Lithium Complexes Derived of Benzylphosphines: Synthesis, Characterization and Evaluation in the ROP of rac-Lactide and ε-Caprolactone


Abstract
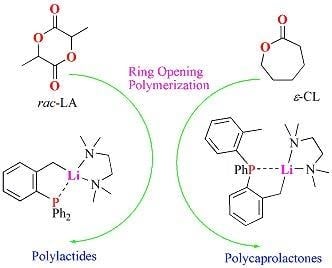
:
1. Introduction
2. Results and Discussion
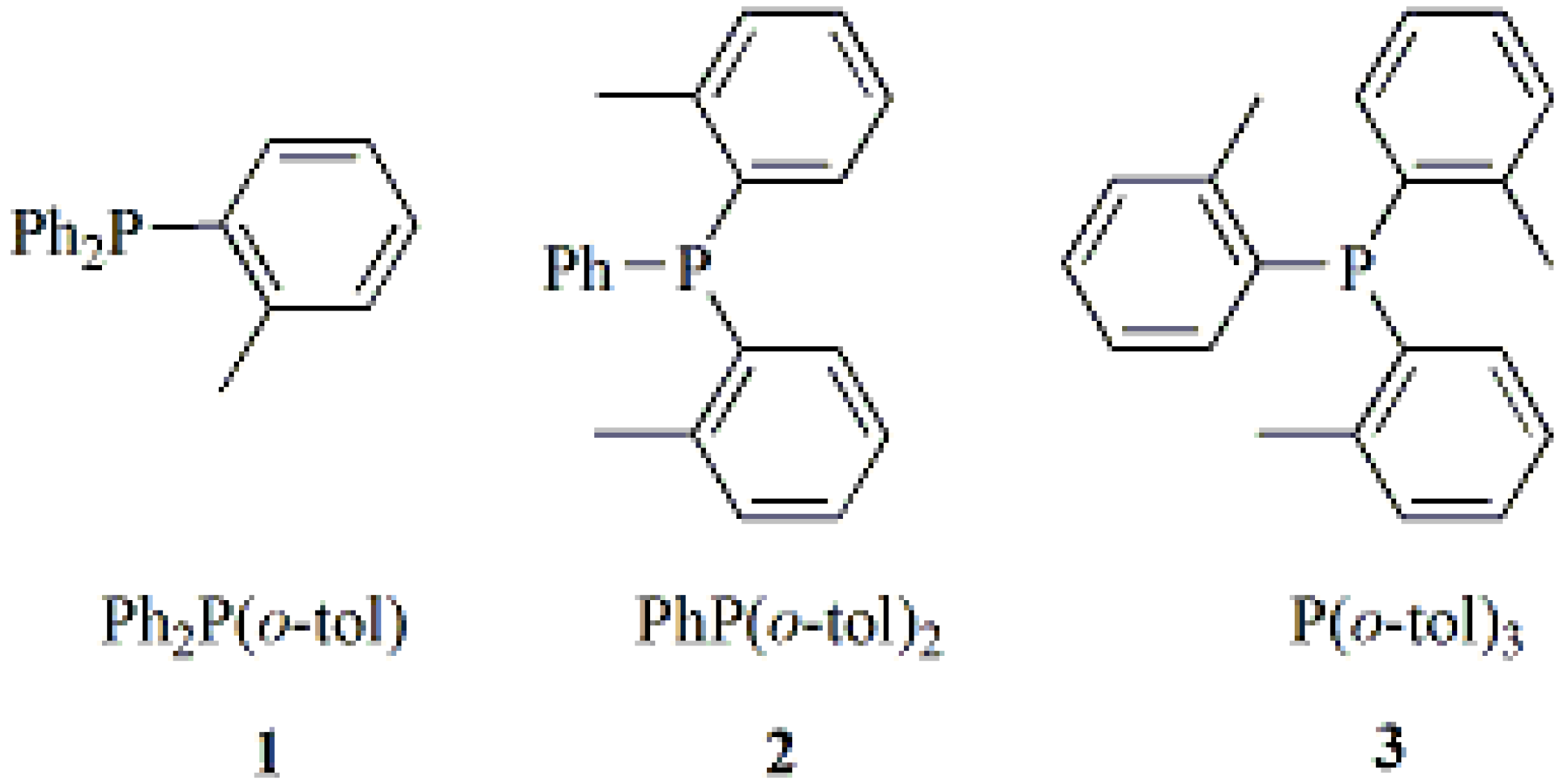
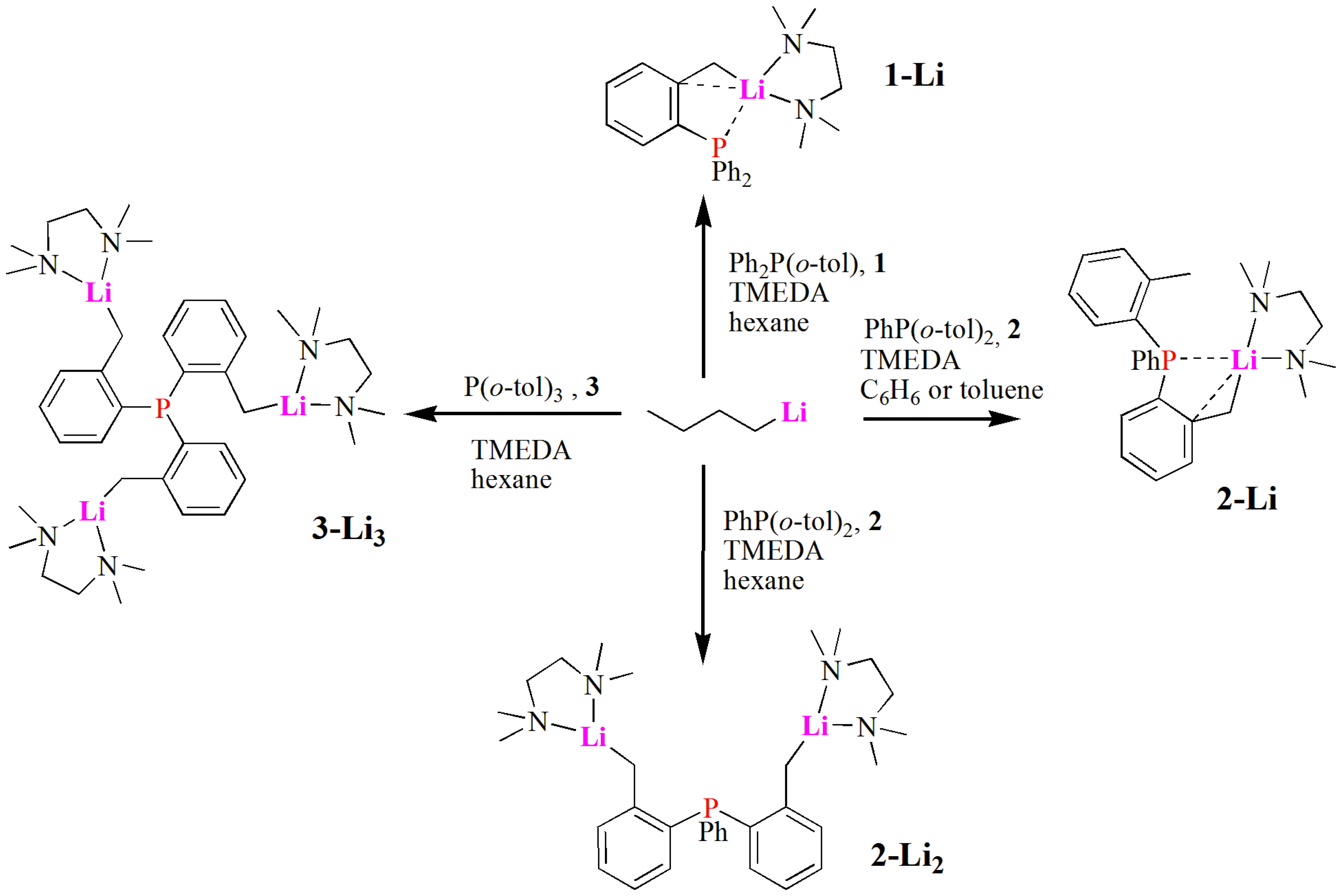
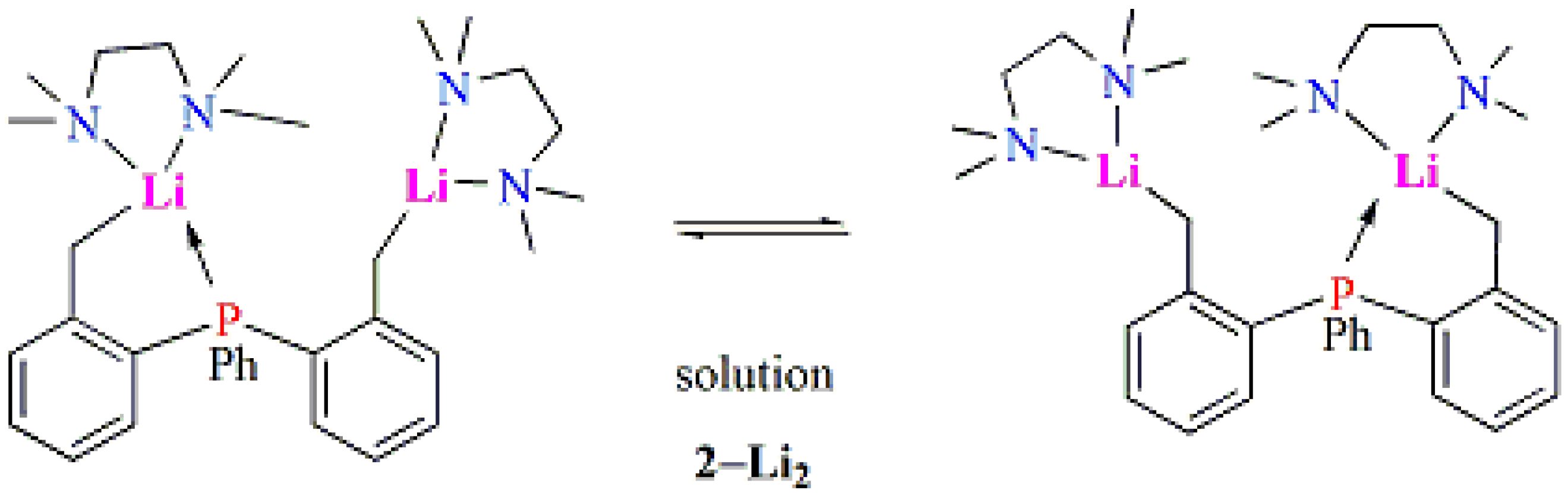
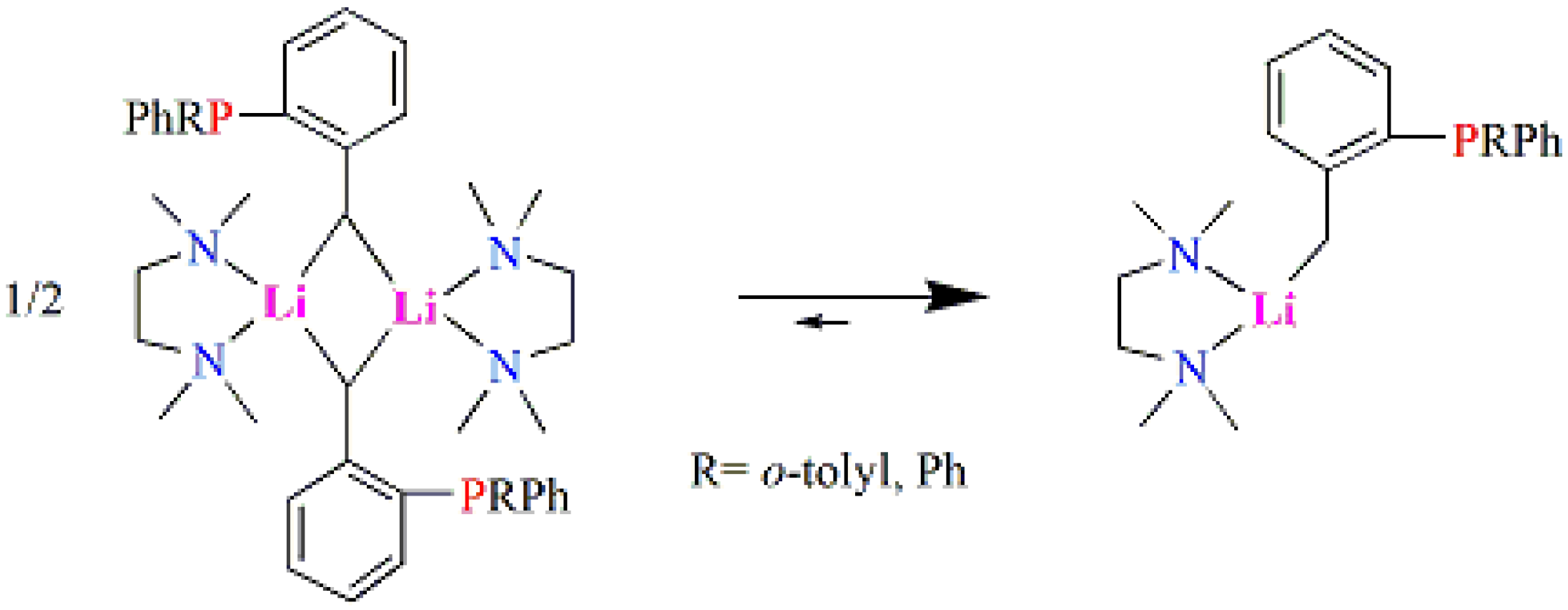
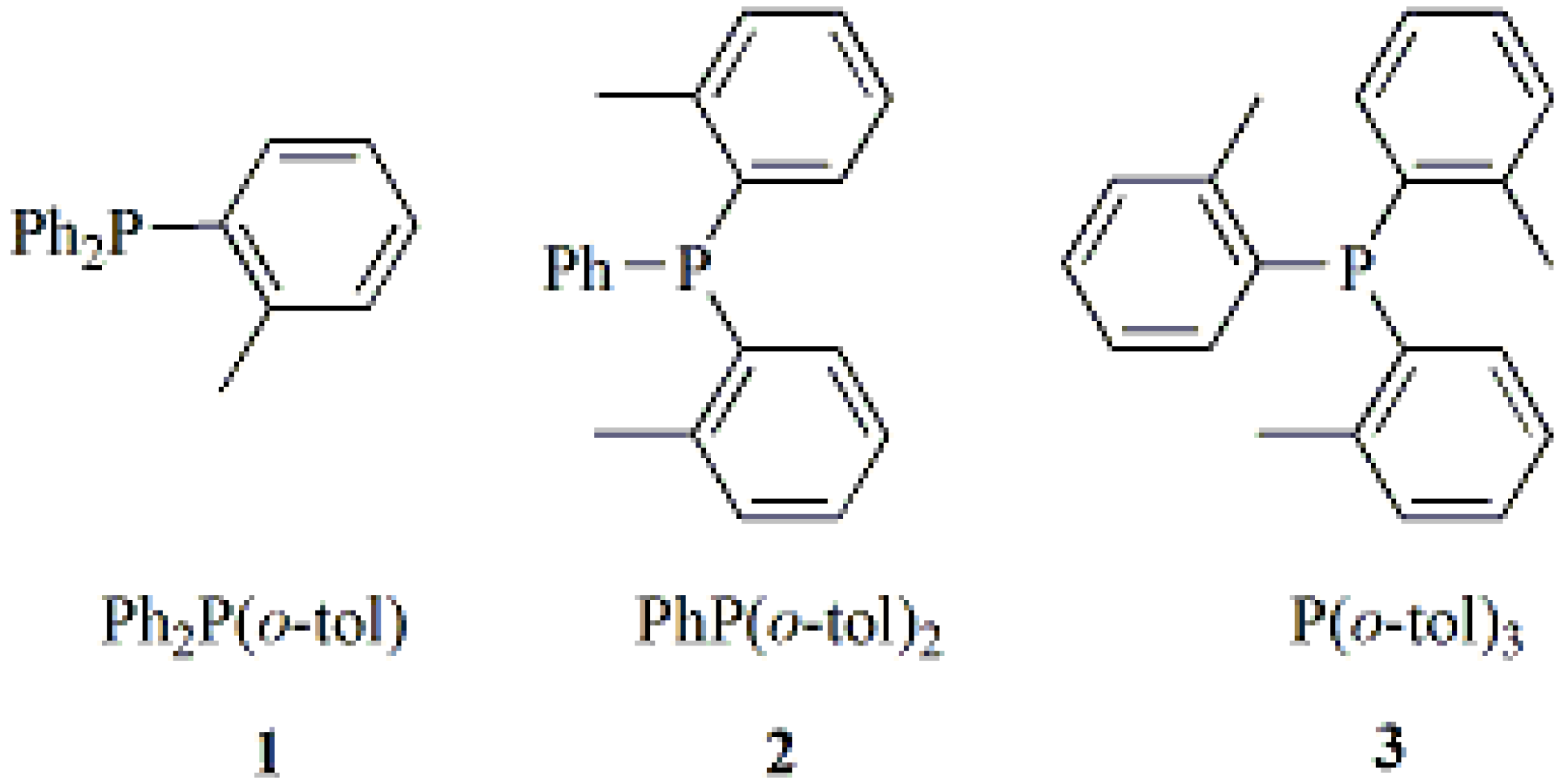
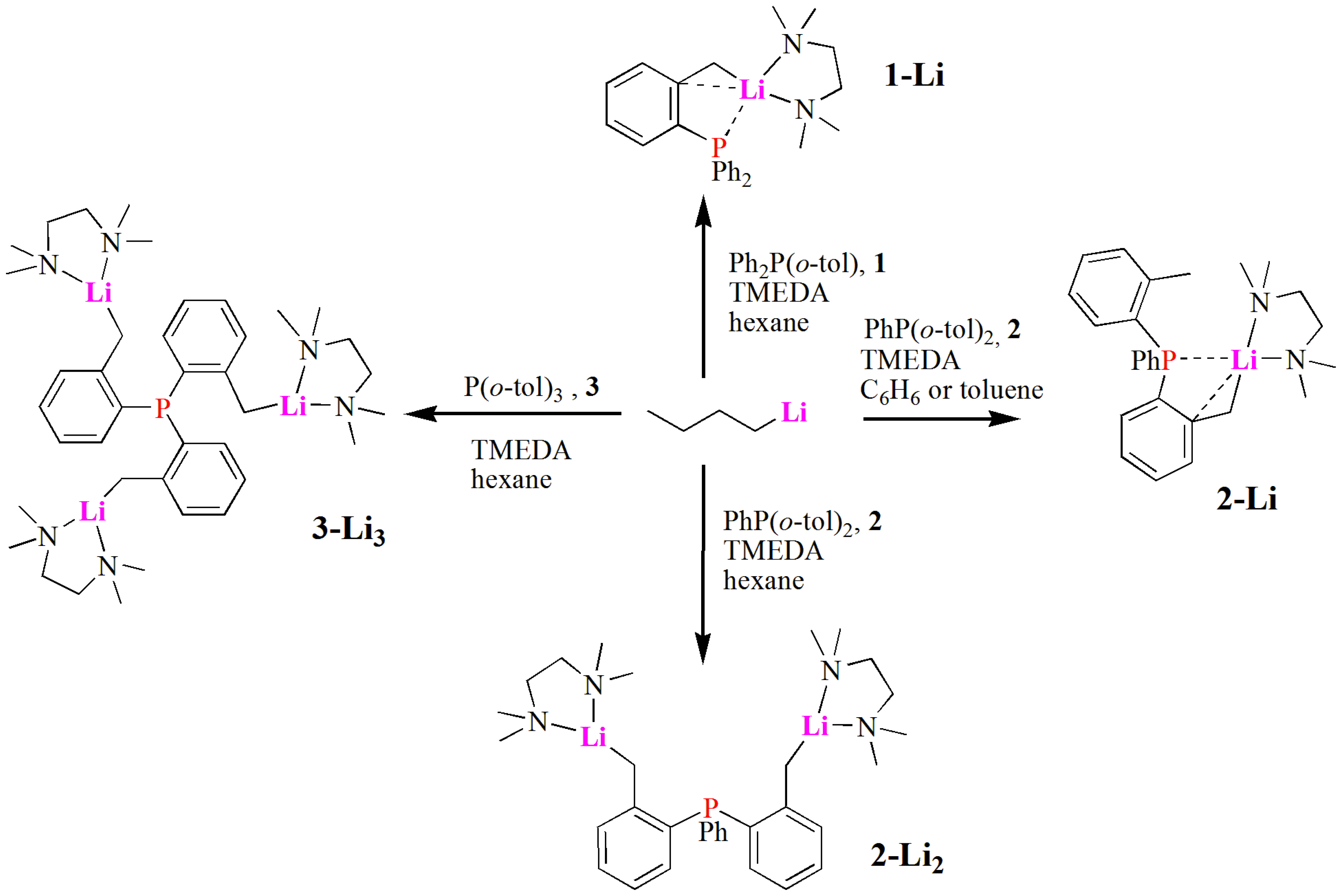
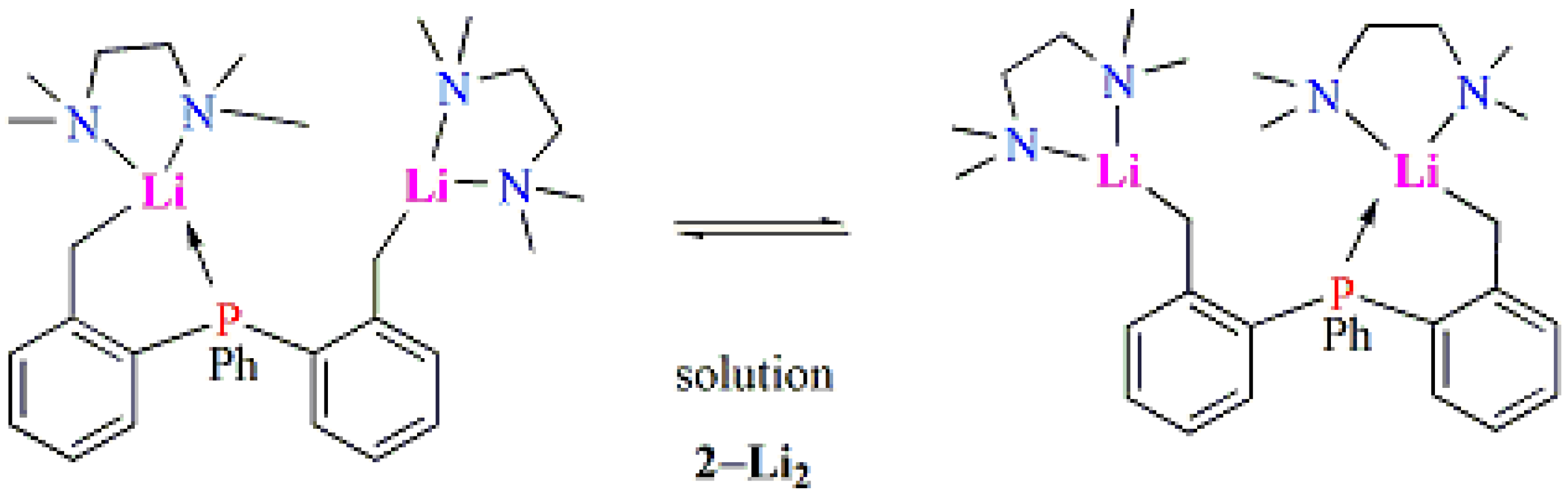
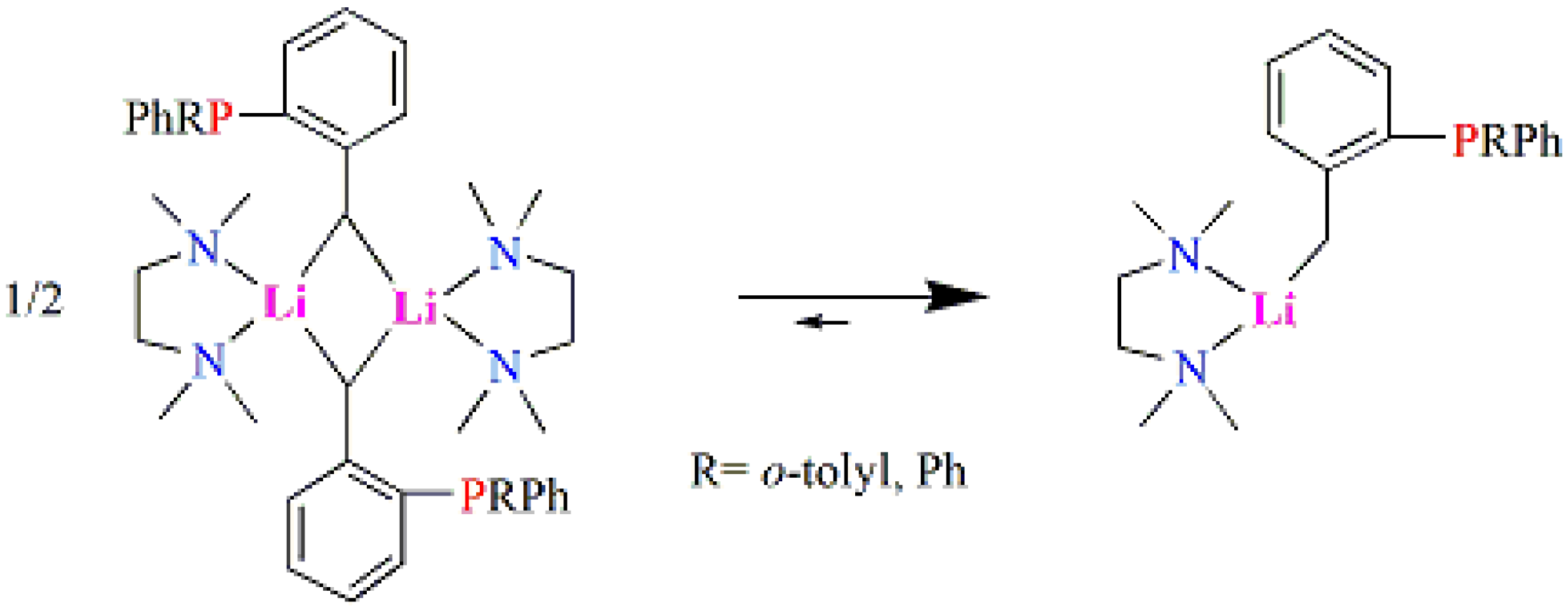
2.1. Synthesis and Characterization of Lithium Complexes Derived of Organophosphines
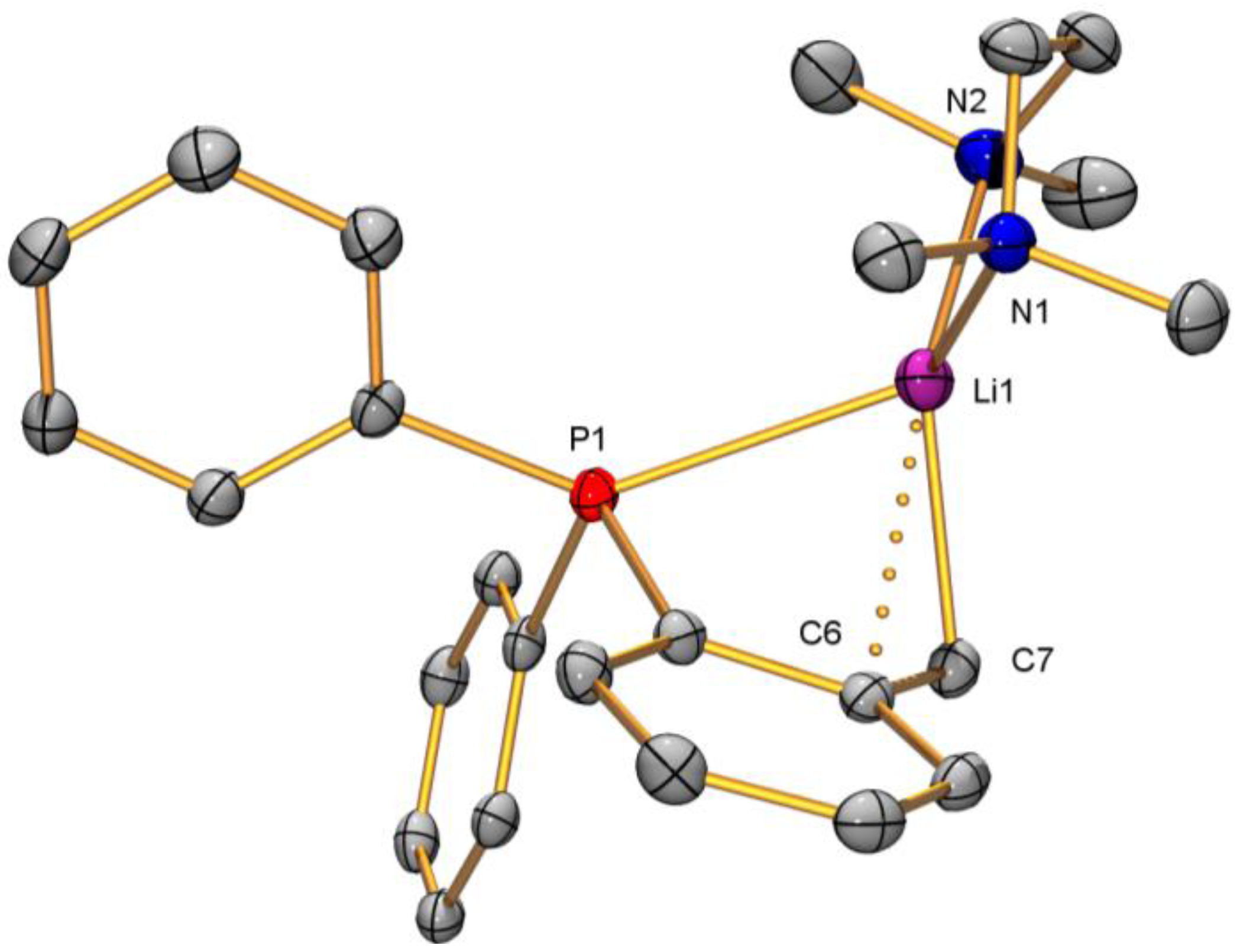
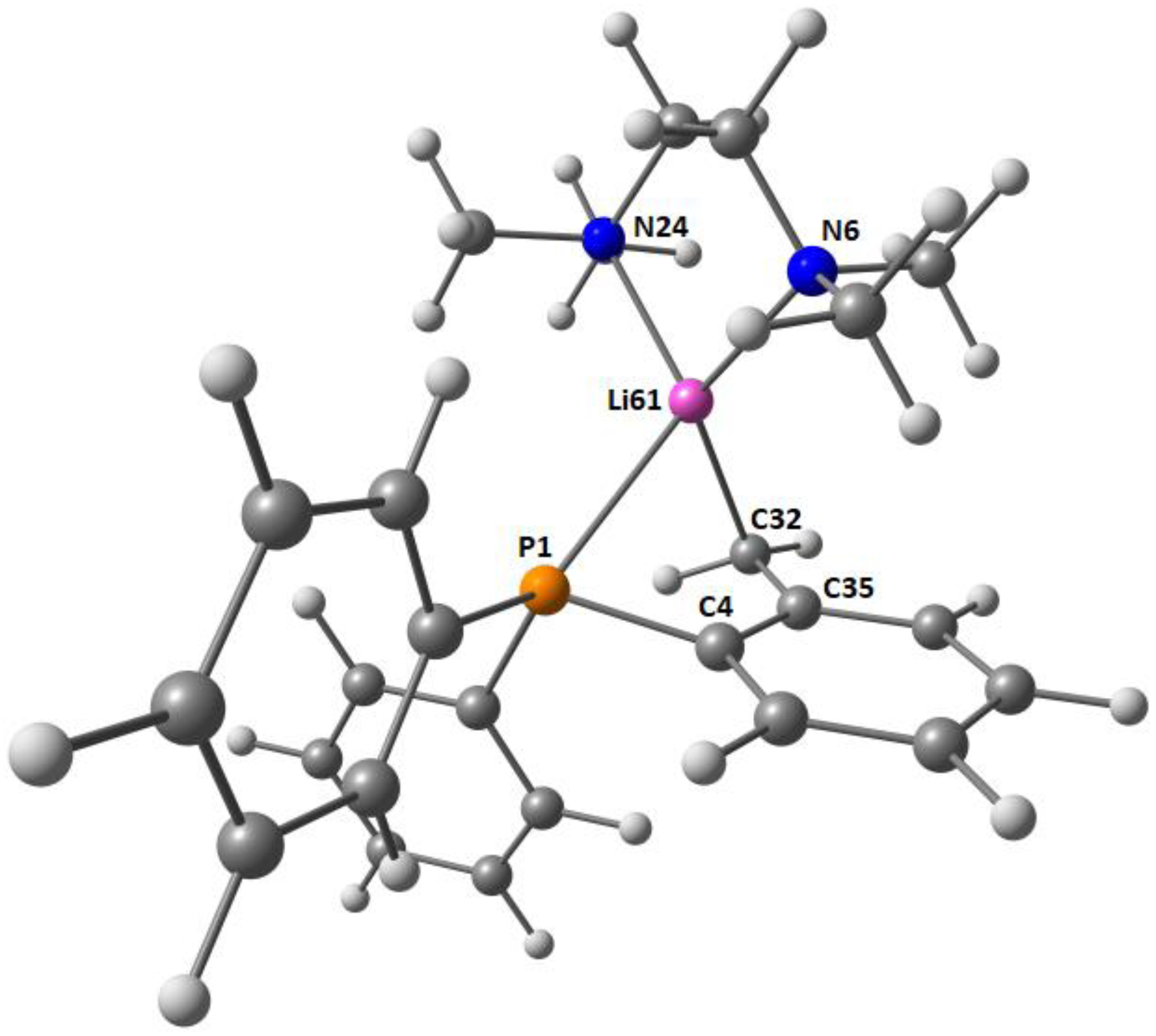
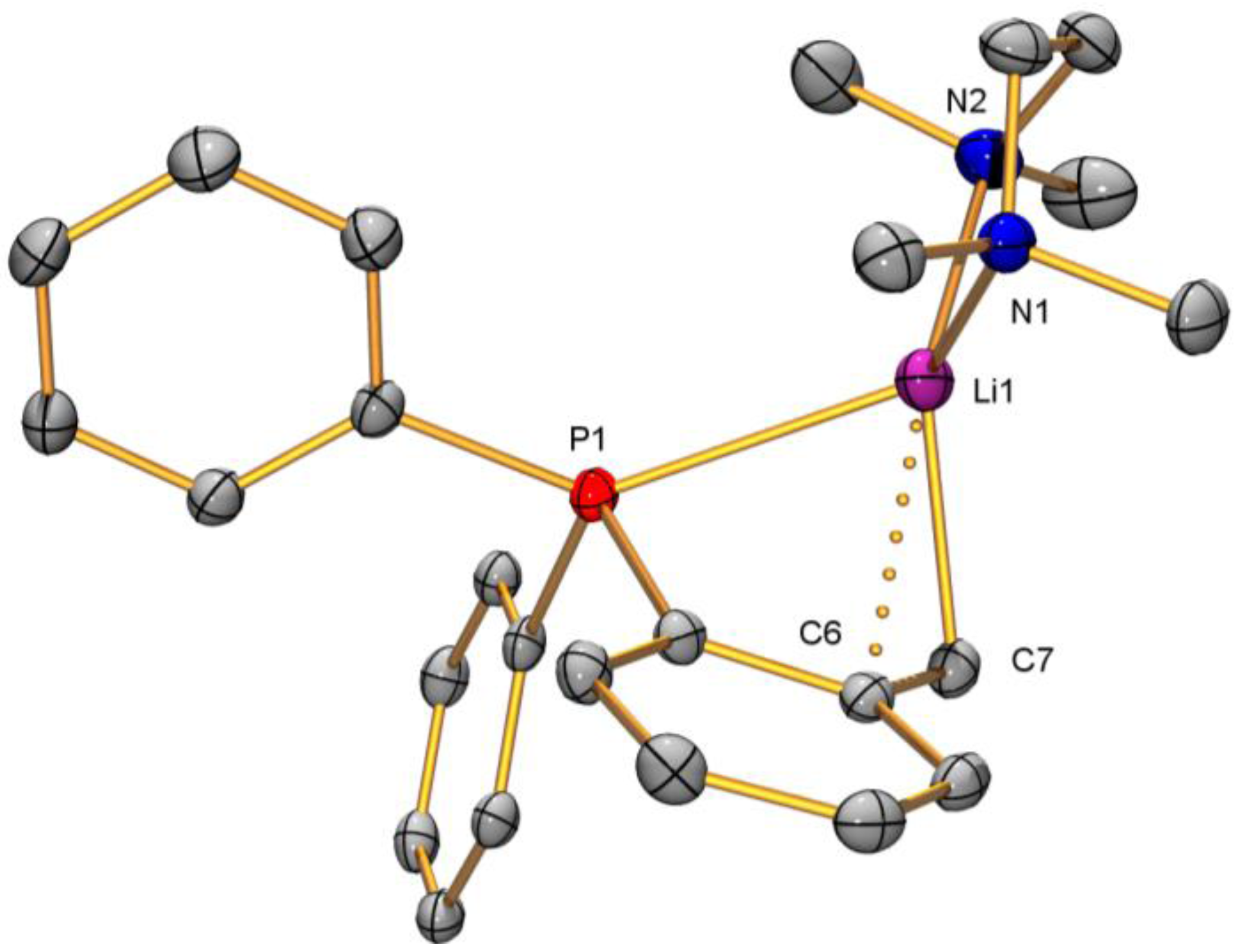
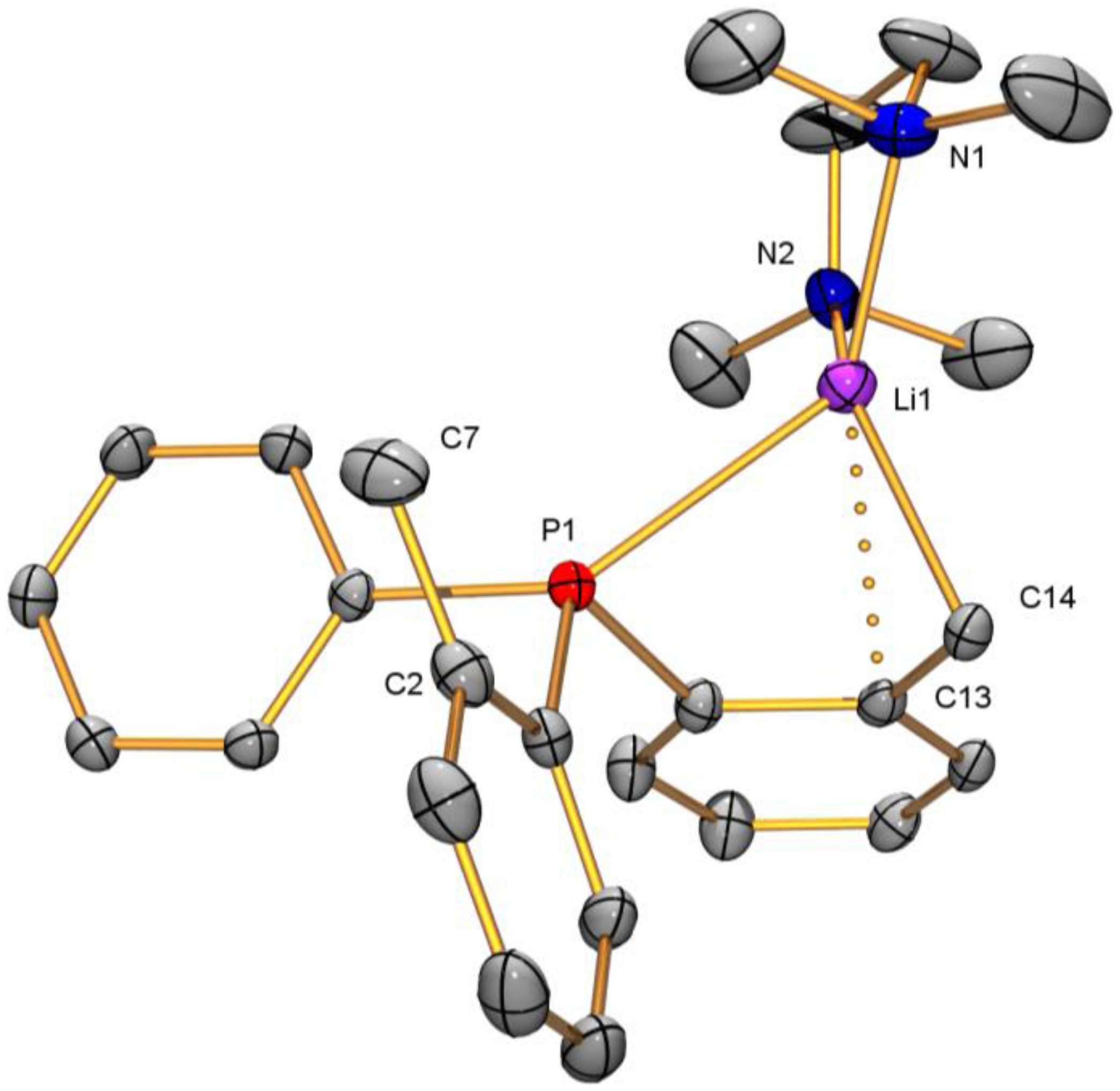
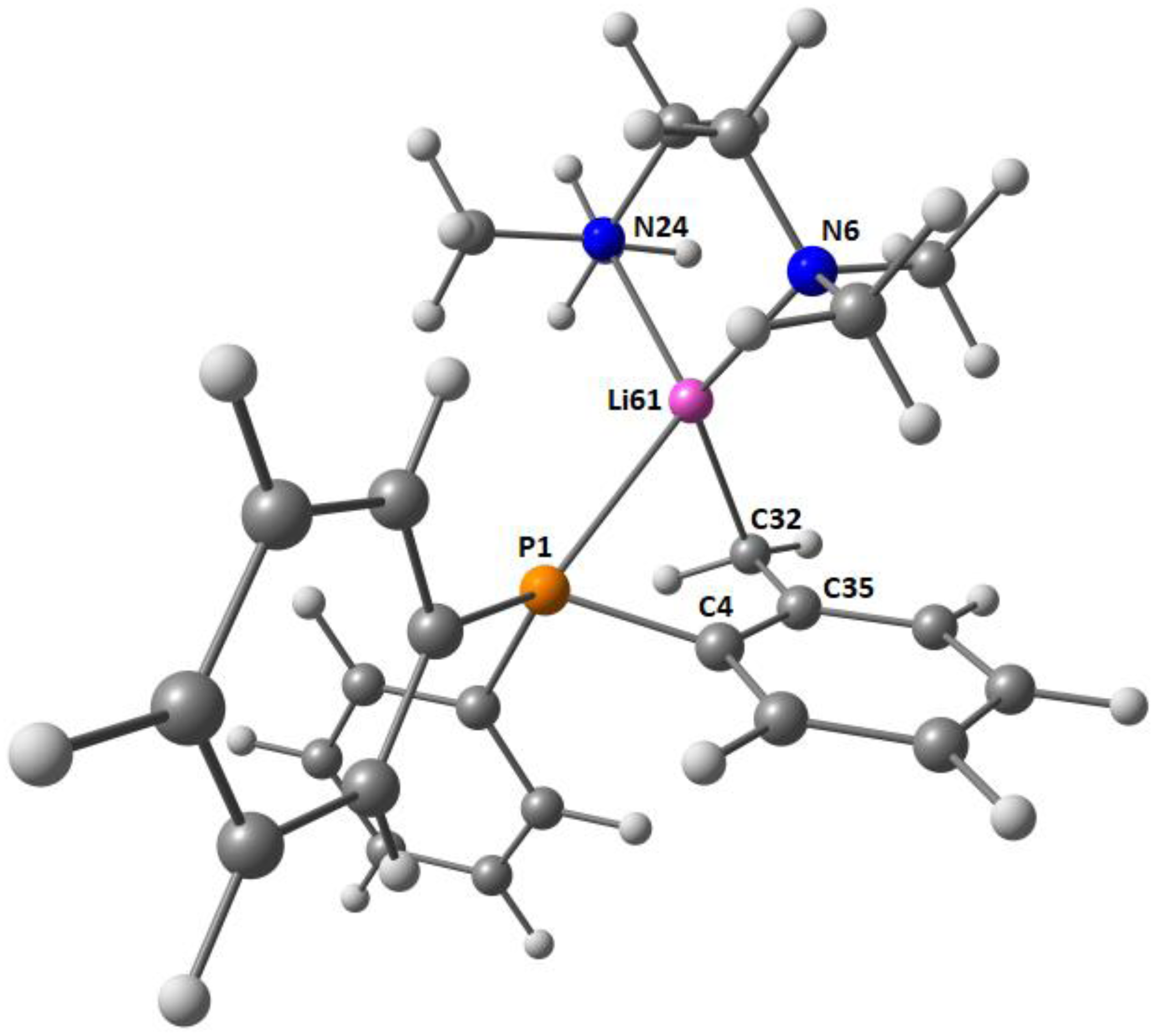
2.2. Solid-State Structure Determination
2.3. DFT Studies
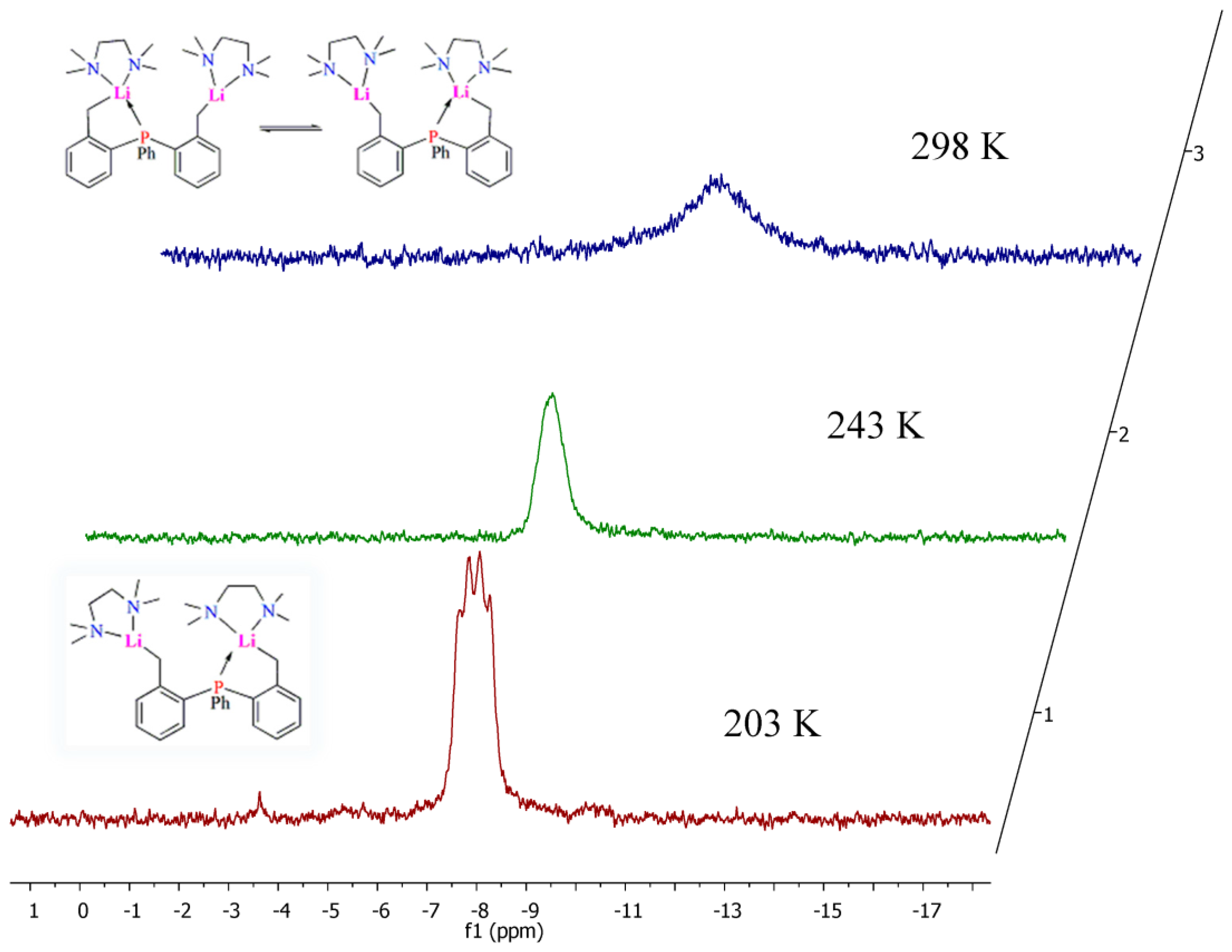
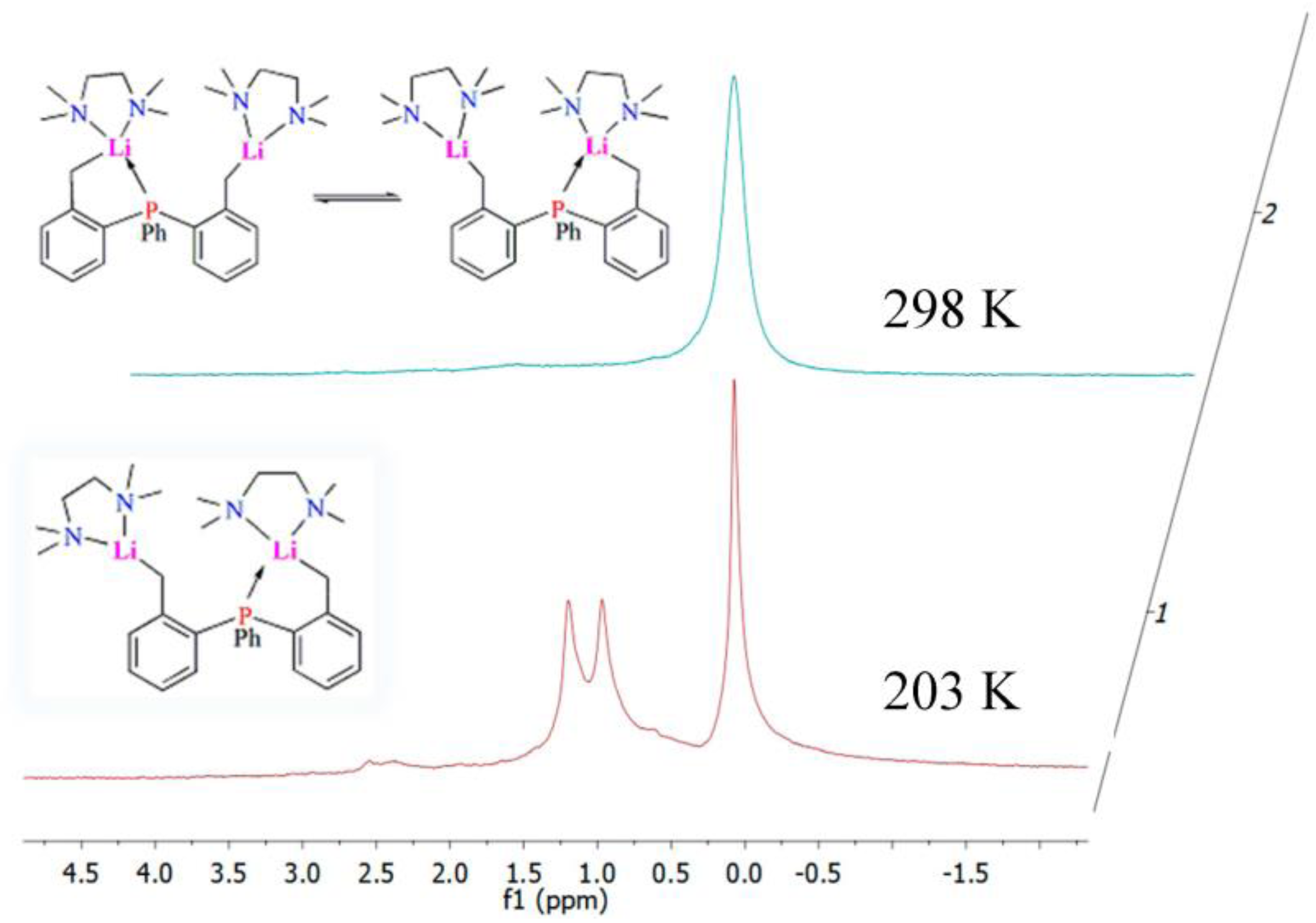
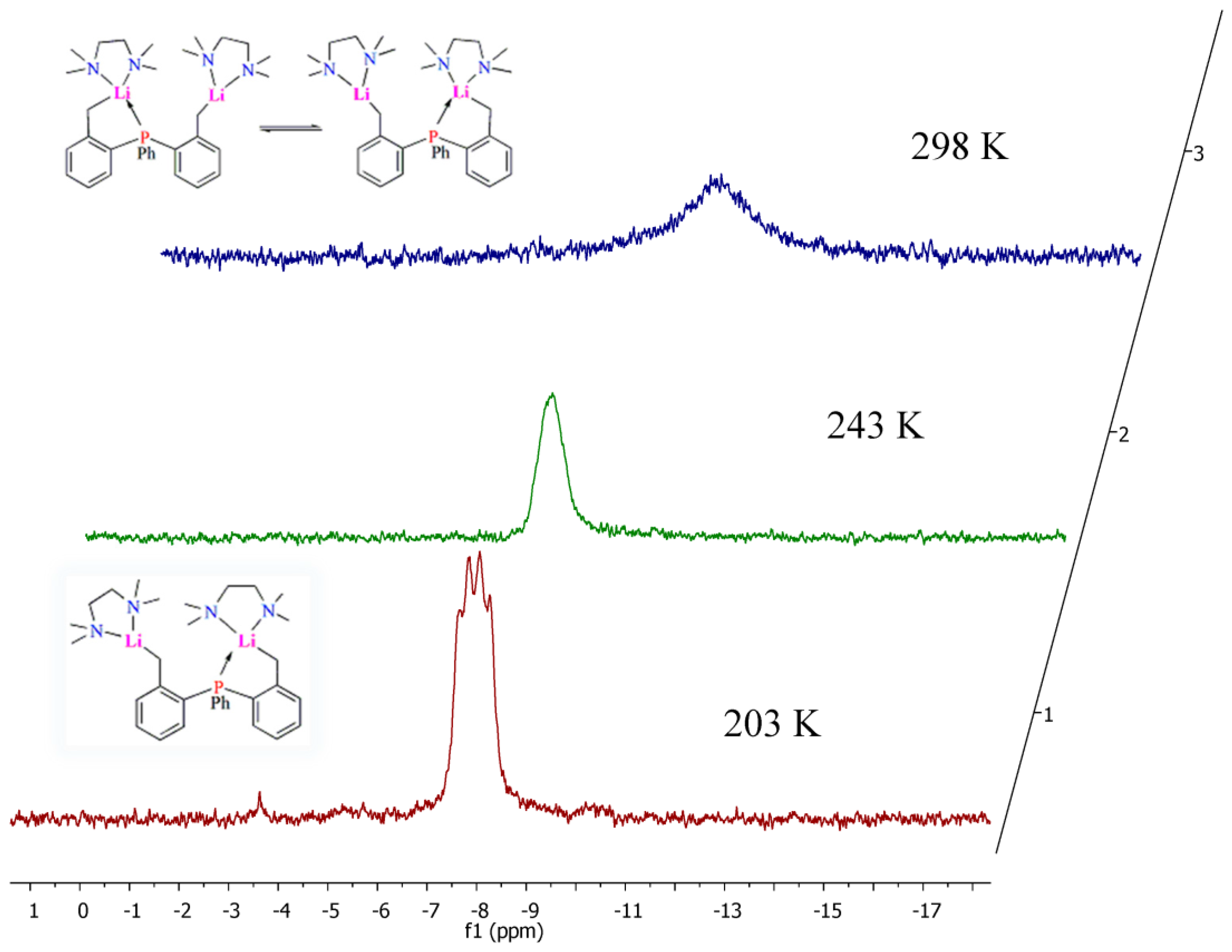
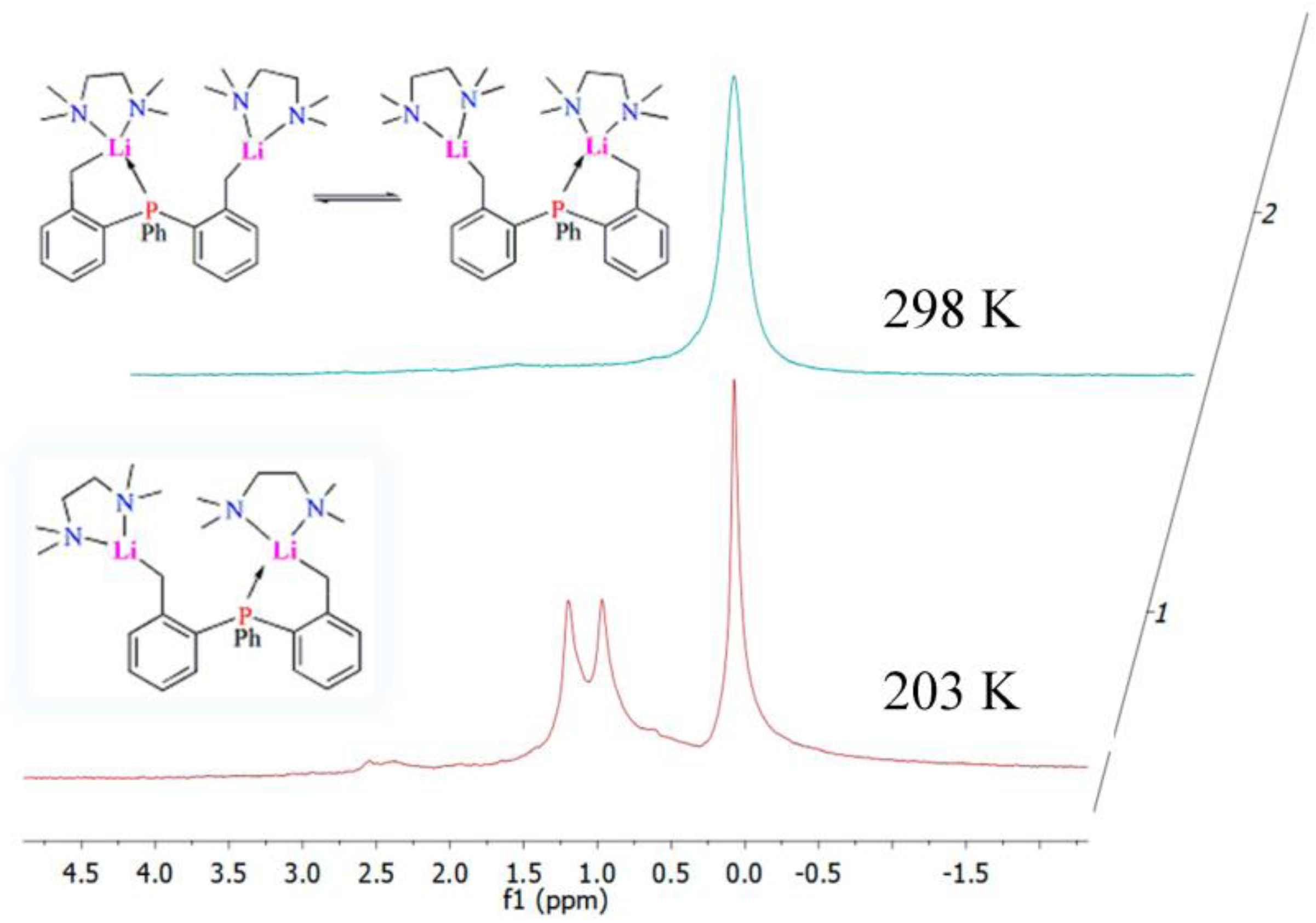
2.4. Solution NMR Spectroscopy
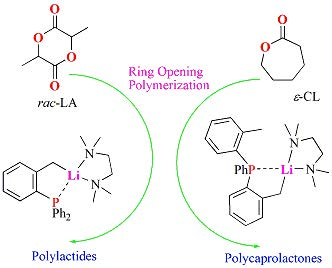
2.5. Preliminary Polymerization Studies
2.5.1. Solvent-Free Ring Opening Polymerizaton of ε-Caprolactone
2.5.2. Solvent-Free Ring-Opening Polymerization of rac-LA
3. Materials and Methods
3.1. General Considerations
3.2. Synthesis of Compounds
3.2.1. Synthesis of Complex [Ph2P(C6H4CH2Li·TMEDA)] (1-Li)
3.2.2. Complex [Ph(o-tolyl)P(C6H4CH2Li·TMEDA)] (2-Li)
3.2.3. Complex [PhP(C6H4CH2Li·TMEDA)2] (2-Li2)
3.2.4. Complex [P(C6H4CH2Li·TMEDA)3] (3-Li3)
3.3. Polymerization Procedure
3.3.1. Solvent-Free Ring-Opening Polymerization of ε-CL
3.3.2. Solvent-Free Ring-Opening Polymerization of rac-LA
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gibson, V.C.; Spitzmesser, S.K. Advances in Non-Metallocene Olefin Polymerization Catalysis. Chem. Rev. 2003, 103, 283–316. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Chen, H.-X.; Huang, D.; Zhang, Y.; Yao, Y.-M. A facile route to lithium complexes supported by β-ketoiminate ligands and their reactivity. J. Organomet. Chem. 2014, 749, 7–12. [Google Scholar] [CrossRef]
- Pretula, J.; Slomkowski, S.; Penczek, S. Polylactides—Methods of synthesis and characterization. Adv. Drug Deliv. Rev. 2016, 107, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Yao, C.; Yang, Y.; Xu, S.; Ma, H. Potassium complexes supported by monoanionic tetradentate amino-phenolate ligands: Synthesis, structure and catalysis in the ring-opening polymerization of rac-lactide. Dalton Trans. 2017, 46, 6087–6097. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, B.M.; Cheng, M.; Moore, D.R.; Ovitt, T.M.; Lobkovsky, E.B.; Coates, G.W. Polymerization of Lactide with Zinc and Magnesium β-Diiminate Complexes: Stereocontrol and Mechanism. J. Am. Chem. Soc. 2001, 123, 3229–3238. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhang, W.; Wang, S.; Solan, G.A.; Liang, T.; Rajendran, N.M.; Sun, W.-H. Sodium iminoquinolates with cubic and hexagonal prismatic motifs: Synthesis, characterization and their catalytic behavior toward the ROP of rac-lactide. Inorg. Chem. Front. 2016, 3, 1178–1189. [Google Scholar] [CrossRef]
- Sun, Y.; Xiong, J.; Dai, Z.; Pan, X.; Tang, N.; Wu, J. Stereoselective Alkali-Metal Catalysts for Highly Isotactic Poly(rac-lactide) Synthesis. Inorg. Chem. 2016, 55, 136–143. [Google Scholar] [CrossRef] [PubMed]
- Fernández-Alvarez, F.J.; Lalrempuia, R.; Oro, L.A. Monoanionic NSiN-type ligands in transition metal coordination chemistry and catalysis. Coord. Chem. Rev. 2017, 350, 49–60. [Google Scholar] [CrossRef]
- Labet, M.; Thielemans, W. Synthesis of polycaprolactone: A review. Chem. Soc. Rev. 2009, 38, 3484–3504. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Palma, V.; Muñoz-Hernandez, M.A.; Cuevas-Chavez, C.A.; Vendier, L.; Grellier, M.; Sabo-Etienne, S. Phosphinodi(benzylsilane) PhP{(o-C6H4CH2)SiMe2H}2: A versatile “PSi2Hx” pincer-type ligand at ruthenium. Inorg. Chem. 2013, 52, 9798–9806. [Google Scholar] [CrossRef] [PubMed]
- Corona-Gonzalez, M.V.; Zamora-Moreno, J.; Cuevas-Chavez, C.A.; Rufino-Felipe, E.; Mothes-Martin, E.; Coppel, Y.; Muñoz-Hernandez, M.A.; Vendier, L.; Flores-Alamo, M.; Grellier, M.; et al. A family of rhodium and iridium complexes with semirigid benzylsilyl phosphines: From bidentate to tetradentate coordination modes. Dalton Trans. 2017, 46, 8827–8838. [Google Scholar] [CrossRef] [PubMed]
- Montiel-Palma, V.; Muñoz-Hernandez, M.A.; Ayed, T.; Barthelat, J.-C.; Grellier, M.; Vendier, L.; Sabo-Etienne, S. Agostic Si-H bond coordination assists C-H bond activation at ruthenium in bis(phosphinobenzylsilane) complexes. Chem. Commun. 2007, 3963–3965. [Google Scholar] [CrossRef] [PubMed]
- Setzer, W.N.; Schleyer, P.V.R. X-Ray Structural Analyses of Organolithium Compounds. In Advances in Organometallic Chemistry; Stone, F.G.A., West, R., Eds.; Academic Press: Cambridge, MA, USA, 1985; Volume 24, pp. 353–451. [Google Scholar]
- Feil, F.; Harder, S. Benzyl Complexes of the Heavier Alkaline-Earth Metals: The First Crystal Structure of a Dibenzylstrontium Complex. Organometallics 2001, 20, 4616–4622. [Google Scholar] [CrossRef]
- Bildmann, U.J.; Müller, G. Synthesis and Structure of the Tmeda Adduct of a Dibenzyl Lithiate Anion Containing Four-Coordinate Lithium. Organometallics 2001, 20, 1689–1691. [Google Scholar] [CrossRef]
- Fraenkel, G.; Martin, K. Benzylic Lithium Compounds: The Missing Link in Carbon-Lithium Covalency. Dynamics of Ion Reorientation, Rotation around the Ring-Benzyl Bond, and Bimolecular C-Li Exchange. J. Am. Chem. Soc. 1995, 117, 10336–10344. [Google Scholar] [CrossRef]
- Arnold, J.; Knapp, V.; Schmidt, J.A.R.; Shafir, A. Reactions of N,N[′],N[′′]-trimethyl-1,4,7-triazacyclononane with butyllithium reagents. J. Chem. Soc. Dalton Trans. 2002, 3273–3274. [Google Scholar] [CrossRef]
- Decken, A.; Cowley, A.H. The isolation and X-ray crystal structure of [Li(Et2O)2(CHPh)PPh2]. J. Organomet. Chem. 1996, 509, 135–137. [Google Scholar] [CrossRef]
- Cordero, B.; Gomez, V.; Platero-Prats, A.E.; Reves, M.; Echeverria, J.; Cremades, E.; Barragan, F.; Alvarez, S. Covalent radii revisited. Dalton Trans. 2008, 2832–2838. [Google Scholar] [CrossRef] [PubMed]
- Pyykkö, P.; Atsumi, M. Molecular Single-Bond Covalent Radii for Elements 1–118. Chem. Eur. J. 2009, 15, 186–197. [Google Scholar] [CrossRef] [PubMed]
- Byrne, L.T.; Engelhardt, L.M.; Jacobsen, G.E.; Leung, W.-P.; Papasergio, R.I.; Raston, C.L.; Skelton, B.W.; Twiss, P.; White, A.H. Synthesis of [small alpha]-, [gamma]-phosphorus functionalized alkyl lithium species; X-ray structures of [{Li(L)(CH2PMeR)}2][L =NNN[′]N[′′]-tetramethylethylenediamine (tmen), R = Me or Ph; L =(-)sparteine, R = Ph] and [Li(tmen){CH(SiMe3)C6H4PPh2-o}]. J. Chem. Soc. Dalton Trans. 1989, 105–113. [Google Scholar] [CrossRef]
- Blair, V.L.; Stevens, M.A.; Thompson, C.D. The importance of the Lewis base in lithium mediated metallation and bond cleavage reaction of allyl amines and allyl phosphines. Chem. Commun. 2016, 52, 8111–8114. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.J.; Barrett, T.; Parsons, S. Lithiation studies of [PhP(CH2Ph)3]Cl; X-ray crystal structure of the phosphoniodiylide [(TMEDA)Li(PhCH)2PPh(CH2Ph)] and its rhodium and chromium complexes. J. Organomet. Chem. 2001, 625, 236–244. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2007, 120, 215–241. [Google Scholar] [CrossRef]
- Assadourian, L.; Faure, R.; Gau, G. Lithium-7 NMR studies of alkylarenelithium salts. J. Organomet. Chem. 1985, 280, 153–158. [Google Scholar] [CrossRef]
- Beno, M.A.; Hope, H.; Olmstead, M.M.; Power, P.P. Isolation and X-ray crystal structures of the solvate complexes [Li(Et2O)benzyl]x and [Li(THF)2mesityl]2. Organometallics 1985, 4, 2117–2121. [Google Scholar] [CrossRef]
- Xiong, J.; Zhang, J.; Sun, Y.; Dai, Z.; Pan, X.; Wu, J. Iso-Selective Ring-Opening Polymerization of rac-Lactide Catalyzed by Crown Ether Complexes of Sodium and Potassium Naphthalenolates. Inorg. Chem. 2015, 54, 1737–1743. [Google Scholar] [CrossRef] [PubMed]
- Coudane, J.; Ustariz-Peyret, C.; Schwach, G.; Vert, M. More about the stereodependence of DD and LL pair linkages during the ring-opening polymerization of racemic lactide. J. Polym. Sci. Part A Polym. Chem. 1997, 35, 1651–1658. [Google Scholar] [CrossRef]
- Agilent Technologies. CrysAlisPro; 1.171.37.35; Agilent Technologies: Santa Clara, CA, USA, 2014. [Google Scholar]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SAINT-Plus Suite; 6.0; Bruker AXS, Inc.: Madison, WI, USA, 2000. [Google Scholar]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. Sect. C 2015, C71, 3–8. [Google Scholar]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal structure determination. Acta Crystallogr. Sect. A. 2015, A71, 3–8. [Google Scholar]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010. [Google Scholar]
Sample Availability: Samples of the compounds are not available from the authors. |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Cat | t (Minute) | Conversion a (%) | Mn,calcd., b × 103 | Mn,obsd c × 103 | Mw/Mn |
|---|---|---|---|---|---|---|
| 1 | 1-Li | 2 | 99 | 11.3 | 32.7 | 1.70 |
| 2 | 2-Li | 2 | 94 | 10.7 | 7.4 | 2.69 |
| 3 | 2-Li2 | 6 | 98 | 11.2 | 28.4 | 2.97 |
| 4 | 3-Li3 | 1 | 98 | 11.2 | 16.0 | 2.89 |
| Entry | Cat | Conversion a (%) | Mn,calcd. b × 103 | Mn,obsd c × 103 | Mw/Mn | Pr d |
|---|---|---|---|---|---|---|
| 1 | 1-Li | 93 | 13.4 | 12.1 | 1.83 | 0.61 |
| 2 | 2-Li | 95 | 13.7 | 3.4 | 2.53 | 0.52 |
| 3 | 2-Li2 | 97 | 14.0 | 5.3 | 2.61 | 0.58 |
| 4 | 3-Li3 | 98 | 14.1 | 6.7 | 2.24 | 0.53 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rufino-Felipe, E.; Muñoz-Hernández, M.-Á.; Montiel-Palma, V. Lithium Complexes Derived of Benzylphosphines: Synthesis, Characterization and Evaluation in the ROP of rac-Lactide and ε-Caprolactone. Molecules 2018, 23, 82. https://doi.org/10.3390/molecules23010082
Rufino-Felipe E, Muñoz-Hernández M-Á, Montiel-Palma V. Lithium Complexes Derived of Benzylphosphines: Synthesis, Characterization and Evaluation in the ROP of rac-Lactide and ε-Caprolactone. Molecules. 2018; 23(1):82. https://doi.org/10.3390/molecules23010082
Chicago/Turabian StyleRufino-Felipe, Ernesto, Miguel-Ángel Muñoz-Hernández, and Virginia Montiel-Palma. 2018. "Lithium Complexes Derived of Benzylphosphines: Synthesis, Characterization and Evaluation in the ROP of rac-Lactide and ε-Caprolactone" Molecules 23, no. 1: 82. https://doi.org/10.3390/molecules23010082