
A Trisbenzimidazole Phosphoramidite Building Block Enables High-Yielding Syntheses of RNA-Cleaving Oligonucleotide Conjugates


Institute of Organic Chemistry and Chemical Biology, Goethe University Frankfurt, Max-von-Laue-Str. 7, D-60438 Frankfurt am Main, Germany
*
Author to whom correspondence should be addressed.
Molecules 2020, 25(8), 1842; https://doi.org/10.3390/molecules25081842
Submission received: 19 March 2020
/
Revised: 13 April 2020
/
Accepted: 14 April 2020
/
Published: 16 April 2020
(This article belongs to the Special Issue Synthesis and Applications of Oligonucleotide Conjugate II)
Abstract
:The RNA cleaving catalyst tris(2-aminobenzimidazole) when attached to the 5’ terminus of oligonucleotides cuts complementary RNA strands in a highly site-specific manner. Conjugation was previously achieved by the acylation of an amino linker by an active ester of the catalyst. However, this procedure was low yielding and not reliable. Here, a phosphoramidite building block is described that can be coupled to oligonucleotides by manual solid phase synthesis in total yields around 85%. Based on this chemistry, we have now studied the impact of LNA (locked nucleic acids) nucleotides on the rates and the site-specificities of RNA cleaving conjugates. The highest reaction rates and the most precise cuts can be expected when the catalyst is attached to a strong 5’ closing base pair and when the oligonucleotide contains several LNA units that are equally distributed in the strand. However, when placed in the 5’ position, LNA building blocks tend to diminish the specificity of RNA cleavage.
1. Introduction
The interest in artificial ribonucleases was triggered some decades ago by the early work on antisense therapeutics. Structurally modified oligonucleotides were developed in these years in order to achieve sufficient levels of RNA affinity and stability against enzymatic degradation. Hybrids of heavily modified strands and their target RNAs, on the other hand, often lost their ability to recruit RNAse H [1,2]. Synthetic RNA cleavers were designed and attached to oligonucleotides to render their activity sequence-specific to compensate this disadvantage [3,4,5,6,7]. Since then, complexes of lanthanide and of other cations have been most often used for this purpose [8,9,10,11,12,13]. Although in modern life sciences gapmers [2,14,15] and siRNAs [16] are the preferred tools for achieving the site-specific cleavage of RNA strands, there is still interest in synthetic ribonucleases and recent years have seen important progress in the field. Effective catalysts that are based on metal ions [17,18,19,20,21,22,23], guanidines or polyamines [24,25,26,27], peptide conjugates [28,29,30,31,32], and deoxyribozymes [33,34] have been described. Starting from heterocyclic guanidine analogs [35], we have investigated the conjugates of tris(2-aminobenzimidazoles) and different types of oligonucleotides, such as DNA [36], PNA [37,38], or DNA-LNA mixmers [39], which were shown to act as sequence-specific metal-free RNA cleavers. Product inhibition prevented multiple turnover when the catalyst was attached to a terminal position of the conjugates and the reaction was run under isothermal conditions, because the number of base pairs in the conjugate-substrate hybrid does not change in the cleavage step [36]. However, this problem could be resolved by placing the catalyst in a central position. Such conjugates were shown to act with multiple substrate turnover [38].
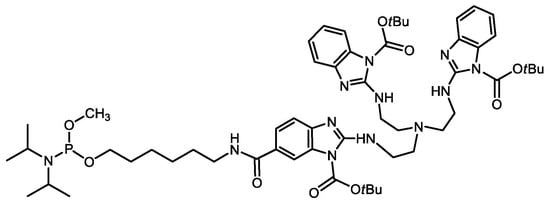
In a recent study [39], the 15mer DNA conjugates 3 and 5 were synthesized and then used to cleave 22mer model RNAs 1 and 2, derived from the PIM1 3’-UTR (Figure 1). The sequences of RNAs 1 and 2 correspond to a validated siRNA cleavage site (nt 1495–1509) and a binding site for miR-33a (nt 2099–2113) [40]. When 150 nM of RNA 1 were incubated with increasing amounts of conjugate 3, concentrations of 3 µM were required to obtain rate saturation (50 mM Tris buffer, pH 8.0, 37 °C). A substrate half-life of 14 h was observed under such conditions and a value of 15 h for the cleavage of RNA 2 by conjugate 5. We then replaced the 15mer DNA part by DNA-LNA mixmers to improve substrate affinities (Figure 1). As expected, rate saturation with conjugates 4 and 6 was now observed at lower concentrations of 750 nM. To our surprise, the incorporation of LNA building blocks also had a major impact on kinetics. The half-life of RNA 2, when incubated with conjugate 6, dropped from 15 h to 10 h. The half-life of RNA 1 even dropped to 3.5 h in the presence of mixmer conjugate 4. Apart from cleaving the 22mer model RNA 1, this conjugate was also shown to precisely cut PIM1 derived 155mer and 412mer transcripts [39]. In the present study, we investigated the influence of LNA nucleotides on cleavage patterns and kinetics in a more systematic way. However, our previous method of catalyst attachment was not fully satisfying: a tritylated 5’ amino linker was deprotected, while the oligonucleotide strand was still bound to the support. Catalyst 8 (Scheme 1), activated with DIC and HOBt, was then added and it formed a stable amide bond. The conjugation yield did not exceed 30% and could occasionally drop to zero due to accidental acylation of the amino linker by capping reagents or by protecting group migration. Here, we describe two phosphoramidite building blocks of the catalyst (Scheme 1, compounds 11 and 12) that can be manually attached to the 5’ end of DNA or DNA-LNA mixmers in excellent yield.
2. Results
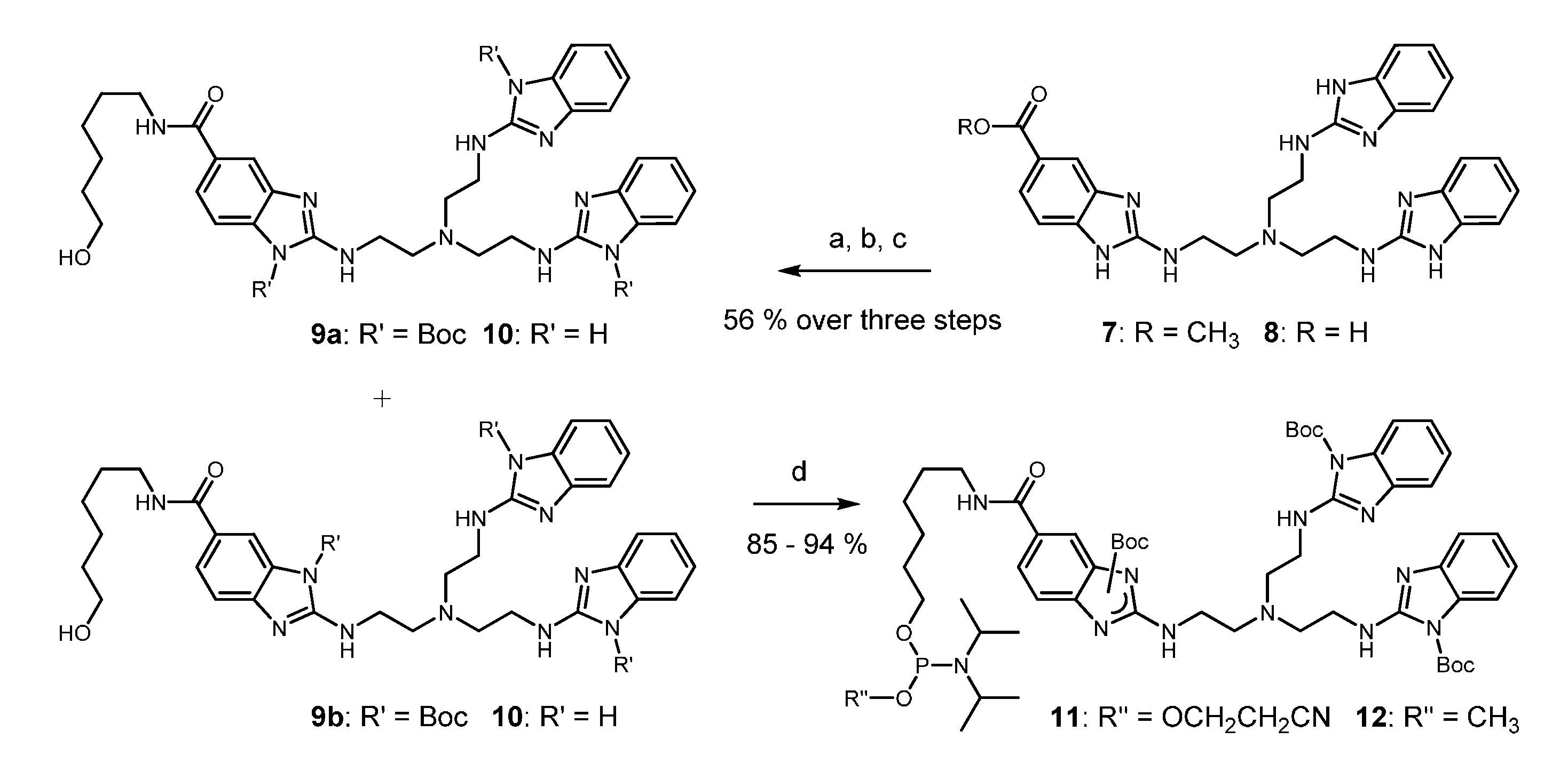
2-Aminobenzimidazoles are nucleophilic species that must be protected before phosphitylation by an electron withdrawing group. Starting from the readily accessible ester 7 [39], acid induced hydrolysis (8) and reaction with di-tert-butyl dicarbonate converted the compound into a triply acylated intermediate that was subsequently coupled with 6-aminohexanol. Steps a–c could be performed in a one-pot procedure. A mixture of two constitutional isomers 9a and 9b was isolated in 56% yield and in batches of 11.5 g. Boc residues are normally stable against nucleophilic attack. However, in the present case, standard conditions of oligonucleotide deprotection (aqueous ammonia/EtOH 3: 1, 55 °C, 16 h) completely removed all Boc groups from 9a and 9b producing compound 10 without any visible degradation of the trisbenzimidazole (for details, see Supplementary Materials). Furthermore, compounds 9a and 9b were fully compatible with typical reagents for oligonucleotide synthesis, such as 0.5 M 5-ethylthio-1H-tetrazole (ETT) in acetonitrile and commercial oxidizer solution. Before testing Boc as protecting group, we experimented with acetyl, pivaloyl, and ethoxycarbonyl. With the high reactivity of N-acyl imidazoles in mind, it was not unexpected to find all of these groups insufficiently stable. In the last step, the phosphitylation of alcohols 9a and 9b gave access to the phosphoramidites 11 and 12 (each consisting of two constitutional isomers) in good to excellent yields (Scheme 1). However, the cyanoethoxy amidite 11 showed significant formation of H-phosphonate during evaporation of the solvent at 30 °C after column chromatography and also after storage at −20 °C for a couple of days. Although the reason is not yet clear, decomposition might result from some residual Brønsted basicity of the protected trisbenzimidazole. The methoxy amidite 12, in contrast, was stable and it could be stored for several months. Manual coupling to the 5’ end of resin-bound oligonucleotides and standard deprotection resulted in excellent total yields of conjugates around 85% (determined by HPLC). Samples of amidite 11 containing some H-phosphonate, nevertheless coupled with comparable efficiency. Unfortunately, we failed to transfer this reaction to an automated DNA synthesizer under standard conditions when amidite and tetrazole are pre-mixed and then pumped into the reaction column (PerSeptive Expedite, 1.0 μmol scale). It seems that after activation the phosphoramidite rapidly undergoes some unknown side reactions. By increasing the pump rate, coupling yields up to 60% could be obtained, but they never reached the almost quantitative conversion that was seen in the manual method. Thus, conjugates 13–20 were prepared by the manual conjugation protocol, in which phosphoramidite activation directly occurs on the solid support.
The 22mer RNA parts of substrates 1 and 2 are both embedded into a chain of 14 thymidine residues. The strands are labeled with a fluorescent dye allowing for us to detect them and their cleavage fragments in an ALFexpress sequencer. Upon base induced fragmentation, only linkages between ribonucleotides can be cleaved. Thus, 22 well resolved peaks became visible in the hydrolysis ladder. The 3’ terminal thymidine tetramer helped to separate the peaks of the longest fragment from the substrate peak (Figure 2a). When RNA 1 was cleaved by conjugate 3, a comparison of the cleavage pattern and the base ladder immediately showed that the reaction mainly occurred between nucleotides 15 and 16, directly at the end of the duplex. The mixmer conjugate 4 induced a very similar pattern (Figure 2a). However, when the 5’ terminal G was replaced by an LNA analog, additional cleavage between nucleotides 16 and 17 increased. This effect was visible for conjugates 13, 14, and 16, independent from the presence of further LNA building blocks in direct proximity. In contrast, conjugate 15 with a 5’ terminal deoxynucleotide fairly reproduced the cleavage pattern of conjugates 3 and 4.
In rate saturation experiments (Figure 2b), conjugate 13 showed weaker substrate binding as compared to compounds 14–16 containing two or more LNA nucleotides. Cleavage kinetics for compound 13 was determined at concentrations of 3 µM, as in the case of the DNA conjugate 3. For conjugates 14–16, rate saturation, indicating full occupation of the substrate strands, was already attained at concentrations of 750 nM. Nevertheless, we determined the RNA cleavage kinetics for all new conjugates at concentrations of 3 µM (50 mM Tris buffer, pH 8.0, 37 °C). In all cases, the data could be fitted to a first-order rate model (Figure 3).
The single LNA nucleotide that was present in compound 13 did not significantly change reaction rates (kmax = 0.052 h−1, t1/2 = 13.3 h) when compared to the unmodified DNA conjugate 3 (kmax = 0.050 h−1, t1/2 = 14 h), but downgraded the precision of the cut. Conjugates containing two or three LNA nucleotides reacted faster: 15 (kmax = 0.107 h−1, t1/2 = 6.5 h) ≈ 14 (kmax = 0.108 h−1, t1/2 = 6.4 h) < 16 (kmax = 0.124 h−1, t1/2 = 5.6 h) (Figure 3a). However, none of the new conjugates could compete with the LNA-rich conjugate 4 (kmax = 0.20 h−1, t1/2 = 3.5 h). Although the introduction of LNA nucleotides increased the substrate affinities and reaction rates in general, they induced cleavage in the n+1 position when brought to the 5’ terminus.
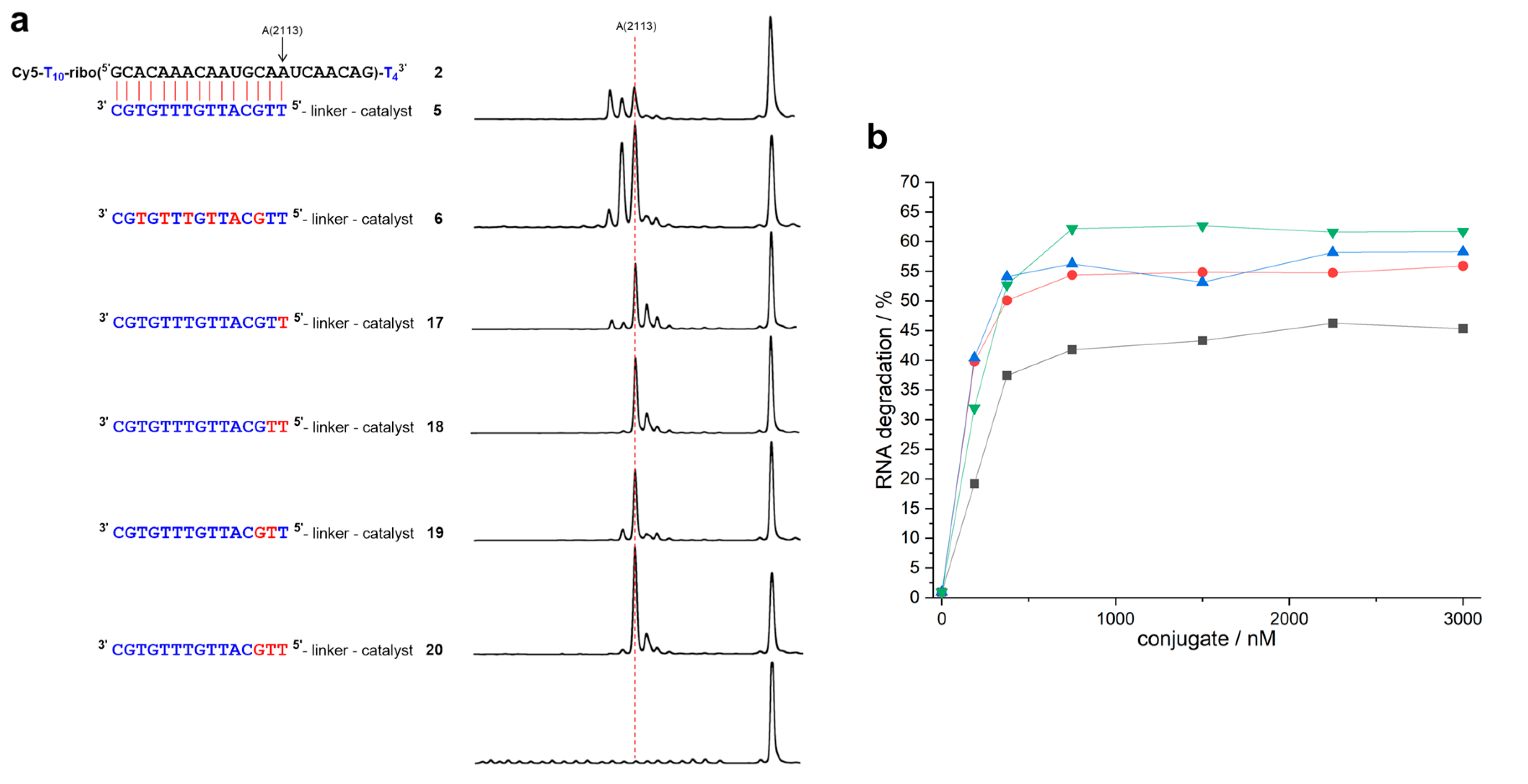
The duplex that formed from RNA 2 and conjugate 5 differs from the previous cases in so far as the two closing base pairs are weak and prone to fraying: In addition to cleavage between nucleotides 15 and 16, the reaction also occurs after nucleotides 13 and 14 (Figure 4a). In conjugate 17 with a single 5’ terminal LNA nucleotide, fraying is reduced, but not fully suppressed. Furthermore, additional cleavage occurs in positions n + 1 (and n + 2), as in the case of conjugate 13. When compared to the DNA conjugate 5 (kmax = 0.046 h−1, t1/2 = 15 h), the LNA building block even retards RNA cleavage (kmax = 0.038 h−1, t1/2 = 18.3 h). Site-selectivity improves in the presence of two LNA building blocks, but, depending on the location, either residual fraying or some additional cleavage in position n + 1 occurs. The reaction rates of conjugates 18 (kmax = 0.049 h−1, t1/2 = 14.2 h) and 19 (kmax = 0.053 h−1, t1/2 = 13.0 h) come close to the value of the DNA conjugate 5. Conjugate 20 (kmax = 0.071 h−1, t1/2 = 9.8 h) with three 5’ terminal LNA nucleotides not only cleaves as fast as the DNA-LNA mixmer 6 (kmax = 0.069 h−1, t1/2 = 10 h), it also shows the best site-selectivity that was observed with RNA 2: More than 70% of the strands are cleaved directly after the duplex region (Table 1).
3. Discussion
The availability of the phosphoramidite building blocks 11 and 12 has greatly facilitated the covalent attachment of oligonucleotides and RNA cleaving trisbenzimidazoles. This approach has allowed us to investigate the impact of LNA nucleotides in a more systematic way. RNA 1 represents the first type of target where cleavage is intended to occur after a strong G-C closing base pair that prevents the fraying of the substrate-conjugate duplex. The highest level of site-specificity is already seen with the unmodified DNA conjugate 3. 89% of the fragments are directly cleaved after the final base pair (Table 1). The introduction of LNA building blocks does not further improve this number. On the contrary, LNA as part of the closing base pair leads to 20–30% cleavage in the n + 1 position. Interestingly, it has been shown in a previous study that duplex stabilization by LNA nucleotides is much less pronounced when brought to the 5’ terminus instead to internal positions [41]. The incorporation of several LNA modifications, although it does not improve the precision of the cut, can accelerate the reaction up to four-fold. The situation is different when the closing base pairs of the RNA-conjugate duplex are weak as in the case of RNA 2. LNA building blocks have a minor impact on cleavage rates, but they considerably prevent fraying and improve site-specificity (Table 1). However, the cleavage rates and specificities both stay behind the numbers found for RNA 1. From these observations, some general conclusions for the design of synthetic RNA cleavers can be drawn: In all conjugates tested so far, the trisbenzimidazole catalyst works best when placed after a 5’ terminal G-C base pair. The stacking of the catalyst on the purine nucleobase might be important. Several LNA modifications evenly distributed in the DNA part of the conjugate can largely increase reaction rates. LNA in the 5’ position, however, is contraproductive and it spoils the site-specificity of the conjugate. The reason, steric hindrance or conformational effects, is not clear at present. The high stability of DNA-LNA mixmers, such as 4 and 6, and their independence from metal ions may allow for them not only to act in buffer solution, but also in living cells.
4. Materials and Methods
4.1. General
All of the chemicals were reagent grade and used as purchased. All of the reactions were performed under an argon atmosphere. The reactions were monitored by TLC using silica gel 60 F-254 aluminum sheets (Merck, Darmstadt, Germany). Compounds were visualized by UV light (254 and 366 nm). Column chromatography was carried out on silica gel 60 (0.04–0.063 mm). Proton nuclear magnetic resonance (1H-NMR) spectra and carbon nuclear magnetic resonance (13C-NMR) were recorded at 300 K with Bruker AV 300 (1H: 300 MHz; 13C: 75.5 MHz) and Bruker AV 500 (1H: 500 MHz; 13C: 125.8 MHz) NMR spectrometers (Bruker, Karlsruhe, Germany). Chemical shifts for protons are reported in parts per million (δ scale) and internally referenced to the proton resonances of the solvent (CDCl3: δ 7.26, DMSO-d6: δ 2.50). The chemical shifts for carbon are reported in parts per million (δ scale) and referenced to the carbon resonances of the solvent (CDCl3: δ 77.16, DMSO-d6: δ 39.52). The data are represented, as follows: chemical shift, multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, quin = quintet, m = multiplet, dd = doublet of doublets, td = triplet of doublets), coupling constants in Hz, and integration. The IR spectra were recorded with a Jasco FT/IR-420 spectrometer (Jasco Germany GmbH, Pfungstadt, Germany) equipped with an ATR unit. ESI-MS spectra were obtained on a Fisons VG Plattform II (Fisons VG Biotech MS, Altrincham, UK). HRMS spectra were recorded on a MALDI LTQ Orbitrap mass spectrometer from Thermo Scientific (Waltham, MA, USA).
Di-tert-butyl-2,2’-((((2-((1-(tert-butoxycarbonyl)-6-((6-hydroxyhexyl)carbamoyl)-1H-benzo[d]imidazol-2-yl)amino)ethyl)azanediyl)bis(ethane-2,1-diyl))bis(azanediyl))bis(1H-benzo[d]imidazole-1-carboxylate) (9a/b): A suspension of the ester 7 [39] (12.4 g, 22.4 mmol) in 6 M hydrochloric acid (150 mL) was refluxed for 6 h. The solvent was removed under reduced pressure and the residue was dried in vacuo to obtain a light brown solid. To a solution of the carboxylic acid 8 and DIPEA (13.7 mL, 78.5 mmol, 3.5 eq) in DMF (250 mL) Boc2O (17.1 g, 78.5 mmol, 3.5 eq) was added. After 3 h at room temperature, HOBt (6.87 g, 44.9 mmol, 2 eq), DIC (5.68 g, 44.9 mmol, 2 eq), and 6-aminohexan-1-ol (5.26 g, 44.9 mmol, 2 eq) were added and the solution was stirred at room temperature over night. The solvent was removed under reduced pressure and the residue was purified via column chromatography (SiO2, DCM/MeOH 20:1) to obtain the mixture of 9a and 9b as a light yellow foam. Yield: 11.7 g (12.5 mmol, 56% over three steps). Rf = 0.37, 0.42 (DCM/MeOH 15:1). 1H NMR (500 MHz, CDCl3, mixture of two isomers): δ [ppm] = 8.12 (d, J = 1.8 Hz, 0.6 H), 7.85 (t, J = 5.2 Hz, 0.6 H), 7.76–7.72 (m, 2.9 H), 7.56 (dd, J = 8.4 Hz, J = 1.7 Hz, 0.6 H), 7.51–7.48 (m, 2.9 H), 7.33 (dd, J = 7.9 Hz, J = 1.2 Hz, 2 H), 7.26 (d, J = 8.1 Hz, 0.6 H), 7.13 (td, J = 7.7 Hz, J = 1.2 Hz, 2 H), 6.97 (tt, J = 7.6 Hz, J = 1.4 Hz, 2 H), 6.56 (t, J = 5.8 Hz, 0.4 H), 6.34 (t, J = 5.8 Hz, 0.6 H), 3.74–3.58 (m, 6 H), 3.60 (q, J = 6.2 Hz, 2 H), 3.42 (q, J = 6.7 Hz, 2 H), 3.01 (q, J = 5.2 Hz, 2 H), 2.96 (t, J = 5.3 Hz, 4 H), 1.61–1.59 (m, 2 H), 1.56–1.52 (m, 2 H), 1.49 (s, 5 H), 1.48 (s, 3.7 H), 1.46 (s, 18 H), 1.40–1.35 (m, 4 H). 13C NMR (126 MHz, CDCl3, mixture of two isomers): δ [ppm] = 167.83, 167.80, 155.7, 155.9, 154.2, 154.1, 151.1, 151.0, 150.8, 150.7, 145.8, 142.5, 142.32, 142.26, 132.8, 131.0, 130.9, 130.52, 130.47, 126.6, 124.39, 124.36, 122.7, 120.50, 120.47, 120.4, 116.26, 116.22, 115.4, 114.2, 114.1, 113.92, 113.90, 113.7, 86.29, 86.26, 85.7, 85.6, 62.5, 62.4, 54.82, 54.76, 54.72, 54.6, 41.7, 41.6, 41.5, 40.0, 39.9, 32.64, 32.57, 29.82, 29.75, 29.6, 28.2, 27.91, 27.87, 26.7, 26.6, 25.43, 25.39. IR: ṽ [cm−1] = 3379 (w), 2976 (w), 2929 (w), 2856 (w), 1720 (s), 1626 (m), 1601 (m), 1574 (s), 1502 (w), 1456 (m), 1433 (w), 1394 (w), 1367 (m), 1352 (m), 1333 (s), 1296 (m), 1281 (m), 1261 (m), 1213 (m), 1334 (s), 1084 (w), 1055 (w), 1018 (w), 922 (w), 904 (w), 866 (w), 833 (m), 768 (m), 756 (m), 739 (s). MS (ESI+): m/z (%) = 938.73 (100) [M + H]+, calcd for C49H68N11O8: 938.52. HRMS (MALDI): m/z = 938.52458 [M + H]+, calcd for C49H67N11O8 + H+: 938.52468.
2-((2-(Bis(2-((1H-benzo[d]imidazol-2-yl)amino)ethyl)amino)ethyl)amino)-N-(6-hydroxy-hexyl)-1H-benzo[d]imidazole-5-carboxamide (10). To a solution of the carboxylic acid 8 (1.00 g, 1.54 mmol, 1 eq) and DIPEA (1.08 mL, 6.17 mmol, 4 eq) in DMF (3 mL) HOBt (0.21 g, 1.54 mmol, 1 eq) and DIC (0.19 g, 1.54 mmol, 1 eq) was added. After 1 h at room temperature, a solution of 6-aminohexan-1-ol (0.18 g, 1.54 mmol, 1 eq) in DMF (1 mL) was added dropwise. After stirring at room temperature overnight, the solvent was removed under reduced pressure and the residue was purified via column chromatography (SiO2, DCM/MeOH 10:1 + 1% NEt3) to obtain the product as a light yellow foam. Yield: 0.45 g (0.71 mmol, 46%). Rf = 0.33 (DCM/MeOH 10:1 + 1% NEt3). 1H NMR (300 MHz, DMSO-d6): δ [ppm] = 8.16 (t, J = 5.7 Hz, 1 H), 7.64 (d, J = 1.6 Hz, 1 H), 7.44 (dd, J = 8.3 Hz, J = 1.7 Hz, 1 H), 7.14–7.08 (m, 5 H), 6.88–6.82 (m, 4 H), 6.81 (bs, 1 H), 6.68 (bs, 2 H), 3.44–3.35 (m, 8 H), 3.22 (q, J = 6.6 Hz, 2 H), 2.78 (t, J = 6.5 Hz, 6 H), 1.50 (quin, J = 6.6 Hz, 2 H), 1.41 (quin, J = 6.4 Hz, 2 H), 1.32–1.27 (m, 4 H). 13C NMR (75 MHz, DMSO-d6): δ [ppm] = 172.5, 167.0, 156.9, 155.6, 125.8, 119.2, 111.6, 110.3, 60.7, 57.9, 53.7, 40.6, 39.2, 32.5, 29.4, 26.5, 25.3, 21.4. MS (ESI+): m/z (%) = 638.45 (100) [M + H]+, calcd for C34H44N11O2: 638.37. HRMS (MALDI): m/z = 638.36654 [M + H]+, calcd for C34H43N11O2 + H+: 638.36740.
Di-tert-butyl-2,2’-((((2-((1-(tert-butoxycarbonyl)-6-((6-(((2-cyanoethoxy)(diisopropylamino)-phosphino)oxy)hexyl)carbamoyl)-1H-benzo[d]imidazol-2-yl)amino)ethyl)azanediyl)bis-(ethane-2,1-diyl))bis(azanediyl))bis(1H-benzo[d]imidazole-1-carboxylate) (11). To a solution of the alcohols 9a/b (1.0 g, 1.07 mmol, 1 eq) and NEt3 (0.74 mL, 5.33 mmol, 5 eq) in dry DCM (10 mL) N,N-diisopropylcyanoethylphosphonamidic chloride (0.50 g, 2.13 mmol, 2 eq) was added at 0 °C. After 15 min. at 0 °C and 30 min. at room temperature, saturated NaHCO3 solution was added. The phases were separated and the aqueous layer was extracted with DCM. The extracts were dried over MgSO4, the solvent was removed under reduced pressure at 30 °C, and the residue was purified via column chromatography (SiO2, EtOAc/MeOH 25:1 + 2% NEt3). Yield: 1.03 g (0.905 mmol, 85%). Rf = 0.27, 0.32 (EtOAc/MeOH 25:1 + 2% NEt3). 1H NMR (300 MHz, DMSO-d6, mixture of two isomers): δ [ppm] = 8.29 (t, J = 5.7 Hz, 0.4 H), 8.22 (t, J = 5.6 Hz, 0.6 H), 8.06 (d, J = 1.4 Hz, 0.6 H), 7.72–7.71 (m, 0.5 H), 7.68 (t, J = 5.5 Hz, 0.6 H), 7.61 (dd, J = 8.3 Hz, J = 1.5 Hz, 0.6 H), 7.55–7.49 (m, 4.8 H), 7.20–7.16 (m, 2.6 H), 7.10 (td, J = 7.7 Hz, J = 0.8 Hz, 2 H), 6.97 (t, J = 7.6 Hz, 2 H), 3.78–3.64 (m, 2 H), 3.62–3.48 (m, 10 H), 3.26–3.20 (m, 2 H), 2.90–2.85 (m, 6 H), 6.97 (t, J = 5.9 Hz, 2 H), 1.59–1.54 (m, 4 H), 1.50–1.46 (m, 27 H), 1.37–1.30 (m, 4 H), 1.13–1.10 (m, 12 H). 13C NMR (75 MHz, DMSO-d6, mixture of two isomers): δ [ppm] = 166.5, 166.2, 155.0, 153.7, 150.24, 150.21, 150.0, 149.9, 145.2, 142.8, 142.6, 132.4, 130.3, 130.2, 128.3, 126.7, 123.9, 123.23, 123.19, 119.9, 119.0, 115.7, 113.7, 85.8, 85.7, 85.3, 62.9, 58.2, 58.0, 53.6, 42.5, 42.3, 41.1, 30.7, 30.64, 30.61, 29.2, 27.4, 27.3, 26.2, 25.2, 24.4, 24.3, 22.6, 19.9, 19.8. 31P NMR (121 MHz, DMSO-d6, mixture of two isomers): δ [ppm] = 146.36, 147.35. IR: ṽ [cm−1] = 3383 (w), 2970 (w), 2931 (w), 2866 (w), 1720 (s), 1628 (m), 1601 (m), 1574 (s), 1537 (w), 1500 (w), 1456 (m), 1433 (w), 1394 (w), 1352 (m), 1335 (s), 1296 (w), 1281 (w), 1261 (m), 1215 (m), 1136 (s), 1080 (w), 1053 (w), 976 (w), 924 (w), 876 (w), 833 (w), 768 (m), 758 (m), 741 (m), 717 (m), 652 (m). MS (ESI+): m/z (%) = 1138.59 (100) [M + H]+, calcd for C58H85N13O9P: 1138.63.
Di-tert-butyl-2,2’-((((2-((1-(tert-butoxycarbonyl)-6-((6-(((diisopropylamino)(methoxy)-phosphino)oxy)hexyl)carbamoyl)-1H-benzo[d]imidazol-2-yl)amino)ethyl)azanediyl)bis-(ethane-2,1-diyl))bis-(azanediyl))bis(1H-benzo[d]imidazole-1-carboxylate) (12). To a solution of the alcohols 9a/b (1.0 g, 1.07 mmol, 1 eq) and NEt3 (0.74 mL, 5.33 mmol, 5 eq) in dry DCM (15 mL) N,N-diisopropylmethylphosphonamidic chloride (0.32 g, 1.61 mmol, 1.5 eq) was added at 0 °C. After 15 min. at 0 °C and 30 min. at room temperature saturated NaHCO3 solution was added. The phases were separated and the aqueous layer was extracted with DCM. The extracts were dried over MgSO4, the solvent was removed under reduced pressure at 30 °C and the residue was purified via column chromatography (SiO2, EtOAc/MeOH 25:1 + 2% NEt3). Yield: 1.11 g (1.01 mmol, 94%). Rf = 0.31, 0.38 (EtOAc/MeOH 25:1 + 2% NEt3). 1H NMR (300 MHz, DMSO-d6, mixture of two isomers): δ [ppm] = 8.29 (t, J = 5.5 Hz, 0.4 H), 8.22 (t, J = 5.7 Hz, 0.6 H), 8.06 (d, J = 1.8 Hz, 0.6 H), 7.72 (t, J = 1.1 Hz, 0.4 H), 7.68 (t, J = 5.2 Hz, 0.6 H), 7.62 (dd, J = 8.4 Hz, J = 1.8 Hz, 1 H), 7.55–7.48 (m, 4.8 H), 7.20–7.16 (m, 2.6 H), 7.10 (td, J = 7.6 Hz, J = 1.2 Hz, 2 H), 6.97 (td, J = 7.6 Hz, J = 1.2 Hz, 2 H), 3.60–3.49 (m, 10 H), 3.27 (d, J = 13.0 Hz, 3 H), 3.27–3.20 (m, 2 H), 2.87 (t, J = 5.1 Hz, 6 H), 1.58–1.51 (m, 4 H), 1.50–1.46 (m, 27 H), 1.37–1.30 (m, 4 H), 1.10 (d, J = 6.8 Hz, 12 H). 13C NMR (75 MHz, DMSO-d6, mixture of two isomers): δ [ppm] = 166.5, 166.2, 154.9, 153.7, 150.2, 149.9, 145.2, 142.8, 142.6, 132.4, 130.7, 130.3, 130.2, 126.7, 123.9, 119.8, 115.7, 114.5, 114.3, 113.7, 85.7, 85.6, 85.3, 62.9, 62.6, 53.54, 53.47, 50.0, 49.8, 42.3, 42.1, 41.07, 41.04, 30.81, 30.78, 30.72, 29.3, 29.1, 27.4, 27.3, 26.2, 25.23, 25.20, 24.47, 24.41, 24.38, 24.31. 31P NMR (121 MHz, DMSO-d6): δ [ppm] = 146.98, 146.97. IR: ṽ [cm−1] = 3383 (w), 2970 (w), 2931 (w), 2866 (w), 1720 (s), 1628 (m), 1601 (m), 1574 (s), 1537 (w), 1500 (w), 1456 (m), 1433 (w), 1394 (w), 1352 (m), 1335 (s), 1296 (w), 1281 (w), 1261 (m), 1215 (m), 1136 (s), 1080 (w), 1053 (w), 976 (w), 924 (w), 876 (w), 833 (w), 768 (m), 758 (m), 741 (m), 717 (m), 652 (m). MS (ESI+): m/z (%) = 1099.60 (100) [M + H]+, calcd for C56H84N12O9P: 1099.62.
4.2. Synthesis, Purification, Quantification and Analysis of Conjugates
All of the DNA-LNA mixmers required to prepare conjugates 13–20 were purchased from Eurogentec, Seraing, Belgium and delivered when still bound to the solid support. Reversed Phase (RP) HPLC was executed on a Jasco LC-900 HPLC system (Jasco Germany GmbH, Pfungstadt, Germany) equipped with a Jasco UV-975 detector (detection at 254 nm) and a semi preparative column Phenomenex Jupiter 4 μm Proteo 90 Å (250 × 10 mm). All of the conjugates were lyophilized. Water was treated with DEPC and autoclaved.
4.2.1. Manual Phosphoramidite Coupling
Phosphoramidite (40 mg per 1 µmol of oligonucleotide) and oligo on CPG were placed in a syringe and dried in vacuo. The syringe was repeatedly flushed with argon and 1.5 mL of a 0.5 M solution of 5-ethylthio-1H-tetrazole (ETT) in dry MeCN was added. After shaking at room temperature for 15 min., the solution was discarded and the CPG was washed several times with MeCN. 200 µL oxidizer (2% I2 in pyridine/water) were added and then shaken for 45 s. Excess oxidizer was removed by repeatedly washing with MeCN.
4.2.2. Isolation and Purification
For cleavage from solid support, the CPG was treated with NH4OH/EtOH (3:1) twice for 30 min. at room temperature and the solutions were incubated at 50 °C over night. RP-HPLC: A: 1 M TEAA buffer pH 7.0, B: acetonitrile, C: DEPC-H2O. Gradient: constant 10% A, 3% B from 0–1 min, 3–33% from 1–20 min.; flow 4 mL/min.; and, column temperature 50 °C.
4.2.3. Quantification
Oligonucleotide concentrations were determined via UV spectrometry on a nanodrop2000 (Thermo Scientific, Waltham, MA, USA) using Lambert–Beer´s law. Extinction coefficients were calculated by a nearest neighbor model according to literature [42]. For simplification, influences of the tris(2-aminobenzimidazole) part were neglected.
4.2.4. Mass spectrometry
Oligonucleotides were analyzed via ESI mass spectrometry while using a LCMS instrument with microTOF-Q II analyser (Bruker Daltonics, Bremen, Germany). An Agilent 1200 Series HPLC using methanol/0.005 M TEAA buffer (gradient 0–60%) was applied as LC system.
4.2.5. Cleavage Studies
For saturation experiments [36] a solution containing 150 nM Cy5 labeled RNA, 0–3 µM conjugate and 50 mM Tris buffer (pH 8.0) was mixed in an Eppendorf tube (DNA LoBind Tubes) to reach a final volume of 10 µL. This tube was kept at 37 °C for 20 h. The cleavage reactions were stopped by addition of 15 µL loading buffer (8 M urea, 20 mM EDTA, 0.2% crocein orange) and samples were kept at −196 °C. For analysis, 1 µL of this solution was added to an ALFexpress sequencer (see Supplementary Materials).
For kinetic measurements, a solution containing 150 nM Cy5 labeled RNA, 3 µM conjugate, and 50 mM Tris buffer (pH 8.0) was mixed in an Eppendorf tube (DNA LoBind Tubes) to reach a final volume of 200 µL. This tube was kept at 37 °C and samples of 10 µL were drawn after indicated time intervals. The cleavage reactions were stopped by addition of 15 µL loading buffer (8 M urea, 20 mM EDTA, 0.2% crocein orange) and samples were kept at −196 °C. For analysis, 1 µL of this solution was added to an ALFexpress sequencer.
To prepare an RNA hydrolysis ladder, a solution of 375 nM RNA and 0.25 mM Na2CO3 was mixed in an Eppendorf tube (DNA LoBind Tubes) to reach a final volume of 8 µL. This tube was kept at 90 °C for 12 min. After cooling in an ice bath, the reaction was stopped by addition of 2 µL 1 M AcOH and 15 µL loading buffer (8 M urea, 20 mM EDTA, 0.2% crocein orange). The samples were stored at −20 °C until usage.
Supplementary Materials
The following are available online, HPLC plots and mass spectra of conjugates 13–20. Removal of Boc by ammonolysis. Description of the ALFexpress sequencer. NMR spectra.
Author Contributions
F.Z. performed the experiments and analyzed the data. F.Z. and M.W.G. designed the study and wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding
Acknowledgments
The authors would like to thank Elisabeth Kalden for running the ALFexpress sequencer.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Furdon, P.J.; Dominski, Z.; Ryszard Kole, R. RNase H cleavage of RNA hybridized to oligonudeotides containing methylphosphonate, phosphorothioate and phosphodiester bonds. Nucleic Acids Res. 1989, 17, 9193–9204. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, S.; Mayrand, S.H.; Zamecnik, P.C.; Pederson, T. Site-specific excision from RNA by RNase H and mixed-phosphate-backbone oligodeoxynucleotides. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 1401–1405. [Google Scholar] [CrossRef] [Green Version]
- Hall, J.; Hüsken, D.; Pieles, U.; Moser, H.E.; Häner, R. Efficient sequence-specific cleavage of RNA using novel europium complexes conjugated to oligonucleotides. Chem. Biol. 1994, 1, 185–190. [Google Scholar] [CrossRef]
- Bashkin, J.K.; Frolova, E.I.; Sampath, U. Sequence-specific cleavage of HIV mRNA by a ribozyme mimic. J. Am. Chem. Soc. 1994, 116, 5981–5982. [Google Scholar] [CrossRef]
- Matsumura, K.; Endo, M.; Komiyama, M. Lanthanide complex–oligo-DNA hybrid for sequence-selective hydrolysis of RNA. J. Chem. Soc. Chem. Commun. 1994, 2019–2020. [Google Scholar] [CrossRef]
- Magda, D.; Miller, R.A.; Sessler, J.L.; Iverson, B.L. Site-specific hydrolysis of RNA by europium(III) texaphyrin conjugated to a synthetic oligodeoxyribonucleotide. J. Am. Chem. Soc. 1994, 116, 7439–7440. [Google Scholar] [CrossRef]
- Hovinen, J.; Guzaev, A.; Azhayeva, E.; Azhayev, A.; Lönnberg, H. Imidazole Tethered Oligodeoxyribonucleotides: Synthesis and RNA Cleaving Activity. J. Org. Chem. 1995, 60, 2205–2209. [Google Scholar] [CrossRef]
- Trawick, B.N.; Daniher, A.T.; Bashkin, J.K. Inorganic mimics of ribonucleases and ribozymes: From random cleavage to sequence-specific chemistry to catalytic antisense drugs. Chem. Rev. 1998, 98, 939–960. [Google Scholar] [CrossRef]
- Häner, R. Artificial ribonucleases. Chimia 2001, 55, 1035–1037. [Google Scholar]
- Komiyama, M.; Sumaoka, J.; Kuzuya, A.; Yamamoto, Y. Sequence-selective artificial ribonucleases. Methods Enzymol. 2001, 341, 455–468. [Google Scholar]
- Morrow, J.R.; Iranzo, O. Synthetic metallonucleases for RNA cleavage. Curr. Opin. Chem. Biol. 2004, 8, 192–200. [Google Scholar] [CrossRef]
- Niittymäki, T.; Lönnberg, H. Artificial ribonucleases. Org. Biomol. Chem. 2006, 4, 15–25. [Google Scholar] [CrossRef]
- Lönnberg, H. Cleavage of RNA phosphodiester bonds by small molecular entities: A mechanistic insight. Org. Biomol. Chem. 2011, 9, 1687–1703. [Google Scholar] [CrossRef]
- Monia, B.P.; Lesnik, E.A.; Gonzalez, C.; Lima, W.F.; McGee, D.; Guinosso, C.J.; Kawasaki, A.M.; Cook, P.D.; Freier, S.M. Evaluation of 2‘-Modified Oligonucleotides Containing 2’-Deoxy Gaps as Antisense Inhibitors of Gene Expression. J. Biol. Chem. 1993, 268, 14514–14522. [Google Scholar]
- Jepsen, J.S.; Wengel, J. LNA-antisense rivals siRNA for gene silencing. Curr. Opin. Drug Discov. Devel. 2004, 7, 188–194. [Google Scholar]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide RNAs mediate RNA interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar] [CrossRef]
- Murtola, M.; Wenska, M.; Strömberg, R. PNAzymes that are artificial RNA restriction enzymes. J. Am. Chem. Soc. 2010, 132, 8984–8990. [Google Scholar] [CrossRef]
- Ghidini, A.; Steunenberg, P.; Murtola, M.; Strömberg, R. Synthesis of PNA Oligoether Conjugates. Molecules 2014, 19, 3135–3148. [Google Scholar] [CrossRef] [Green Version]
- Ghidini, A.; Murtola, M.; Strömberg, R. Oligonucleotide based artificial ribonucleases (OBANs). In DNA in supramolecular chemistry and nanotechnology; Stulz, E., Clever, G.H., Eds.; John Wiley: Chichester, UK, 2015; pp. 158–171. ISBN 9781118696880. [Google Scholar]
- Ghidini, A.; Murtola, M.; Strömberg, R. Influence of conjugation and other structural changes on the activity of Cu2+ based PNAzymes. Org. Biomol. Chem. 2016, 14, 2768–2773. [Google Scholar] [CrossRef]
- Murtola, M.; Ghidini, A.; Virta, P.; Strömberg, R. Zinc Ion-Dependent Peptide Nucleic Acid-Based Artificial Enzyme that Cleaves RNA—Bulge Size and Sequence Dependence. Molecules 2017, 22, 1856. [Google Scholar] [CrossRef] [Green Version]
- Luige, O.; Murtola, M.; Ghidini, A.; Strömberg, R. Further Probing of Cu2+-Dependent PNAzymes Acting as Artificial RNA Restriction Enzymes. Molecules 2019, 24, 672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuzuya, A.; Machida, K.; Shi, Y.; Tanaka, K.; Komiyama, M. Site-Selective RNA Activation by Acridine-Modified Oligodeoxynucleotides in Metal-Ion Catalyzed Hydrolysis: A Comprehensive Study. ACS Omega 2017, 2, 5370–5377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvio, R.; Volpi, S.; Folcarelli, T.; Casnati, A.; Cacciapaglia, R. A calix[4]arene with acylguanidine units as an efficient catalyst for phosphodiester bond cleavage in RNA and DNA model compounds. Org. Biomol. Chem. 2019, 17, 7482–7492. [Google Scholar] [CrossRef] [PubMed]
- Czescik, J.; Zamolo, S.; Darbre, T.; Mancin, F.; Scrimin, P. Factors Influencing the Activity of Nanozymes in the Cleavage of an RNA Model Substrate. Molecules 2019, 24, 2814. [Google Scholar] [CrossRef] [Green Version]
- Niittymäki, T.; Virta, P.; Ketomäki, K.; Lönnberg, H. Di(azacrown) Conjugates of 2‘-O-Methyl Oligoribonucleotides as Sequence-Selective Artificial Ribonucleases. Bioconjugate Chem. 2007, 18, 1583–1592. [Google Scholar] [CrossRef]
- Gaglione, M.; Milano, G.; Chambery, A.; Moggio, L.; Romanelli, A.; Messere, A. PNA-based artificial nucleases as antisense and anti-miRNA oligonucleotide agents. Mol. BioSyst. 2011, 7, 2490–2499. [Google Scholar] [CrossRef] [Green Version]
- Mironova, N.L.; Pyshnyi, D.V.; Shtadler, D.V.; Fedorova, A.A.; Vlassov, V.V.; Zenkova, M.A. RNase T1 mimicking artificial ribonuclease. Nucleic Acids Res. 2007, 35, 2356–2367. [Google Scholar] [CrossRef] [Green Version]
- Williams, A.; Staroseletz, Y.; Zenkova, M.A.; Jeannin, L.; Aojula, H.; Bichenkova, E.V. Peptidyl−Oligonucleotide Conjugates Demonstrate Efficient Cleavage of RNA in a Sequence-Specific Manner. Bioconjugate Chem. 2015, 26, 1129–1143. [Google Scholar] [CrossRef]
- Patutina, O.A.; Bichenkova, E.V.; Miroshnichenko, S.K.; Mironova, N.L.; Trivoluzzi, L.T.; Burusco, K.K.; Bryce, R.A.; Vlassov, V.V.; Zenkova, M.A. Novel peptide-oligonucleotide bioconjugates that silence miR-21 in lymphosarcoma cells. Biomaterials 2017, 122, 163–178. [Google Scholar] [CrossRef] [Green Version]
- Patutina, O.A.; Bazhenov, M.A.; Miroshnichenko, S.K.; Mironova, N.L.; Pyshnyi, D.V.; Vlassov, V.V.; Zenkova, M.A. Peptide-oligonucleotide conjugates exhibiting pyrimidine-X cleavage specificity efficiently silence miRNA target acting synergistically with RNase H. Sci. Rep. 2018, 8, 14990. [Google Scholar] [CrossRef] [Green Version]
- Pavlova, A.S.; Ogurtsova, P.A.; Koroleva, L.S.; Serpokrylova, I.Y.; Lomzov, A.A.; Pyshnaya, I.A.; Silnikov, V.N.; Pyshnyi, D.V. Novel Bisimidazole-Containing Peptidomimetic Molecules for Мetal-Independent RNA Cleavage: Synthesis and Solid-Phase Screening Method. Russ. J. Bioorg. Chem. 2019, 45, 813–824. [Google Scholar] [CrossRef]
- Trepanier, J.B.; Tanner, J.E.; Alfieri, C. Reduction in intracellular HCV RNA and virus protein expression in human hepatoma cells following treatment with 2-O-methyl-modified anti-core deoxyribozyme. Virology 2008, 377, 339–344. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Liu, E.; Lam, C.H.; Perrin, D.M. A densely modified M2+-independent DNAzyme that cleaves RNA efficiently with multiple catalytic turnover. Chem. Sci. 2018, 9, 1813–1821. [Google Scholar] [CrossRef] [PubMed]
- Scheffer, U.; Strick, A.; Ludwig, V.; Peter, S.; Kalden, E.; Göbel, M.W. Metal-free catalysts for the hydrolysis of RNA derived from guanidines, 2-aminopyridines, and 2-aminobenzimidazoles. J. Am. Chem. Soc. 2005, 127, 2211–2217. [Google Scholar] [CrossRef]
- Gnaccarini, C.; Peter, S.; Scheffer, U.; Vonhoff, S.; Klussmann, S.; Göbel, M.W. Site-specific cleavage of RNA by a metal-free artificial nuclease attached to antisense oligonucleotides. J. Am. Chem. Soc. 2006, 128, 8063–8067. [Google Scholar] [CrossRef] [PubMed]
- Danneberg, F.; Ghidini, A.; Dogandzhiyski, P.; Kalden, E.; Strömberg, R.; Göbel, M.W. Sequence-specific RNA cleavage by PNA conjugates of the metal-free artificial ribonuclease tris(2-aminobenzimidazole). Beilstein J. Org. Chem. 2015, 11, 493–498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dogandzhiyski, P.; Ghidini, A.; Danneberg, F.; Strömberg, R.; Göbel, M.W. Studies on tris(2-aminobenz- imidazole)-PNA based artificial nucleases: A comparison of two analytical techniques. Bioconjugate Chem. 2015, 26, 2514–2519. [Google Scholar] [CrossRef]
- Zellmann, F.; Thomas, L.; Scheffer, U.; Hartmann, R.K.; Göbel, M.W. Site-Specific Cleavage of RNAs Derived from the PIM1 3‘-UTR by a Metal-Free Artificial Ribonuclease. Molecules 2019, 24, 807. [Google Scholar] [CrossRef] [Green Version]
- Thomas, M.; Lange-Grünweller, K.; Weirauch, U.; Gutsch, D.; Aigner, A.; Grünweller, A.; Hartmann, R.K. The proto-oncogene Pim-1 is a target of miR-33a. Oncogene 2012, 31, 918–928. [Google Scholar] [CrossRef] [Green Version]
- Kierzek, E.; Ciesielska, A.; Pasternak, K.; Mathews, D.H.; Turner, D.H.; Kierzek, R. The influence of locked nucleic acid residues on the thermodynamic properties of 2’-O-methyl-RNA/RNA heteroduplexes. Nucleic Acids Res. 2005, 33, 5082–5093. [Google Scholar] [CrossRef] [Green Version]
- Nanodrop: Gray, D.M.; Hung, S.H.; Johnson, K.H. Absorption and Circular-Dichroism Spectroscopy of Nucleic-Acid Duplexes and Triplexes. Methods in Enzymology 1995, 246, 19. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |
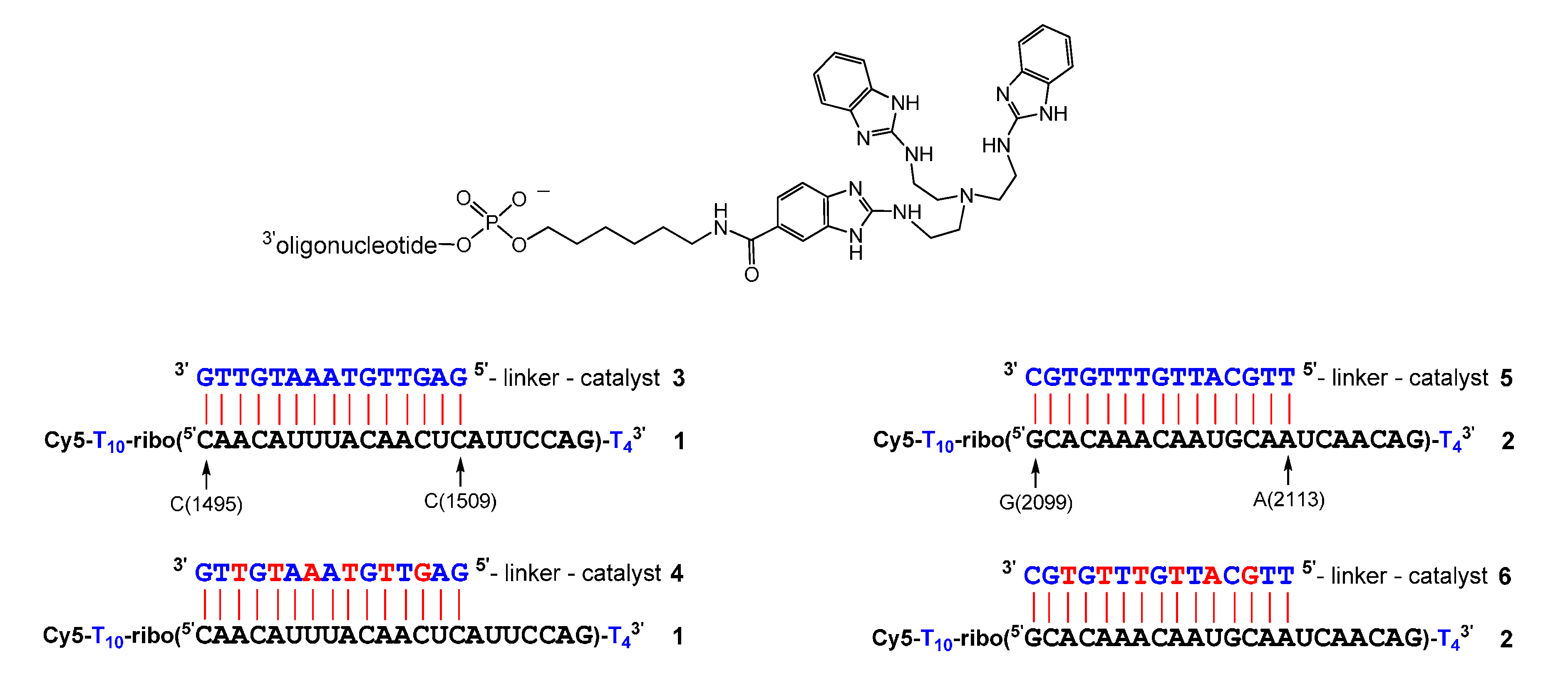
Figure 1.
Sequence of RNAs 1 and 2 and of conjugates 3–6. DNA and RNA nucleotides are depicted in blue and black, LNA nucleotides in red. The general structure of the conjugates is shown on top.
Figure 1.
Sequence of RNAs 1 and 2 and of conjugates 3–6. DNA and RNA nucleotides are depicted in blue and black, LNA nucleotides in red. The general structure of the conjugates is shown on top.
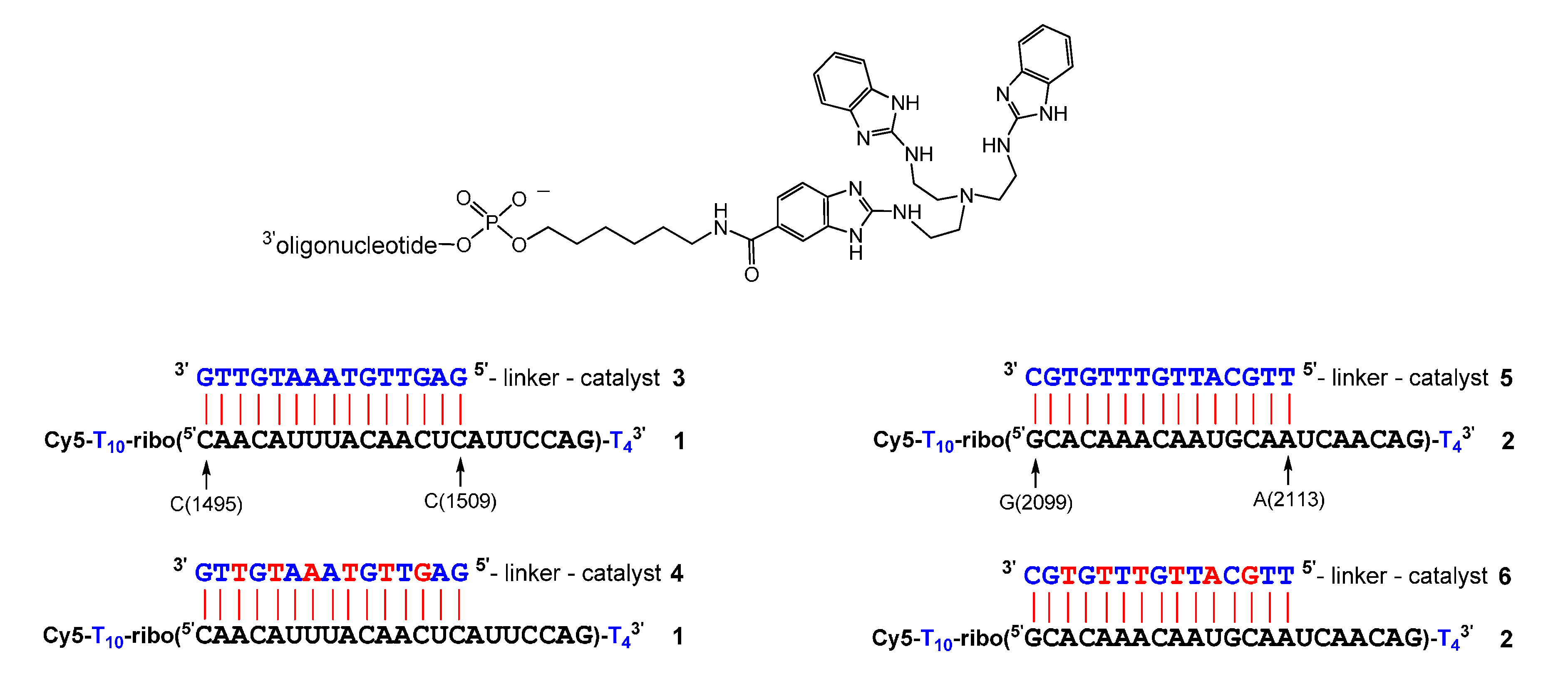
Scheme 1.
Syntheses of tris(2-aminobenzimidazole) phosphoramidites 11 and 12. Conditions: (a): 6 M HCl(aq), reflux, 6 h; (b): Boc2O, DIPEA, DMF, rt, 3 h; (c): 6-aminohexan-1-ol, DIC, HOBt, DMF, rt, 16 h; (d): N,N-diisopropylcyanoethylphosphonamidic chloride or N,N-diisopropylmethylphosphon-amidic chloride, NEt3, CH2Cl2, 0 °C to rt, 45 min.
Scheme 1.
Syntheses of tris(2-aminobenzimidazole) phosphoramidites 11 and 12. Conditions: (a): 6 M HCl(aq), reflux, 6 h; (b): Boc2O, DIPEA, DMF, rt, 3 h; (c): 6-aminohexan-1-ol, DIC, HOBt, DMF, rt, 16 h; (d): N,N-diisopropylcyanoethylphosphonamidic chloride or N,N-diisopropylmethylphosphon-amidic chloride, NEt3, CH2Cl2, 0 °C to rt, 45 min.

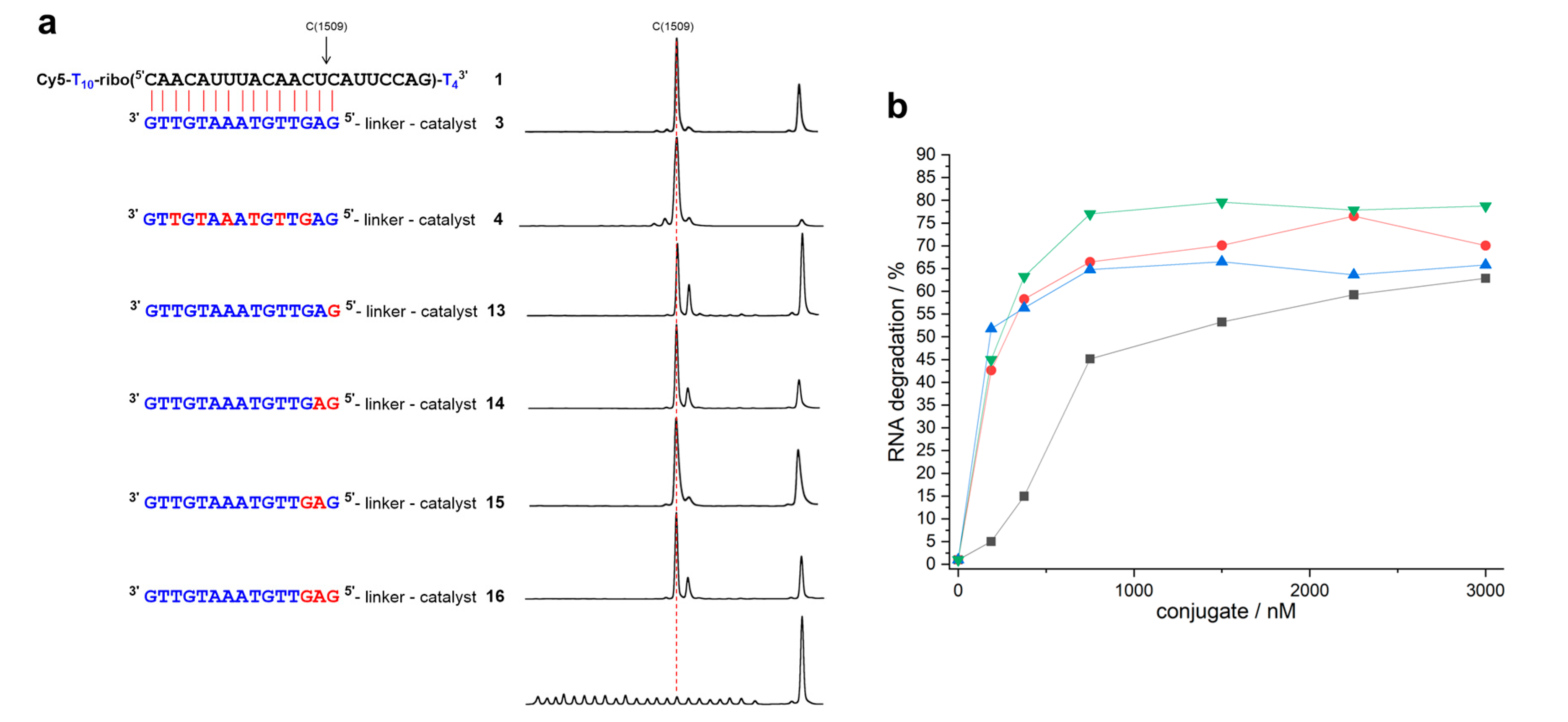
Figure 2.
(a) Structures of RNA 1 and complementary mixmer conjugates 13–16 with their corresponding cleavage patterns and hydrolysis ladder of RNA 1. DNA, LNA, and RNA nucleotides are shown in blue, red, and black letters, respectively. The red dashed line indicates position 15, the last nucleotide in the RNA-conjugate duplex (150 nM of RNA 1, 3 µM of conjugate, 50 mM Tris buffer (pH 8.0), 37 °C, 19–21 h). (b) Percentage of RNA cleavage after constant time as a function of conjugate concentration (150 nM of RNA 1, 50 mM Tris buffer (pH 8.0), 37 °C, 20 h). Black squares: conjugate 13. Red circles: conjugate 14. Blue triangles: conjugate 15. Green triangles: conjugate 16. Data points are connected by lines for the sake of clarity.
Figure 2.
(a) Structures of RNA 1 and complementary mixmer conjugates 13–16 with their corresponding cleavage patterns and hydrolysis ladder of RNA 1. DNA, LNA, and RNA nucleotides are shown in blue, red, and black letters, respectively. The red dashed line indicates position 15, the last nucleotide in the RNA-conjugate duplex (150 nM of RNA 1, 3 µM of conjugate, 50 mM Tris buffer (pH 8.0), 37 °C, 19–21 h). (b) Percentage of RNA cleavage after constant time as a function of conjugate concentration (150 nM of RNA 1, 50 mM Tris buffer (pH 8.0), 37 °C, 20 h). Black squares: conjugate 13. Red circles: conjugate 14. Blue triangles: conjugate 15. Green triangles: conjugate 16. Data points are connected by lines for the sake of clarity.
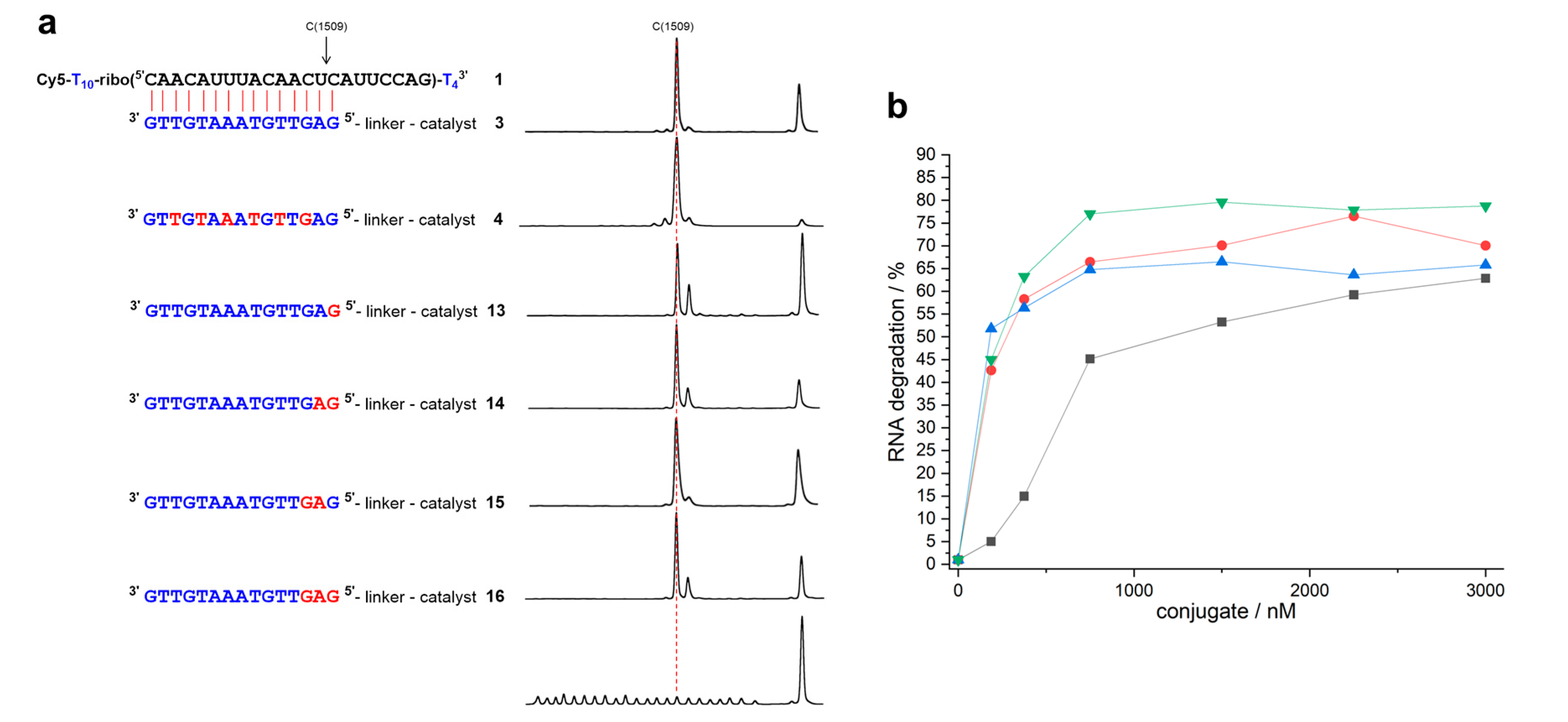
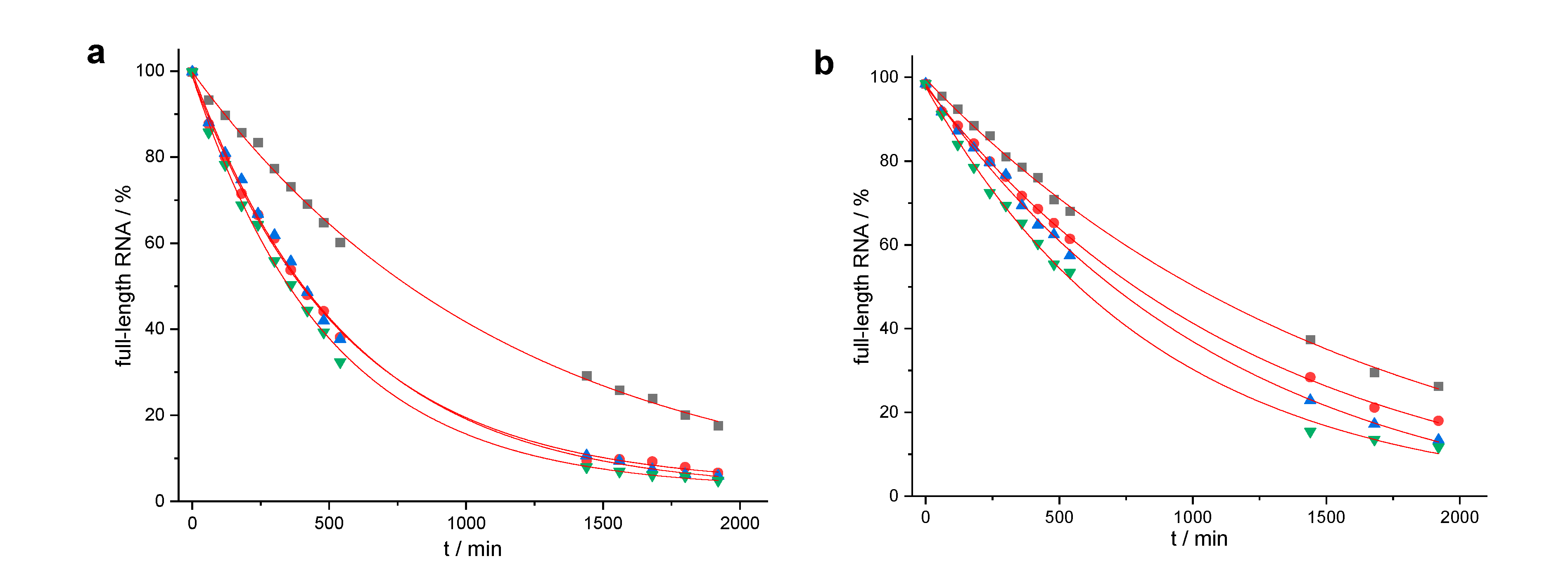
Figure 3.
Determination of rate constants: (a) 150 nM of RNA 1 were incubated with 3 µM of 13, 14, 15 or 16 (50 mM Tris buffer (pH 8.0), 37 °C); (b) 150 nM of RNA 2 were incubated with 3 µM of 17, 18, 19, or 20 (50 mM Tris buffer (pH 8.0), 37 °C) The amount of residual full-length RNA is shown as a function of time. Representative examples of single experiments are shown, and the data were fitted to a first-order rate model (single exponential decay). Mean values of kmax (calculated from three independent experiments) were accurate within ± 15%. (a) Reaction of RNA 1 with conjugates 13 (black squares), 14 (red circles), 15 (blue triangles), 16 (green triangles). (b) Reaction of RNA 2 with conjugates 17 (black squares), and 18 (red circles), 19 (blue triangles), 20 (green triangles).
Figure 3.
Determination of rate constants: (a) 150 nM of RNA 1 were incubated with 3 µM of 13, 14, 15 or 16 (50 mM Tris buffer (pH 8.0), 37 °C); (b) 150 nM of RNA 2 were incubated with 3 µM of 17, 18, 19, or 20 (50 mM Tris buffer (pH 8.0), 37 °C) The amount of residual full-length RNA is shown as a function of time. Representative examples of single experiments are shown, and the data were fitted to a first-order rate model (single exponential decay). Mean values of kmax (calculated from three independent experiments) were accurate within ± 15%. (a) Reaction of RNA 1 with conjugates 13 (black squares), 14 (red circles), 15 (blue triangles), 16 (green triangles). (b) Reaction of RNA 2 with conjugates 17 (black squares), and 18 (red circles), 19 (blue triangles), 20 (green triangles).
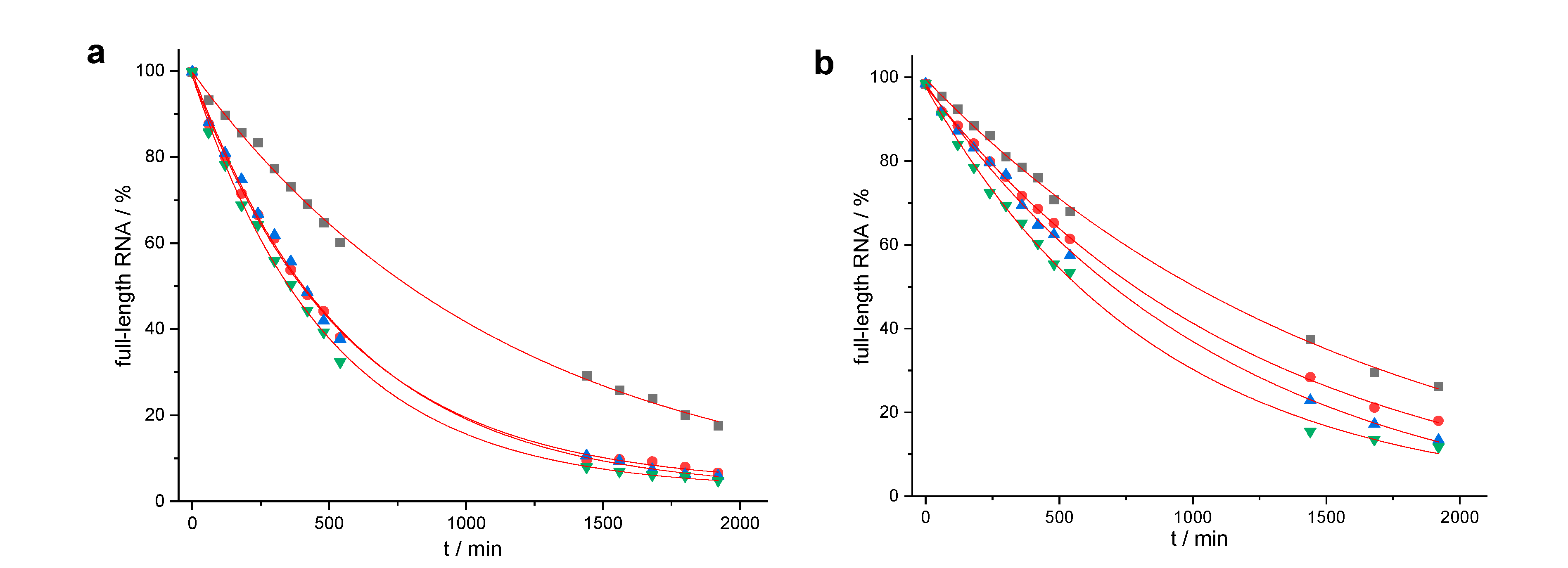
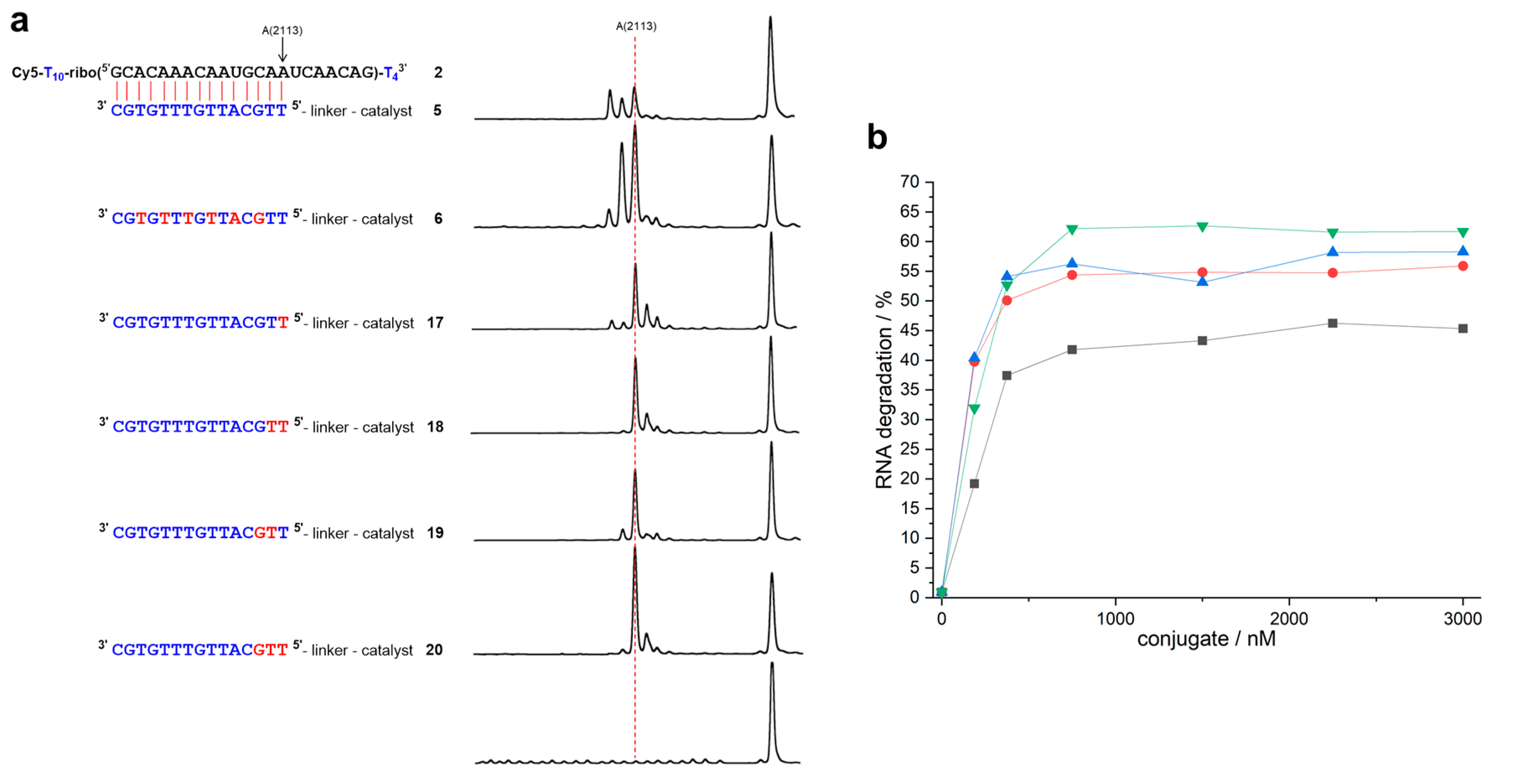
Figure 4.
(a) Structures of RNA 2 and of complementary mixmer conjugates 17–20 with their corresponding cleavage patterns. DNA, LNA, and RNA nucleotides are shown in blue, red, and black letters, respectively. The red dashed line indicates position 15, the last nucleotide in the RNA-conjugate duplex (150 nM of RNA 2, 3 µM of conjugate, 50 mM Tris buffer (pH 8.0), 37 °C, 19–21 h). (b) Percentage of RNA cleavage after constant time as a function of conjugate concentration (150 nM of RNA 2, 50 mM Tris buffer (pH 8.0), 37 °C, 20 h). Black squares: conjugate 17. Red circles: conjugate 18. Blue triangles: conjugate 19. Green triangles: conjugate 20. Data points are connected by lines for the sake of clarity.
Figure 4.
(a) Structures of RNA 2 and of complementary mixmer conjugates 17–20 with their corresponding cleavage patterns. DNA, LNA, and RNA nucleotides are shown in blue, red, and black letters, respectively. The red dashed line indicates position 15, the last nucleotide in the RNA-conjugate duplex (150 nM of RNA 2, 3 µM of conjugate, 50 mM Tris buffer (pH 8.0), 37 °C, 19–21 h). (b) Percentage of RNA cleavage after constant time as a function of conjugate concentration (150 nM of RNA 2, 50 mM Tris buffer (pH 8.0), 37 °C, 20 h). Black squares: conjugate 17. Red circles: conjugate 18. Blue triangles: conjugate 19. Green triangles: conjugate 20. Data points are connected by lines for the sake of clarity.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Kinetics and site-selectivity of RNA cleavage by conjugates 3–20.
| Conjugate | t1/2 | n − 2 | n − 1 | n | n + 1 | n + 2 | n + 3 |
|---|---|---|---|---|---|---|---|
| 31,3 | 14 ± 0.9 h | 1.4% | 2.5% | 89.0% | 7.1% | ||
| 41,3 | 3.5 ± 0.4 h | 1.6% | 5.8% | 83.9% | 8.6% | ||
| 131 | 13.3 ± 0.8 h | 1.5% | 65.2% | 30.2% | 3.1% | ||
| 141 | 6.4 ± 0.2 h | 1.2% | 75.3% | 22.1% | 1.4% | ||
| 151 | 6.5 ± 0.5 h | 1.5% | 88.3% | 10.1% | |||
| 161 | 5.6 ± 0.1 h | 1.2% | 74.9% | 22.1% | 1.7% | ||
| 52,3 | 15.0 ± 1.7 h | 29.0% | 22.7% | 35.8% | 5.1% | 3.6% | |
| 62,3 | 10.0 ± 0.3 h | 6.4% | 35.0% | 45.9% | 6.9% | 4.5% | 1.3% |
| 172 | 18.3 ± 1.2 h | 5.2% | 4.1% | 52.1% | 22.5% | 9.6% | 2.3% |
| 182 | 14.2 ± 0.7 h | 2.4% | 67.4% | 21.8% | 6.1% | 2.2% | |
| 192 | 13.0 ± 1.5 h | 10.7% | 71.3% | 9.8% | 6.4% | 1.8% | |
| 202 | 9.8 ± 1.0 h | 3.2% | 72.4% | 17.2% | 3.4% | 1.1% |
Determined by peak integration of all cleavage products. n means cleavage between nucleotides 15 and 16, directly after the duplex region. 1 Reaction with RNA 1. 2 Reaction with RNA 2. 3 Taken from reference 39.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zellmann, F.; Göbel, M.W. A Trisbenzimidazole Phosphoramidite Building Block Enables High-Yielding Syntheses of RNA-Cleaving Oligonucleotide Conjugates. Molecules 2020, 25, 1842. https://doi.org/10.3390/molecules25081842
AMA Style
Zellmann F, Göbel MW. A Trisbenzimidazole Phosphoramidite Building Block Enables High-Yielding Syntheses of RNA-Cleaving Oligonucleotide Conjugates. Molecules. 2020; 25(8):1842. https://doi.org/10.3390/molecules25081842
Chicago/Turabian StyleZellmann, Felix, and Michael W. Göbel. 2020. "A Trisbenzimidazole Phosphoramidite Building Block Enables High-Yielding Syntheses of RNA-Cleaving Oligonucleotide Conjugates" Molecules 25, no. 8: 1842. https://doi.org/10.3390/molecules25081842