
Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters



Abstract
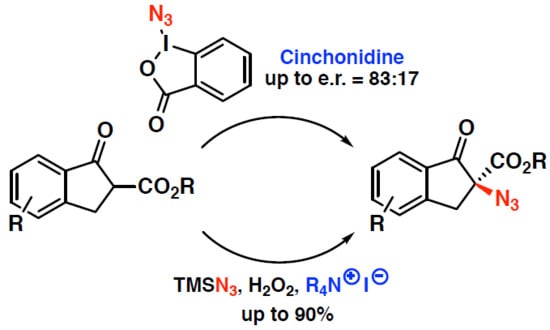
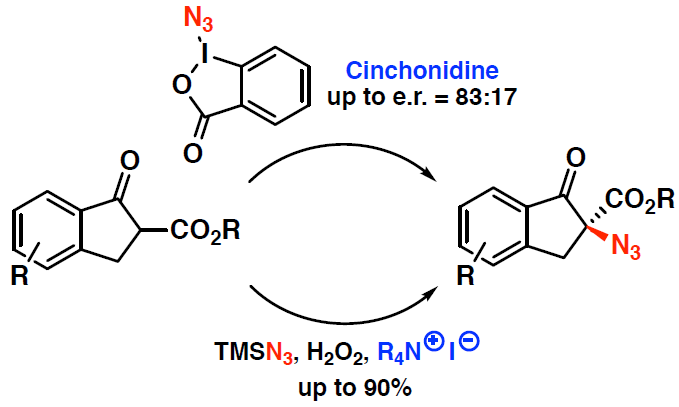
:
1. Introduction
2. Results and Discussion
3. Experimental Section
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Bräse, S.; Banert, K. Organic Azides: Syntheses and Applications; John Wiley & Sons: New York, NY, USA, 2009; ISBN 9780470519981. [Google Scholar]
- Bräse, S.; Gil, C.; Knepper, K.; Zimmermann, V. Organic Azides: An Exploding Diversity of a Unique Class of Compounds. Angew. Chem. Int. Ed. 2005, 44, 5188–5240. [Google Scholar] [CrossRef] [PubMed]
- Bock, V.D.; Hiemstra, H.; van Maarseveen, J.H. CuI-Catalyzed Alkyne–Azide “Click” Cycloadditions from a Mechanistic and Synthetic Perspective. Eur. J. Org. Chem. 2006, 51–68. [Google Scholar] [CrossRef]
- Liang, L.; Astruc, D. The Copper(I)-Catalyzed Alkyne-Azide Cycloaddition (CuAAC) “Click” Reaction and its Applications. An Overview. Coord. Chem. Rev. 2011, 255, 2933–2945. [Google Scholar] [CrossRef]
- Singh, M.S.; Chowdhury, S.; Koley, S. Advances of Azide-Akyne Cycloaddition-Click Chemistry Over the Recent Decade. Tetrahedron 2016, 72, 5257–5283. [Google Scholar] [CrossRef]
- Gololobov, Y.G.; Kasukhin, L.F. Recent Advances in the Staudinger Reaction. Tetrahedron 1992, 48, 1353–1406. [Google Scholar] [CrossRef]
- Scriven, E.F.V.; Turnbull, K. Azides: Their Preparation and Synthetic Uses. Chem. Rev. 1988, 88, 297–368. [Google Scholar] [CrossRef]
- Huang, D.; Yan, G. Recent Advances in Reactions of Azides. Adv. Synth. Catal. 2017, 359, 1600–1619. [Google Scholar] [CrossRef]
- Goswami, M.; de Bruin, B. Metal-Catalyed Azidation of Organic Molecules. Eur. J. Org. Chem. 2017, 2017, 1152–1176. [Google Scholar] [CrossRef] [PubMed]
- Ding, P.-G.; Hu, X.-S.; Zhou, F.; Zhou, J. Catalytic Enantioselective Synthesis of a-Chiral Azides. Org. Chem. Front. 2018. [Google Scholar] [CrossRef]
- Phan, T.B.; Mayr, H. Nucleophilic Reactivity of the Azide Ion in Various Solvents. J. Phys. Org. Chem. 2006, 19, 706–713. [Google Scholar] [CrossRef]
- Harschneck, T.; Hummel, S.; Kirsch, S.F.; Klahn, P. Practical Azidation of 1,3-Dicarbonyls. Chem. Eur. J. 2012, 18, 1187–1193. [Google Scholar] [CrossRef] [PubMed]
- Galligan, M.J.; Akula, R.; Ibrahim, H. Unified Strategy for Iodine(III)-Mediated Halogenation and Azidation of 1,3-Dicarbonyl Compounds. Org. Lett. 2014, 16, 600–603. [Google Scholar] [CrossRef] [PubMed]
- Dhineshkumar, J.; Prabhu, K.R. An Efficient Tertiary Azidation of 1,3-Dicarbonyl Compounds in Water Catalyzed by Tetrabutylammonium Iodide. Eur. J. Org. Chem. 2016, 2016, 447–452. [Google Scholar] [CrossRef]
- Yasui, K.; Kojima, K.; Kato, T.; Odagi, M.; Kato, M.; Nagasawa, K. Guanidinium iodide/urea hydrogen peroxide-catalyzed azidation of β-dicarbonyl compounds with trimethylsilyl azide. Tetrahedron 2016, 72, 5350–5354. [Google Scholar] [CrossRef]
- Zhdankin, V. Hypervalent Iodine Chemistry, Preparation, Structure, and Synthetic Applications of Polyvalent Iodine Compounds; Wiley-VCH: Weinheim, Germany, 2013; ISBN 978-1-118-34103-2. [Google Scholar]
- Yoshimura, A.; Zhdankin, V. Advances in synthetic applications of hypervalent iodine compounds. Chem. Rev. 2016, 116, 3328–3435. [Google Scholar] [CrossRef] [PubMed]
- Zhdankin, V.V.; Krasutsky, A.P.; Kuehl, C.J.; Simonsen, A.J.; Woodward, J.K.; Mismash, B.; Bolz, J.T. Preparation, X-ray Crystal Structure, and Chemistry of Stable Azidoiodinaness Derivatives of Benziodoxole. J. Am. Chem. Soc. 1996, 118, 5192–5197. [Google Scholar] [CrossRef]
- Vita, M.V.; Waser, J. Azidation of β-Keto Esters and Silyl Enol Ethers with a Benziodoxole Reagent. Org. Lett. 2013, 15, 3246–3249. [Google Scholar] [CrossRef] [PubMed]
- Deng, Q.-H.; Bleith, T.; Wadepohl, H.; Gade, L.H. Enantioselective Iron-Catalyzed Azidation of b-Ketoesters and Oxindoles. J. Am. Chem. Soc. 2013, 135, 5356–5359. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Shirakawa, S.; Maruoka, K. Efficient asymmetric synthesis of spiro-2(3H)-furanones via phase-transfer-catalyzed alkynylation. Org. Biomol. Chem. 2014, 12, 5388–5392. [Google Scholar] [CrossRef] [PubMed]
- Fernandez Gonzalez, D.; Brand, J.P.; Mondiere, R.; Waser, J. Ethynylbenziodoxolones (EBX) as Reagents for the Ethynylation of Stabilized Enolates. Adv. Synth. Catal. 2013, 355, 1631–1639. [Google Scholar] [CrossRef]
- Chen, M.; Huang, Z.-T.; Zheng, Q.-Y. Organic base-promoted enantioselective electrophilic cyanation of β-keto esters by using chiral phase-transfer catalysts. Org. Biomol. Chem. 2015, 13, 8812–8816. [Google Scholar] [CrossRef] [PubMed]
- Chowdhury, R.; Schörgenhumer, J.; Novacek, J.; Waser, M. Towards an asymmetric organocatalytic α-cyanation of β-ketoesters. Tetrahedron Lett. 2015, 56, 1911–1914. [Google Scholar] [CrossRef] [PubMed]
- Schörgenhumer, J.; Tiffner, M.; Waser, M. Chiral phase-transfer catalysis in the asymmetric α-heterofunctionalization of prochiral nucleophiles. Beilstein J. Org. Chem. 2017, 13, 1753–1769. [Google Scholar] [CrossRef] [PubMed]
- Novacek, J.; Waser, M. Syntheses and Applications of (Thio)Urea-Containing Chiral Quaternary Ammonium Salt Catalysts. Eur. J. Org. Chem. 2014, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Novacek, J.; Monkowius, U.; Himmelsbach, M.; Waser, M. Asymmetric α-chlorination of β-ketoesters using bifunctional ammonium salt catalysis. Monatsh. Chem. 2016, 147, 533–538. [Google Scholar] [CrossRef]
- Novacek, J.; Izzo, J.A.; Vetticatt, M.J.; Waser, M. Bifunctional Ammonium Salt Catalyzed Asymmetric α-Hydroxylation of β-Ketoesters by Simultaneous Resolution of Oxaziridines. Chem. Eur. J. 2016, 22, 17339–17344. [Google Scholar] [CrossRef] [PubMed]
- Shirakawa, S.; Maruoka, K. Recent Developments in Asymmetric Phase-Transfer Reactions. Angew. Chem. Int. Ed. 2013, 52, 4312–4348. [Google Scholar] [CrossRef] [PubMed]
- Song, C.E. Cinchona Alkaloids in Synthesis & Catalysis; Wiley-VCH: Weinheim, Germany, 2009; ISBN 9783527324163. [Google Scholar]
- Okino, T.; Hoashi, Y.; Takemoto, Y. Enantioselective Michael Reaction of Malonates to Nitroolefins Catalyzed by Bifunctional Organocatalysts. J. Am. Chem. Soc. 2003, 125, 12672–12673. [Google Scholar] [CrossRef] [PubMed]
- Bandar, J.S.; Lambert, T.H. Enantioselective Brønsted Base Catalysis with Chiral Cyclopropenimines. J. Am. Chem. Soc. 2012, 134, 5552–5555. [Google Scholar] [CrossRef] [PubMed]
- Uyanik, M.; Okamoto, H.; Yasui, T.; Ishihara, K. Quaternary Ammonium (Hypo)iodite Catalysis for Enantioselective Oxidative Cycloetherification. Science 2010, 328, 1376–1379. [Google Scholar] [CrossRef] [PubMed]
- Shibatomi, K.; Soga, Y.; Narayama, A.; Fujisawa, I.; Iwasa, S. Highly Enantioselective Chlorination of β-Keto Esters and Subsequent SN2 Displacement of Tertiary Chlorides: A Flexible Method for the Construction of Quaternary Stereogenic Centers. J. Am. Chem. Soc. 2012, 134, 9836–9839. [Google Scholar] [CrossRef] [PubMed]
- Tiffner, M.; Novacek, J.; Busillo, A.; Gratzer, K.; Massa, A.; Waser, M. Design of chiral urea-quaternary ammonium salt hybrid catalysts for asymmetric reactions of glycine Schiff bases. RSC Adv. 2015, 5, 78941–78949. [Google Scholar] [CrossRef] [PubMed]
- Varga, S.; Jakab, G.; Drahos, L.; Holczbauer, T.; Czugler, M.; Soos, T. Double Diastereocontrol in Bifunctional Thiourea Organocatalysis: Iterative Michael–Michael–Henry Sequence Regulated by the Configuration of Chiral Catalysts. Org. Lett. 2011, 13, 5416–5419. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Not available. |

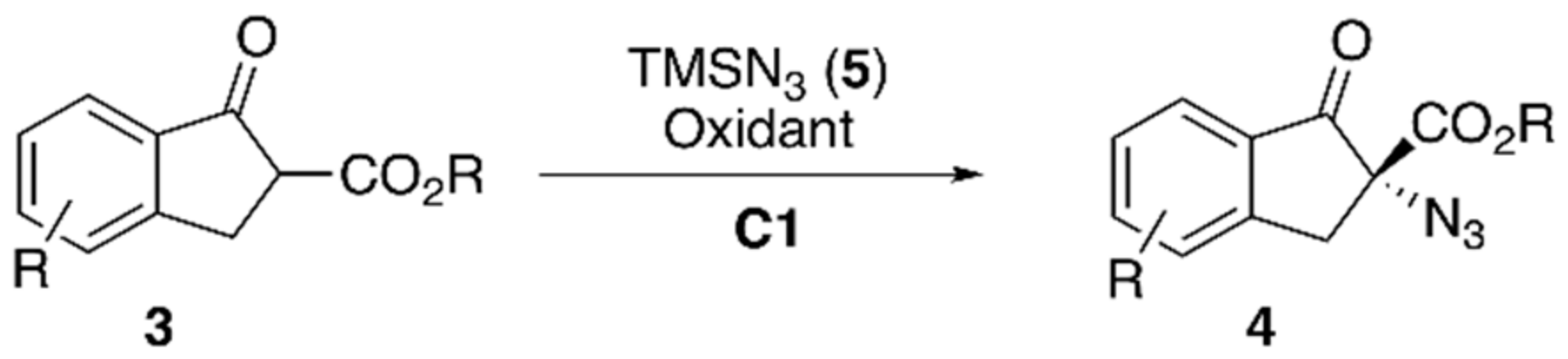
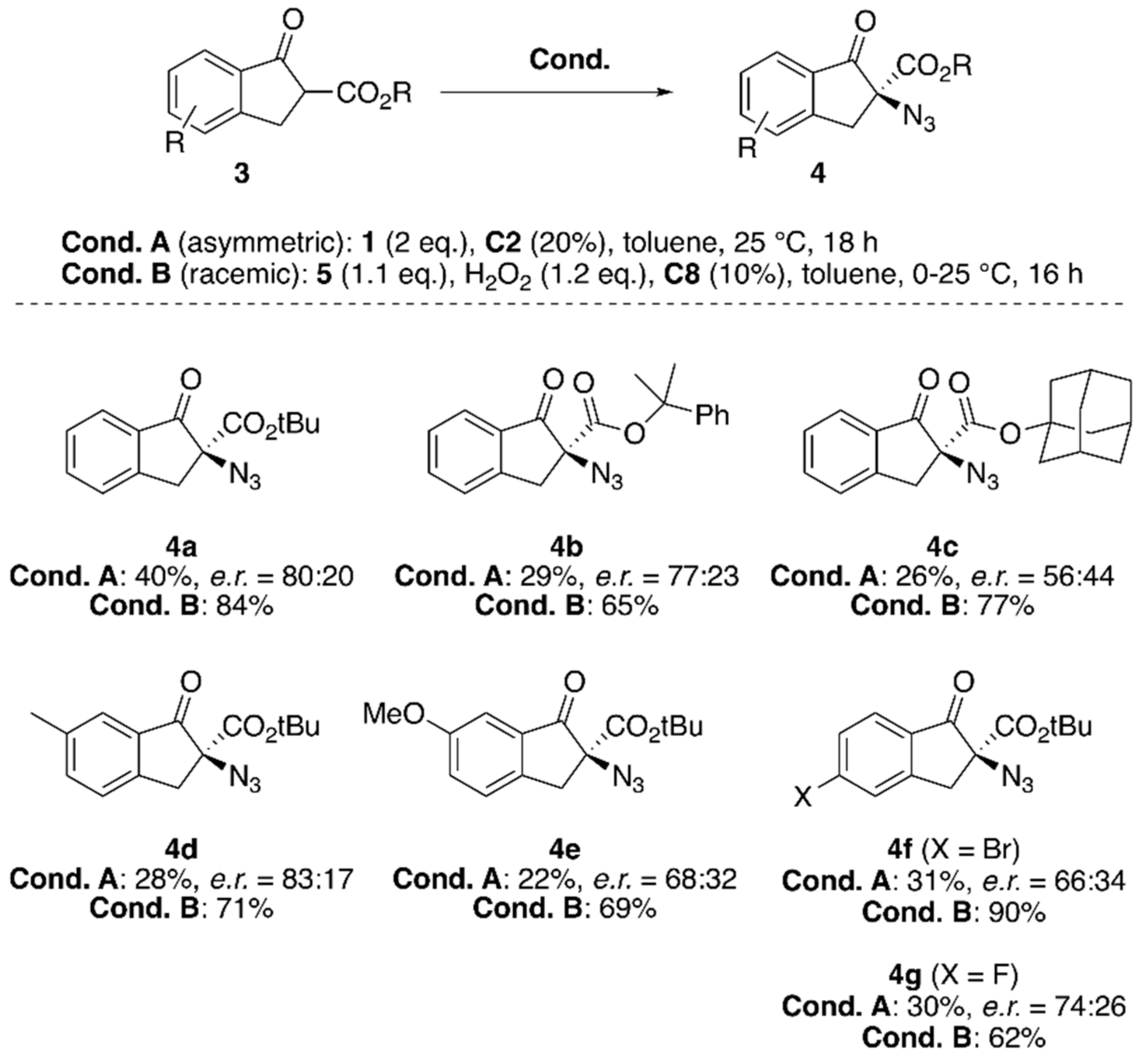

{kind=link}
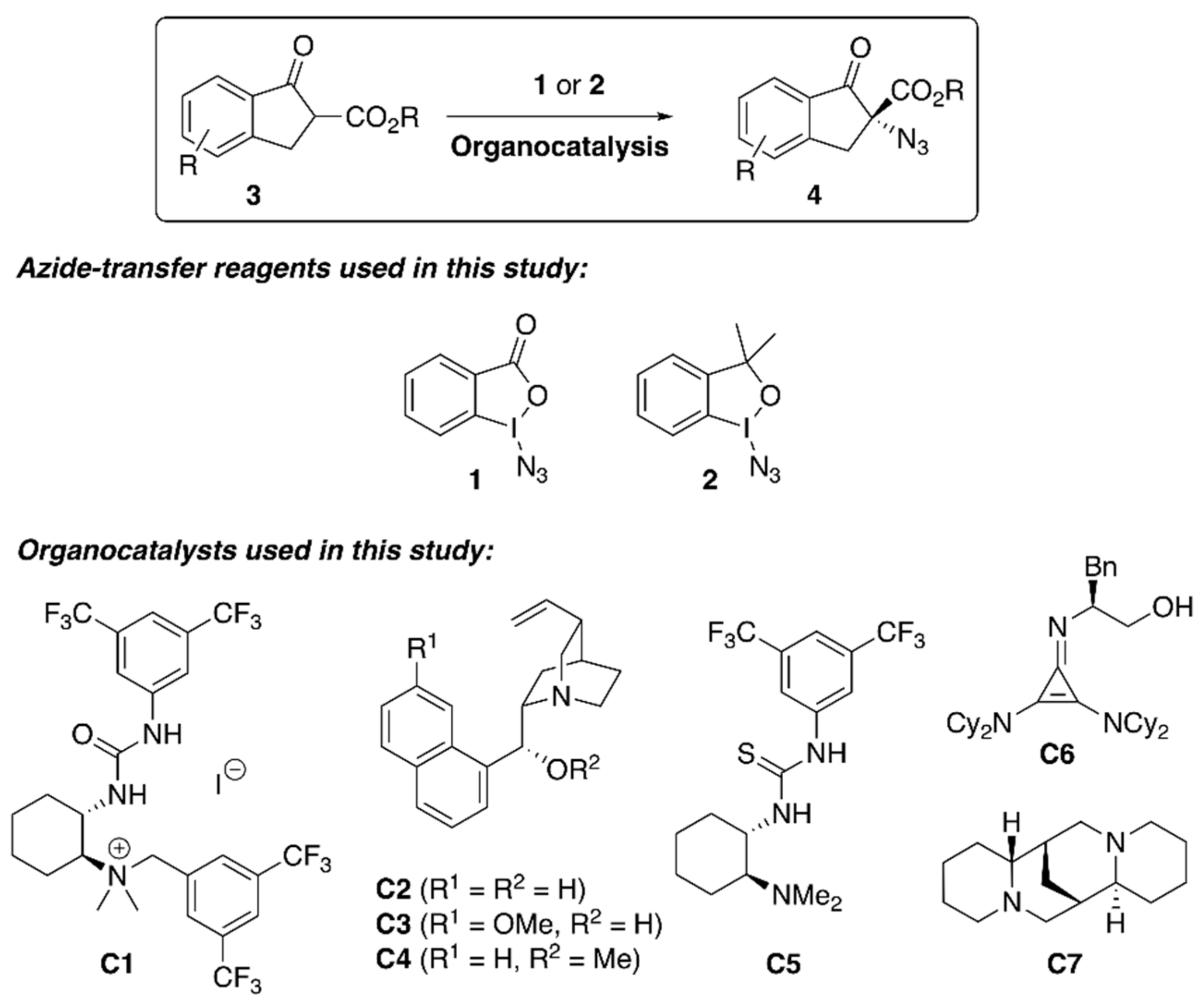{kind=link}
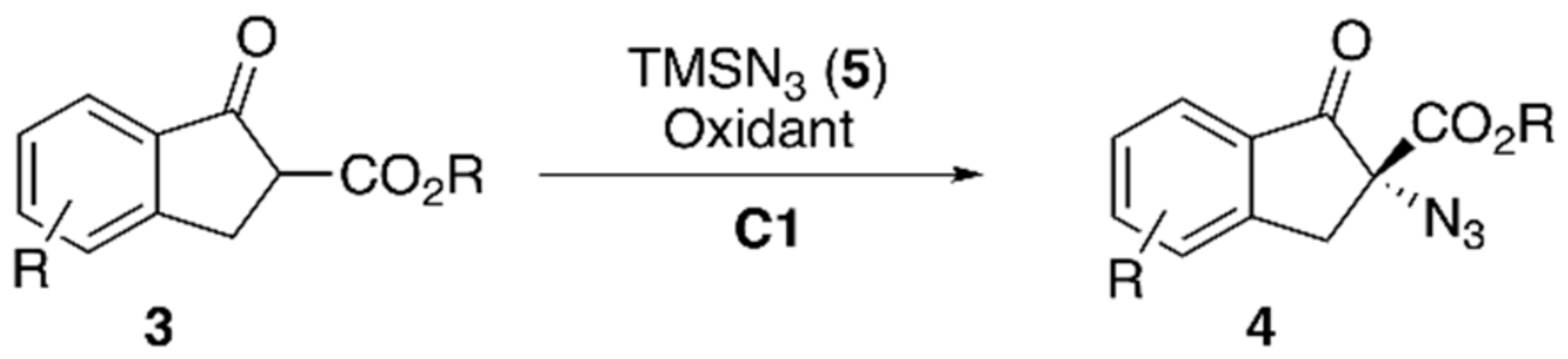{kind=link}
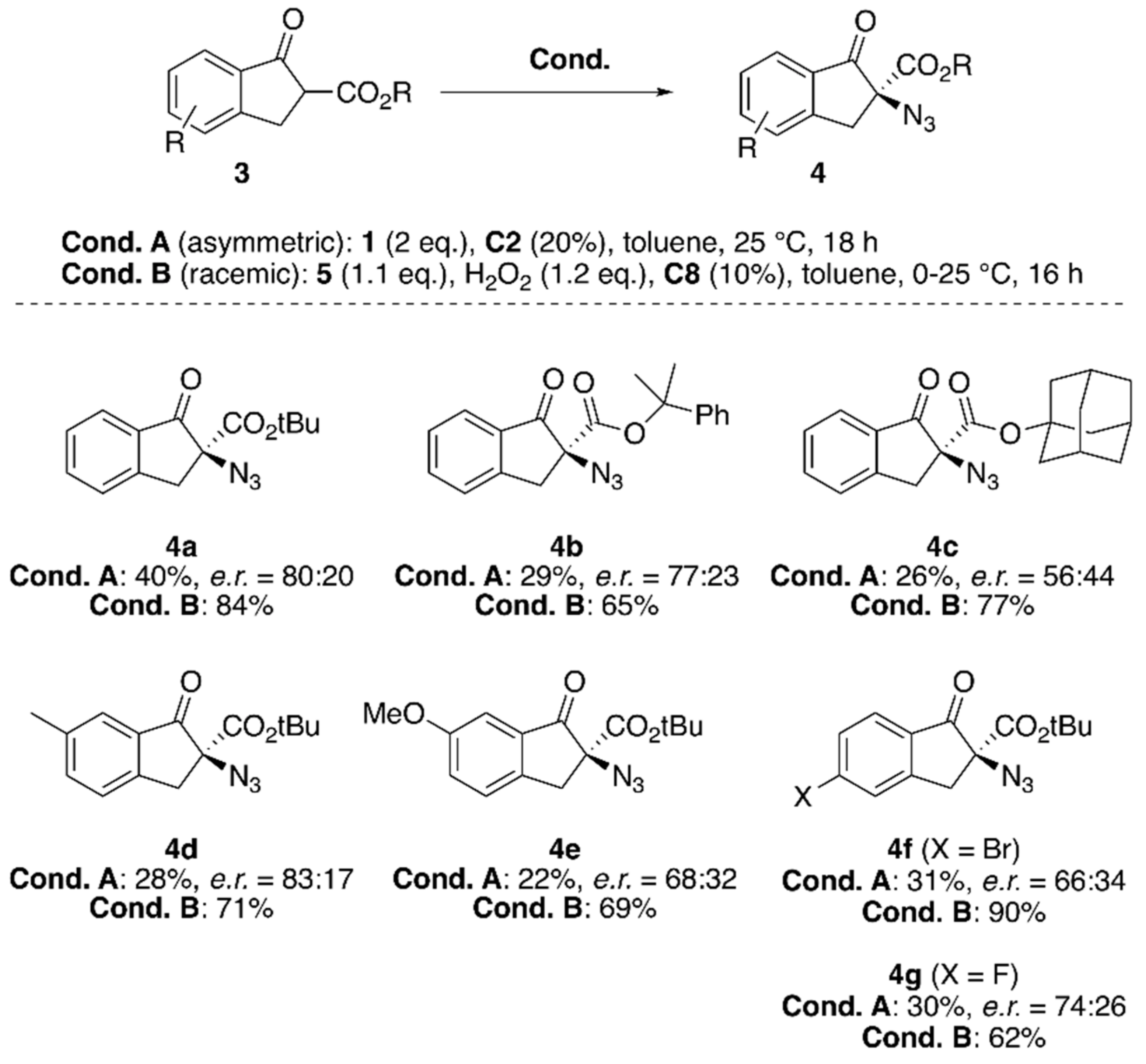{kind=link}

| Entry 1 | Cat. | N3+ | Base | Solv. | T (°C) | t (h) | Yield 2 (%) | e.r. (S:R) 3 |
|---|---|---|---|---|---|---|---|---|
| 1 | C1 (10%) | 1 (1.1 eq.) | K2CO3 (1.1 eq.) | toluene | 25 | 3 | 80 | 56:44 |
| 2 | C1 (10%) | 1 (1.1 eq.) | - | toluene | 25 | 16 | 50 | 55:45 |
| 3 | C1 (10%) | 1 (1.1 eq.) | K2CO3 (1.1 eq.) | MTBE | 25 | 0.5 | 85 | 55:45 |
| 4 | C1 (10%) | 1 (1.1 eq.) | DMAP (1.1 eq.) | MTBE | 25 | 0.5 | 88 | 52:48 |
| 5 | C2 (20%) | 1 (1.1 eq.) | - | toluene | 25 | 18 | 18 | 65:35 |
| 6 | C2 (40%) | 1 (1.1 eq.) | - | toluene | 25 | 40 | 44 | 80:20 |
| 7 | C2 (40%) | 1 (1.1 eq.) | - | toluene | 50 | 40 | 39 | 67:33 |
| 8 | C2 (40%) | 1 (1.1 eq.) | K2CO3 (1.1 eq.) | toluene | 25 | 1.5 | 40 | 77:23 |
| 9 | C2 (100%) | 1 (1.1 eq.) | - | toluene | 25 | 1.5 | 52 | 81:19 |
| 10 | C2 (100%) | 1 (1.1 eq.) | - | MTBE | 25 | 1.5 | 66 | 67:33 |
| 11 | C2 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | 40 | 80:20 |
| 12 | C2 (20%) | 1 (2 eq.) | - | toluene | 25 | 72 | 41 | 80:20 |
| 13 | C2 (20%) | 2 (2 eq.) | - | toluene | 25 | 18 | 24 | 56:44 |
| 14 | C2 (20%) | 1 (2 eq.) | - | CH2Cl2 | 25 | 18 | 39 | 75:25 |
| 15 | C2 (20%) | 1 (2 eq.) | - | THF | 25 | 18 | 19 | 55:45 |
| 16 | C3 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | 35 | 63:37 |
| 17 | C4 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | 15 | 58:42 |
| 18 | C5 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | traces | n.d. |
| 19 | C6 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | 7 | 54:46 |
| 20 | C7 (20%) | 1 (2 eq.) | - | toluene | 25 | 18 | 28 | 46:54 |
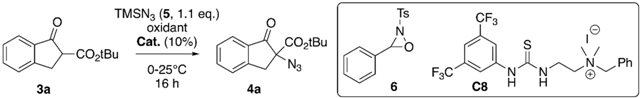
| Entry | Oxidant | Solvent | Cat. | Yield [%] 1 | e.r. [S:R] 2 |
|---|---|---|---|---|---|
| 1 | H2O2 (35%) (1.2 eq.) | toluene | C1 | 81 | 55:45 |
| 2 | H2O2 (35%) (1.2 eq.) | toluene | - | 0 | - |
| 3 | H2O2 (35%) (1.2 eq.) | AcN | C1 | 86 | 53:47 |
| 4 | t-BuOOH (1.2 eq.) | toluene | C1 | 76 | 54:46 |
| 5 | 6 (1.2 eq.) | toluene | C1 | 80 | 52:48 |
| 6 | H2O2 (35%) (1.2 eq.) | toluene | C8 | 84 | - |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tiffner, M.; Stockhammer, L.; Schörgenhumer, J.; Röser, K.; Waser, M. Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters. Molecules 2018, 23, 1142. https://doi.org/10.3390/molecules23051142
Tiffner M, Stockhammer L, Schörgenhumer J, Röser K, Waser M. Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters. Molecules. 2018; 23(5):1142. https://doi.org/10.3390/molecules23051142
Chicago/Turabian StyleTiffner, Maximilian, Lotte Stockhammer, Johannes Schörgenhumer, Katharina Röser, and Mario Waser. 2018. "Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters" Molecules 23, no. 5: 1142. https://doi.org/10.3390/molecules23051142
APA StyleTiffner, M., Stockhammer, L., Schörgenhumer, J., Röser, K., & Waser, M. (2018). Towards an Asymmetric Organocatalytic α-Azidation of β-Ketoesters. Molecules, 23(5), 1142. https://doi.org/10.3390/molecules23051142