Andrographolide Suppresses MV4-11 Cell Proliferation through the Inhibition of FLT3 Signaling, Fatty Acid Synthesis and Cellular Iron Uptake

 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
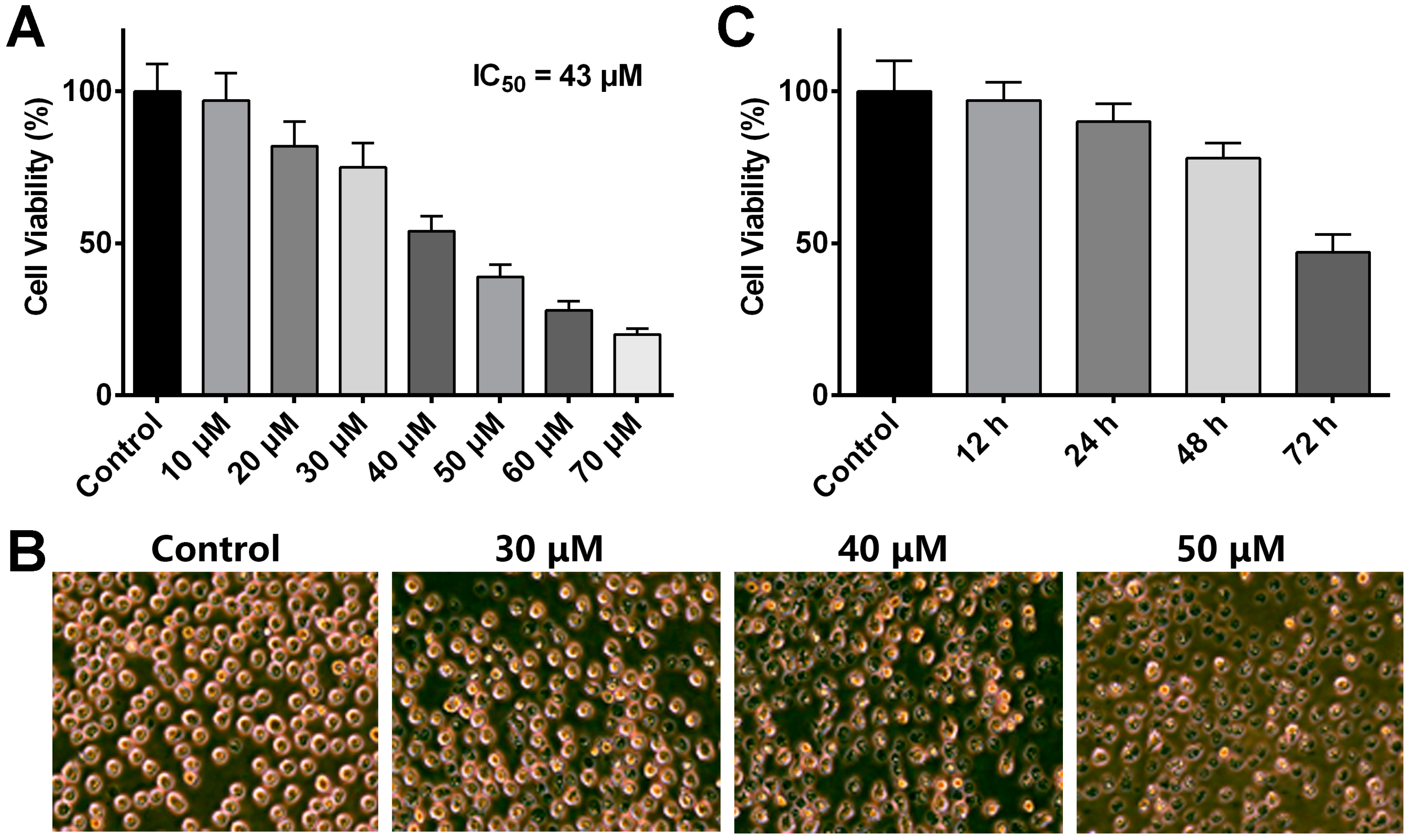
2.1. ADR Inhibits MV4-11 Cell Proliferation in a Dose- and Time-Dependent Manner
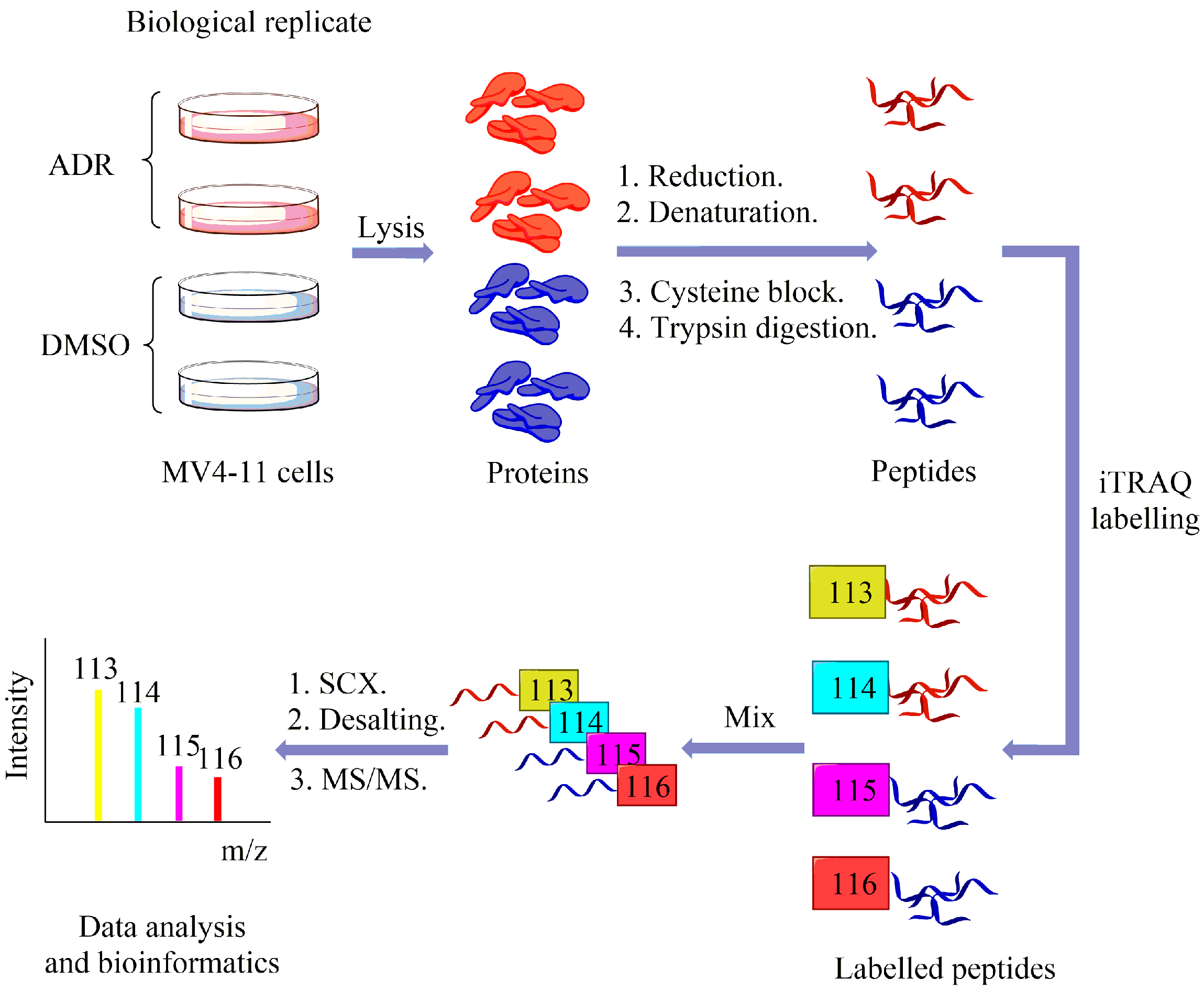
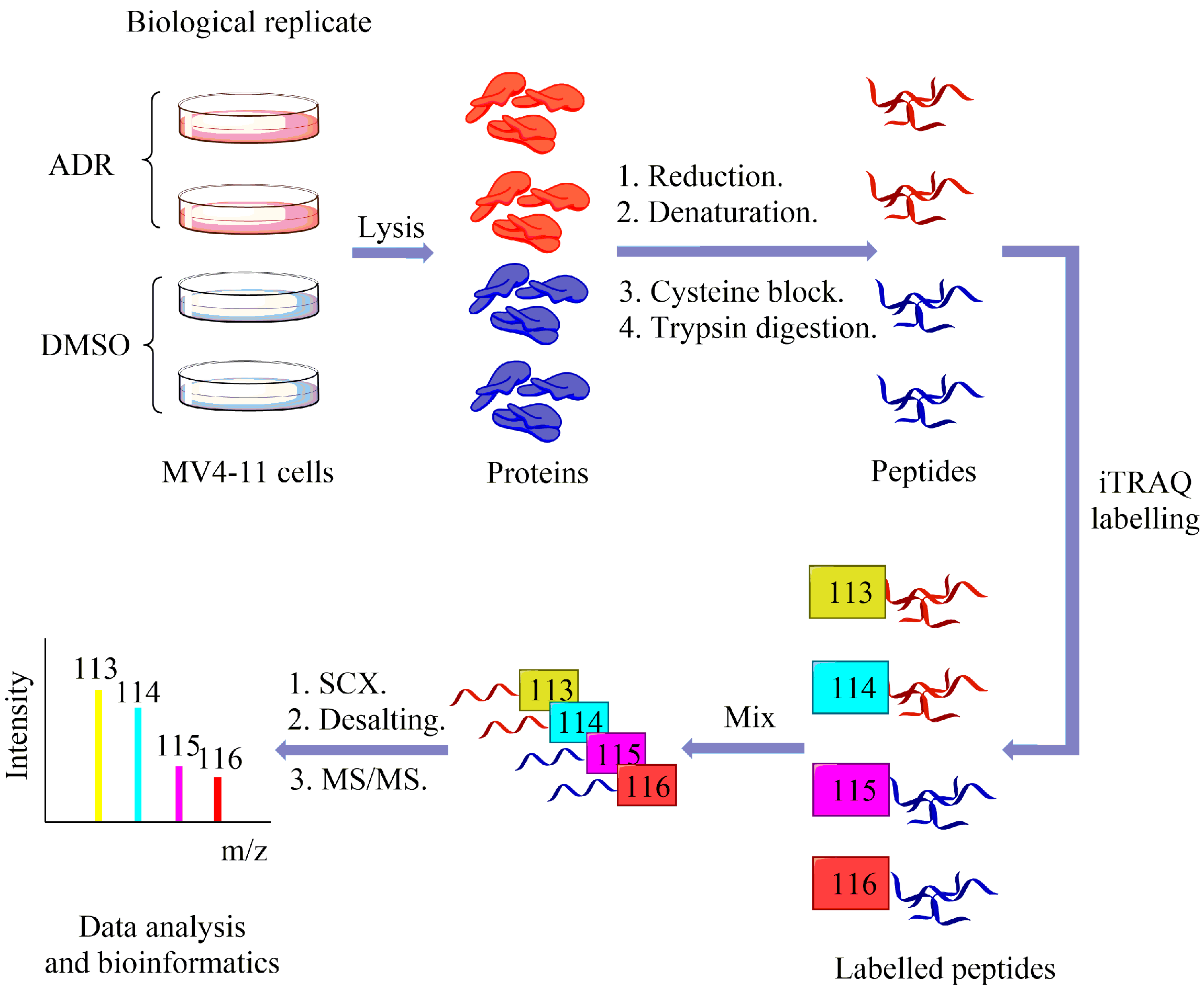
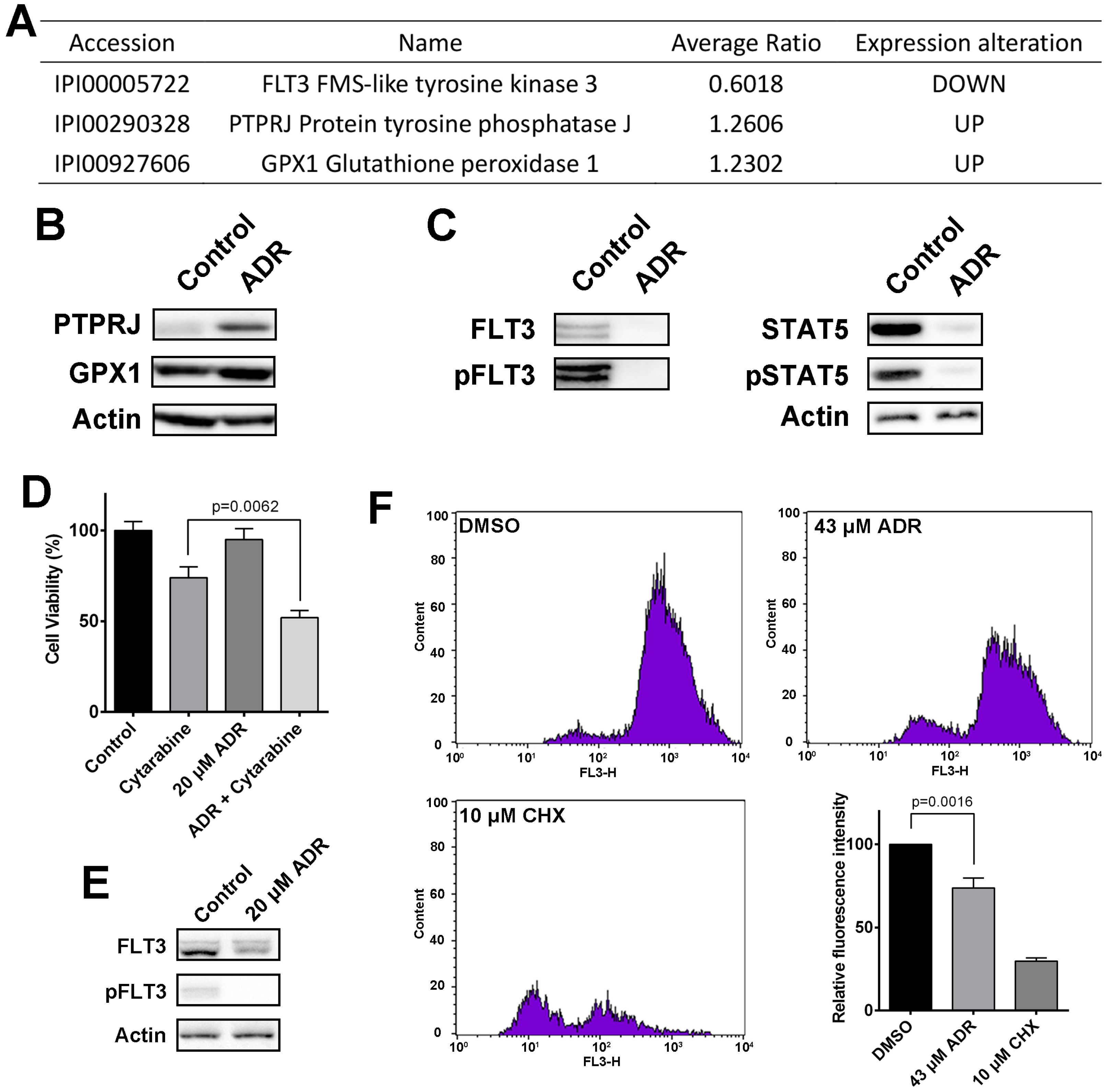
2.2. Using Quantitative Proteomics Approach to Identify Differentially Expressed Proteins in ADR-Treated MV4-11 Cells
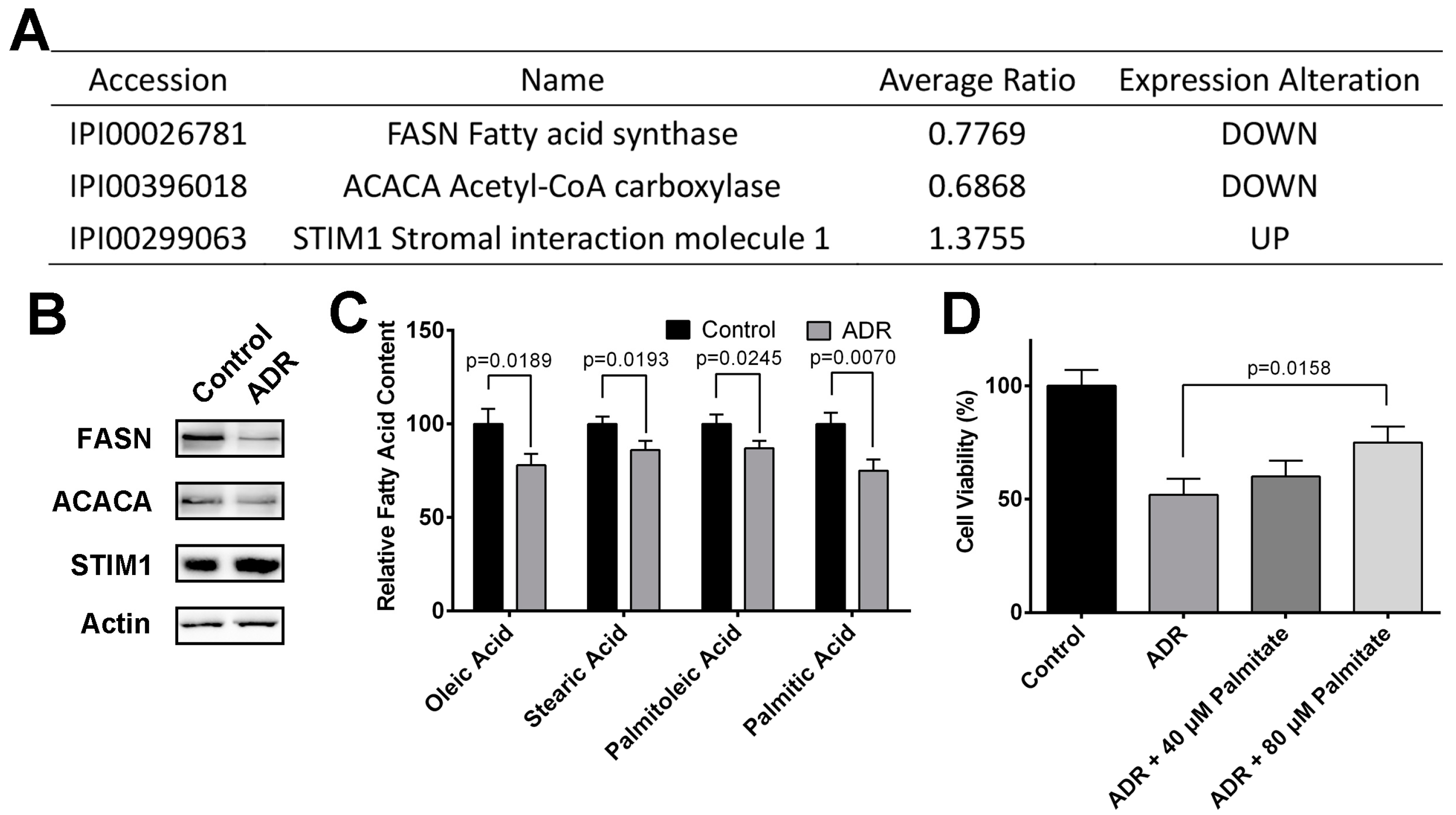
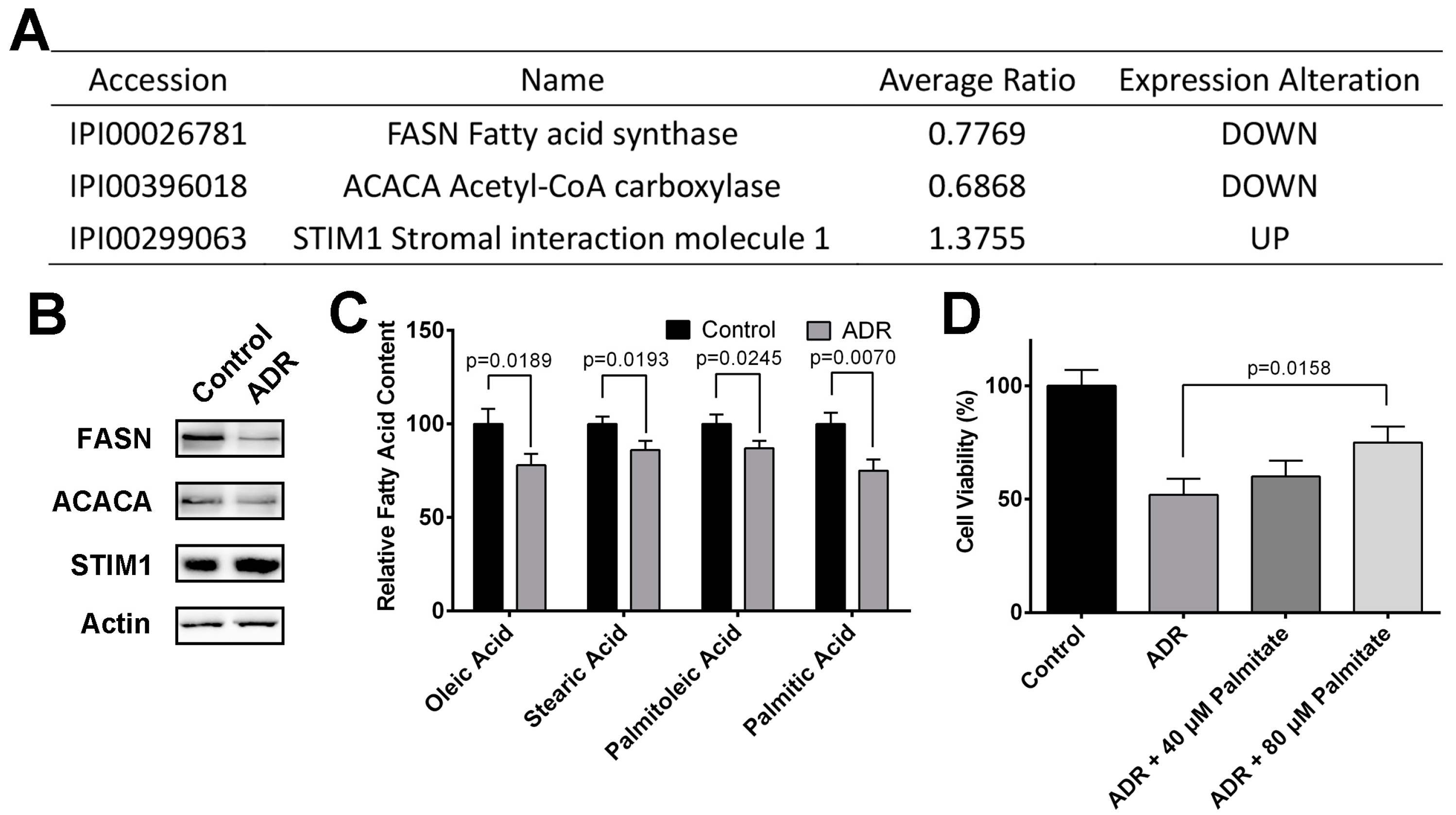
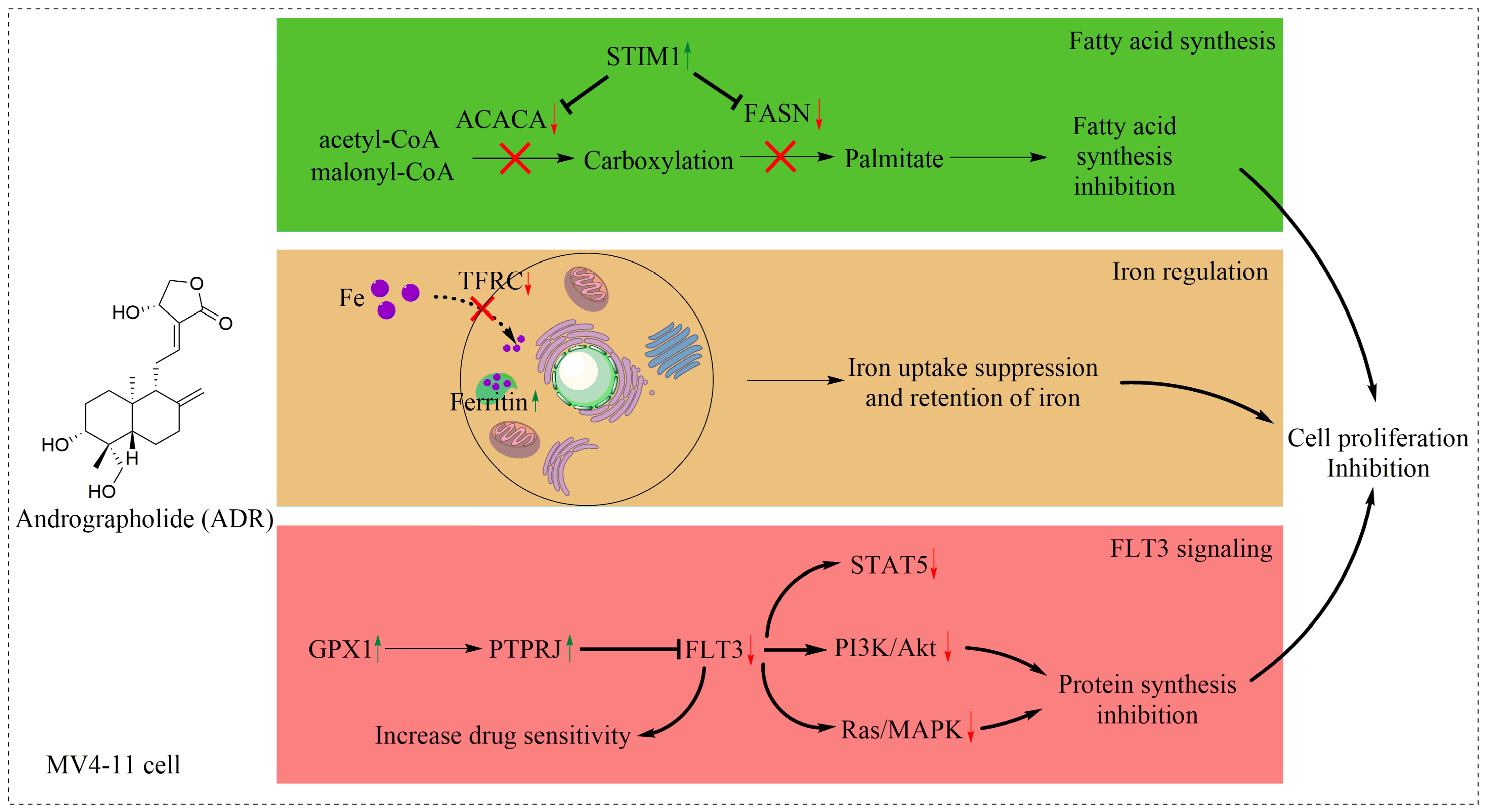
2.3. ADR Inhibits MV4-11 Proliferation Through Suppressing Fatty Acid Synthesis
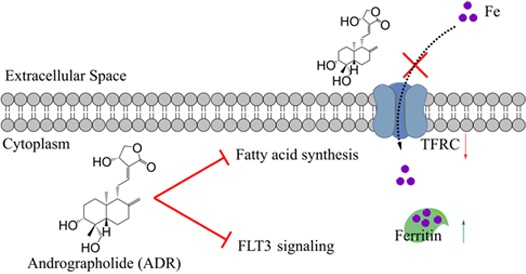
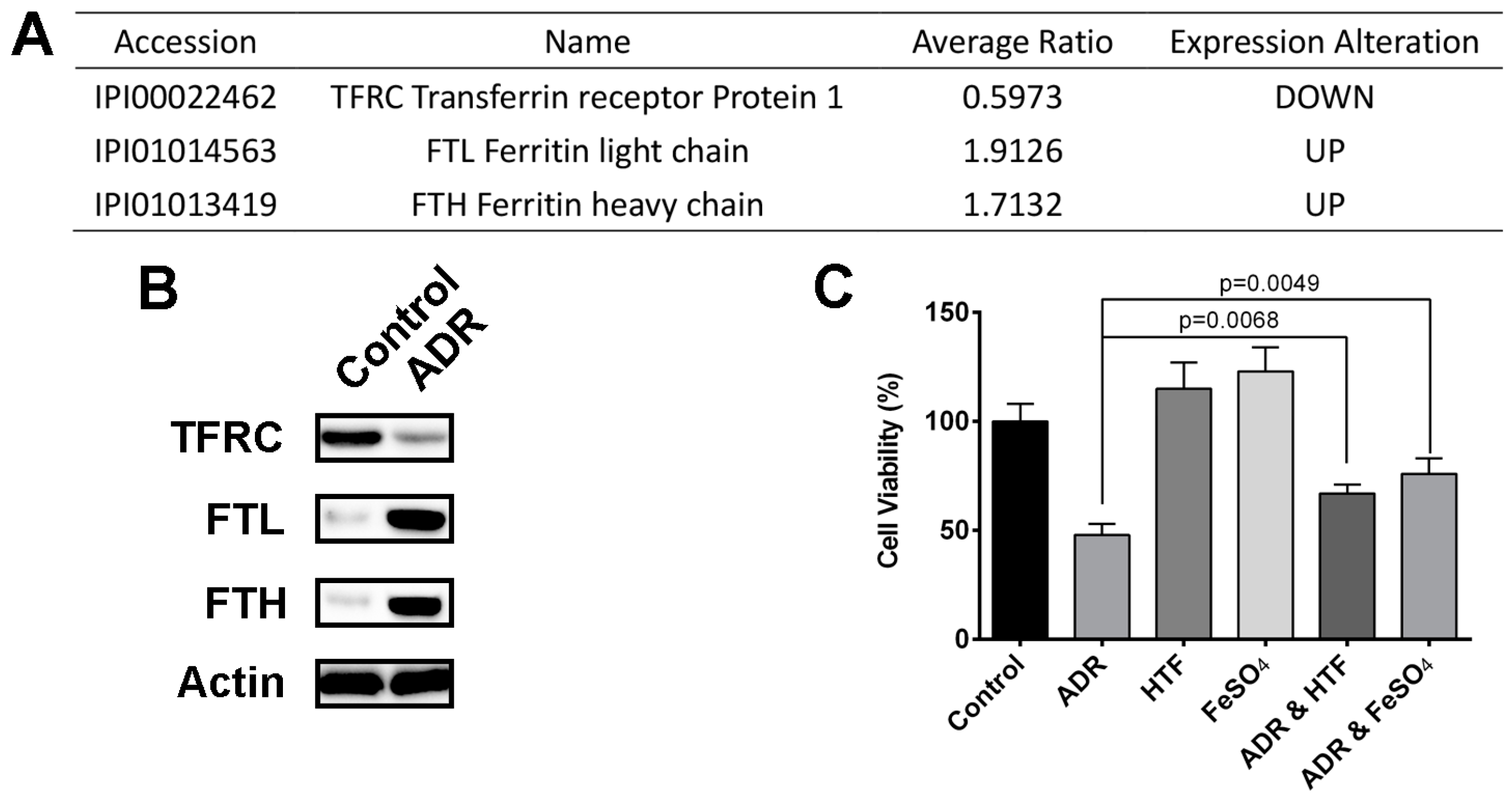
2.4. ADR Suppresses Iron Uptake and Induces Retention of Iron in Storage Repositories to INHIBIT MV4-11 Cells Proliferation
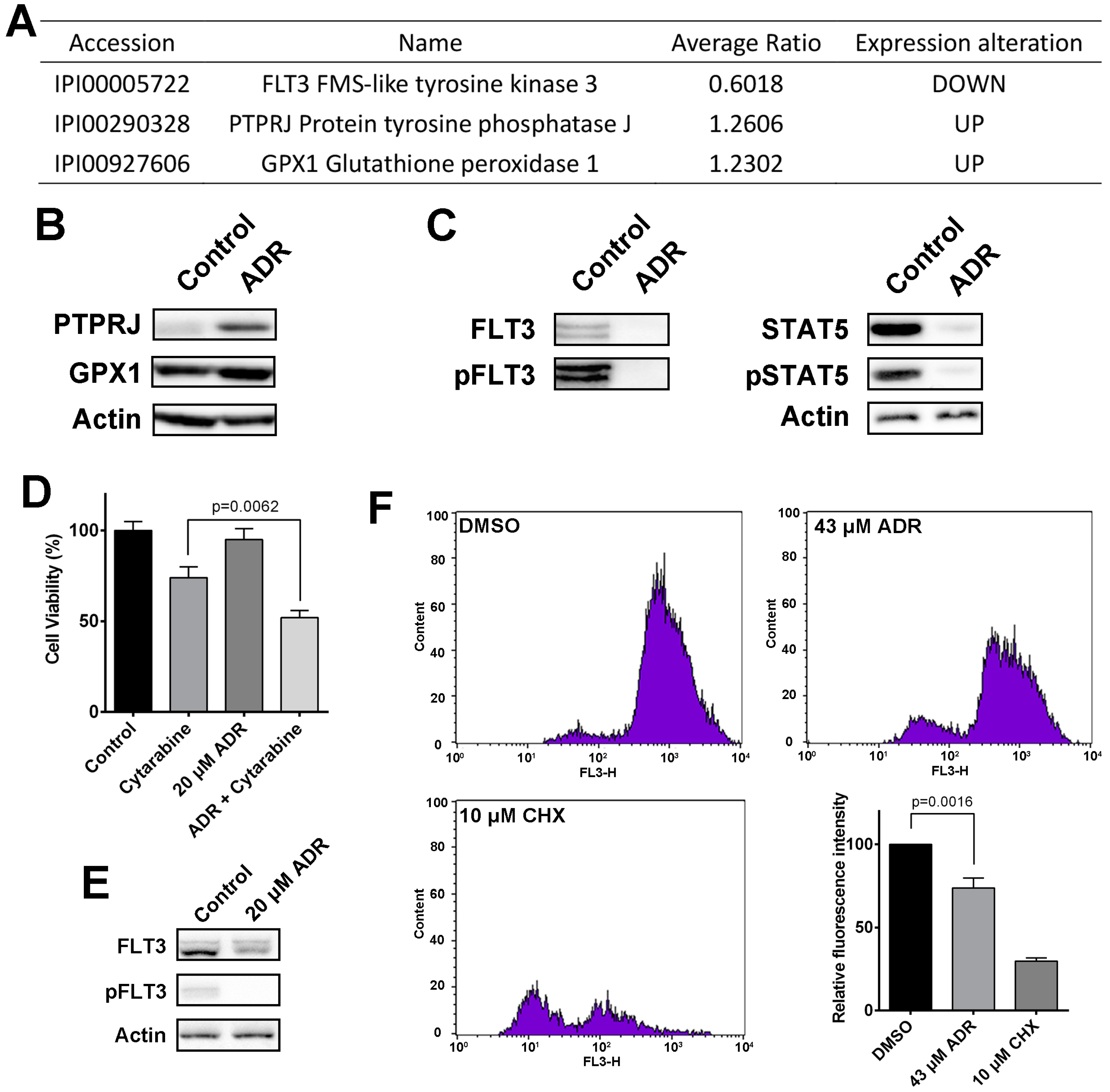
2.5. ADR Blocks FLT3 Signaling Pathway and Suppresses Protein Synthesis in MV4-11 Cells
2.6. ADR Inhibits Fatty Acid Synthesis, Cellular Iron Pool and FLT3 Signaling in NB4 Cells
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Antibodies
4.3. CCK-8 Cell Viability Assay
4.4. iTRAQ Labelling
4.5. 1D LC-MS/MS Analysis
4.6. Peptide and Protein Identification, Data Analysis
4.7. Western Blotting Assay
4.8. GC/MS Analysis for Fatty Acid Content
4.9. Nascent Protein Synthesis Quantification Using Click Chemistry
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Herrmann, H.; Blatt, K.; Shi, J.; Gleixner, K.V.; Cerny-Reiterer, S.; Müllauer, L.; Vakoc, C.R.; Sperr, W.R.; Horny, H.-P.; Bradner, J.E.; et al. Small-molecule inhibition of BRD4 as a new potent approach to eliminate leukemic stem-and progenitor cells in acute myeloid leukemia (AML). Oncotarget 2012, 3, 1588–1599. [Google Scholar] [CrossRef] [PubMed]
- Tenen, D.G. Abnormalities of the CEBP alpha transcription factor: A major target in acute myeloid leukemia. Leukemia 2001, 15, 688–689. [Google Scholar] [CrossRef] [PubMed]
- Boissel, N.; Leroy, H.; Brethon, B.; Philippe, N.; De Botton, S.; Auvrignon, A.; Raffoux, E.; Leblanc, T.; Thomas, X.; Hermine, O.; et al. Incidence and prognostic impact of c-Kit, FLT3, and Ras gene mutations in core binding factor acute myeloid leukemia (CBF-AML). Leukemia 2006, 20, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Tauro, S.; Craddock, C.; Peggs, K.; Begum, G.; Mahendra, P.; Cook, G.; Marsh, J.; Milligan, D.; Goldstone, A.; Hunter, A.; et al. Allogeneic stem-cell transplantation using a reduced-intensity conditioning regimen has the capacity to produce durable remissions and long-term disease-free survival in patients with high-risk acute myeloid leukemia and myelodysplasia. J. Clin. Oncol. 2005, 23, 9387–9393. [Google Scholar] [CrossRef] [PubMed]
- Zeng, Z.; Shi, Y.X.; Samudio, I.J.; Wang, R.-Y.; Ling, X.; Frolova, O.; Levis, M.; Rubin, J.B.; Negrin, R.R.; Estey, E.H.; et al. Targeting the leukemia microenvironment by CXCR4 inhibition overcomes resistance to kinase inhibitors and chemotherapy in AML. Blood 2009, 113, 6215–6224. [Google Scholar] [CrossRef] [PubMed]
- Van den Heuvel-Eibrink, M.; Wiemer, E.; Prins, A.; Meijerink, J.; Vossebeld, P.; VAN DER, H.; Pieters, R.; Sonneveld, P. Increased expression of the breast cancer resistance protein (BCRP) in relapsed or refractory acute myeloid leukemia (AML). Leukemia 2002, 16, 833–839. [Google Scholar] [CrossRef] [PubMed]
- Griffin, J.D. Point mutations in the FLT3 gene in AML. Blood 2001, 97, 2193. [Google Scholar] [CrossRef]
- Brandts, C.H.; Sargin, B.; Rode, M.; Biermann, C.; Lindtner, B.; Schwäble, J.; Buerger, H.; Müller-Tidow, C.; Choudhary, C.; McMahon, M.; et al. Constitutive activation of Akt by Flt3 internal tandem duplications is necessary for increased survival, proliferation, and myeloid transformation. Cancer Res. 2005, 65, 9643–9650. [Google Scholar] [CrossRef] [PubMed]
- Chapuis, N.; Tamburini, J.; Cornillet-Lefebvre, P.; Gillot, L.; Bardet, V.; Willems, L.; Park, S.; Green, A.S.; Ifrah, N.; Dreyfus, F.; et al. Autocrine IGF-1/IGF-1R signaling is responsible for constitutive PI3K/Akt activation in acute myeloid leukemia: Therapeutic value of neutralizing anti-IGF-1R antibody. Haematologica 2010, 95, 415–423. [Google Scholar] [CrossRef] [PubMed]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of FLT3-activating mutations in 979 patients with acute myelogenous leukemia: Association with FAB subtypes and identification of subgroups with poor prognosis. Blood 2002, 99, 4326–4335. [Google Scholar] [CrossRef] [PubMed]
- Shimada, A.; Taki, T.; Tabuchi, K.; Tawa, A.; Horibe, K.; Tsuchida, M.; Hanada, R.; Tsukimoto, I.; Hayashi, Y. KIT mutations, and not FLT3 internal tandem duplication, are strongly associated with a poor prognosis in pediatric acute myeloid leukemia with t (8; 21): A study of the Japanese Childhood AML Cooperative Study Group. Blood 2006, 107, 1806–1809. [Google Scholar] [CrossRef] [PubMed]
- O’Farrell, A.-M.; Abrams, T.J.; Yuen, H.A.; Ngai, T.J.; Louie, S.G.; Yee, K.W. H.; Wong, L.M.; Hong, W.; Lee, L.B.; Town, A.; et al. SU11248 is a novel FLT3 tyrosine kinase inhibitor with potent activity in vitro and in vivo. Blood 2003, 101, 3597–3605. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small-molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Greig, N.H.; Soncrant, T.T.; Shetty, H.U.; Momma, S.; Smith, Q.R.; Rapoport, S.I. Brain uptake and anticancer activities of vincristine and vinblastine are restricted by their low cerebrovascular permeability and binding to plasma constituents in rat. Cancer Chemother. Pharmacol. 1990, 26, 263–268. [Google Scholar] [CrossRef] [PubMed]
- Giri, A.; Narasu, M.L. Production of podophyllotoxin from Podophyllum hexandrum: A potential natural product for clinically useful anticancer drugs. Cytotechnology 2000, 34, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, Y.-H.; Liu, L.F. Identification of mammalian DNA topoisomerase I as an intracellular target of the anticancer drug camptothecin. Cancer Res. 1988, 48, 1722–1726. [Google Scholar] [PubMed]
- Zhang, C.J.; Wang, J.; Zhang, J.; Lee, Y.M.; Feng, G.; Lim, T.K.; Shen, H.M.; Lin, Q.; Liu, B. Mechanism-Guided Design and Synthesis of a Mitochondria-Targeting Artemisinin Analogue with Enhanced Anticancer Activity. Angew. Chem. Int. Ed. 2016, 55, 13770–13774. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, C.J.; Chia, W.N.; Loh, C.C.; Li, Z.; Lee, Y.M.; He, Y.; Yuan, L.X.; Lim, T.K.; Liu, M.; et al. Haem-activated promiscuous targeting of artemisinin in Plasmodium falciparum. Nat. Commun. 2015, 6, 10111. [Google Scholar] [CrossRef] [PubMed]
- Neto, C.C. Cranberry and its phytochemicals: A review of in vitro anticancer studies. J. Nutr. 2007, 137, 186S–193S. [Google Scholar] [PubMed]
- Neto, C.C.; Amoroso, J.W.; Liberty, A.M. Anticancer activities of cranberry phytochemicals: An update. Mol. Nutr. Food Res. 2008, 52, S18–S27. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.-F.; Ye, B.-Q.; Li, Y.-D.; Wang, J.-G.; He, X.-J.; Lin, X.; Yao, X.; Ma, D.; Slungaard, A.; Hebbel, R.P.; et al. Andrographolide attenuates inflammation by inhibition of NF-κB activation through covalent modification of reduced cysteine 62 of p50. J. Immunol. 2004, 173, 4207–4217. [Google Scholar] [CrossRef] [PubMed]
- Shen, Y.-C.; Chen, C.-F.; Chiou, W.-F. Andrographolide prevents oxygen radical production by human neutrophils: Possible mechanism (s) involved in its anti-inflammatory effect. Br. J. Pharmacol. 2002, 135, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Calabrese, C.; Berman, S.H.; Babish, J.G.; Ma, X.; Shinto, L.; Dorr, M.; Wells, K.; Wenner, C.A.; Standish, L.J.; et al. A phase I trial of andrographolide in HIV positive patients and normal volunteers. Phyther. Res. 2000, 14, 333–338. [Google Scholar] [CrossRef]
- Handa, S.S.; Sharma, A. Hepatoprotective activity of andrographolide from Andrographis paniculata against carbontetrachloride. Indian J. Med. Res. 1990, 92, 276–283. [Google Scholar] [PubMed]
- Wang, J.; Tan, X.F.; Nguyen, V.S.; Yang, P.; Zhou, J.; Gao, M.; Li, Z.; Lim, T.K.; He, Y.; Ong, C.S.; et al. A quantitative chemical proteomics approach to profile the specific cellular targets of andrographolide, a promising anticancer agent that suppresses tumor metastasis. Mol. Cell. Proteomics 2014, 13, 876–886. [Google Scholar] [CrossRef] [PubMed]
- Jada, S.R.; Subur, G.S.; Matthews, C.; Hamzah, A.S.; Lajis, N.H.; Saad, M.S.; Stevens, M.F.G.; Stanslas, J. Semisynthesis and in vitro anticancer activities of andrographolide analogues. Phytochemistry 2007, 68, 904–912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harjotaruno, S.; Widyawaruyanti, A.; Sismindari, S.; Zaini, N.C. Apoptosis inducing effect of andrographolide on TF-47 human breast cancer cell line. Afr. J. Tradit. Complem. 2007, 4, 345–351. [Google Scholar] [CrossRef]
- Cheung, H.-Y.; Cheung, S.-H.; Li, J.; Cheung, C.-S.; Lai, W.-P.; Fong, W.-F.; Leung, F.-M. Andrographolide isolated from Andrographis paniculata induces cell cycle arrest and mitochondrial-mediated apoptosis in human leukemic HL-60 cells. Planta Med. 2005, 71, 1106–1111. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Cheung, H.-Y.; Zhang, Z.; Chan, G.K.L.; Fong, W.-F. Andrographolide induces cell cycle arrest at G2/M phase and cell death in HepG2 cells via alteration of reactive oxygen species. Eur. J. Pharmacol. 2007, 568, 31–44. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-H.; Hsiao, G.; Lee, A.-R.; Wu, C.-C.; Yen, M.-H. Andrographolide suppresses endothelial cell apoptosis via activation of phosphatidyl inositol-3-kinase/Akt pathway. Biochem. Pharmacol. 2004, 67, 1337–1345. [Google Scholar] [CrossRef] [PubMed]
- Michelsen, K.S.; Wong, M.H.; Ko, B.; Thomas, L.S.; Dhall, D.; Targan, S.R. HMPL-004 (Andrographis paniculata extract) prevents development of murine colitis by inhibiting T cell proliferation and TH1/TH17 responses. Inflamm. Bowel Dis. 2013, 19, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Shen, K.; Ji, L.; Lu, B.; Xu, C.; Gong, C.; Morahan, G.; Wang, Z. Andrographolide inhibits tumor angiogenesis via blocking VEGFA/VEGFR2-MAPKs signaling cascade. Chem. Biol. Interact. 2014, 218, 99–106. [Google Scholar] [CrossRef] [PubMed]
- Han, Y.; Bu, L.M.; Ji, X.; Liu, C.Y.; Wang, Z.H. Modulation of multidrug resistance by andrographolid in a HCT-8/5-FU multidrug-resistant colorectal cancer cell line. J. Dig. Dis. 2005, 6, 82–86. [Google Scholar] [CrossRef] [PubMed]
- Quentmeier, H.; Reinhardt, J.; Zaborski, M.; Drexler, H.G. FLT3 mutations in acute myeloid leukemia cell lines. Leukemia 2003, 17, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.R.; Schulze, A. Lipid metabolism in cancer. FEBS J. 2012, 279, 2610–2623. [Google Scholar] [CrossRef] [PubMed]
- Zhang, F.; Du, G. Dysregulated lipid metabolism in cancer. World J. Biol. Chem. 2012, 3, 167–174. [Google Scholar] [CrossRef] [PubMed]
- Lupu, R.; Menendez, J.A. Pharmacological inhibitors of fatty acid synthase (FASN)-catalyzed endogenous fatty acid biogenesis: A new family of anti-cancer agents? Curr. Pharm. Biotechnol. 2006, 7, 483–494. [Google Scholar] [CrossRef] [PubMed]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Lang, F.; Eylenstein, A.; Shumilina, E. Regulation of Orai1/STIM1 by the kinases SGK1 and AMPK. Cell Calcium 2012, 52, 347–354. [Google Scholar] [CrossRef] [PubMed]
- Lovley, D.R.; Phillips, E.J.P. Novel mode of Microbial energy metabolism: Organic carbon oxidation coupled to dissimilatory reduction of iron or manganese. Appl. Environ. Microbiol. 1988, 54, 1472–1480. [Google Scholar] [PubMed]
- Richardson, D.R. Molecular mechanisms of iron uptake by cells and the use of iron chelators for the treatment of cancer. Curr. Med. Chem. 2005, 12, 2711–2729. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Shawron, K.M.; Rendina, E.; Peterson, S.K.; Lucas, E.A.; Smith, B.J.; Clarke, S.L. Hypoxia inducible factor-2α is translationally repressed in response to dietary iron deficiency in sprague-dawley rats. J. Nutr. 2011, 141, 1590–1596. [Google Scholar] [CrossRef] [PubMed]
- Jayavelu, A.K.; Moloney, J.N.; Böhmer, F.-D.; Cotter, T.G. NOX-driven ROS formation in cell transformation of FLT3-ITD-positive AML. Exp. Hematol. 2016, 44, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Di Simplicio, P.; Rossi, R.; Falcinelli, S.; Ceserani, R.; Formento, M.L. Antioxidant status in various tissues of the mouse after fasting and swimming stress. Eur. J. Appl. Physiol. Occup. Physiol. 1997, 76, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Levis, M.; Pham, R.; Smith, B.D.; Small, D. In vitro studies of a FLT3 inhibitor combined with chemotherapy: Sequence of administration is important to achieve synergistic cytotoxic effects. Blood 2004, 104, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Bibollet-Bahena, O.; Almazan, G. IGF-1-stimulated protein synthesis in oligodendrocyte progenitors requires PI3K/mTOR/Akt and MEK/ERK pathways. J. Neurochem. 2009, 109, 1440–1451. [Google Scholar] [CrossRef] [PubMed]
- Dreyer, H.C.; Fujita, S.; Cadenas, J.G.; Chinkes, D.L.; Volpi, E.; Rasmussen, B.B. Resistance exercise increases AMPK activity and reduces 4E-BP1 phosphorylation and protein synthesis in human skeletal muscle. J. Physiol. 2006, 576, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Lee, Y.M.; Ng, S.; Shi, Y.; Hua, Z.-C.; Lin, Q.; Shen, H.-M. Nonradioactive quantification of autophagic protein degradation with L-azidohomoalanine labeling. Nat. Protoc. 2017, 12, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Lee, Y.-M.; Koh, P.-L.; Ng, S.; Bao, F.; Lin, Q.; Shen, H.-M. Quantitative chemical proteomics profiling of de novo protein synthesis during starvation-mediated autophagy. Autophagy 2016, 12, 1931–1944. [Google Scholar] [CrossRef] [PubMed]
- Lanotte, M.; Martin-Thouvenin, V.; Najman, S.; Balerini, P.; Valensi, F.; Berger, R. NB4, a maturation inducible cell line with t (15; 17) marker isolated from a human acute promyelocytic leukemia (M3). Blood 1991, 77, 1080–1086. [Google Scholar] [PubMed]
- Gordaliza, M. Natural products as leads to anticancer drugs. Clin. Transl. Oncol. 2007, 9, 767–776. [Google Scholar] [CrossRef] [PubMed]
- Da Rocha, A.B.; Lopes, R.M.; Schwartsmann, G. Natural products in anticancer therapy. Curr. Opin. Pharmacol. 2001, 1, 364–369. [Google Scholar] [CrossRef]
- Rajagopal, S.; Kumar, R.A.; Deevi, D.S.; Satyanarayana, C.; Rajagopalan, R. Andrographolide, a potential cancer therapeutic agent isolated from Andrographis paniculata. J. Exp. Ther. Oncol. 2003, 3, 147–158. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.C.W.; Chan, T.K.; Ng, D.S.W.; Sagineedu, S.R.; Stanslas, J.; Wong, W.S. Andrographolide and its analogues: Versatile bioactive molecules for combating inflammation and cancer. Clin. Exp. Pharmacol. Physiol. 2012, 39, 300–310. [Google Scholar] [CrossRef] [PubMed]
- Abu-Ghefreh, A.A.; Canatan, H.; Ezeamuzie, C.I. In vitro and in vivo anti-inflammatory effects of andrographolide. Int. Immunopharmacol. 2009, 9, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Chandrasekaran, C.V.; Gupta, A.; Agarwal, A. Effect of an extract of Andrographis paniculata leaves on inflammatory and allergic mediators in vitro. J. Ethnopharmacol. 2010, 129, 203–207. [Google Scholar] [CrossRef] [PubMed]
- Kumar, R.A.; Sridevi, K.; Kumar, N.V.; Nanduri, S.; Rajagopal, S. Anticancer and immunostimulatory compounds from Andrographis paniculata. J. Ethnopharmacol. 2004, 92, 291–295. [Google Scholar] [CrossRef] [PubMed]
- Wen, L.; Xia, N.; Chen, X.; Li, Y.; Hong, Y.; Liu, Y.; Wang, Z. Activity of antibacterial, antiviral, anti-inflammatory in compounds andrographolide salt. Eur. J. Pharmacol. 2014, 740, 421–427. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.Y.; Tan, B.K.H. Hypotensive activity of aqueous extract of Andrographis paniculata in rats. Clin. Exp. Pharmacol. Physiol. 1996, 23, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Jada, S.R.; Matthews, C.; Saad, M.S.; Hamzah, A.S.; Lajis, N.H.; Stevens, M.F.G.; Stanslas, J. Benzylidene derivatives of andrographolide inhibit growth of breast and colon cancer cells in vitro by inducing G1 arrest and apoptosis. Br. J. Pharmacol. 2008, 155, 641–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lim, J.C.W.; Jeyaraj, E.J.; Sagineedu, S.R.; Wong, W.S.F.; Stanslas, J. SRS06, a new semisynthetic andrographolide derivative with improved anticancer potency and selectivity, inhibits nuclear factor-κB nuclear binding in the A549 non-small cell lung cancer cell line. Pharmacology 2015, 95, 70–77. [Google Scholar] [CrossRef] [PubMed]
- Kim, T.G.; Hwi, K.K.; Hung, C.S. Morphological and biochemical changes of andrographolide-induced cell death in human prostatic adenocarcinoma PC-3 cells. In Vivo (Brooklyn) 2005, 19, 551–557. [Google Scholar]
- Song, Y.; Xin, Z.; Wan, Y.; Li, J.; Ye, B.; Xue, X. Synthesis and anticancer activity of some novel indolo[3,2-b]andrographolide derivatives as apoptosis-inducing agents. Eur. J. Med. Chem. 2015, 90, 695–706. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-G.; Li, J.-B.; Liu, F.-R.; Wu, T.; Yu, M.; Xu, H.-M. Andrographolide inhibits the adhesion of gastric cancer cells to endothelial cells by blocking E-selectin expression. Anticancer Res. 2007, 27, 2439–2447. [Google Scholar] [PubMed]
- Su, Y.; Ge, J.; Zhu, B.; Zheng, Y.; Zhu, Q.; Yao, S.Q. Target identification of biologically active small molecules via in situ methods. Curr. Opin. Chem. Biol. 2013, 17, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Wu, M.-L.; Doerksen, R.J.; Ho, C.-T.; Huang, T.-C. Andrographolide induces apoptosis via down-regulation of glyoxalase 1 and HMG-CoA reductase in HL-60 cells. J. Funct. Foods 2015, 14, 226–235. [Google Scholar] [CrossRef]
- Swinnen, J.V.; Brusselmans, K.; Verhoeven, G. Increased lipogenesis in cancer cells: New players, novel targets. Curr. Opin. Clin. Nutr. Metab. Care 2006, 9, 358–365. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Shi, Y.; Xu, C.; Zhang, C.; Wong, Y.K.; Lee, Y.M.; Krishna, S.; He, Y.; Lim, T.K.; et al. Mechanistic investigation of the specific anticancer property of artemisinin and its combination with aminolevulinic acid for enhanced anticolorectal cancer activity. ACS Cent. Sci. 2017, 3, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Brusselmans, K.; Vrolix, R.; Verhoeven, G.; Swinnen, J.V. Induction of cancer cell apoptosis by flavonoids is associated with their ability to inhibit fatty acid synthase activity. J. Biol. Chem. 2005, 280, 5636–5645. [Google Scholar] [CrossRef] [PubMed]
- Shariff, A.; Manna, P.K.; Paranjothy, K.L.K.; Manjula, M. Entrapment of andrographolide in cross-linked aliginate pellets: II. physicochemical characterization to study the pelletization of andrographolide. Pak. J. Pharm. Sci. 2007, 20, 9–15. [Google Scholar] [PubMed]
- Chen, C.-C.; Chuang, W.-T.; Lin, A.-H.; Tsai, C.-W.; Huang, C.-S.; Chen, Y.-T.; Chen, H.-W.; Lii, C.-K. Andrographolide inhibits adipogenesis of 3T3-L1 cells by suppressing C/EBPβ expression and activation. Toxicol. Appl. Pharmacol. 2016, 307, 115–122. [Google Scholar] [CrossRef] [PubMed]
- De Schrijver, E.; Brusselmans, K.; Heyns, W.; Verhoeven, G.; Swinnen, J.V. RNA interference-mediated silencing of the fatty acid synthase gene attenuates growth and induces morphological changes and apoptosis of LNCaP prostate cancer cells. Cancer Res. 2003, 63, 3799–3804. [Google Scholar] [PubMed]
- Pardee, T.; DeFord-Watts, L.M.; Peronto, E.; Levitan, D.A.; Hurd, D.D.; Kridel, S. Altered lipid and mitochondrial metabolism are viable targets in acute leukemia. Blood 2011, 118, 3618. [Google Scholar]
- Seligman, P.A.; Kovar, J.; Gelfand, E.W. Lymphocyte proliferation is controlled by both iron availability and regulation of iron uptake pathways. Pathobiology 1992, 60, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Graziadei, I.; Gaggl, S.; Kaserbacher, R.; Braunsteiner, H.; Vogel, W. The acute-phase protein α 1-antitrypsin inhibits growth and proliferation of human early erythroid progenitor cells (burst-forming units-erythroid) and of human erythroleukemic cells (K562) in vitro by interfering with transferrin iron uptake. Blood 1994, 83, 260–268. [Google Scholar] [PubMed]
- Yang, D.C.; Jiang, X.P.; Elliott, R.L.; Head, J.F. Inhibition of growth of human breast carcinoma cells by an antisense oligonucleotide targeted to the transferrin receptor gene. Anticancer Res. 2001, 21, 1777–1787. [Google Scholar] [PubMed]
- Wu, K.-J.; Polack, A.; Dalla-Favera, R. Coordinated regulation of iron-controlling genes, H-ferritin and IRP2, by c-MYC. Science 1999, 283, 676–679. [Google Scholar] [CrossRef] [PubMed]
- Thweatt, R.; Murano, S.; Fleischmann, R.D.; Goldstein, S. Isolation and characterization of gene sequences overexpressed in Werner syndrome fibroblasts during premature replicative senescence. Exp. Gerontol. 1992, 27, 433–440. [Google Scholar] [CrossRef]
- Zwaan, C.M.; Meshinchi, S.; Radich, J.P.; Veerman, A.J.P.; Huismans, D.R.; Munske, L.; Podleschny, M.; Hählen, K.; Pieters, R.; Zimmermann, M.; et al. FLT3 internal tandem duplication in 234 children with acute myeloid leukemia: Prognostic significance and relation to cellular drug resistance. Blood 2003, 102, 2387–2394. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Sattler, M.; Ray, A.; Griffin, J.D. Drug resistance in mutant FLT3-positive AML. Oncogene 2010, 29, 5120. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, E.; Barrett, R.; Liu, Q.; Stone, R.; Gray, N.; Griffin, J.D. FLT3 inhibition and mechanisms of drug resistance in mutant FLT3-positive AML. Drug Resist. Updat. 2009, 12, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Arora, D.; Stopp, S.; Böhmer, S.-A.; Schons, J.; Godfrey, R.; Masson, K.; Razumovskaya, E.; Rönnstrand, L.; Tänzer, S.; Bauer, R.; et al. Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J. Biol. Chem. 2011, 286, 10918–10929. [Google Scholar] [CrossRef] [PubMed]
- Godfrey, R.; Arora, D.; Bauer, R.; Stopp, S.; Müller, J.P.; Heinrich, T.; Böhmer, S.-A.; Dagnell, M.; Schnetzke, U.; Scholl, S.; et al. Cell transformation by FLT3 ITD in acute myeloid leukemia involves oxidative inactivation of the tumor suppressor protein-tyrosine phosphatase DEP-1/PTPRJ. Blood 2012, 119, 4499–4511. [Google Scholar] [CrossRef] [PubMed]
- Roux, P.P.; Blenis, J. ERK and p38 MAPK-activated protein kinases: A family of protein kinases with diverse biological functions. Microbiol. Mol. Biol. Rev. 2004, 68, 320–344. [Google Scholar] [CrossRef] [PubMed]
- Taylor, W.E.; Bhasin, S.; Artaza, J.; Byhower, F.; Azam, M.; Willard, D.H.; Kull, F.C.; Gonzalez-Cadavid, N. Myostatin inhibits cell proliferation and protein synthesis in C 2 C 12 muscle cells. Am. J. Physiol. Metab. 2001, 280, E221–E228. [Google Scholar]
- Wang, J.; Zhang, J.; Zhang, C.-J.; Wong, Y.K.; Lim, T.K.; Hua, Z.-C.; Liu, B.; Tannenbaum, S.R.; Shen, H.-M.; Lin, Q. In situ proteomic profiling of curcumin targets in HCT116 colon cancer cell line. Sci. Rep. 2016, 6, 22146. [Google Scholar] [CrossRef] [PubMed]
- Martin, S.J.; Lennon, S.V.; Bonham, A.M.; Cotter, T.G. Induction of apoptosis (programmed cell death) in human leukemic HL-60 cells by inhibition of RNA or protein synthesis. J. Immunol. 1990, 145, 1859–1867. [Google Scholar] [PubMed]
- Kang, J.X.; Wang, J. A simplified method for analysis of polyunsaturated fatty acids. BMC Biochem. 2005, 6, 1–4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bligh, E.G.; Dyer, W.J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 1959, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds used in this study are not available from the authors. |


© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Zhang, J.; Yuan, L.; Lay, Y.; Wong, Y.K.; Lim, T.K.; Ong, C.S.; Lin, Q.; Wang, J.; Hua, Z. Andrographolide Suppresses MV4-11 Cell Proliferation through the Inhibition of FLT3 Signaling, Fatty Acid Synthesis and Cellular Iron Uptake. Molecules 2017, 22, 1444. https://doi.org/10.3390/molecules22091444
Chen X, Zhang J, Yuan L, Lay Y, Wong YK, Lim TK, Ong CS, Lin Q, Wang J, Hua Z. Andrographolide Suppresses MV4-11 Cell Proliferation through the Inhibition of FLT3 Signaling, Fatty Acid Synthesis and Cellular Iron Uptake. Molecules. 2017; 22(9):1444. https://doi.org/10.3390/molecules22091444
Chicago/Turabian StyleChen, Xiao, Jianbin Zhang, Lixia Yuan, Yifei Lay, Yin Kwan Wong, Teck Kwang Lim, Chye Sun Ong, Qingsong Lin, Jigang Wang, and Zichun Hua. 2017. "Andrographolide Suppresses MV4-11 Cell Proliferation through the Inhibition of FLT3 Signaling, Fatty Acid Synthesis and Cellular Iron Uptake" Molecules 22, no. 9: 1444. https://doi.org/10.3390/molecules22091444