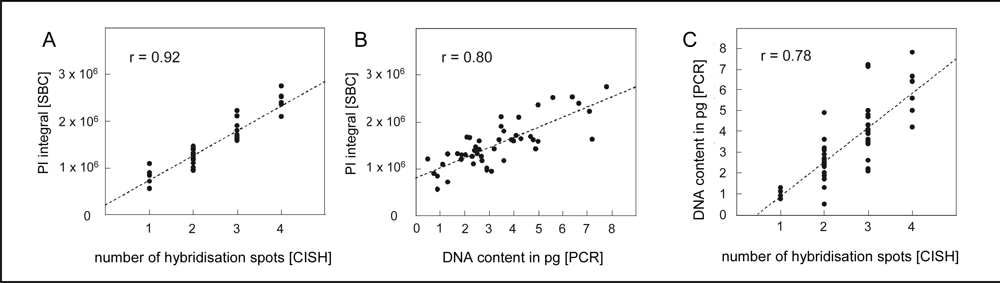
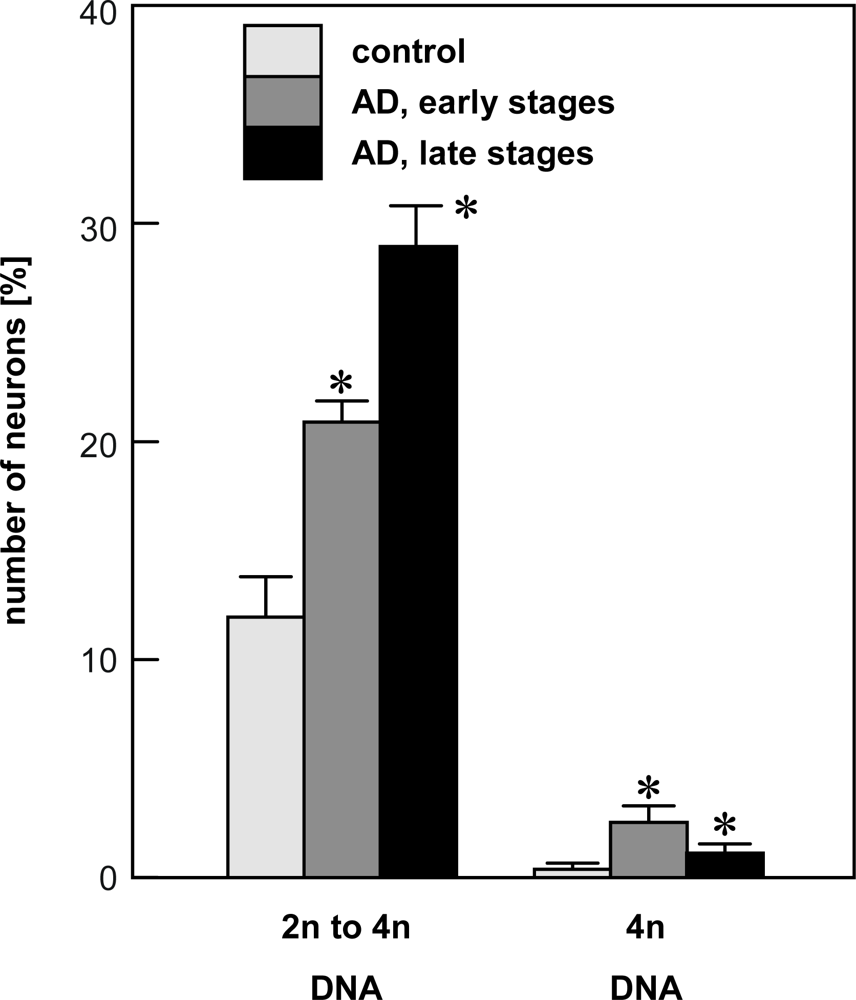
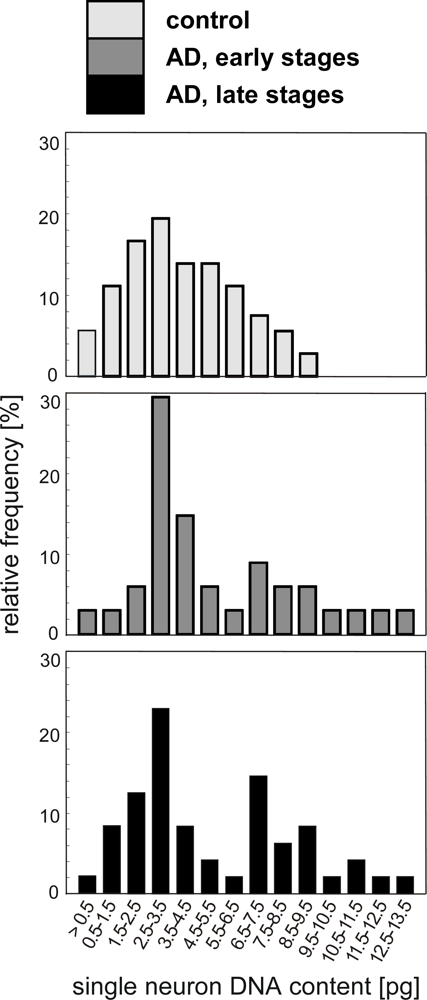
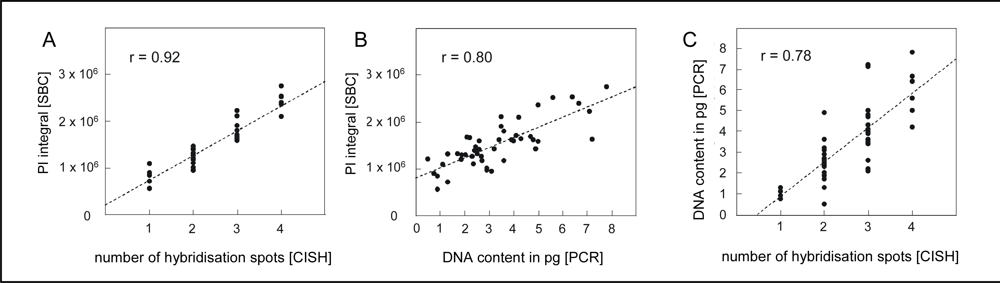
4.1. Quantification of single neuron DNA amount by Slide-based Cytometry (SBC)
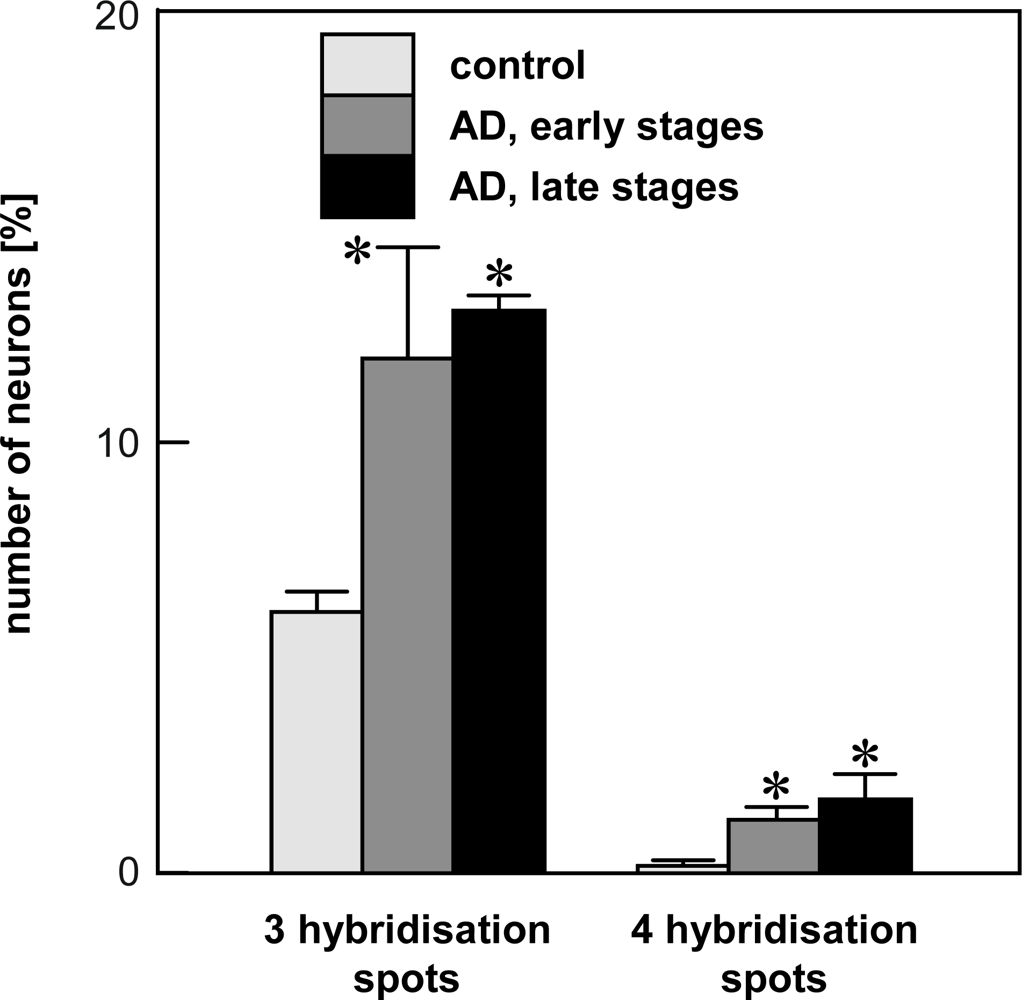
4.2. Chromogenic in situ hybridization
4.3. Laser capture microdissection of identified neurons and PCR amplification of alu-repeats
5. Conclusions
References
- Iourov, IY; Vorsanova, SG; Yurov, YB. Chromosomal mosaicism goes global. Mol. Cytogen 2008, 1, 26. [Google Scholar]
- Iourov, IY; Liehr, T; Vorsanova, SG; Kolotii, AD; Yurov, YB. Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosome Res 2006, 14, 223–229. [Google Scholar]
- Kaushal, D; Contos, JJ; Treuner, K; Yang, AH; Kingsbury, MA; Rehen, SK; McConnell, MJ; Okabe, M; Barlow, C; Chun, J. Alteration of gene expression by chromosome loss in the postnatal mouse brain. J. Neurosci 2003, 23, 5599–5606. [Google Scholar]
- Kingsbury, MA; Friedman, B; McConnell, MJ; Rehen, SK; Yang, AH; Kaushal, D; Chun, J. Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Nat. Acad. Sci. USA 2005, 102, 6143–6147. [Google Scholar]
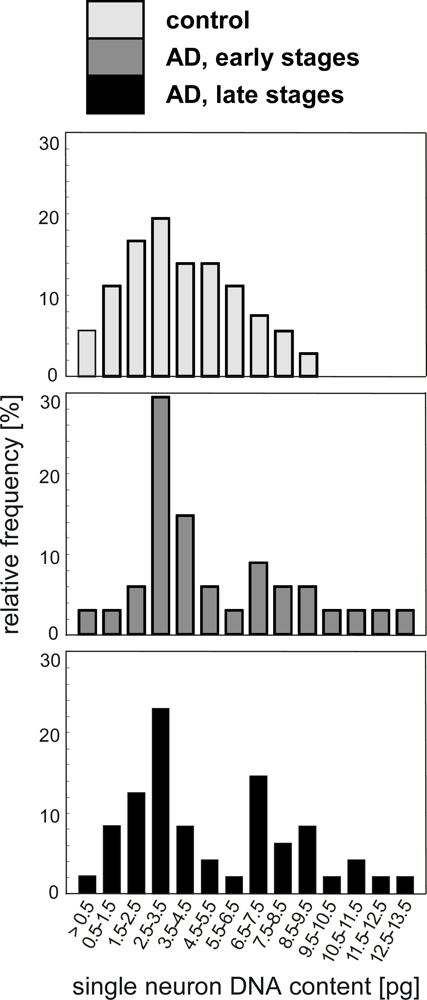
- Mosch, B; Morawski, M; Mittag, A; Lenz, D; Tarnok, A; Arendt, T. Aneuploidy and DNA replication in the normal human brain and Alzheimer's disease. J. Neurosci 2007, 27, 6859–6867. [Google Scholar]
- Pack, SD; Weil, RJ; Vortmeyer, AO; Zeng, W; Li, J; Okamoto, H; Furuta, M; Pak, E; Lubensky, IA; Oldfield, EH; Zhuang, Z. Individual adult human neurons display aneuploidy: Detection by fluorescence in situ hybridization and single neuron PCR. Cell Cycle 2005, 4, 1758–1760. [Google Scholar]
- Rehen, SK; McConnell, MJ; Kaushal, D; Kingsbury, MA; Yang, AH; Chun, J. Chromosomal variation in neurons of the developing and adult mammalian nervous system. Proc. Nat. Acad. Sci. USA 2001, 98, 13361–6. [Google Scholar]
- Rehen, SK; Yung, YC; McCreight, MP; Kaushal, D; Yang, AH; Almeida, BS; Kingsbury, MA; Cabral, KM; McConnell, MJ; Anliker, B; Fontanoz, M; Chun, J. Constitutional aneuploidy in the normal human brain. J. Neurosci 2005, 25, 2176–2180. [Google Scholar]
- Westra, JW; Peterson, SE; Yung, YC; Mutoh, T; Barral, S; Chun, J. Aneuploid mosaicism in the developing and adult cerebellar cortex. J. Comp. Neurol 2008, 507, 1944–1951. [Google Scholar]
- Yang, Y; Geldmacher, DS; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer's disease. J. Neurosci 2001, 21, 2661–2668. [Google Scholar]
- Yurov, YB; Iourov, IY; Monakhov, VV; Soloviev, IV; Vostrikov, VM; Vorsanova, SG. The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J. Histochem. Cytochem 2005, 53, 385–390. [Google Scholar]
- King, RC; Stansfiled, WD. A Dictionary of Genetics, 4th Ed ed; Oxford University Press: New York, NY, USA, 1990. [Google Scholar]
- Lengauer, C; Kinzler, KW; Vogelstein, B. Genetic instabilities in human cancers. Nature 1998, 396, 643–649. [Google Scholar]
- Modi, D; Berde, P; Bhartiya, D. Down syndrome: A study of chromosomal mosaicism. Reprod. Biomed 2003, 6, 499–503. [Google Scholar]
- Palmer, CG; Reichmann, A. Chromosomal and clinical findings in 110 females with Turner syndrome. Hum. Genet 1976, 35, 35–49. [Google Scholar]
- Kawame, H; Sugio, Y; Fuyama, Y; Hayashi, Y; Suzuki, H; Kurosawa, K; Maekawa, K. Syndrome of microcephaly, Dandy-Walker malformation, and Wilms tumor caused by mosaic variegated aneuploidy with premature centromere division (PCD): Report of a new case and review of the literature. J. Hum. Genet 1999, 44, 219–224. [Google Scholar]
- Yurov, YB; Vorsanova, SG; Iourov, IY; Demidova, IA; Beresheva, AK; Kravetz, VS; Monakhov, VV; Kolotii, AD; Voinova-Ulas, VY; Gorbachevskaya, NL. Unexplained autism is frequently associated with low-level mosaic aneuploidy. J. Med. Genet 2007, 44, 521–525. [Google Scholar]
- Kingsbury, MA; Yung, YC; Peterson, SE; Westra, JW; Chun, J. Aneuploidy in the normal and diseased brain. Cell Mol. Life Sci 2006, 63, 2626–2641. [Google Scholar]
- Bregnard, A; Knüsel, A; Kuenzle, CC. Are all the neuronal nuclei polyploid? Histochemistry 1975, 43, 59–61. [Google Scholar]
- Swartz, FJ; Bhatnagar, KP. Are CNS neurons polyploid? A critical analysis based upon cytophotometric study of the DNA content of cerebellar and olfactory bulbar neurons of the bat. Brain Res 1981, 208, 267–281. [Google Scholar]
- Yurov, YB; Iourov, IY; Vorsanova, SG; Liehr, T; Kolotii, AD; Kutsev, SI; Pellestor, F; Beresheva, AK; Demidova, IA; Kravets, VS; Monakhov, VV; Soloviev, IV. Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS ONE 2007, 2, e558. [Google Scholar]
- Yang, AH; Kaushal, D; Rehen, SK; Kriedt, K; Kingsbury, MA; McConnell, MJ; Chun, J. Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J. Neurosci 2003, 23, 10454–10462. [Google Scholar]
- Harrison, RH; Kuo, HC; Scriven, PN; Handyside, AH; Ogilvie, CM. Lack of cell cycle checkpoints in human cleavage stage embryos revealed by a clonal pattern of chromosomal mosaicism analysed by sequential multicolour FISH. Zygote 2000, 8, 217–224. [Google Scholar]
- Voullaire, L; Slater, H; Williamson, R; Wilton, L. Chromosome analysis of blastomeres from human embryos by using comparative genomic hybridization. Hum. Genet 2000, 106, 210–217. [Google Scholar]
- Osada, T; Kusakabe, H; Akutsu, H; Yagi, T; Yanagimachi, R. Adult murine neurons: Their chromatin and chromosome changes and failure to support embryonic development as revealed by nuclear transfer. Cytogenet Genome Res 2002, 97, 7–12. [Google Scholar]
- Blaschke, AJ; Staley, K; Chun, J. Widespread programmed cell death in proliferative and postmitotic regions of the fetal cerebral cortex. Development 1996, 122, 1165–1174. [Google Scholar]
- Thomas, P; Fenech, M. Chromosome 17 and 21 aneuploidy in buccal cells is increased with ageing and in Alzheimer's disease. Mutagenesis 2008, 23, 57–65. [Google Scholar]
- McConnell, MJ; Kaushal, D; Yang, AH; Kingsbury, MA; Rehen, SK; Treuner, K; Helton, R; Annas, EG; Chun, J; Barlow, C. Failed clearance of aneuploid embryonic neural progenitor cells leads to excess aneuploidy in the Atm-deficient but not the Trp53-deficient adult cerebral cortex. J. Neurosci 2004, 24, 8090–8096. [Google Scholar]
- Yurov, YB; Vostrikov, VM; Vorsanova, SG; Monakhov, VV; Iourov, IY. Multicolor fluorescent in situ hybridization on post-mortem brain in schizophrenia as an approach for identification of low-level chromosomal aneuploidy in neuropsychiatric diseases. Brain Dev 2001, 23(Suppl 1), S186–S190. [Google Scholar]
- Yurov, YB; Iourov, IY; Vorsanova, SG; Demidova, IA; Kravetz, VS; Beresheva, AK; Kolotii, AD; Monakchov, VV; Uranova, NA; Vostrikov, VM; Soloviev, IV; Liehr, T. The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr Res 2008, 98, 139–147. [Google Scholar]
- Gage, FH. Mammalian neural stem cells. Science 2000, 287, 1433–1438. [Google Scholar]
- Duesberg, P; Li, R; Fabarius, A; Hehlmann, R. Aneuploidy and cancer: From correlation to causation. Contrib. Microbiol 2006, 13, 16–44. [Google Scholar]
- Pellestor, F; Andréo, B; Arnal, F; Humeau, C; Demaille, J. Maternal aging and chromosomal abnormalities: new data drawn from in vitro unfertilized human oocytes. Hum Genet 2003, 112, 195–203. [Google Scholar]
- Rosenbusch, B. The incidence of aneuploidy in human oocytes assessed by conventional cytogenetic analysis. Hereditas 2004, 141, 97–105. [Google Scholar]
- Gimelbrant, A; Hutchinson, JN; Thompson, BR; Chess, A. Widespread monoallelic expression on human autosomes. Science 2007, 318, 1136–1140. [Google Scholar]
- Sebat, J; Lakshmi, B; Troge, J; Alexander, J; Young, J; Lundin, P; Månér, S; Massa, H; Walker, M; Chi, M; Navin, N; Lucito, R; Healy, J; Hicks, J; Ye, K; Reiner, A; Gilliam, TC; Trask, B; Patterson, N; Zetterberg, A; Wigler, M. Large-scale copy number polymorphism in the human genome. Science 2004, 305, 525–528. [Google Scholar]
- Singleton, AB; Farrer, M; Johnson, J; Singleton, A; Hague, S; Kachergus, J; Hulihan, M; Peuralinna, T; Dutra, A; Nussbaum, R; Lincoln, S; Crawley, A; Hanson, M; Maraganore, D; Adler, C; Cookson, MR; Muenter, M; Baptista, M; Miller, D; Blancato, J; Hardy, J; Gwinn- Hardy, K. alpha-Synuclein locus triplication causes Parkinson's disease. Science 2003, 302, 841. [Google Scholar]
- Hassold, T; Hunt, P. To err (meiotically) is human: The genesis of human aneuploidy. Nat. Rev. Genet 2001, 2, 280–291. [Google Scholar]
- Vorsanova, SG; Kolotii, AD; Iourov, IY; Monakhov, VV; Kirillova, EA; Soloviev, IV; Yurov, YB. Evidence for high frequency of chromosomal mosaicism in spontaneous abortions revealed by interphase FISH analysis. J. Histochem. Cytochem 2005, 53, 375–380. [Google Scholar]
- Rezaie, R; Daly, EM; Cutter, WJ; Murphy, DG; Robertson, DM; Delisi, LE; Mackay, CE; Barrick, TR; Crow, TJ; Roberts, N. The influence of sex chromosome aneuploidy on brain asymmetry. Am J Med Genet B Neuropsychiatr Genet 2008. [Google Scholar]
- Bigner, SH; Mark, J; Bigner, DD. Cytogenetics of human brain tumors. Cancer Genet. Cytogenet 1990, 47, 141–154. [Google Scholar]
- Yamada, K; Kasama, M; Kondo, T; Shinoura, N; Yoshioka, M. Chromosome studies in 70 brain tumors with special attention to sex chromosome loss and single autosomal trisomy. Cancer Genet. Cytogenet 1994, 73, 46–52. [Google Scholar]
- Guttenbach, M; Koschorz, B; Bernthaler, U; Grimm, T; Schmid, M. Sex chromosome loss and aging: In situ hybridization studies on human interphase nuclei. Am J Hum Genet 1995, 57, 1143–50. [Google Scholar]
- Jacobs, PA; Court Brown, WM; Doll, R. Distribution of human chromosome counts in relation to age. Nature 1961, 191, 1178–1180. [Google Scholar]
- Nowinski, GP; Van Dyke, DL; Tilley, BC; Jacobsen, G; Babu, VR; Worsham, MJ; Wilson, GN; Weiss, L. The frequency of aneuploidy in cultured lymphocytes is correlated with age and gender but not with reproductive history. Am. J. Hum. Genet 1990, 46, 1101–1111. [Google Scholar]
- Ly, DH; Lockhart, DJ; Lerner, RA; Schultz, PG. Mitotic misregulation and human aging. Science 2000, 287, 2486–2492. [Google Scholar]
- Buckton, KE; Whalley, LJ; Lee, M; Christie, JE. Chromosome changes in Alzheimer's presenile dementia. J. Med. Genet 1983, 20, 46–51. [Google Scholar]
- Moorhead, PS; Heyman, A. Chromosome studies of patients with Alzheimer disease. Am. J. Med. Genet 1983, 14, 545–556. [Google Scholar]
- Nordenson, I; Adolfsson, R; Beckman, G; Bucht, G; Winblad, B. Chromosomal abnormality in dementia of Alzheimer type. Lancet 1980, 1(8166), 481–482. [Google Scholar]
- Nordensson, I; Beckman, G; Adolfsson, R; Bucht, G; Winblad, B. Cytogenetic changes in patients with senile dementia. Age Ageing 1983, 12, 285–295. [Google Scholar]
- Smith, A; Broe, GA; Williamson, M. Chromosome aneuploidy in Alzheimer's disease. Clin. Genet 1983, 24, 54–57. [Google Scholar]
- Ward, BE; Cook, RH; Robinson, A; Austin, JH. Increased aneuploidy in Alzheimer disease. Am. J. Med. Genet 1979, 3, 137–144. [Google Scholar]
- White, BJ; Crandall, C; Goudsmit, J; Morrow, CH; Alling, DW; Gajdusek, DC; Tijio, JH. Cytogenetic studies of familial and sporadic Alzheimer disease. Am. J. Med. Genet 1981, 10, 77–89. [Google Scholar]
- Gorman, P; Roylance, R. Fluorescence in situ hybridization and comparative genomic hybridization. Methods Mol. Med 2006, 120, 269–295. [Google Scholar]
- Jacobs, PA; Baikie, AG; Court Brown, WM; Strong, JA. The somatic chromosomes in mongolism. Lancet 1959, 1(7075), 710. [Google Scholar]
- Lejeune, J; Turpin, R; Gautier, M. Mongolism; a chromosomal disease (trisomy). Bull. Acad. Natl. Med 1959, 143, 256–265. [Google Scholar]
- Lichter, P; Cremer, T; Tang, CJ; Watkins, PC; Manuelidis, L; Ward, DC. Rapid detection of human chromosome 21 aberrations by in situ hybridization. Proc. Nat. Acad. Sci. USA 1988, 85, 9664–9668. [Google Scholar]
- Zheng, YL; Ferguson-Smith, MA; Warner, JP; Ferguson-Smith, ME; Sargent, CA; Carter, NP. Analysis of chromosome 21 copy number in uncultured amniocytes by fluorescence in situ hybridization using a cosmid contig. Prenat. Diagn 1992, 12, 931–943. [Google Scholar]
- Kumra, S; Wiggs, E; Krasnewich, D; Meck, J; Smith, AC; Bedwell, J; Fernandez, T; Jacobsen, LK; Lenane, M; Rapoport, JL. Brief report: association of sex chromosome anomalies with childhood-onset psychotic disorders. J. Am. Acad. Child. Adolesc. Psy 1998, 37, 292–296. [Google Scholar]
- Kunugi, H; Lee, KB; Nanko, S. Cytogenetic findings in 250 schizophrenics: Evidence confirming an excess of the X chromosome aneuploidies and pericentric inversion of chromosome 9. Schizophr. Res 1999, 40, 43–47. [Google Scholar]
- Kaplan, AR. Chromosomal mosaicisms and occasional acentric chromosomal fragments in schizophrenic patients. Biol. Psy 1970, 2, 89–94. [Google Scholar]
- DeLisi, LE; Friedrich, U; Wahlstrom, J; Boccio-Smith, A; Forsman, A; Eklund, K; Crow, TJ. Schizophrenia and sex chromosome anomalies. Schizophr. Bull 1994, 20, 495–505. [Google Scholar]
- Toyota, T; Shimizu, H; Yamada, K; Yoshitsugu, K; Meerabux, J; Hattori, E; Ichimiya, T; Yoshikawa, T. Karyotype analysis of 161 unrelated schizophrenics: no increased rates of X chromosome mosaicism or inv(9), using ethnically matched and age-stratified controls. Schizophr. Res 2001, 52, 171–179. [Google Scholar]
- Mors, O; Mortensen, PB; Ewald, H. No evidence of increased risk for schizophrenia or bipolar affective disorder in persons with aneuploidies of the sex chromosomes. Psychol. Med 2001, 31, 425–430. [Google Scholar]
- Jacquemont, S; Bocéno, M; Rival, JM; Méchinaud, F; David, A. High risk of malignancy in mosaic variegated aneuploidy syndrome. Am. J. Med. Genet 2002, 109, 17–21. [Google Scholar]
- Kajii, T; Ikeuchi, T; Yang, ZQ; Nakamura, Y; Tsuji, Y; Yokomori, K; Kawamura, M; Fukuda, S; Horita, S; Asamoto, A. Cancer-prone syndrome of mosaic variegated aneuploidy and total premature chromatid separation: report of five infants. Am. J. Med. Genet 2001, 104, 57–64. [Google Scholar]
- Limwongse, C; Schwartz, S; Bocian, M; Robin, NH. Child with mosaic variegated aneuploidy and embryonal rhabdomyosarcoma. Am. J. Med. Genet 1999, 82, 20–24. [Google Scholar]
- Callier, P; Faivre, L; Cusin, V; Marle, N; Thauvin-Robinet, C; Sandre, D; Rousseau, T; Sagot, P; Lacombe, E; Faber, V; Mugneret, F. Microcephaly is not mandatory for the diagnosis of mosaic variegated aneuploidy syndrome. Am. J. Med. Genet. A 2005, 137, 204–207. [Google Scholar]
- Thiel, G; Losanowa, T; Kintzel, D; Nisch, G; Martin, H; Vorpahl, K; Witkowski, R. Karyotypes in 90 human gliomas. Cancer Genet. Cytogenet 1992, 58, 109–120. [Google Scholar]
- Galli, R; Binda, E; Orfanelli, U; Cipelletti, B; Gritti, A; De Vitis, S; Fiocco, R; Foroni, C; Dimeco, F; Vescovi, A. Isolation and characterization of tumorigenic, stem-like neural precursors from human glioblastoma. Cancer Res 2004, 64, 7011–7021. [Google Scholar]
- Singh, SK; Clarke, ID; Terasaki, M; Bonn, VE; Hawkins, C; Squire, J; Dirks, PB. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar]
- Singh, SK; Hawkins, C; Clarke, ID; Squire, JA; Bayani, J; Hide, T; Henkelman, RM; Cusimano, MD; Dirks, PB. Identification of human brain tumour initiating cells. Nature 2004, 432, 396–401. [Google Scholar]
- St George-Hyslop, PH; Tanzi, RE; Polinsky, RJ; Neve, RL; Pollen, D; Drachman, D; Growdon, J; Cupples, LA; Nee, L; Myers, RH; O’Sullivan, D; Watkins, PC; Amos, JA; Deutsch, CK; Bodfish, JW; Kinsbourne, M; Feldman, RG; Bruni, A; Amaducci, L; Foncin, JF; Gusella, JF. Absence of duplication of chromosome 21 genes in familial and sporadic Alzheimer's disease. Science 1987, 238, 664–666. [Google Scholar]
- Tanzi, RE; Bird, ED; Latt, SA; Neve, RL. The amyloid beta protein gene is not duplicated in brains from patients with Alzheimer's disease. Science 1987a, 238, 666–669. [Google Scholar]
- Tanzi, RE; St George-Hyslop, PH; Haines, JL; Polinsky, RJ; Nee, L; Foncin, JF; Neve, RL; McClatchey, AI; Conneally, PM; Gusella, JF. The genetic defect in familial Alzheimer's disease is not tightly linked to the amyloid beta-protein gene. Nature 1987b, 329, 156–157. [Google Scholar]
- Rovelet-Lecrux, A; Hannequin, D; Raux, G; Le Meur, N; Laquerrière, A; Vital, A; Dumanchin, C; Feuillette, S; Brice, A; Vercelletto, M; Dubas, F; Frebourg, T; Campion, D. APP locus duplication causes autosomal dominant early-onset Alzheimer disease with cerebral amyloid angiopathy. Nat. Genet 2006, 38, 24–26. [Google Scholar]
- Braak, H; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991, 82, 239–259. [Google Scholar]
- Pearson, RC; Esiri, MM; Hiorns, RW; Wilcock, GK; Powell, TP. Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc. Nat. Acad. Sci. USA 1985, 82, 4531–4534. [Google Scholar]
- Esiri, MM; Chance, SA. Vulnerability to Alzheimer's pathology in neocortex: The roles of plasticity and columnar organization. J Alzheim Dis 2006, 9(3 Suppl), 79–89. [Google Scholar]
- Nagy, Z; Esiri, MM; Jobst, KA; Morris, JH; King, EM; McDonald, B; Litchfield, S; Barnetson, L. Clustering of pathological features in Alzheimer's disease: Clinical and neuroanatomical aspects. Dementia 1996, 7, 121–127. [Google Scholar]
- Kosik, KS; Rogers, J; Kowall, NW. Senile plaques are located between apical dendritic clusters. J. Neuropathol. Exp. Neurol 1987, 46, 1–11. [Google Scholar]
- Buldyrev, SV; Cruz, L; Gomez-Isla, T; Gomez-Tortosa, E; Havlin, S; Le, R; Stanley, HE; Urbanc, B; Hyman, BT. Description of microcolumnar ensembles in association cortex and their disruption in Alzheimer and Lewy body dementias. Proc. Nat. Acad. Sci. USA 2000, 97, 5039–5043. [Google Scholar]
- Terry, RD; Peck, A; DeTeresa, R; Schechter, R; Horoupian, DS. Some morphometric aspects of the brain in senile dementia of the Alzheimer type. Ann. Neurol 1981, 10, 184–192. [Google Scholar]
- Duyckaerts, C; Hauw, JJ; Piette, F; Rainsard, C; Poulain, V; Berthaux, P; Escourolle, R. Cortical atrophy in senile dementia of the Alzheimer type is mainly due to a decrease in cortical length. Acta Neuropathol 1985, 66, 72–74. [Google Scholar]
- Chance, SA; Casanova, MF; Switala, AE; Crow, TJ; Esiri, MM. Minicolumn thinning in temporal lobe association cortex but not primary auditory cortex in normal human ageing. Acta Neuropathol 2006, 111, 459–464. [Google Scholar]
- Buxhoeveden, D; Fobbs, A; Roy, E; Casanova, M. Quantitative comparison of radial cell columns in children with Down's syndrome and controls. J Intellect Disabil Res 2002, 46, 76–81. [Google Scholar]
- Colombo, JA; Quinn, B; Puissant, V. Disruption of astroglial interlaminar processes in Alzheimer's disease. Brain Res. Bull 2002, 58, 235–242. [Google Scholar]
- Armstrong, RA; Cairns, NJ; Lantos, PL. Dementia with Lewy bodies: clustering of Lewy bodies in human patients. Neurosci. Lett 1997, 224, 41–44. [Google Scholar]
- Armstrong, RA; Cairns, NJ; Lantos, PL. Clustering of Pick bodies in patients with Pick's disease. Neurosci. Lett 1998, 242, 81–84. [Google Scholar]
- Casanova, MF. Modular concepts of brain organization and the neuropathology of psychiatric conditions. Psychiatry Res 2003, 118, 101–102. [Google Scholar]
- Rapoport, SI. Brain evolution and Alzheimer's disease. Rev. Neurol. (Paris) 1988, 144, 79–90. [Google Scholar]
- Arendt, T. Synaptic plasticity and cell cycle activation in neurons are alternative effector pathways: The Dr. Jekyll and Mr. Hyde Concept of Alzheimer’s disease or The yin and yang of Neuroplasticity. Prog. Neurobiol 2003, 71, 83–248. [Google Scholar]
- Marin, O; Rubenstein, JL. A long, remarkable journey: tangential migration in the telencephalon. Nat. Rev. Neurosci 2001, 2, 780–790. [Google Scholar]
- Molyneaux, BJ; Arlotta, P; Menezes, JR; Macklis, JD. Neuronal subtype specification in the cerebral cortex. Nat. Rev. Neurosci 2007, 8, 427–437. [Google Scholar]
- Sidman, RL; Rakic, P. Neuronal migration, with special reference to developing human brain: A review. Brain Res 1973, 62, 1–35. [Google Scholar]
- Luskin, MB; Parnavelas, JG; Barfield, JA. Neurons, astrocytes, and oligodendrocytes of the rat cerebral cortex originate from separate progenitor cells: an ultrastructural analysis of clonally related cells. J. Neurosci 1993, 13, 1730–1750. [Google Scholar]
- Kornack, DR; Rakic, P. Radial and horizontal deployment of clonally related cells in the primate neocortex: relationship to distinct mitotic lineages. Neuron 1995, 15, 311–321. [Google Scholar]
- Rakic, P. Specification of cerebral cortical areas. Science 1988, 241, 170–176. [Google Scholar]
- Mountcastle, VB. Modality and topographic properties of single neurons of cat's somatic sensory cortex. J. Neurophysiol 1957, 20, 408–434. [Google Scholar]
- Mountcastle, VB. An organizing principle for cerebral function. In The Mindful Brain; Edelman, GM, Mountcastle, VB, Eds.; MIT Press: Cambridge, MA, USA, 1978; pp. 7–50. [Google Scholar]
- Mountcastle, VB. The columnar organization of the neocortex. Brain 1997, 120, 701–722. [Google Scholar]
- Kriegstein, A; Noctor, S; Martinez-Cerdeno, V. Patterns of neural stem and progenitor cell division may underlie evolutionary cortical expansion. Nat. Rev. Neurosci 2006, 7, 883–890. [Google Scholar]
- Krubitzer, L; Kahn, DM. Nature versus nurture revisited: an old idea with a new twist. Prog. Neurobiol 2003, 70, 33–52. [Google Scholar]
- Kornack, DR; Rakic, P. Changes in cell-cycle kinetics during the development and evolution of primate neocortex. Proc. Nat. Acad. Sci. USA 1998, 95, 1242–1246. [Google Scholar]
- Takahashi, T; Nowakowski, RS; Caviness, VS, Jr. The cell cycle of the pseudostratified ventricular epithelium of the embryonic murine cerebral wall. J. Neurosci 1995, 15, 6046–57. [Google Scholar]
- Hill, RS; Walsh, CA. Molecular insights into human brain evolution. Nature 2005, 437, 64–67. [Google Scholar]
- Iourov, IY; Liehr, T; Vorsanova, SG; Yurov, YB. Interphase chromosome-specific multicolor banding (ICS-MCB): A new tool for analysis of interphase chromosomes in their integrity. Biomol. Eng 2007, 24, 415–417. [Google Scholar]
- Mosch, B; Mittag, A; Lenz, D; Arendt, T; Tárnok, A. Laser scanning cytometry in human brain slices. Cytometry A 2006, 69, 135–138. [Google Scholar]
- Kriete, A. Cytomics in the realm of systems biology. Cytometry A 2005, 68, 19–20. [Google Scholar]
- Valet, G; Leary, JF; Tarnok, A. Cytomics-new technologies: Towards a human cytome project. Cytometry A 2004, 59, 167–171. [Google Scholar]


© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Arendt, T.; Mosch, B.; Morawski, M. Neuronal Aneuploidy in Health and Disease:A Cytomic Approach to Understand the Molecular Individuality of Neurons. Int. J. Mol. Sci. 2009, 10, 1609-1627. https://doi.org/10.3390/ijms10041609
Arendt T, Mosch B, Morawski M. Neuronal Aneuploidy in Health and Disease:A Cytomic Approach to Understand the Molecular Individuality of Neurons. International Journal of Molecular Sciences. 2009; 10(4):1609-1627. https://doi.org/10.3390/ijms10041609
Chicago/Turabian StyleArendt, Thomas, Birgit Mosch, and Markus Morawski. 2009. "Neuronal Aneuploidy in Health and Disease:A Cytomic Approach to Understand the Molecular Individuality of Neurons" International Journal of Molecular Sciences 10, no. 4: 1609-1627. https://doi.org/10.3390/ijms10041609