5-Oxo-hexahydroquinoline Derivatives and Their Tetrahydroquinoline Counterparts as Multidrug Resistance Reversal Agents


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
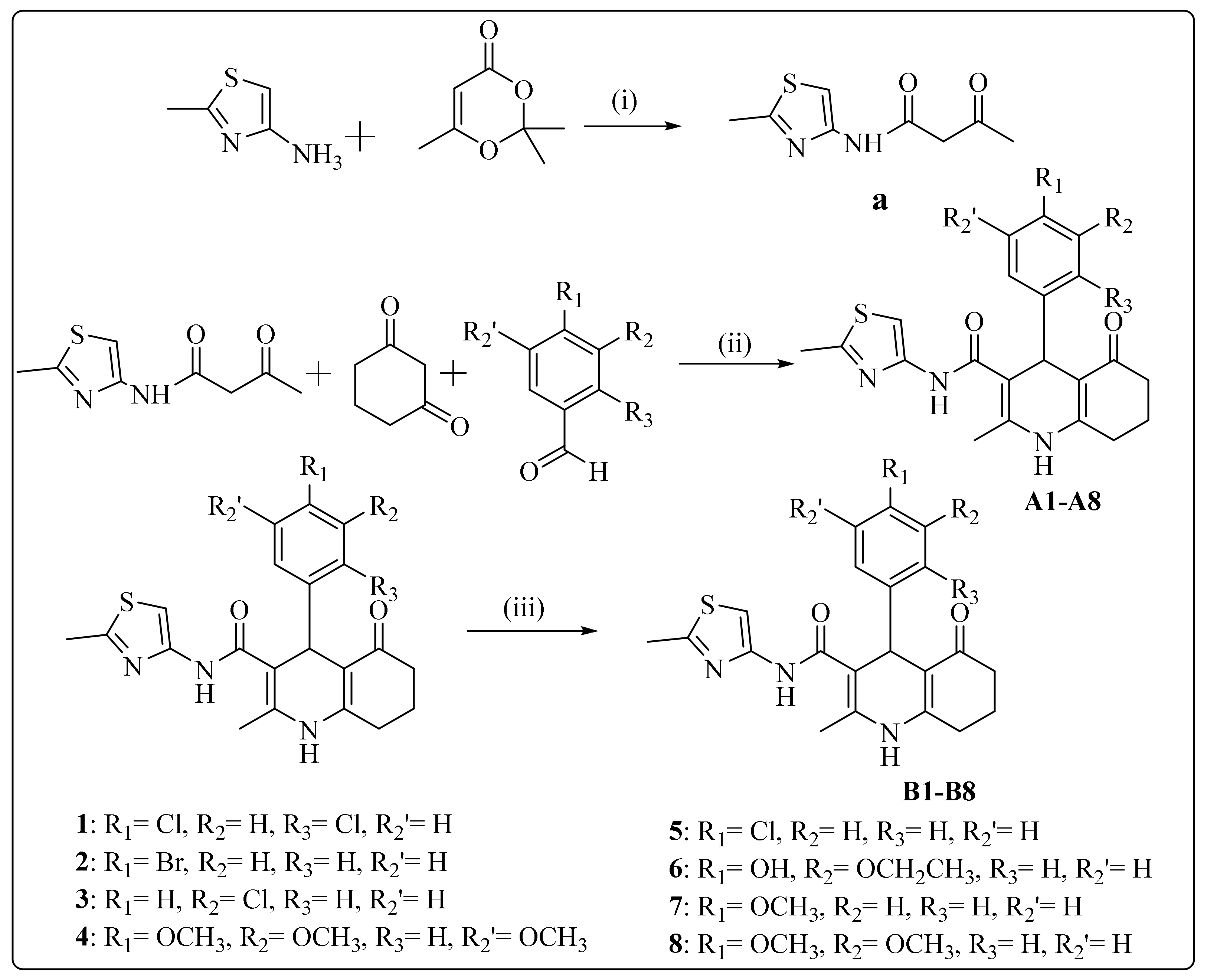
2.1. Chemistry
General Procedure of the Synthesis
2.2. Biological Evaluation
2.2.1. Cell Lines
2.2.2. Reagents
2.2.3. MDR Reversal Assay
2.2.4. Cell Cycle Analysis
2.2.5. Cell Viability Assay
2.3. Statistical Analysis
3. Results and Discussion
3.1. Synthesis
3.2. Biological Evaluations
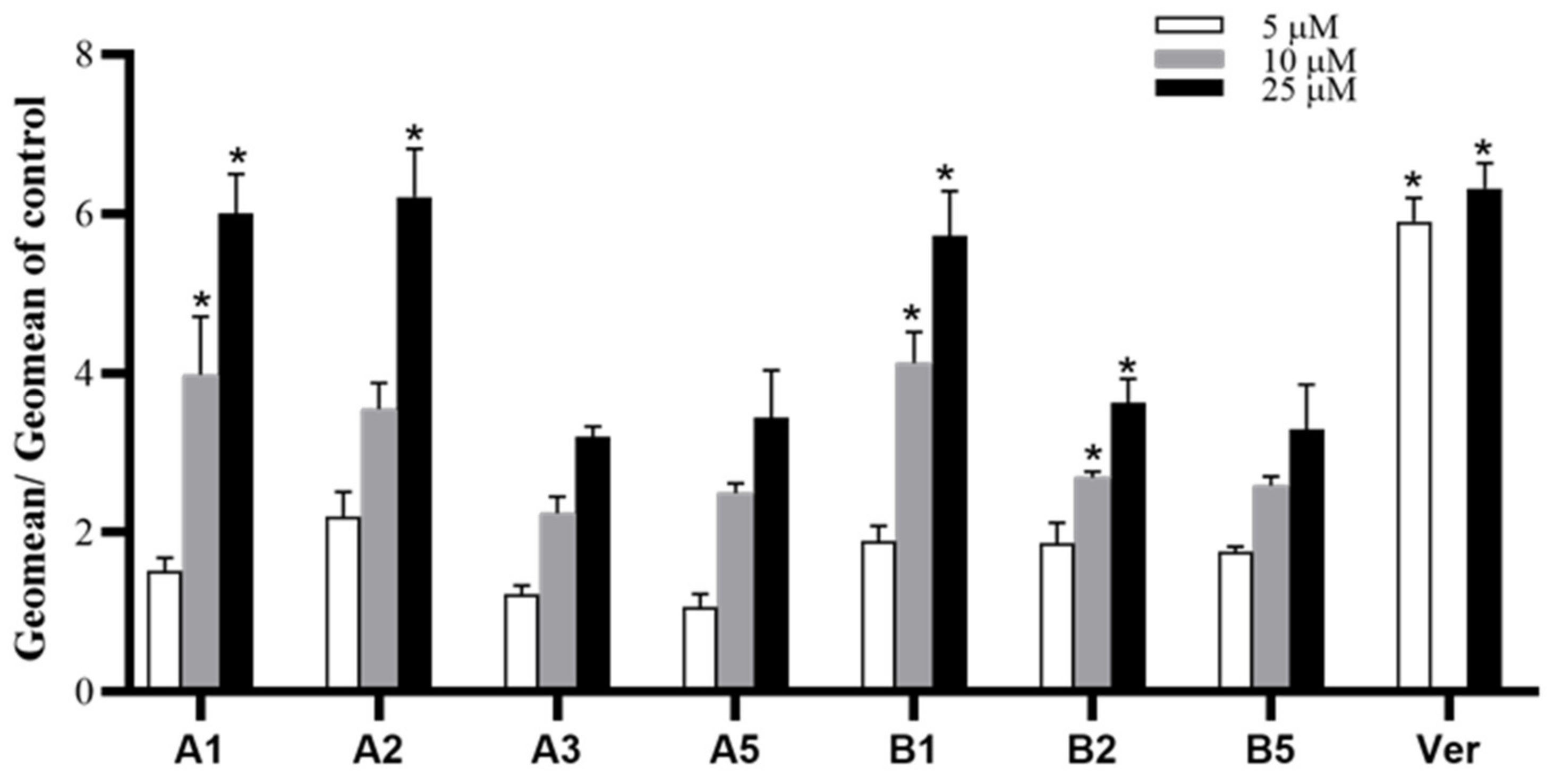
3.2.1. MDR Reversal Assay
3.2.2. Effect of Synthesized Derivatives on Cell Cycle
3.2.3. Cell Viability Assay
3.2.4. Structure-Activity Relationships
3.2.5. Comparison between 1,4-Dihydropyridine and Pyridine Structures
3.2.6. Influence of the Substituted Group at C4 Position of the Phenyl Ring
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations:
References
- Fitzmaurice, C.; Akinyemiju, T.F.; Al Lami, F.H.; Alam, T.; Alizadeh-Navaei, R.; Allen, C.; Alsharif, U.; Alvis-Guzman, N.; Amini, E.; Anderson, B.O.; et al. Global, Regional, and National Cancer Incidence, Mortality, Years of Life Lost, Years Lived with Disability, and Disability-Adjusted Life-Years for 29 Cancer Groups, 1990 to 2016: A Systematic Analysis for the Global Burden of Disease Study. JAMA Oncol. 2018, 4, 1553–1568. [Google Scholar] [PubMed]
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP–dependent transporters. Nat. Rev. Cancer. 2002, 2, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Revisiting the role of ABC transporters in multidrug-resistant cancer. Nat. Rev. Cancer. 2018, 18, 452–464. [Google Scholar] [CrossRef] [PubMed]
- Breier, A.; Gibalova, L.; Seres, M.; Barancik, M.; Sulova, Z. New insight into p-glycoprotein as a drug target. Anticancer Agents Med Chem. 2013, 13, 159–170. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochimica et Biophysica Acta (BBA)-Biomembranes 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Teodori, E.; Dei, S.; Martelli, C.; Scapecchi, S.; Gualtieri, F. The functions and structure of ABC transporters: Implications for the design of new inhibitors of Pgp and MRP1 to control multidrug resistance (MDR). Curr. Drug Targets 2006, 7, 893–909. [Google Scholar] [CrossRef]
- Li, X.; Yuan, H.; Wu, J.; Li, J.; Qu, X.; Xu, W.; Tang, W. Strategies to overcome or circumvent P-glycoprotein mediated multidrug resistance. Curr. Med. Chem. 2008, 15, 470–476. [Google Scholar] [CrossRef]
- Kuppens, I.E.; Witteveen, E.O.; Jewell, R.C.; Radema, S.A.; Paul, E.M.; Mangum, S.G.; Beijnen, J.H.; Voest, E.E.; Schellens, J.H. A phase I, randomized, open-label, parallel-cohort, dose-finding study of elacridar (GF120918) and oral topotecan in cancer patients. Clin. Cancer Res. 2007, 13, 3276–3285. [Google Scholar] [CrossRef] [Green Version]
- Robey, R.W.; Pluchino, K.M.; Hall, M.D.; Fojo, A.T.; Bates, S.E.; Gottesman, M.M. Inhibition of P-glycoprotein (ABCB1)-and multidrug resistance-associated protein 1 (ABCC1)-mediated transport by the orally administered inhibitor, CBT-1®. Biochem. Pharmacol. 2008, 75, 1302–1312. [Google Scholar] [CrossRef] [Green Version]
- Miri, R.; Javidnia, K.; Mirkhani, H.; Hemmateenejad, B.; Sepeher, Z.; Zalpour, M.; Behzad, T.; Khoshneviszadeh, M.; Edraki, N.; Mehdipour, A.R. Synthesis, QSAR and calcium channel modulator activity of new hexahydroquinoline derivatives containing nitroimidazole. Chem. Biol. Drug Des. 2007, 70, 329–336. [Google Scholar] [CrossRef]
- Miri, R.; Javidnia, K.; Sarkarzadeh, H.; Hemmateenejad, B. Synthesis, study of 3D structures, and pharmacological activities of lipophilic nitroimidazolyl-1, 4-dihydropyridines as calcium channel antagonist. Bioorg. Med. Chem. 2006, 14, 4842–4849. [Google Scholar] [CrossRef] [PubMed]
- Edraki, N.; Mehdipour, A.R.; Khoshneviszadeh, M.; Miri, R. Dihydropyridines: Evaluation of their current and future pharmacological applications. Drug Discov. Today 2009, 14, 1058–1066. [Google Scholar] [CrossRef] [PubMed]
- Tomassoni, D.; Lanari, A.; Silvestrelli, G.; Traini, E.; Amenta, F. Nimodipine and its use in cerebrovascular disease: Evidence from recent preclinical and controlled clinical studies. Clin. Exp. Hypertens 2008, 30, 744–766. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, S.A.; Auti, P.B. 1,4-Dihydropyridines: A class of pharmacologically important molecules. Mini Rev. Med. Chem. 2014, 14, 282–390. [Google Scholar] [CrossRef]
- Viradiya, D.; Mirza, S.; Shaikh, F.; Kakadiya, R.; Rathod, A.; Jain, N.; Rawal, R.; Shah, A. Design and Synthesis of 1,4-dihydropyridine Derivatives as Anti-Cancer Agent. Anticancer Agents Med. Chem. 2017, 17, 1003–1013. [Google Scholar] [CrossRef]
- Steiger, S.A.; Li, C.; Backos, D.S.; Reigan, P.; Natale, N. Dimeric isoxazolyl-1, 4-dihydropyridines have enhanced binding at the multi-drug resistance transporter. Bioorg. Med. Chem. 2017, 25, 3223–3234. [Google Scholar] [CrossRef]
- Krauze, A.; Grinberga, S.; Krasnova, L.; Adlere, I.; Sokolova, E.; Domracheva, I.; Shestakova, I.; Andzans, Z.; Duburs, G. Thieno [2, 3-b] pyridines—a new class of multidrug resistance (MDR) modulators. Bioorg. Med. Chem. 2014, 22, 5860–5870. [Google Scholar] [CrossRef]
- Shekari, F.; Sadeghpour, H.; Javidnia, K.; Saso, L.; Nazari, F.; Firuzi, O.; Miri, R. Cytotoxic and multidrug resistance reversal activities of novel 1, 4-dihydropyridines against human cancer cells. Eur. J. Pharmacol. 2015, 746, 233–244. [Google Scholar] [CrossRef]
- Radadiya, A.; Khedkar, V.; Bavishi, A.; Vala, H.; Thakrar, S.; Bhavsar, D.; Shah, A.; Coutinho, E. Synthesis and 3D-QSAR study of 1, 4-dihydropyridine derivatives as MDR cancer reverters. Eur. J. Med. Chem. 2014, 74, 375–387. [Google Scholar] [CrossRef]
- Ranjbar, S.; Firuzi, O.; Edraki, N.; Shahraki, O.; Saso, L.; Khoshneviszadeh, M.; Miri, R. Tetrahydroquinolinone derivatives as potent P-glycoprotein inhibitors: Design, synthesis, biological evaluation and molecular docking analysis. MedChemComm 2017, 8, 1919–1933. [Google Scholar] [CrossRef]
- Firuzi, O.; Javidnia, K.; Mansourabadi, E.; Saso, L.; Mehdipour, A.R.; Miri, R. Reversal of multidrug resistance in cancer cells by novel asymmetrical 1,4-dihydropyridines. Arch. Pharm. Res. 2013, 36, 1392–1402. [Google Scholar] [CrossRef]
- Shahraki, O.; Edraki, N.; Khoshneviszadeh, M.; Zargari, F.; Ranjbar, S.; Saso, L.; Firuzi, O.; Miri, R. Novel 5-oxo-hexahydroquinoline derivatives: Design, synthesis, in vitro P-glycoprotein-mediated multidrug resistance reversal profile and molecular dynamics simulation study. Drug Des. Dev. Ther. 2017, 11, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranjbar, S.; Khonkarn, R.; Moreno, A.; Baubichon-Cortay, H.; Miri, R.; Khoshneviszadeh, M.; Saso, L.; Edraki, N.; Falson, P.; Firuzi, O. 5-Oxo-hexahydroquinoline derivatives as modulators of P-gp, MRP1 and BCRP transporters to overcome multidrug resistance in cancer cells. Toxicol. Appl. Pharmacol. 2019, 362, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, A.; Mohabbati, M.; Cagide, F.; Razzaghi-Asl, N.; Miri, R.; Firuzi, O.; Borges, F. Searching for new cytotoxic agents based on chromen-4-one and chromane-2,4-dione scaffolds. Res. Pharm. Sci. 2019, 14, 74–83. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
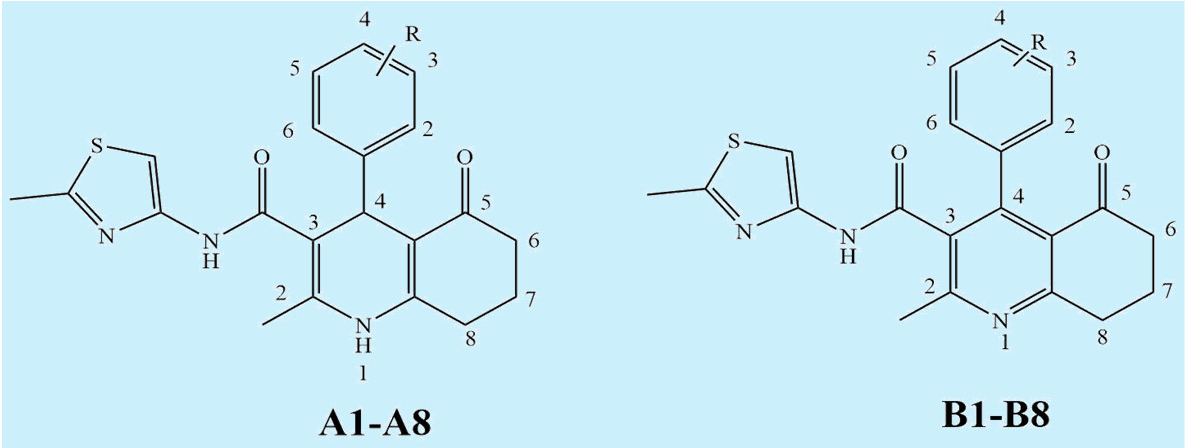
| Compound | Ar | MW | Compound | Ar | MW |
|---|---|---|---|---|---|
| A1 | 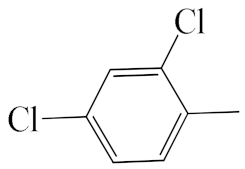 | 448.36 | B1 | 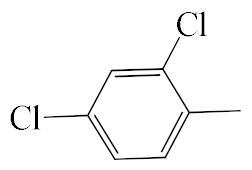 | 446.35 |
| A2 | 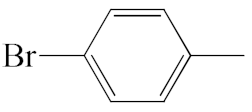 | 458.37 | B2 | 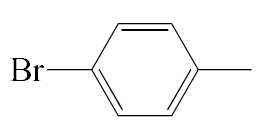 | 456.36 |
| A3 | 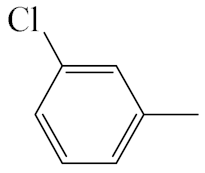 | 413.92 | B3 |  | 411.90 |
| A4 | 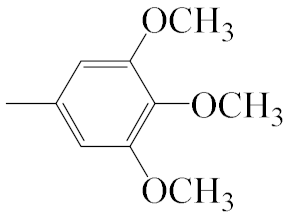 | 469.56 | B4 |  | 467.54 |
| A5 | 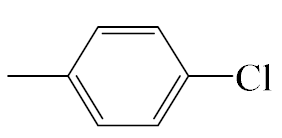 | 413.92 | B5 | 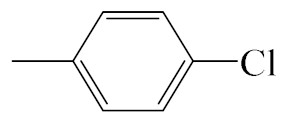 | 411.90 |
| A6 | 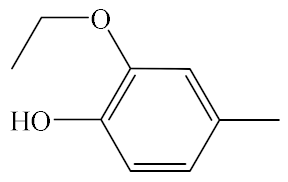 | 439.53 | B6 |  | 437.51 |
| A7 | 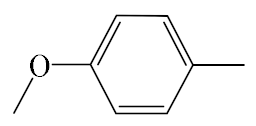 | 409.50 | B7 | 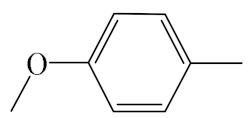 | 407.49 |
| A8 |  | 439.53 | B8 |  | 437.51 |
| Compound | Sub G1 | G0/G1 | S | G2/M |
|---|---|---|---|---|
| Control | 1.99 ± 1.2 | 52.71 ± 0.8 | 20.29 ± 0.7 | 24.99 ± 0.6 |
| A1 (10 µM) | 0.61 ± 0.2 | 52.32 ± 1.8 | 21.85 ± 1.0 | 25.12 ± 1.7 |
| A1 (25 µM) | 0.71 ± 0.2 | 53.30 ± 0.9 | 20.96 ± 1.2 | 25.01 ± 0.7 |
| A1 (100 µM) | 4.23 ± 0.6* | 39.18 ± 1.7 | 21.12 ± 1.6 | 35.48 ± 2.7 |
| A2 (10 µM) | 1.21 ± 0.7 | 56.45 ± 1.1 | 18.90 ± 0.2 | 23.45 ± 0.2 |
| A2 (25 µM) | 1.71 ± 1.1 | 59.47 ± 3.0 | 18.53 ± 1.7 | 20.23 ± 0.7 |
| A2 (100 µM) | 6.88 ± 3.0* | 50.57 ± 2.8 | 19.27 ± 0.3 | 18.37 ± 2.6 |
| A3 (10 µM) | 0.51 ± 0.1 | 50.35 ± 4.5 | 18.23 ± 2.6 | 23.25 ± 1.6 |
| A3 (25 µM) | 0.97 ± 0.2 | 56.18 ± 1.2 | 19.70 ± 2.0 | 23.08 ± 1.0 |
| A3 (100 µM) | 3.59 ± 0.7* | 53.64 ± 1.4 | 13.31 ± 1.0 | 29.46 ± 1.8 |
| A5 (10 µM) | 0.65 ± 0.2 | 56.07 ± 0.3 | 19.03 ± 0.9 | 24.20 ± 1.2 |
| A5 (25 µM) | 0.70 ± 0.3 | 58.03 ± 0.7 | 18.90 ± 0.9 | 22.33 ± 0.5 |
| A5 (100 µM) | 3.17 ± 0.6* | 48.92 ± 1.4 | 18.58 ± 0.6 | 29.32 ± 1.2 |
| B1 (10 µM) | 0.64 ± 0.1 | 49.10 ± 3.7 | 21.27 ± 2.1 | 29.03 ± 2.1 |
| B1 (25 µM) | 0.91 ± 0.0 | 51.13 ± 2.4 | 20.90 ± 2.0 | 27.03 ± 0.6 |
| B1 (100 µM) | 5.08 ± 0.7* | 24.00 ± 4.1 | 25.17 ± 0.7 | 45.77 ± 3.4 |
| B2 (10 µM) | 0.96 ± 0.1 | 53.00 ± 3.6 | 21.80 ± 1.5 | 24.13 ± 2.4 |
| B2 (25 µM) | 1.57 ± 0.4 | 55.83 ± 2.6 | 20.30 ± 1.7 | 22.23 ± 1.4 |
| B2 (100 µM) | 4.25 ± 0.5* | 47.00 ± 0.6 | 19.48 ± 1.8 | 29.27 ± 1.8 |
| B5 (10 µM) | 0.62 ± 0.1 | 53.87 ± 0.7 | 18.13 ± 1.9 | 27.27 ± 2.6 |
| B5 (25 µM) | 0.69 ± 0.1 | 55.43 ± 1.3 | 17.73 ± 1.3 | 26.07 ± 2.7 |
| B5 (100 µM) | 1.91 ± 0.3* | 57.58 ± 0.9 | 13.62 ± 0.8 | 26.87 ± 0.9 |
| Doxorubicin (2.5 µM) | 1.08 ± 0.2 | 34.23 ± 3.3 | 12.57 ± 1.4 | 52.10 ± 4.4 |
| Doxorubicin (5 µM) | 1.81 ± 0.3 | 21.50 ± 2.5 | 12.57 ± 1.7 | 64.13 ± 4.2 |
| Compound | IC50 (µM) in Cultured Cells | |||||
|---|---|---|---|---|---|---|
| MCF-7 | A-549 | K562 | MES-SA-DX5 | MES-SA | HEK-293 | |
| A1 | 72.5 ± 11.9 | 48.8 ± 1.3 | 15.0 ± 0.5 | 40.1 ± 4.7 | 64.1 ± 19.4 | 28.6 ± 4.3 |
| A2 | 56.7 ± 3.6 | 87.1 ± 6.7 | 31.0 ± 2.7 | 31.5 ± 1.5 | >100 | 63.1 ± 7.4 |
| A3 | >100 | >100 | 56.4 ± 7.0 | 78.0 ± 4.0 | >100 | 93.7 ± 11.9 |
| A4 | >100 | >100 | >100 | >100 | - | - |
| A5 | 72.9 ± 12.3 | 87.4 ± 1.5 | 31.5 ± 3.3 | 40.3 ± 3.0 | 69.5 ± 12.5 | 71.1 ± 10.1 |
| A6 | >100 | >100 | >100 | >100 | - | - |
| A7 | >100 | >100 | >100 | >100 | - | - |
| A8 | >100 | >100 | >100 | >100 | - | - |
| B1 | 47.1 ± 2.1 | 60.0 ± 3.0 | 6.7 ± 0.6 | 39.8 ± 1.6 | 55.6 ± 2.4 | 39.9 ± 5.4 |
| B2 | 77.7 ± 8.7 | 86.8 ± 5.0 | 10.1 ± 1.5 | 43.6 ± 2.8 | >100 | 83. 2 ± 10.6 |
| B3 | >100 | >100 | 50.2 ± 2.6 | 85.2 ± 7.4 | >100 | >100 |
| B4 | >100 | >100 | >100 | >100 | - | - |
| B5 | 89.5 ± 3.5 | 91.0 ± 4.6 | 10.4 ± 1.5 | 62.3 ± 5.5 | >100 | 64.2 ± 1.1 |
| B6 | >100 | >100 | >100 | >100 | - | - |
| B7 | >100 | >100 | 32.9 ± 5.1 | >100 | - | - |
| B8 | >100 | >100 | 67.5 ± 8.0 | >100 | - | - |
| Doxorubicin | 0.115 ± 0.042 | 0.575 ± 0.127 | 0.042 ± 0.006 | 2.2 ± 0.3 | 0.010 ± 0.002 | 0.009 ± 0.002 |
| Cisplatin | 26.3 ±7.9 | 14.0 ± 0.5 | 3.3 ± 2.3 | 8.6 ± 0.5 | 1.2 ± 0.1 | 1.1 ± 0.5 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shahraki, O.; Khoshneviszadeh, M.; Dehghani, M.; Mohabbati, M.; Tavakkoli, M.; Saso, L.; Edraki, N.; Firuzi, O. 5-Oxo-hexahydroquinoline Derivatives and Their Tetrahydroquinoline Counterparts as Multidrug Resistance Reversal Agents. Molecules 2020, 25, 1839. https://doi.org/10.3390/molecules25081839
Shahraki O, Khoshneviszadeh M, Dehghani M, Mohabbati M, Tavakkoli M, Saso L, Edraki N, Firuzi O. 5-Oxo-hexahydroquinoline Derivatives and Their Tetrahydroquinoline Counterparts as Multidrug Resistance Reversal Agents. Molecules. 2020; 25(8):1839. https://doi.org/10.3390/molecules25081839
Chicago/Turabian StyleShahraki, Omolbanin, Mehdi Khoshneviszadeh, Mojtaba Dehghani, Maryam Mohabbati, Marjan Tavakkoli, Luciano Saso, Najmeh Edraki, and Omidreza Firuzi. 2020. "5-Oxo-hexahydroquinoline Derivatives and Their Tetrahydroquinoline Counterparts as Multidrug Resistance Reversal Agents" Molecules 25, no. 8: 1839. https://doi.org/10.3390/molecules25081839