
Fluoroalkyl Amino Reagents (FARs): A General Approach towards the Synthesis of Heterocyclic Compounds Bearing Emergent Fluorinated Substituents


Abstract
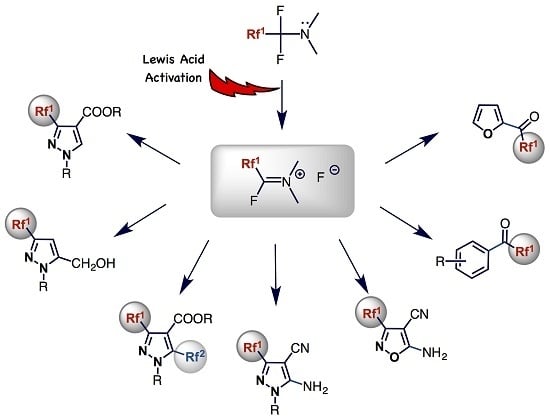
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
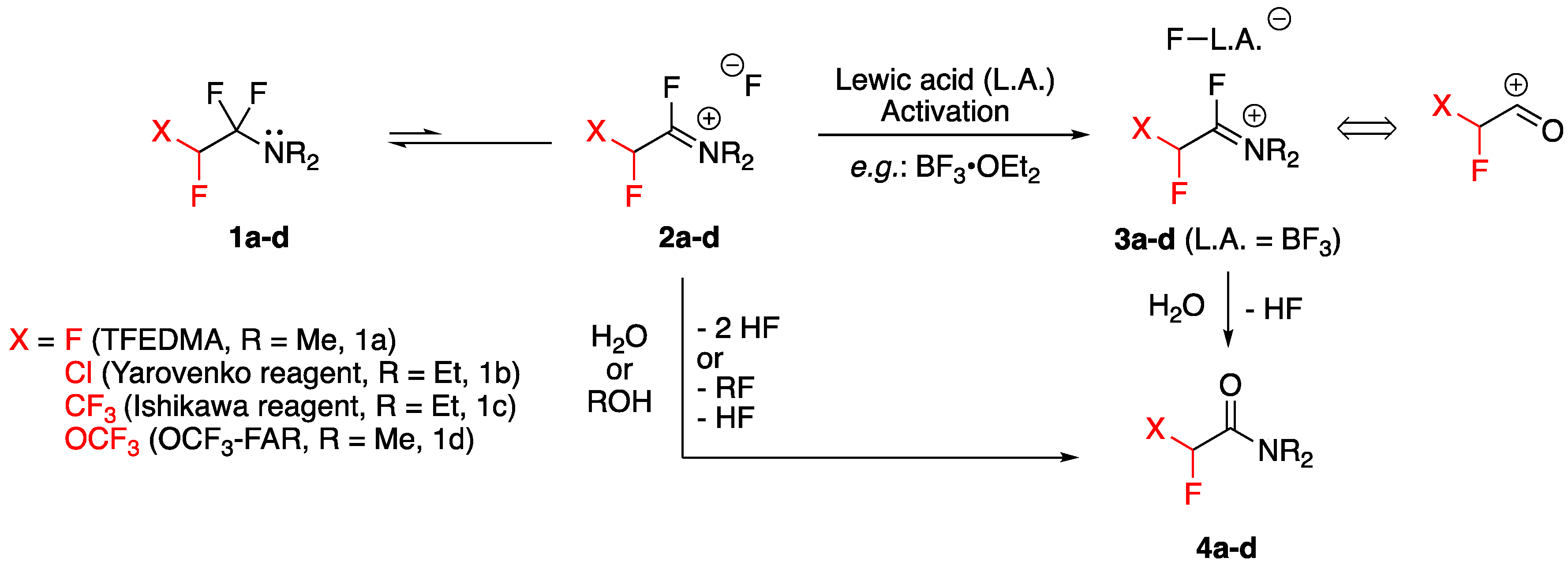
2. Preparation and Properties of Fluoroalkyl Amino Reagents
2.1. Preparation and Availability
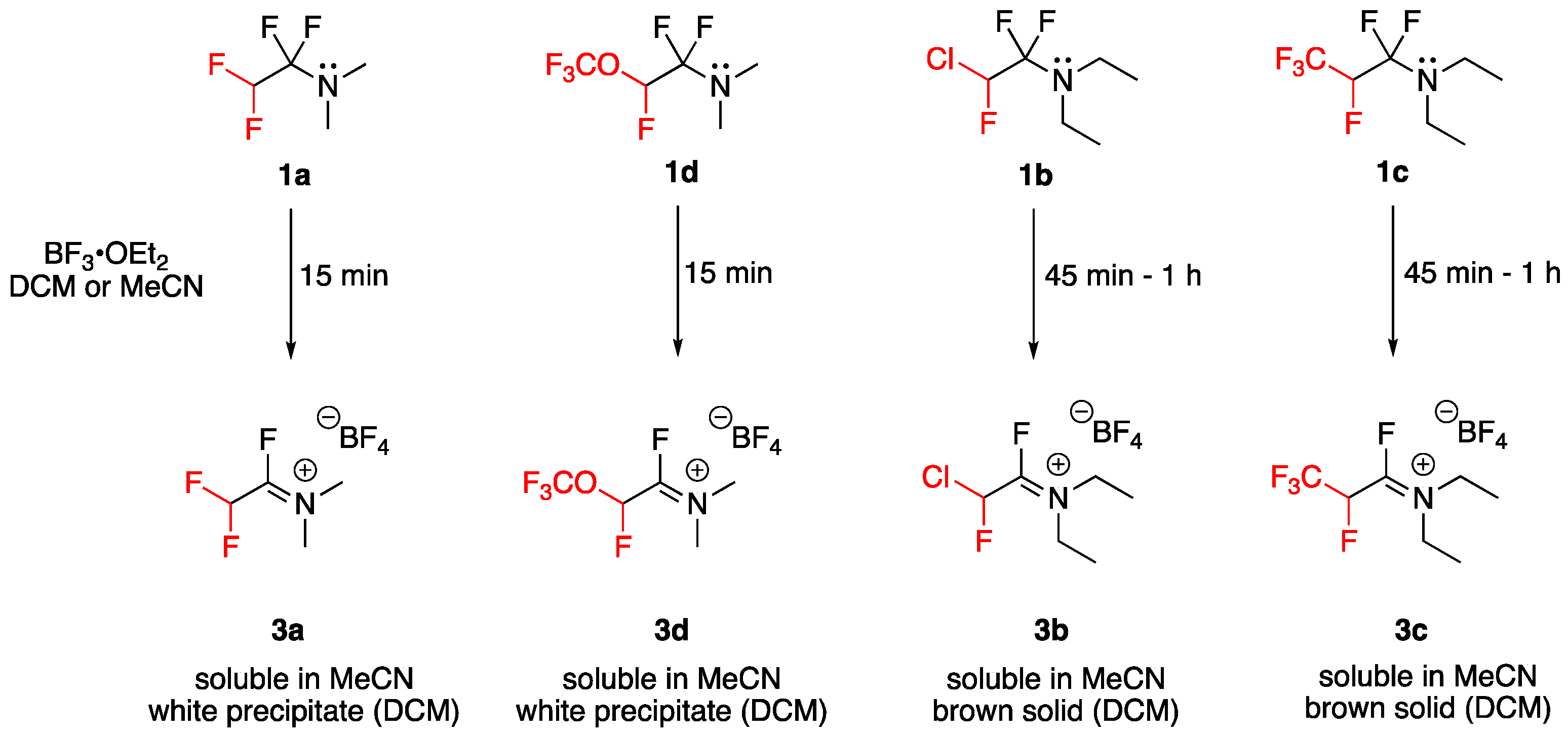
2.2. Lewis Acid Activation of Fluoroalkyl Amino Reagents
3. Fluoroalkyl Amino Reagents: Efficient Tools for Fluorination and for the Transfer of Fluoroalkyl Groups
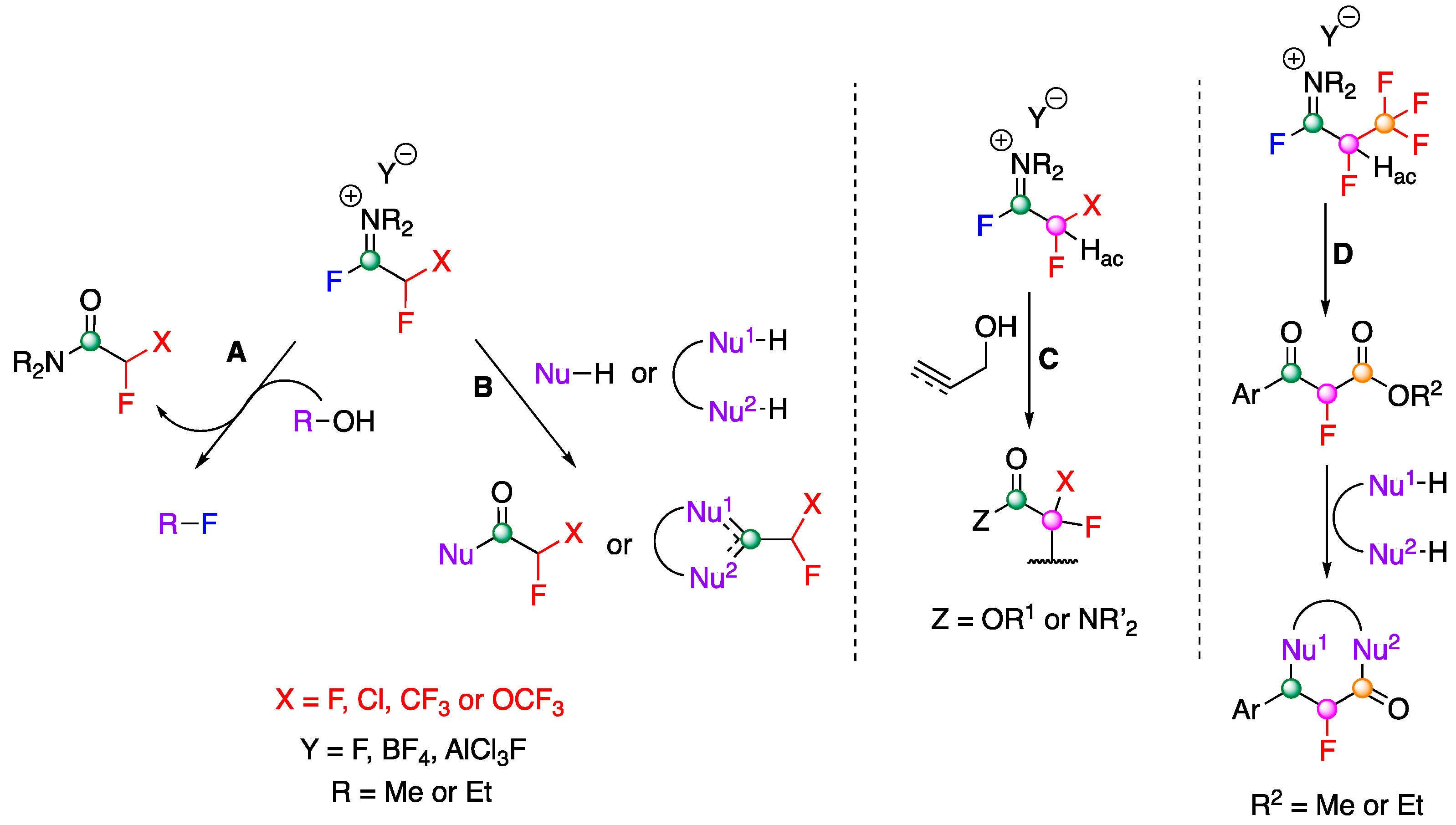
3.1. General Reactivity Modes of FARs
- (A)
- No carbon of the FAR is incorporated in the desired product of the reaction. The FAR acts as an activator of hydroxyl groups, leading to their replacement by fluorine (with release of the hydrolysed FAR as a fluorinated acetamide) or another intramolecular nucleophile as in an example of Beckmann rearrangement. Aldehydes can also be deoxofluorinated. (Section 3.2).
- (B)
- All carbons of the FAR are present in the desired product of the reaction but only one, the carbon of the iminium, undergoes transformations via one or two nucleophilic attack(s). This reactivity mode concerns the acylation of aromatic derivatives (Section 3.3.) and the synthesis of fluorinated heterocycles by ring-closing attacks of heteroatomic nucleophiles (Section 3.4).
- (C)
- All carbons of the FAR are present in the desired product of the reaction and 2 carbons, the carbon of the iminium and the methine in α position, undergo transformations. This kind of reactivity is observed when nucleophiles are either allylic or propargylic alcohols (Section 3.5).
- (D)
- All carbons of the FAR are present in the desired product of the reaction and all of them, namely the carbon of the iminium, the α-methine and the carbon in β position (CF3) undergo transformations. Accordingly, this reactivity is observed only with the Ishikawa reagent (Section 3.6).
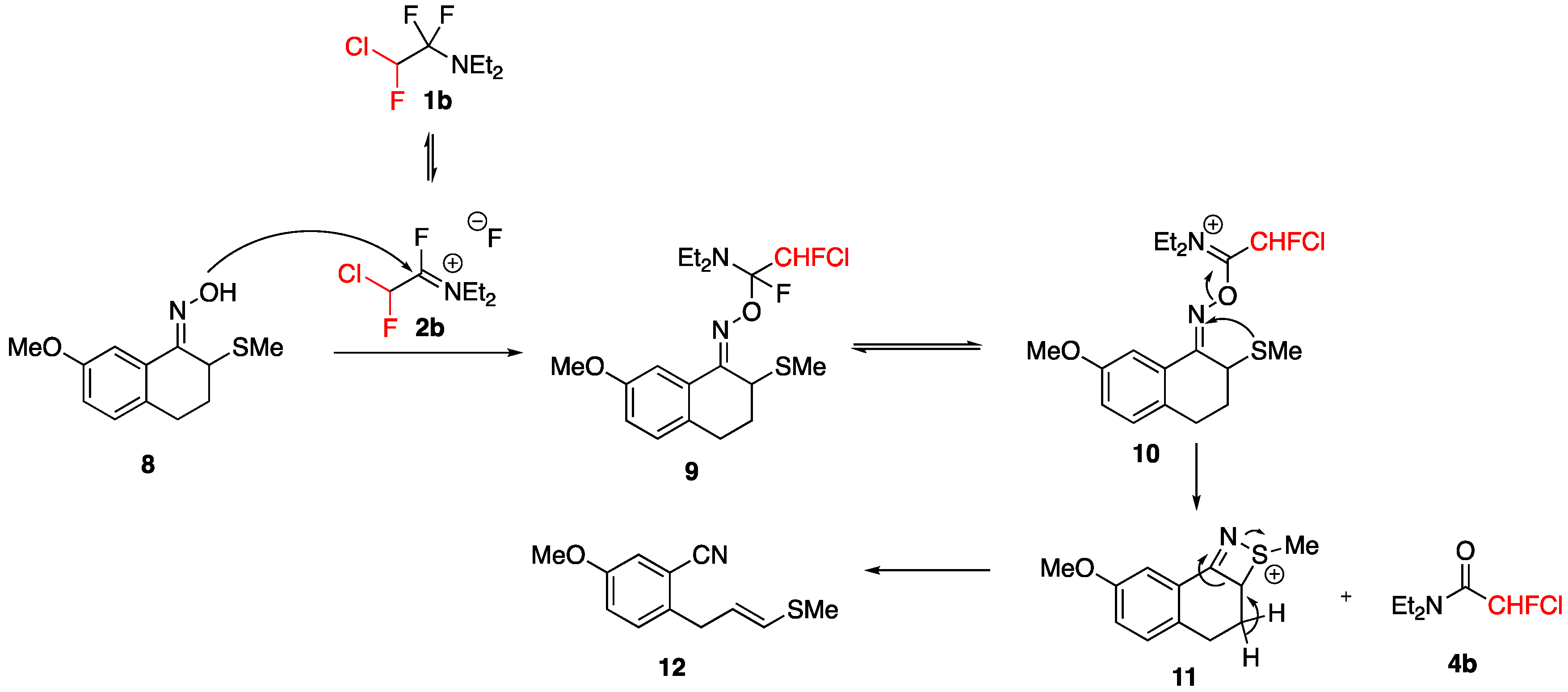
3.2. Nucleophilic Fluorination of the Hydroxyl or Carbonyl Functions
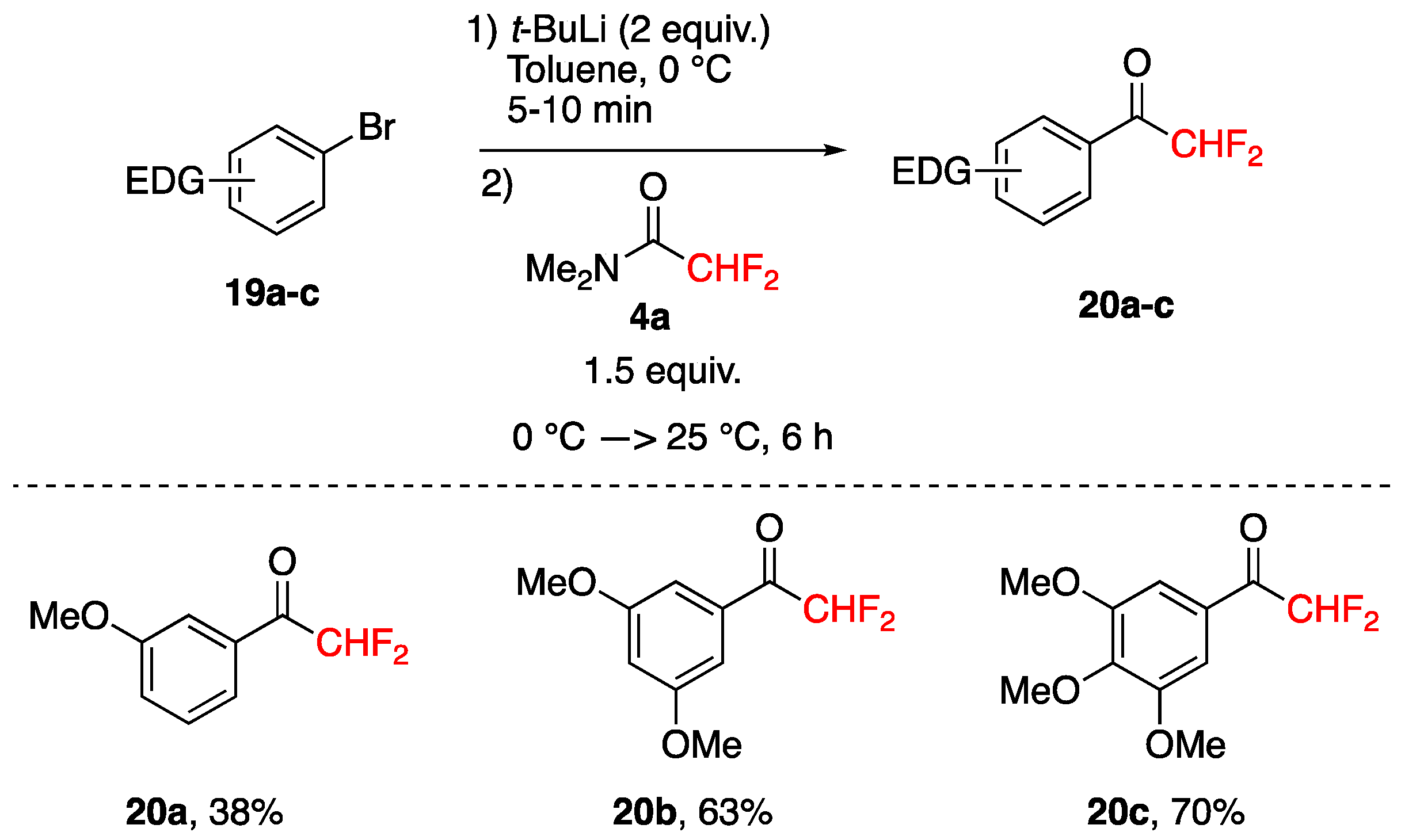
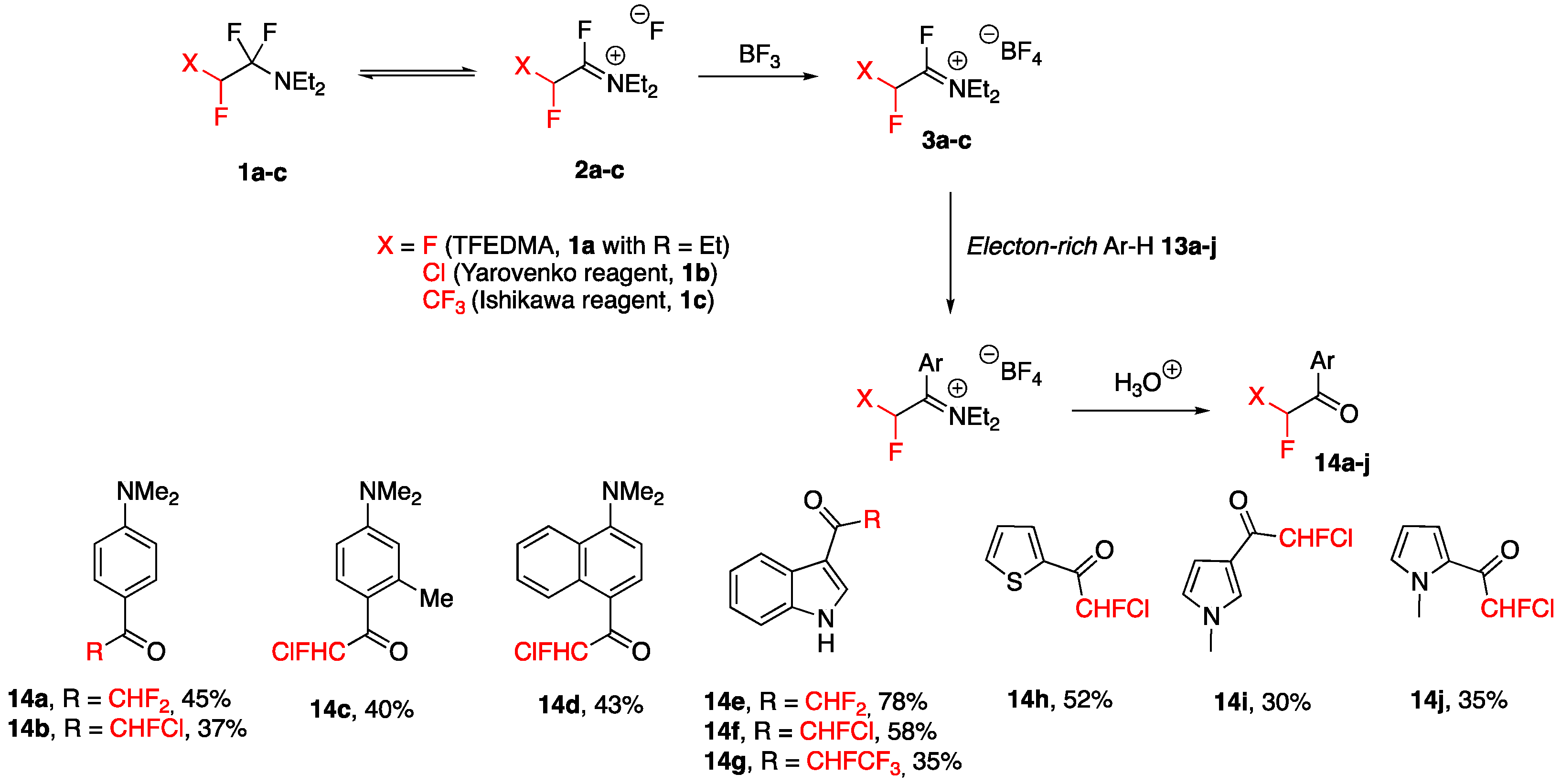
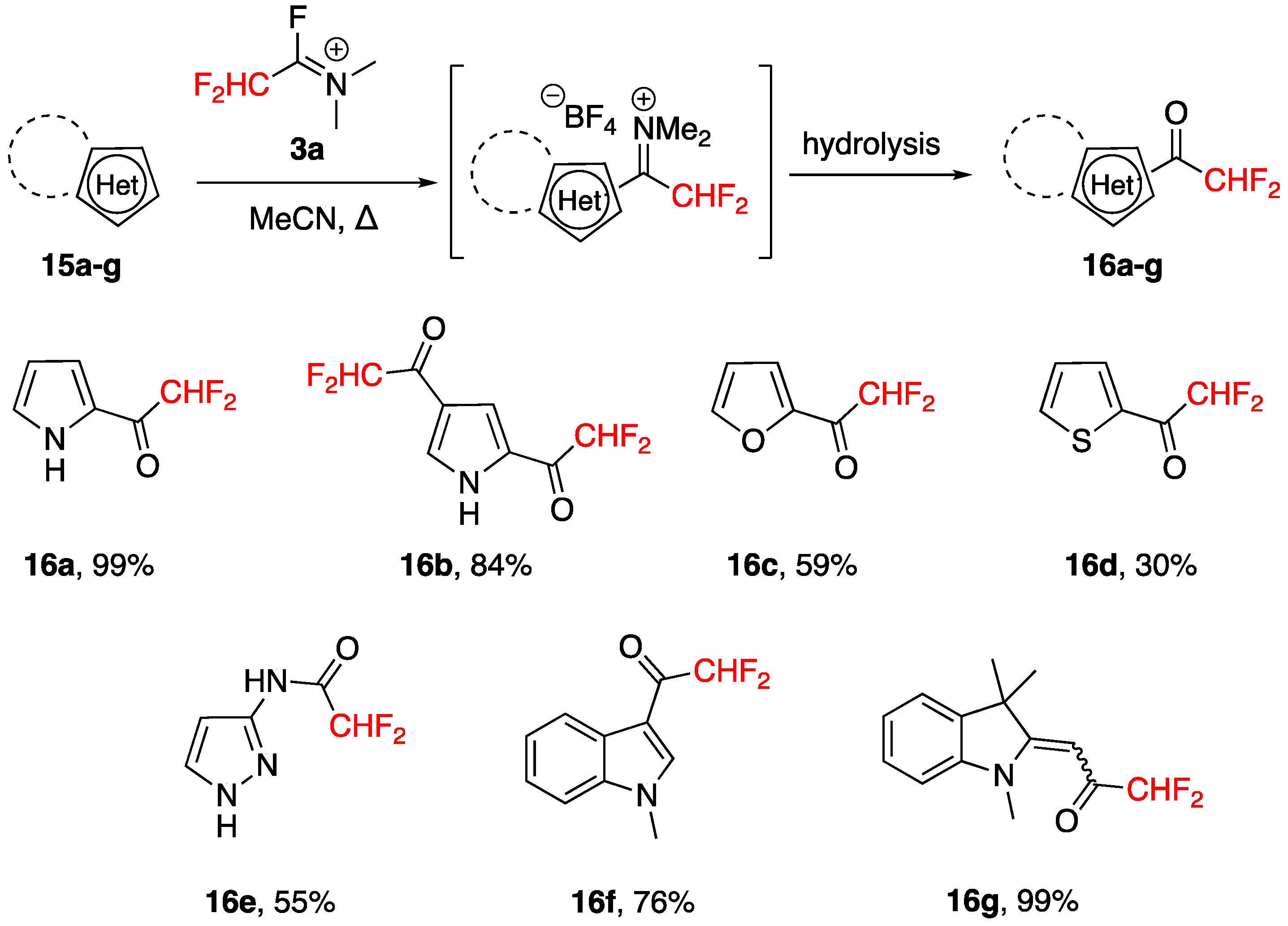
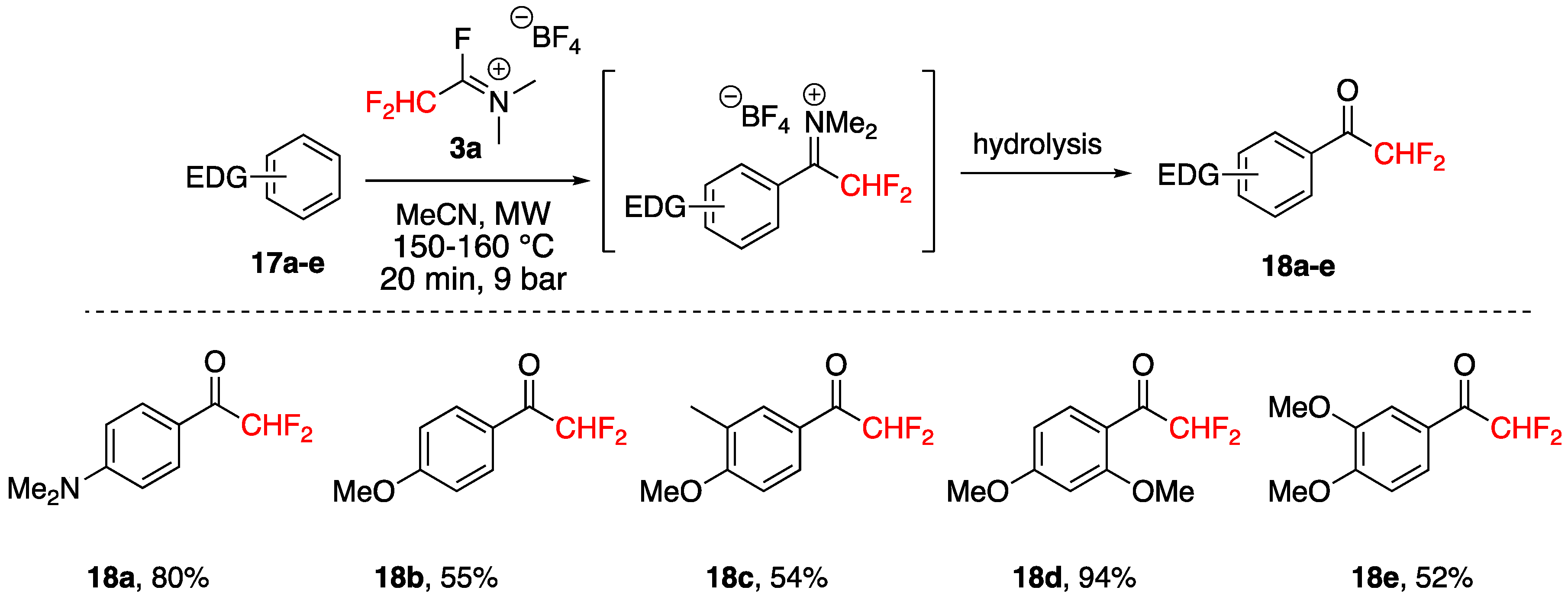
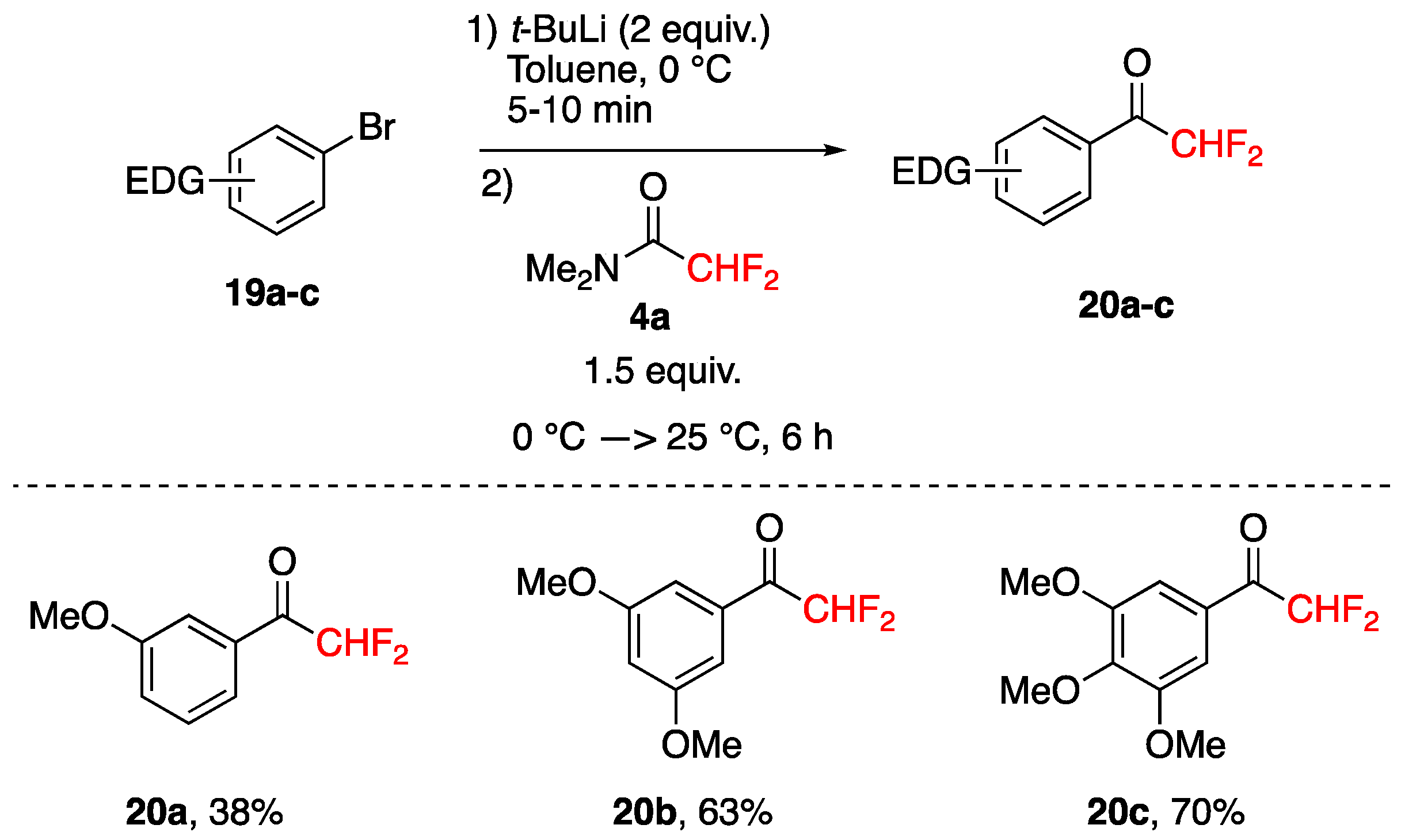
3.3. Acylation of Aromatics
3.4. Synthesis of Fluoroalkylated Heterocycles
3.4.1. Synthesis of Mono-Fluoroalkylated Benzo-Fused Heterocycles from 1,2-Diheteroatom-functionalized Arenes
3.4.2. Synthesis of Mono-Fluoroalkylated Pyrazoles
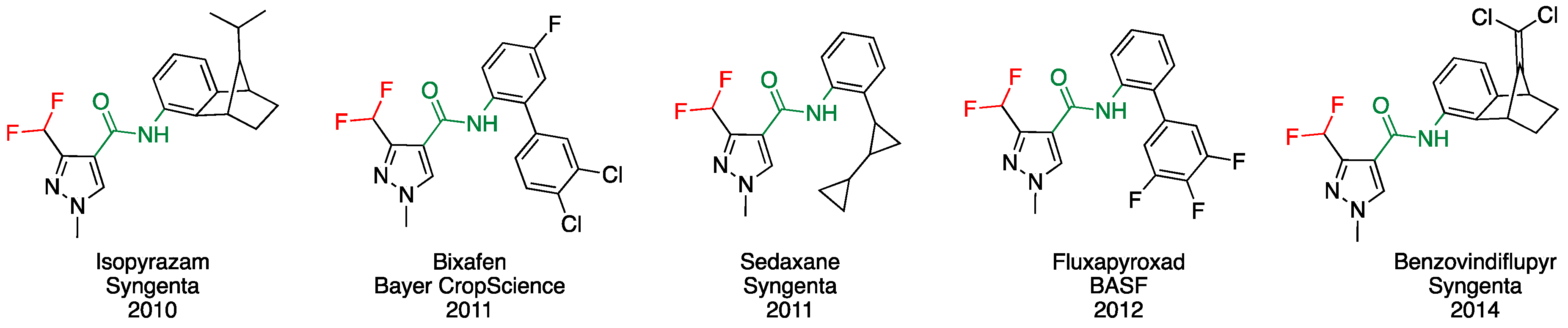
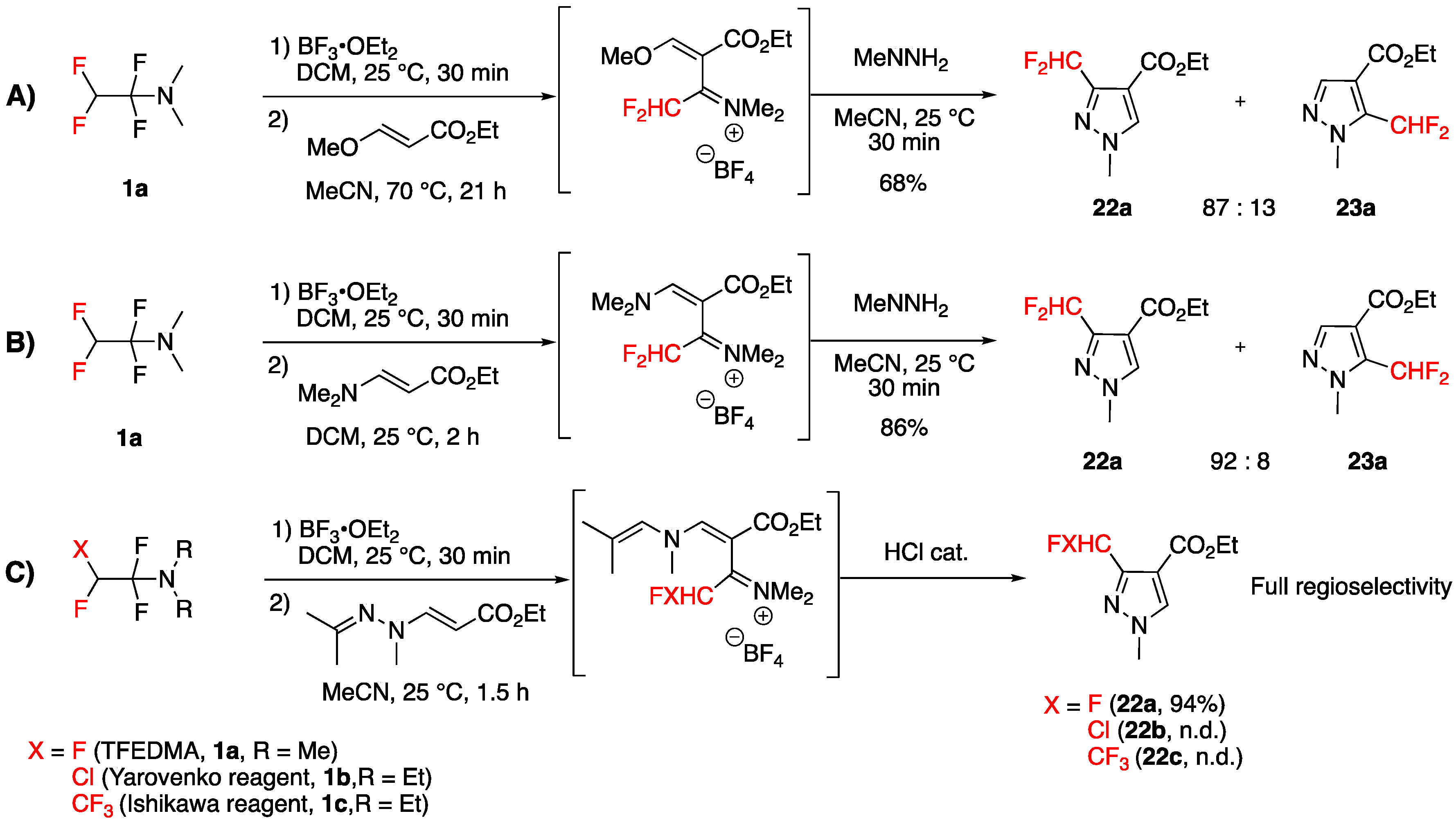
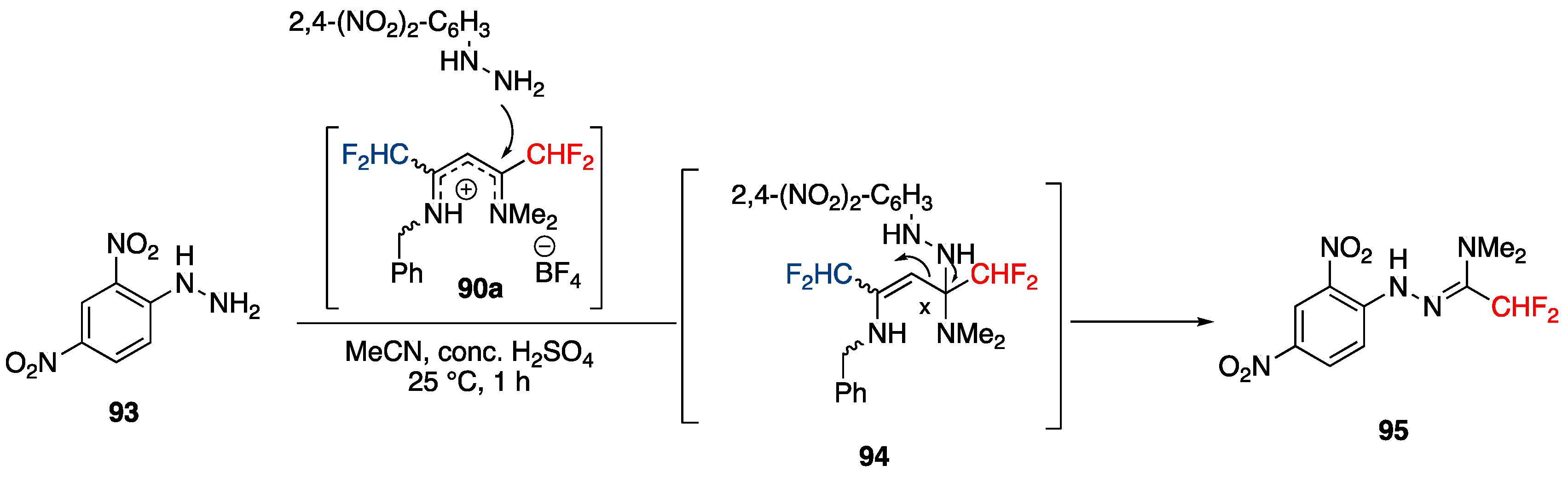
Towards the 3-CHF2-Pyrazolecarboxamide Motif
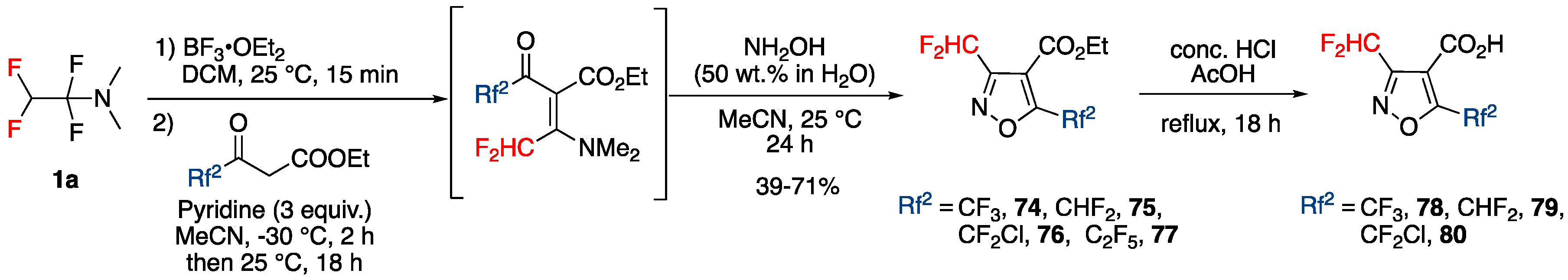
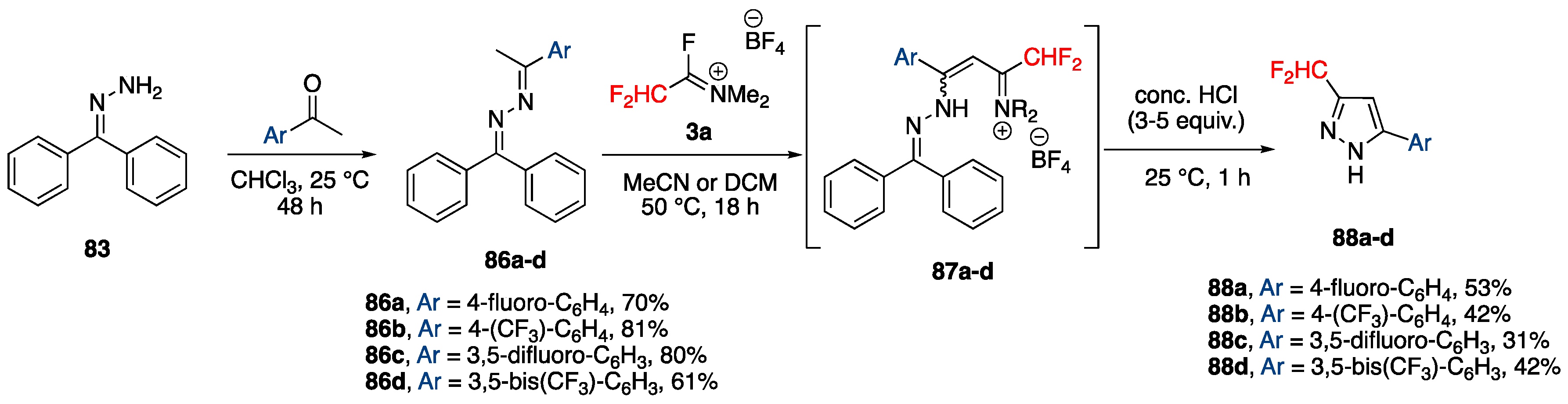
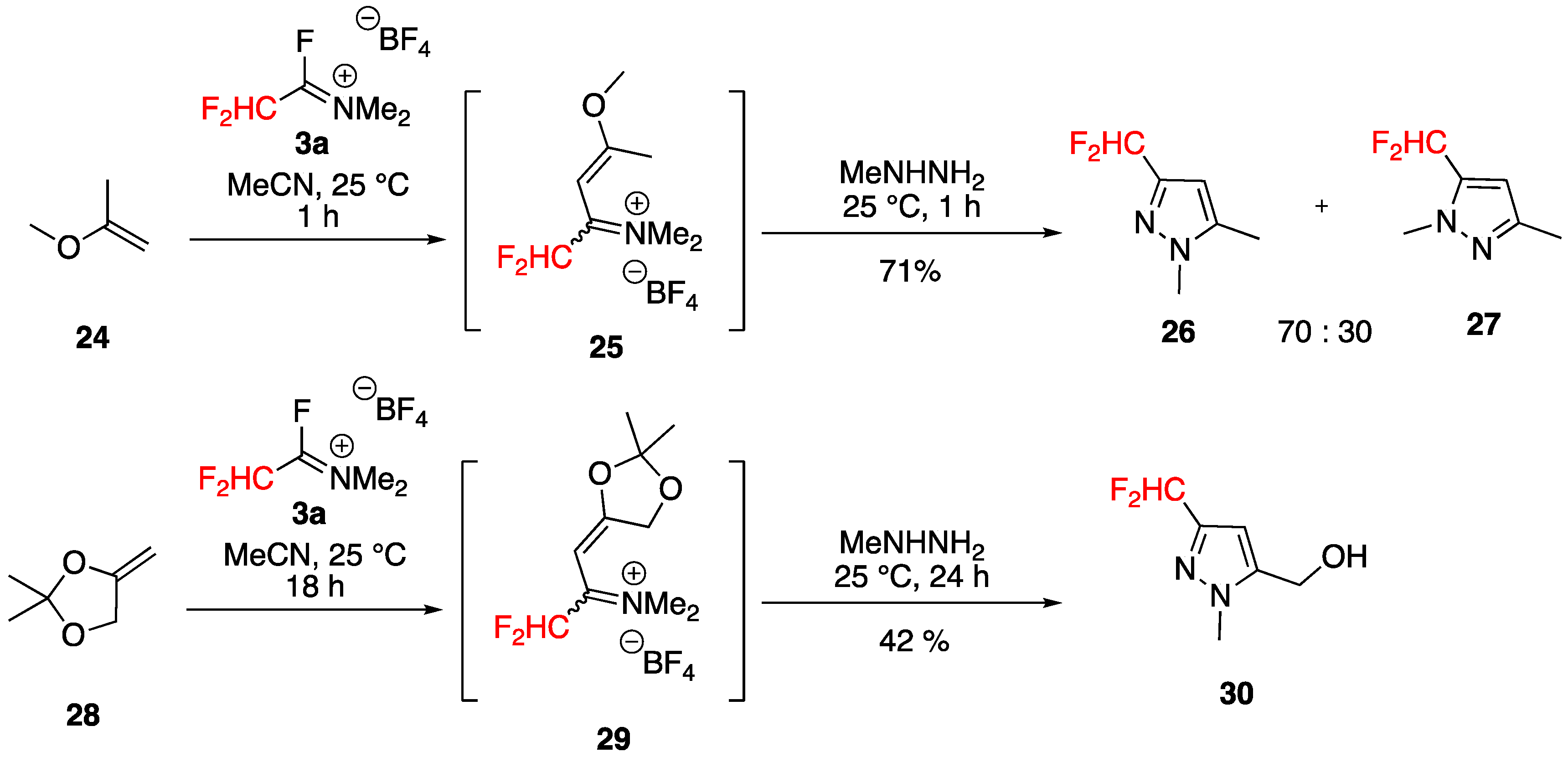
Synthesis of Various Substituted Mono (Fluoroalkyl)pyrazoles and Isoxazoles
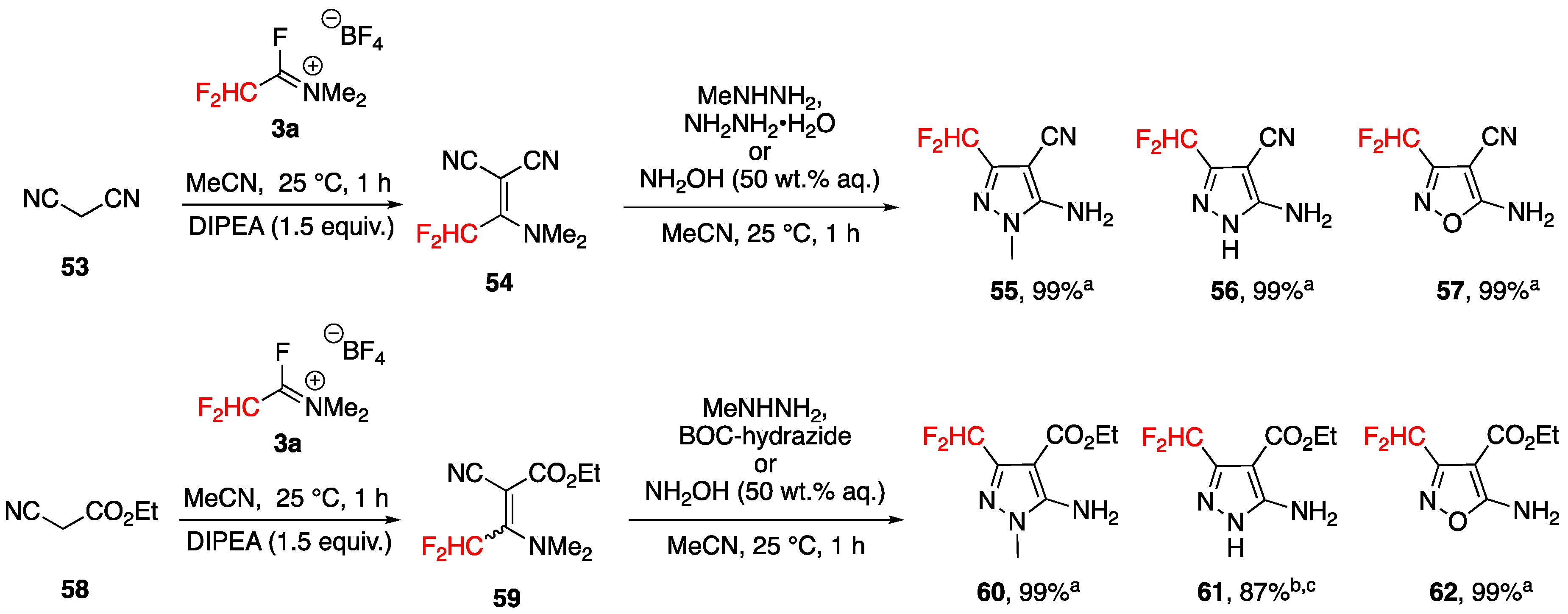
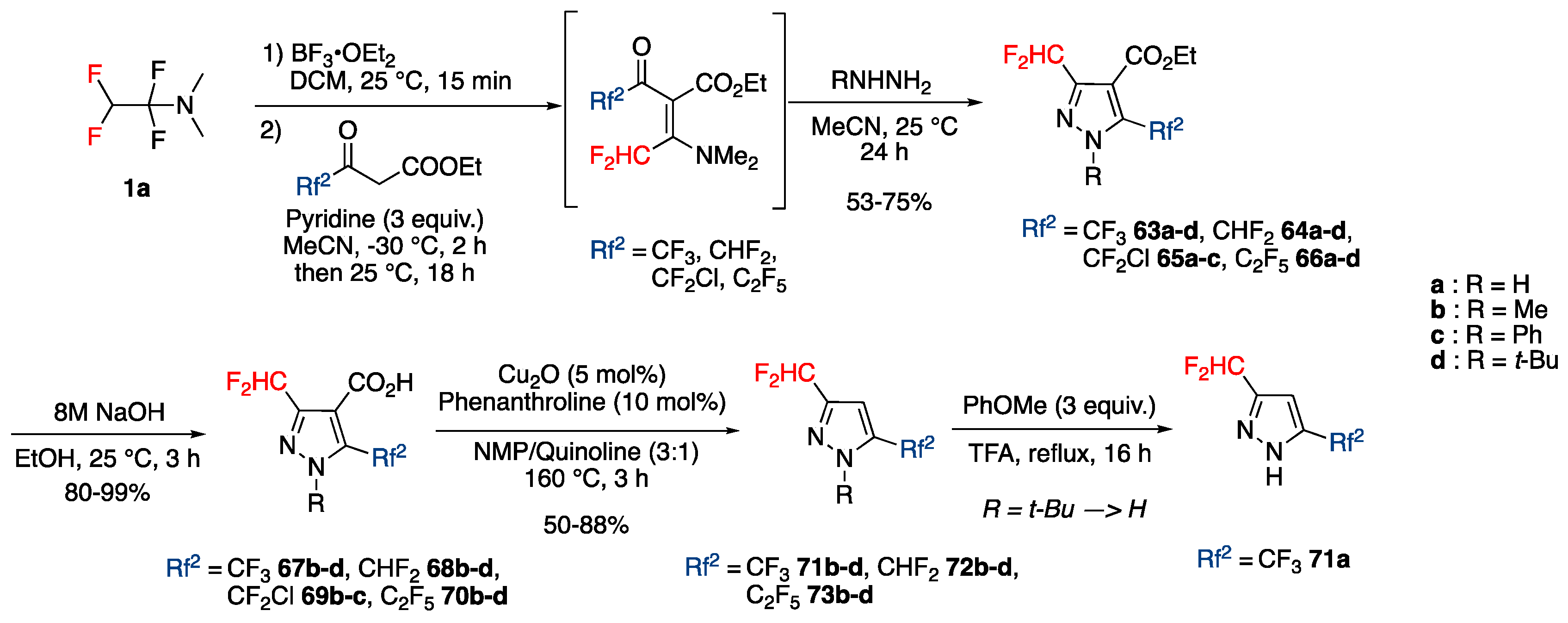
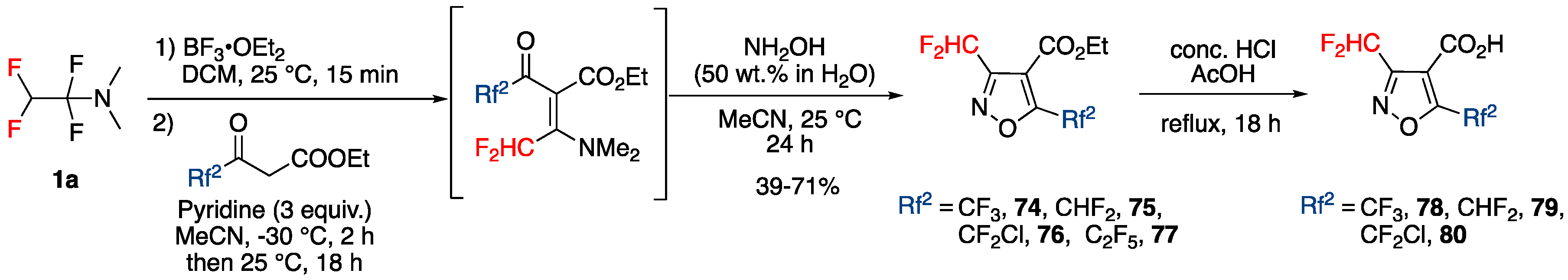
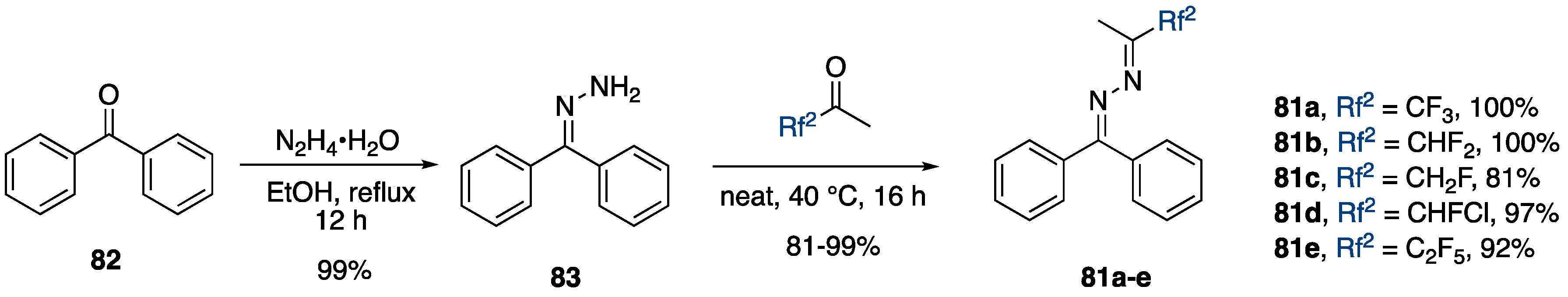
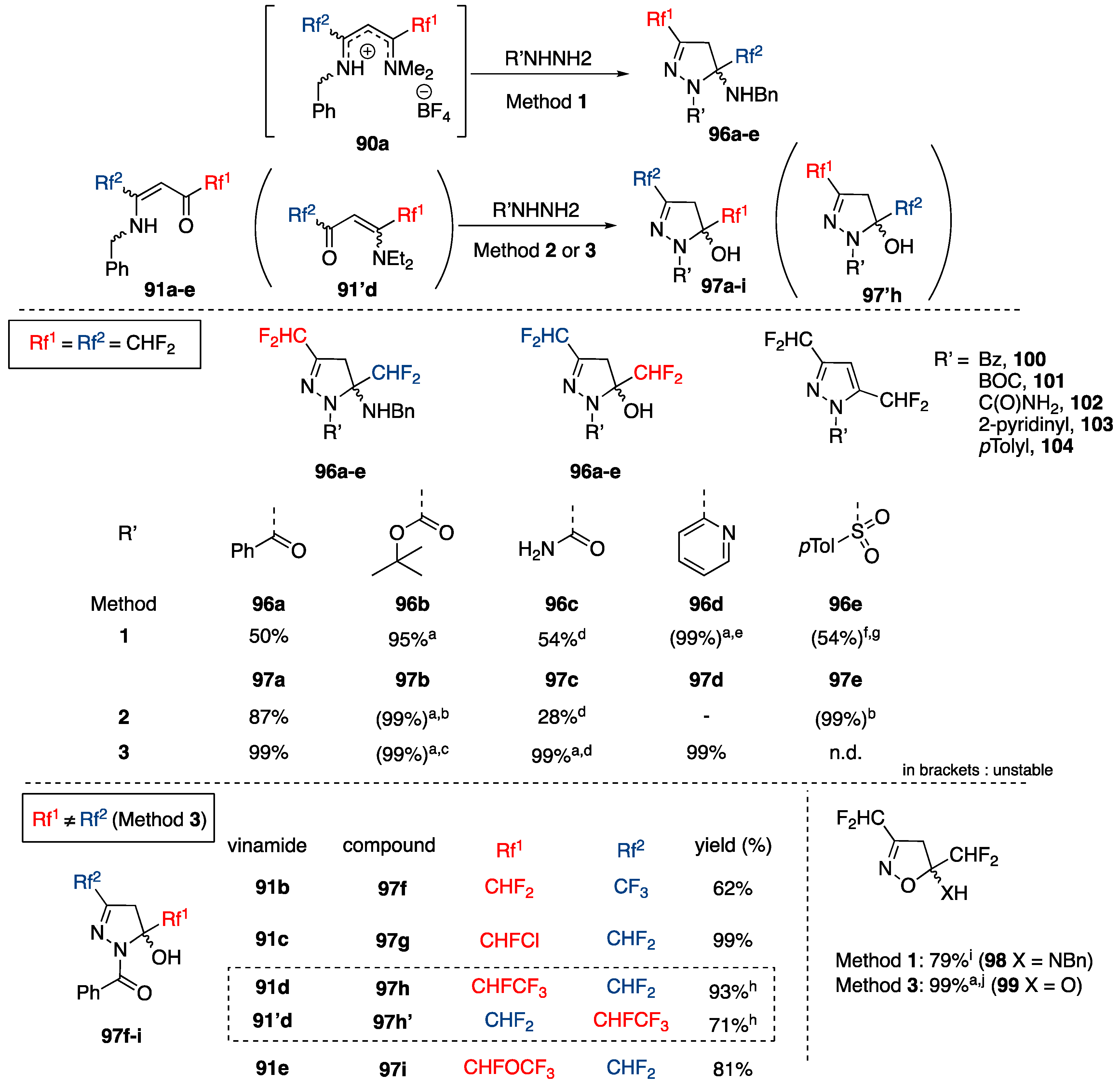
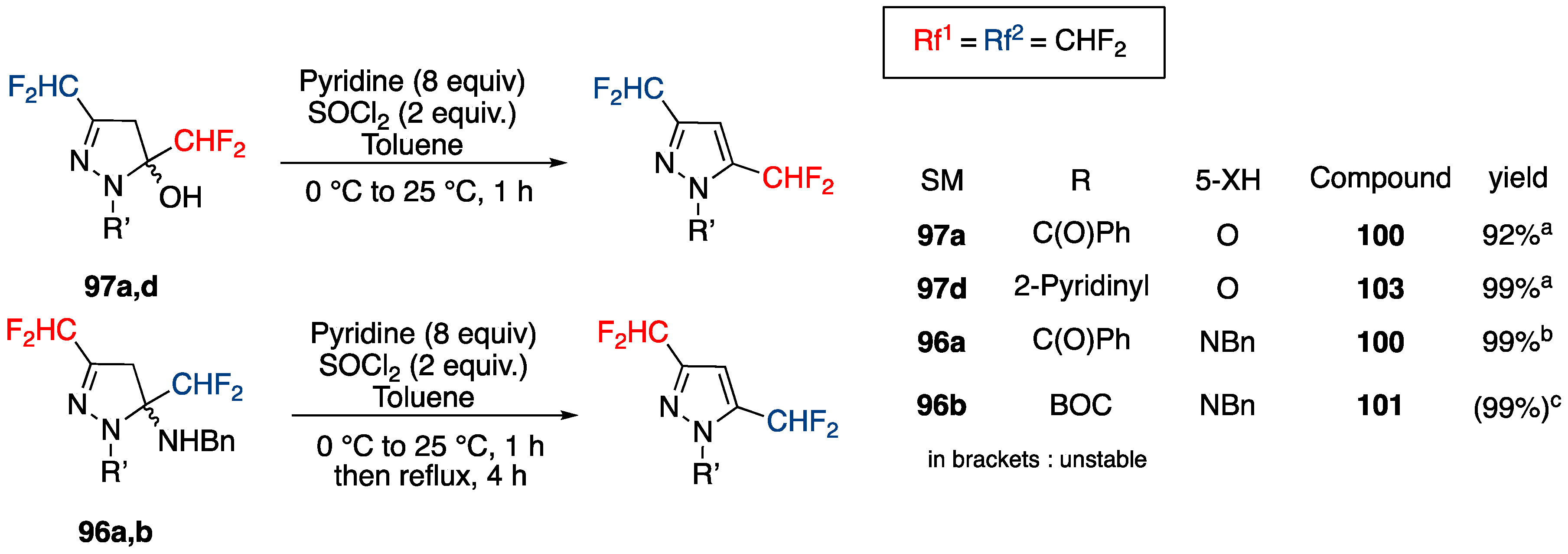
3.4.3. Synthesis of Bis-fluoroalkylated Pyrazoles
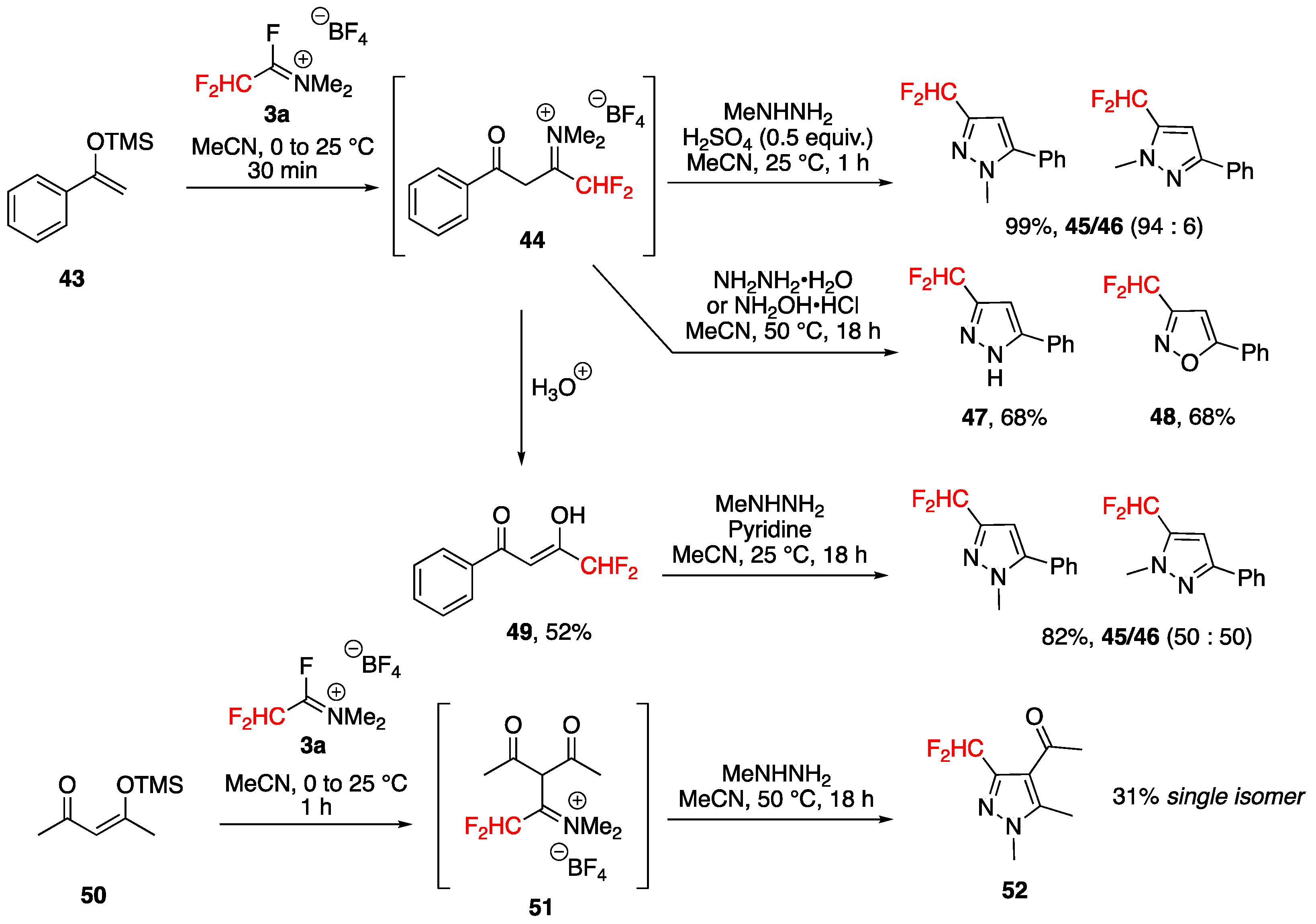
Synthesis of 3,5-Bis(fluoroalkyl)pyrazoles from Fluoroacetoacetates
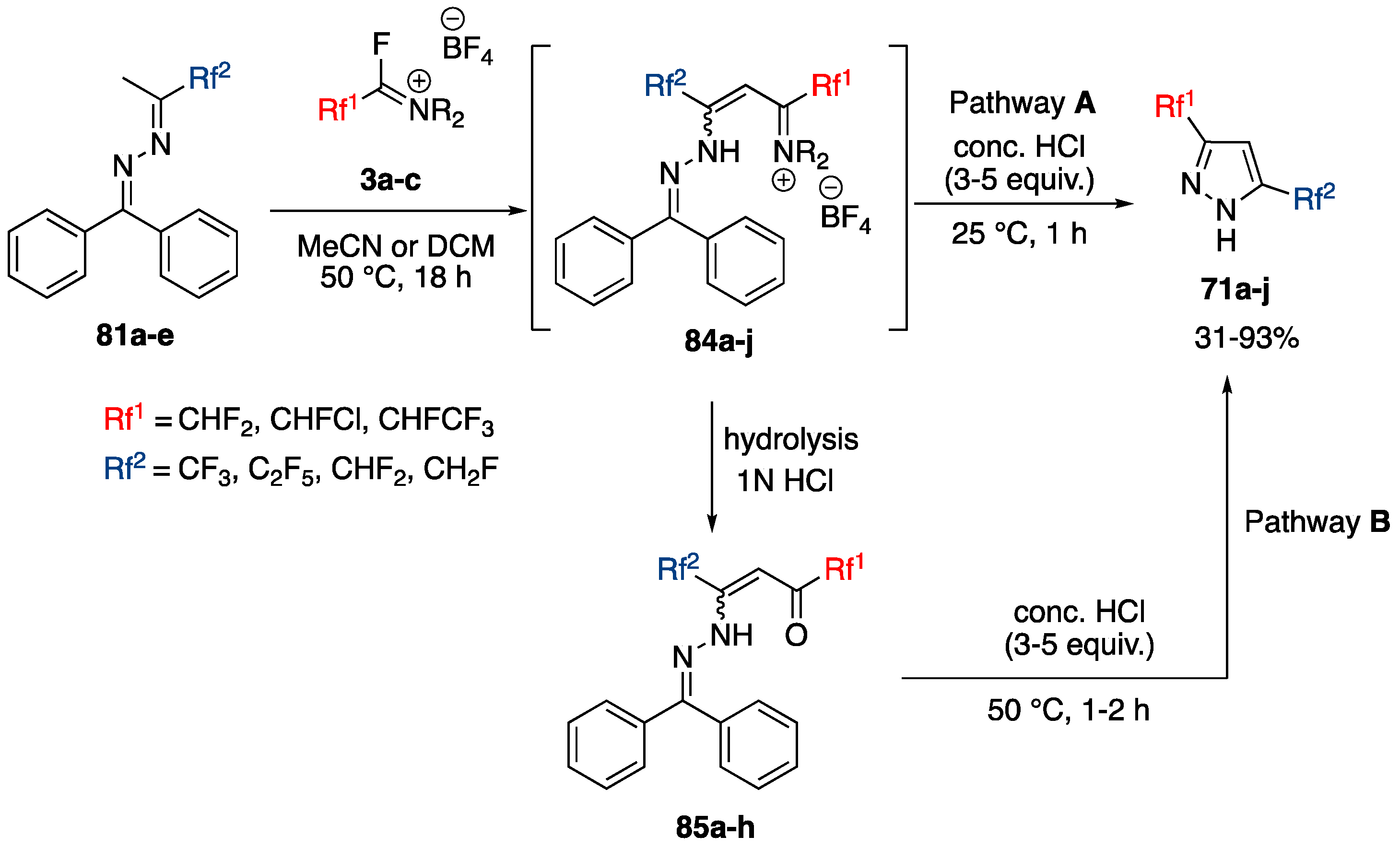
Synthesis of 3,5-Bis(fluoroalkyl)-NH-pyrazoles from Azines
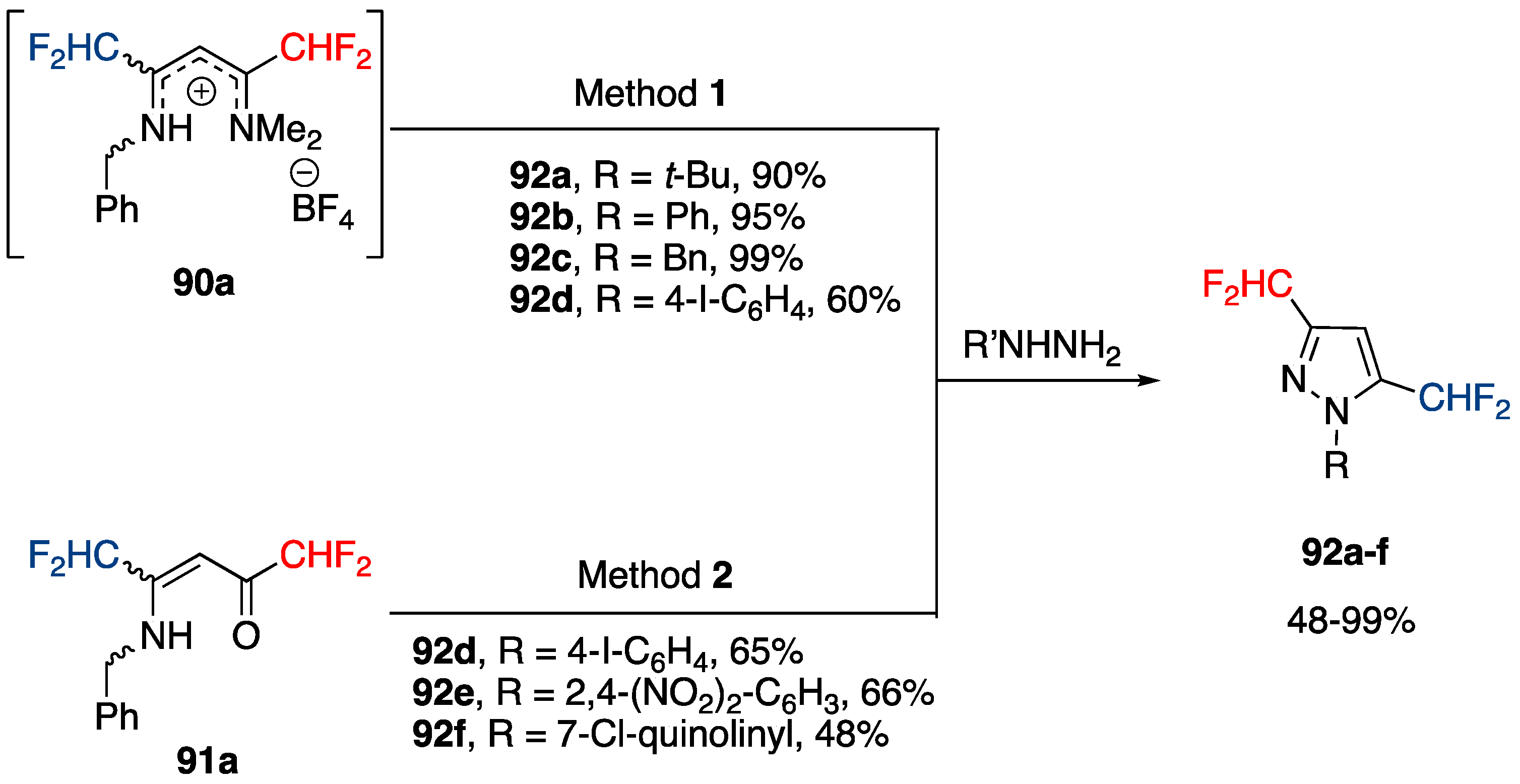
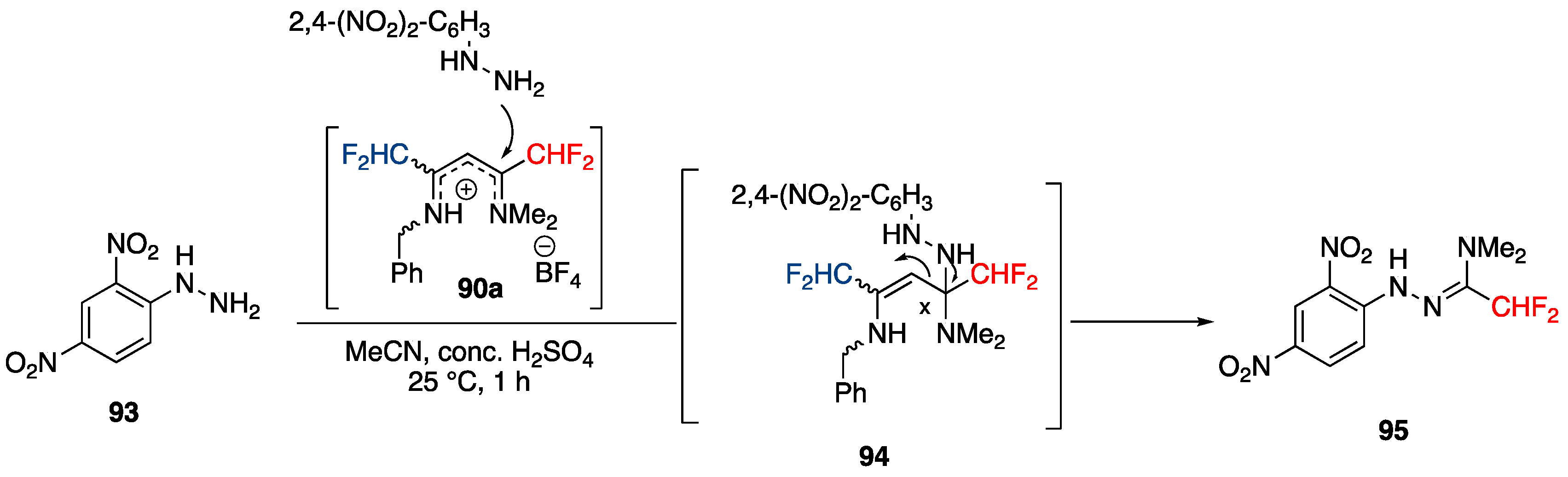
Synthesis of 3,5-Bis(fluoroalkyl)-NH-pyrazoles from Ketimines
Synthesis of 3,5-Bis(fluoroalkyl)-NMe-pyrazoles from Ketimines
Synthesis of 3,5-Bis(fluoroalkyl)-N-substituted-pyrazoles from Ketimines
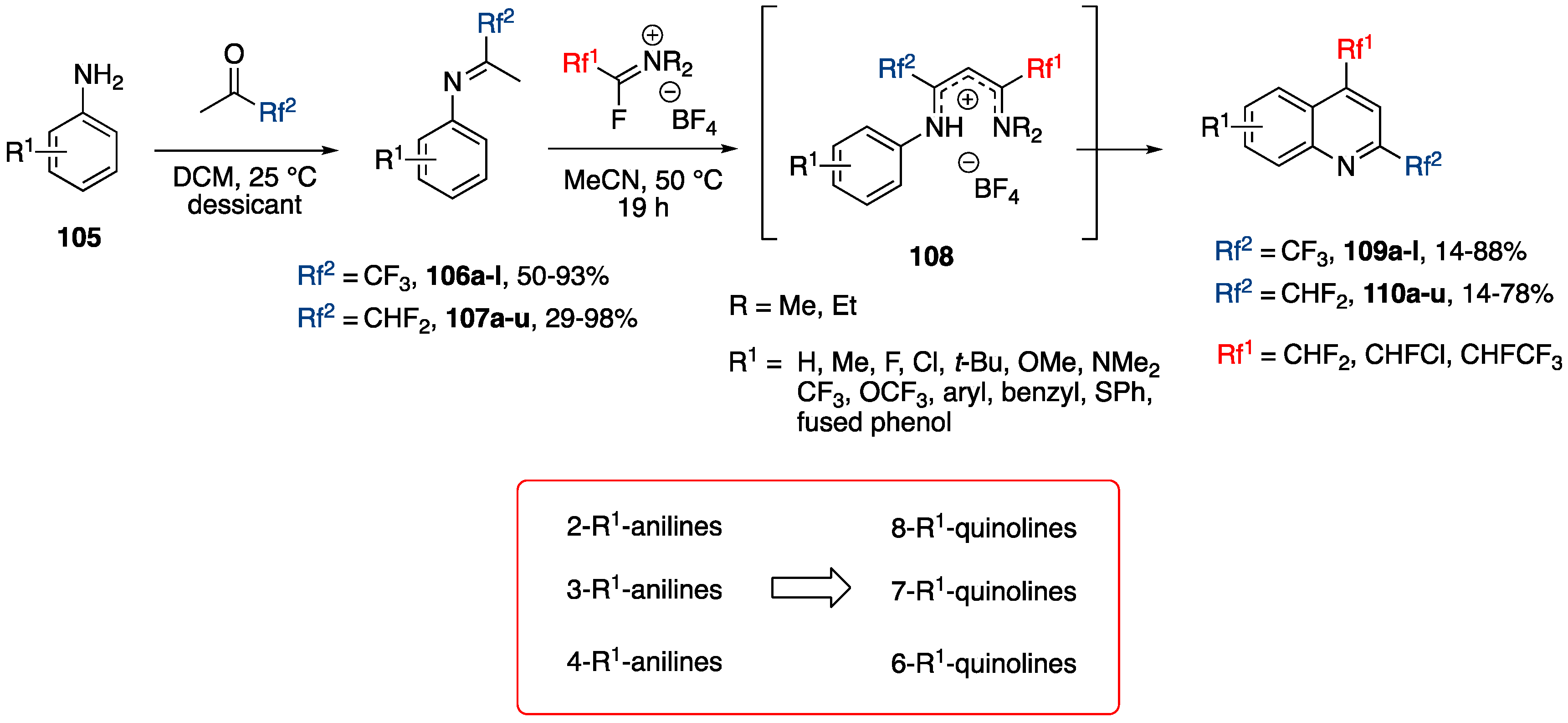
3.4.4. Synthesis of 2,4-Bis(fluoroalkyl)-substituted Quinoline Derivatives
3.5. Reaction with Allylic and Propargylic Alcohols
3.6. Transformation of the Three Carbons of the Ishikawa Reagent
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Bégué, J.-P.; Bonnet-Delpon, D. Fluorinated Drugs. In Bioorganic and Medicinal Chemistry of Fluorine; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2008; pp. 279–351. [Google Scholar]
- Jeschke, P. The unique role of fluorine in the design of active ingredients for modern crop protection. ChemBioChem 2004, 5, 571–589. [Google Scholar] [CrossRef] [PubMed]
- Leroux, F.; Jeschke, P.; Schlosser, M. α-Fluorinated Ethers, Thioethers, and Amines: Anomerically Biased Species. Chem. Rev. 2005, 105, 827–856. [Google Scholar] [CrossRef] [PubMed]
- Hong, W. Agricultural Products Based on Fluorinated Heterocyclic Compounds. In Fluorinated Heterocyclic Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2009; pp. 397–418. [Google Scholar]
- Cartwright, D. Recent Developments in Fluorine-Containing Agrochemicals. In Organofluorine Chemistry: Principles and Commercial Applications; Banks, R.E., Smart, B.E., Tatlow, J.C., Eds.; Springer: Boston, MA, USA, 1994; pp. 237–262. [Google Scholar]
- Fujiwara, T.; O’Hagan, D. Successful fluorine-containing herbicide agrochemicals. J. Fluorine Chem. 2014, 167, 16–29. [Google Scholar] [CrossRef]
- Theodoridis, G. Fluorine-Containing Agrochemicals: An Overview of Recent Developments. In Advances in Fluorine Science; Alain, T., Ed.; Elsevier: Amsterdam, The Netherlands, 2006; Volume 2, pp. 121–175. [Google Scholar]
- Jeschke, P. The unique role of halogen substituents in the design of modern agrochemicals. Pest Manag. Sci. 2010, 66, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Muller, K.; Faeh, C.; Diederich, F. Fluorine in pharmaceuticals: Looking beyond intuition. Science 2007, 317, 1881–1886. [Google Scholar] [CrossRef] [PubMed]
- Purser, S.; Moore, P.R.; Swallow, S.; Gouverneur, V. Fluorine in medicinal chemistry. Chem. Soc. Rev. 2008, 37, 320–330. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Fluorine in health care: Organofluorine containing blockbuster drugs. J. Fluorine Chem. 2010, 131, 1071–1081. [Google Scholar] [CrossRef]
- Landelle, G.; Panossian, A.; Leroux, F.R. Trifluoromethyl ethers and -thioethers as tools for medicinal chemistry and drug discovery. Curr. Top. Med. Chem. 2014, 14, 941–951. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Wang, J.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J.L.; Soloshonok, V.A.; Izawa, K.; Liu, H. Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of Major Pharmaceutical Companies: New Structural Trends and Therapeutic Areas. Chem. Rev. 2016, 116, 422–518. [Google Scholar] [CrossRef] [PubMed]
- Pruett, R.L.; Barr, J.T.; Rapp, K.E.; Bahner, C.T.; Gibson, J.D.; Lafferty, R.H. Reactions of Polyfluoro Olefins. II. Reactions with Primary and Secondary Amines. J. Am. Chem. Soc. 1950, 72, 3646–3650. [Google Scholar] [CrossRef]
- Knunyants, I.L.; German, L.S.; Dyatkin, B.L. Reactions of fluoro olefins Communication 6. Reactions of perfluoroisobutylene and perfluoropropene with nucleophilic reagents. Russ. Chem. Bull. 1956, 5, 1387–1394. [Google Scholar] [CrossRef]
- Yarovenko, N.N.; Raksha, M.A. Fluorination with α-fluorinated amines. Zh. Obshch. Khim. 1959, 29, 2159–2163. [Google Scholar]
- England, D.C.; Melby, L.R.; Dietrich, M.A.; Lindsey, R.V. Nucleophilic Reactions of Fluoroölefins. J. Am. Chem. Soc. 1960, 82, 5116–5122. [Google Scholar] [CrossRef]
- Takaoka, A.; Iwakiri, H.; Ishikawa, N. F-Propene-Dialkylamine Reaction-Products as Fluorinating Agents. Bull. Chem. Soc. Jpn. 1979, 52, 3377–3380. [Google Scholar] [CrossRef]
- Petrov, V.A.; Swearingen, S.; Hong, W.; Chris Petersen, W. 1,1,2,2-Tetrafluoroethyl-N,N-dimethylamine: A new selective fluorinating agent. J. Fluorine Chem. 2001, 109, 25–31. [Google Scholar] [CrossRef]
- Walkowiak, J.; Koroniak, H. Preparation of α-Fluoro Amino and α-Fluoro Enamino Reagents. In Efficient Preparations of Fluorine Compounds; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 379–384. [Google Scholar]
- Schmitt, E.; Panossian, A.; Vors, J.P.; Funke, C.; Lui, N.; Pazenok, S.; Leroux, F.R. A Major Advance in the Synthesis of Fluoroalkyl Pyrazoles: Tuneable Regioselectivity and Broad Substitution Patterns. Chem. Eur. J. 2016, 22, 11239–11244. [Google Scholar] [CrossRef] [PubMed]
- Pazenok, S.; Vors, J.-P.; Leroux, F.R.; Schmitt, E. Process for Preparing Substituted Pyrazoles Containing Haloalkoxy-and Haloalkylthio Groups from α,α-Dihaloalkylamines and Ketimines. WO2016207167, 21 June 2016. [Google Scholar]
- Viehe, H.G.; Janousek, Z. The Chemistry of Dichloromethylenammonium Salts (“Phosgenimonium Salts”). Angew. Chem. Int. Ed. Engl. 1973, 12, 806–818. [Google Scholar] [CrossRef]
- Ishikawa, N.; Kitazume, T.; Takaoka, A. Fluorinating Agents for O-Functional Groups. J. Synth. Org. Chem. Jpn. 1979, 37, 606–611. [Google Scholar] [CrossRef]
- Hamman, S.; Barrelle, M.; Tetaz, F.; Beguin, C.G. Acide fluoro-2 phenyl-2 acetique: Synthesis, configuration absolute et emploi comme agent chiral de derivation. J. Fluorine Chem. 1987, 37, 85–94. [Google Scholar] [CrossRef]
- Spero, G.B.; Pike, J.E.; Lincoln, F.H.; Thompson, J.L. A new approach to 16α-halo corticoids. II The synthesis of 16α-fluoro and 16α-chloro corticoids. Steroids 1968, 11, 769–786. [Google Scholar] [CrossRef]
- Knox, L.H.; Velarde, E.; Berger, S.; Cuadriello, D.; Cross, A.D. Steroids. CCXL. The Reaction of Steroidal Alcohols with 2-Chloro-1,1,2-trifluorotriethylamine. J. Org. Chem. 1964, 29, 2187–2195. [Google Scholar] [CrossRef]
- Allen, G.R.; Weiss, M.J. New Progestational Agents. Nonclassical 17-Alkylpregnene Structures. J. Med. Chem. 1964, 7, 684–686. [Google Scholar] [CrossRef] [PubMed]
- Ayer, D.E. The Synthesis of 15β-Fluoro Corticoids. J. Med. Chem. 1963, 6, 608–610. [Google Scholar] [CrossRef] [PubMed]
- Ayer, D.E. A new method for the preparation of fluoro steroids. Tetrahedron Lett. 1962, 3, 1065–1069. [Google Scholar] [CrossRef]
- Knox, L.H.; Velarde, E.; Berger, S.; Cuadriello, D.; Cross, A.D. The Reactions of Steroidal Alcohols with 2-chloro-1,1,2-trifluorotriethylamine. Tetrahedron Lett. 1962, 3, 1249–1255. [Google Scholar] [CrossRef]
- Crabbe, P.; Carpio, H.; Velarde, E.; Fried, J.H. Chemistry of difluorocyclopropenes. Application to the synthesis of steroidal allenes. J. Org. Chem. 1973, 38, 1478–1483. [Google Scholar] [CrossRef]
- Knox, L.H.; Velarde, E.; Berger, S.; Delfín, I.; Grezemkovsky, R.; Cross, A.D. Steroids. CCLXXXIV. Reactions of 19-Hydroxy-Δ5-3-acetoxy Steroids with Diethyl(2-chloro-1,1,2-trifluoroethyl)amine. J. Org. Chem. 1965, 30, 4160–4165. [Google Scholar] [CrossRef] [PubMed]
- Wood, K.R.; Fisher, D.; Kent, P.W. Fluorocarbohydrates. Part XIV. Reaction of N-(2-chloro-1,1,2-trifluoroethyl)diethylamine with some O-isopropylidene sugars. J. Chem. Soc. 1966, 21, 1994–1997. [Google Scholar] [CrossRef]
- Bergmann, E.D.; Cohen, A.M. Organic Fluorine Compounds. Part 43. Applications of (1,1,2-Trifluoro-2-Chloroethyl)-Diethylamine as Fluorinating Agent. Isr. J. Chem. 1970, 8, 925–933. [Google Scholar] [CrossRef]
- Bateson, J.H.; Cross, B.E. Reactions of 2-chloro-NN-diethyl-1,1,2-trifluoroethylamine with alcohols. Part I. Preparation of 2β- and 4β-fluorogibberellins. J. Chem. Soc., Perkin Trans. 1 1974, 2409–2413. [Google Scholar] [CrossRef]
- Müller, B.; Peter, H.; Schneider, P.; Bickel, H. New β-Lactam-Antibiotics. Fluorinated Cephalosporins. Preliminary Communication. Modifikationen von Antibiotika. 15. Mitteilung. Helv. Chim. Acta 1975, 58, 2469–2473. [Google Scholar] [CrossRef] [PubMed]
- O’Hagan, D. Preparation of monofluorocarboxylic acids using N,N-diethyl-1,1.2,3,3,3-hexafluoropropylamine. J. Fluorine Chem. 1989, 43, 371–377. [Google Scholar] [CrossRef]
- Watanabe, S.; Fujita, T.; Usui, Y.; Kitazume, T. Fluorination of hydroxyesters with N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine. J. Fluorine Chem. 1986, 31, 247–253. [Google Scholar] [CrossRef]
- Watanabe, S.; Fujita, T.; Sakamoto, M.; Kuramochi, T.; Kitazume, T. Reactions of monoesters of ethylene glycol with N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine. J. Fluorine Chem. 1987, 36, 361–372. [Google Scholar] [CrossRef]
- Watanabe, S.; Fujita, T.; Sakamoto, M.; Endo, H.; Kitazume, T. Fluorination of aromatic α-hydroxyesters with N,N-diethyl-1,1,2,3,3,3-hexafluoropropaneamine. J. Fluorine Chem. 1990, 47, 187–192. [Google Scholar] [CrossRef]
- Bresciani, S.; Slawin, A.M. Z.; O’Hagan, D. A regio- and stereoisomeric study of allylic alcohol fluorination with a range of reagents. J. Fluorine Chem. 2009, 130, 537–543. [Google Scholar] [CrossRef]
- Araki, K.; Katagiri, T.; Inoue, M. Facile synthesis of 1,7,8-trifluoro-2-naphthol via DMAP catalyzed cycloaromatization. J. Fluorine Chem. 2014, 157, 41–47. [Google Scholar] [CrossRef]
- Tan, X.; Soualmia, F.; Furio, L.; Renard, J.-F.; Kempen, I.; Qin, L.; Pagano, M.; Pirotte, B.; El Amri, C.; Hovnanian, A.; et al. Toward the First Class of Suicide Inhibitors of Kallikreins Involved in Skin Diseases. J. Med. Chem. 2015, 58, 598–612. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.H.; Zheng, C.; Li, F.; Zhao, L.; Chen, F.E.; He, Q.Q. An Efficient Enantioselective Synthesis of Florfenicol Based on Sharpless Asymmetric Dihydroxylation. Synthesis 2012, 44, 699–704. [Google Scholar]
- Ando, T.; Koseki, N.; Yasuhara, I.; Matsuo, N.; Ishiwatari, T. Synthesis of Fluorinated Pyrethroids: Conversion of Pyrethroid Metabolites into Some Insecticidal Fluorinated Derivatives. Biosci. Biotechnol. Biochem. 1992, 56, 1581–1583. [Google Scholar] [CrossRef]
- Tanaka, M.; Moriguchi, T.; Kizuka, M.; Ono, Y.; Miyakoshi, S.I.; Ogita, T. Microbial hydroxylation of zofimarin, a sordarin-related antibiotic. J. Antibiot. 2002, 55, 437–441. [Google Scholar] [CrossRef] [PubMed]
- Schumacher, D.P.; Clark, J.E.; Murphy, B.L.; Fischer, P.A. An efficient synthesis of florfenicol. J. Org. Chem. 1990, 55, 5291–5294. [Google Scholar] [CrossRef]
- Cantrell, G.L.; Filler, R. Further studies on the synthesis of α-fluoro carbonyl compounds. J. Fluorine Chem. 1985, 27, 35–45. [Google Scholar] [CrossRef]
- Hudlicky, M.; Merola, J.S. New stereospecific syntheses and X-ray diffraction structures of (−)-d-erythro- and (+)-l-threo-4-fluoroglutamic acid. Tetrahedron Lett. 1990, 31, 7403–7406. [Google Scholar] [CrossRef]
- Hudlický, M. Stereospecific syntheses of all four stereoisomers of 4-fluoroglutamic acid. J. Fluorine Chem. 1993, 60, 193–210. [Google Scholar] [CrossRef]
- Gerus, I.I.; Mironets, R.V.; Shaitanova, E.N.; Kukhar, V.P. Synthesis of new β-trifluoromethyl containing GABA and β-fluoromethyl containing N-benzylpyrrolidinones. J. Fluorine Chem. 2010, 131, 224–228. [Google Scholar] [CrossRef]
- Kitamoto, T.; Ozawa, T.; Abe, M.; Marubayashi, S.; Yamazaki, T. Incorporation of fluoroprolines to proctolin: Study on the effect of a fluorine atom toward peptidic conformation. J. Fluorine Chem. 2008, 129, 286–293. [Google Scholar] [CrossRef]
- Takahashi, Y.; Ogawa, T. Total synthesis of cyclomaltohexaose. Carbohydr. Res. 1987, 164, 277–296. [Google Scholar] [CrossRef]
- Van der Steen, R.; Groesbeek, M.; van Amsterdam, L.J.P.; Lugtenburg, J.; van Oostrum, J.; de Grip, W.J. All E-10,20-methanoretinoylopsin, light-stable rhodopsin. Synthesis and spectroscopy of all E-10,20-methano- and all-E-retinoyl fluoride and their reaction with bovine opsin. Recl. Trav. Chim. Pays-Bas 1989, 108, 20–27. [Google Scholar] [CrossRef]
- Hansen, P.E.; Nicolaisen, F.M.; Schaumburg, K. Deuterium isotope effects on nuclear shielding. Directional effects and nonadditivity in acyl derivatives. J. Am. Chem. Soc. 1986, 108, 625–629. [Google Scholar] [CrossRef]
- Fokin, A.V.; Studnev, Y.N.; Rapkin, A.I.; Sultanbekov, D.A.; Potarina, T.M. Reaction of 1,1,2-trifluoro-2-chloroethyldiethylamine with fluorocarboxylic acids. Bull. Acad. Sci. USSR Div. Chem. Sci. 1984, 33, 372–375. [Google Scholar] [CrossRef]
- Cox, D.G.; Sprague, L.G.; Burton, D.J. The Facile Preparation of HF Free Polyfluorinated Acyl Fluorides. J. Fluorine Chem. 1983, 23, 383–388. [Google Scholar] [CrossRef]
- Haas, A.; Plümer, R.; Schiller, A. Fluorierung ungesättigter Aldehyde mit Schwefeltetrafluorid. Chem. Ber. 1985, 118, 3004–3010. [Google Scholar] [CrossRef]
- Anderson, G.L.; Burks, W.A.; Harruna, I.I. Novel Synthesis of 3-Fluoro-1-Aminoadamantane and Some of its Derivatives. Synth. Commun. 1988, 18, 1967–1974. [Google Scholar] [CrossRef]
- Suzuki, M.; Nishida, Y.; Ohguro, Y.; Miura, Y.; Tsuchida, A.; Kobayashi, K. Synthesis and characterization of asymmetric o- and m-nitrobenzoic acids with a 1,3-benzodioxole skeleton. Tetrahedron Asymmetry 2004, 15, 159–165. [Google Scholar] [CrossRef]
- Kitazume, T.; Ishikawa, N. A Convenient synthesis of acetylenic ketones from β-diketones using α,α-difluoroalkylamines and freeze-dried potassium fluoride. Chem. Lett. 1980, 9, 1327–1328. [Google Scholar] [CrossRef]
- Grieco, L.M.; Halliday, G.A.; Junk, C.P.; Lustig, S.R.; Marshall, W.J.; Petrov, V.A. Reactions of 1,1,2,2-tetrafluoroethyl-N,N-dimethylamine with linear and cyclic 1,3-diketones. J. Fluorine Chem. 2011, 132, 1198–1206. [Google Scholar] [CrossRef]
- Autrey, R.L.; Scullard, P.W. The Second-Order Beckmann Reaction of an α-(Methylthio) Ketone Oxime. J. Am. Chem. Soc. 1965, 87, 3284–3285. [Google Scholar] [CrossRef]
- Autrey, R.L.; Scullard, P.W. Beckmann fragmentation of an α-methylthio ketoxime. J. Am. Chem. Soc. 1968, 90, 4924–4929. [Google Scholar] [CrossRef]
- Wakselman, C.; Tordeux, M. Acylation of electron-rich aromatic nucleus with fluorinated immonium salts. J. Chem. Soc. Chem. Commun. 1975, 956. [Google Scholar] [CrossRef]
- Schmitt, E.; Rugeri, B.; Panossian, A.; Vors, J.P.; Pazenok, S.; Leroux, F.R. In Situ Generated Fluorinated Iminium Salts for Difluoromethylation and Difluoroacetylation. Org. Lett. 2015, 17, 4510–4513. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, S.; Fujita, T.; Sakamoto, M.; Kitazume, T. Reaction of alcohols with N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine in the presence of diisopropylethylamine. J. Fluorine Chem. 1988, 39, 17–22. [Google Scholar] [CrossRef]
- Watanabe, S.; Fujita, T.; Sakamoto, A.; Endo, H.; Kitazume, T. Reactions of nitro alcohols with N,N-diethyl-1,1,2,3,3,3-hexafluoropropylamine. J. Fluorine Chem. 1988, 38, 243–248. [Google Scholar] [CrossRef]
- Watanabe, S.; Fujita, T.; Usui, Y.; Kimura, Y.; Kitazume, T. Fluorination of Halogeno Alcohols with 1,1,2,3,3,3-Hexafluoropropyl Diethylamine. J. Fluorine Chem. 1986, 31, 135–141. [Google Scholar] [CrossRef]
- Takaoka, A.; Iwamoto, K.; Kitazume, T.; Ishikawa, N. Preparation of Benzoheterocycles Containing a Chlorofluoromethyl Group Using the Yarovenko Reagent. J. Fluorine Chem. 1979, 14, 421–428. [Google Scholar] [CrossRef]
- Du, S.; Tian, Z.; Yang, D.; Li, X.; Li, H.; Jia, C.; Che, C.; Wang, M.; Qin, Z. Synthesis, Antifungal Activity and Structure-Activity Relationships of Novel 3-(Difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylic Acid Amides. Molecules 2015, 20, 8395–8408. [Google Scholar] [CrossRef] [PubMed]
- Leroux, P.; Gredt, M.; Leroch, M.; Walker, A.S. Exploring mechanisms of resistance to respiratory inhibitors in field strains of Botrytis cinerea, the causal agent of gray mold. Appl. Environ. Microbiol. 2010, 76, 6615–6630. [Google Scholar] [CrossRef] [PubMed]
- Walter, H. Pyrazole Carboxamide Fungicides Inhibiting Succinate Dehydrogenase. In Bioactive Heterocyclic Compound Classes; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 175–193. [Google Scholar]
- Sierotzki, H.; Scalliet, G. A review of current knowledge of resistance aspects for the next-generation succinate dehydrogenase inhibitor fungicides. Phytopathology 2013, 103, 880–887. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.-B.; Zhou, X.; Ye, Y.-Q.; Wang, P.-Y.; Yang, S. Design, synthesis and insecticidal activities of novel 1-substituted-5-(trifluoromethyl)-1H-pyrazole-4-carboxamide derivatives. Chin. Chem. Lett. 2017, 2, 121–125. [Google Scholar] [CrossRef]
- Veloukas, T.; Karaoglanidis, G.S. Biological activity of the succinate dehydrogenase inhibitor fluopyram against Botrytis cinerea and fungal baseline sensitivity. Pest Manag. Sci. 2012, 68, 858–864. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Simón-Fuentes, A.; Delgado, O.; Román, R. Fluorinated Pyrazoles and Indazoles. In Fluorine in Heterocyclic Chemistry Volume 1: 5-Membered Heterocycles and Macrocycles; Nenajdenko, V., Ed.; Springer: Cham, Switzerland, 2014; pp. 279–321. [Google Scholar]
- Pazenok, S.; Lui, N.; Neeff, A. Process for Preparing 3-Dihalomethylpyrazole-4-Carboxylic Acid Derivatives. WO2008022777, 17 April 2008. [Google Scholar]
- Giornal, F.; Pazenok, S.; Rodefeld, L.; Lui, N.; Vors, J.P.; Leroux, F.R. Synthesis of diversely fluorinated pyrazoles as novel active agrochemical ingredients. J. Fluorine Chem. 2013, 152, 2–11. [Google Scholar] [CrossRef]
- Jaunzems, J.; Braun, M. An Atom-Efficient Route to Ethyl 3-(difluoromethyl)-1-methyl-1H-pyrazole-4-carboxylate (DFMMP)—A Key Building Block for a Novel Fungicide Family. Org. Process Res. Dev. 2014, 18, 1055–1059. [Google Scholar] [CrossRef]
- Graneto, M.J.; Phillips, W.G. 3-Difluoromethyl Pyrazole Carboxamide Fungicides. WO9212970, 6 August 1992. [Google Scholar]
- Nett, M.; Grote, T.; Lohmann, J.K.; Dietz, J.; Smidt, S.P.; Rack, M.; Zierke, T. Method for Producing Difluoromethyl-Substituted Pyrazole Compounds. WO2008152138, 13 June 2008. [Google Scholar]
- Pazenok, S.; Lui, N.; Heinrich, J.D.; Wollner, T. Method for the Regioselective Synthesis of 1-Alkyl-3-Haloalkyl–Pyroazole-4-Carboxylic Acid Derivatives. WO2009106230, 12 February 2009. [Google Scholar]
- Pashkevich, K.I.; Saloutin, V.I.; Fomin, A.N.; Berenblit, V.V.; Plashkin, V.S.; Postovskii, I.Y. Fluoroalkyl-Containing Monopyrazoles and Bispyrazoles. Zh. Vses. Khim. Ova+ 1981, 26, 105–107. [Google Scholar]
- Claire, P.P. K.; Coe, P.L.; Jones, C.J.; Mccleverty, J.A. 3,5-Bis(Trifluoromethyl)Pyrazole and Some N-Substituted Derivatives. J. Fluorine Chem. 1991, 51, 283–289. [Google Scholar] [CrossRef]
- Threadgill, M.D.; Heer, A.K.; Jones, B.G. The Reaction of 1,1,1,5,5,5-Hexafluoropentane-2,4-Dione with Hydrazines—A Reinvestigation. J. Fluorine Chem. 1993, 65, 21–23. [Google Scholar] [CrossRef]
- Sloop, J.C.; Bumgardner, C.L.; Loehle, W.D. Synthesis of fluorinated heterocycles. J. Fluorine Chem. 2002, 118, 135–147. [Google Scholar] [CrossRef]
- Obermayer, D.; Glasnov, T.N.; Kappe, C.O. Microwave-Assisted and Continuous Flow Multistep Synthesis of 4-(Pyrazol-1-yl)carboxanilides. J. Org. Chem. 2011, 76, 6657–6669. [Google Scholar] [CrossRef] [PubMed]
- Maspero, A.; Giovenzana, G.B.; Monticelli, D.; Tagliapietra, S.; Palmisano, G.; Penoni, A. Filling the gap: Chemistry of 3,5-bis(trifluoromethyl)-1H-pyrazoles. J. Fluorine Chem. 2012, 139, 53–57. [Google Scholar] [CrossRef]
- Pazenok, S.; Giornal, F.; Landelle, G.; Lui, N.; Vors, J.P.; Leroux, F.R. A New Life for an Old Reagent: Fluoroalkyl Amino Reagents as Efficient Tools for the Synthesis of Diversely Fluorinated Pyrazoles. Eur. J. Org. Chem. 2013, 2013, 4249–4253. [Google Scholar] [CrossRef]
- Giornal, F.; Landelle, G.; Lui, N.; Vors, J.P.; Pazenok, S.; Leroux, F.R. A New Synthesis and Process Development of Bis(fluoroalkyl)pyrazoles As Novel Agrophores. Org. Process Res. Dev. 2014, 18, 1002–1009. [Google Scholar] [CrossRef]
- Schmitt, E.; Landelle, G.; Vors, J.P.; Lui, N.; Pazenok, S.; Leroux, F.R. A General Approach towards NH-Pyrazoles That Bear Diverse Fluoroalkyl Groups by Means of Fluorinated Iminium Salts. Eur. J. Org. Chem. 2015, 2015, 6052–6060. [Google Scholar] [CrossRef]
- Norris, T.; Colon-Cruz, R.; Ripin, D.H.B. New hydroxy-pyrazoline intermediates, subtle regio-selectivity and relative reaction rate variations observed during acid catalyzed and neutral pyrazole cyclization. Org. Biomol. Chem. 2005, 3, 1844–1849. [Google Scholar] [CrossRef] [PubMed]
- Fustero, S.; Roman, R.; Sanz-Cervera, J.F.; Simon-Fuentes, A.; Bueno, J.; Villanova, S. Synthesis of New Fluorinated Tebufenpyrad Analogs with Acaricidal Activity Through Regioselective Pyrazole Formation. J. Org. Chem. 2008, 73, 8545–8552. [Google Scholar] [CrossRef] [PubMed]
- Bonacorso, H.G.; Porte, L.M.F.; Cechinel, C.A.; Paim, G.R.; Deon, E.D.; Zanatta, N.; Martins, M.A.P. DAST promotes the synthesis of new 5-(trifluoromethyl)-3-(1,1-difluoroethan-2-yl)-1H-pyrazoles. Tetrahedron Lett. 2009, 50, 1392–1394. [Google Scholar] [CrossRef]
- Volochnyuk, D.M.; Pushechnikov, A.O.; Krotko, D.G.; Sibgatulin, D.A.; Kovalyova, S.A.; Tolmachev, A.A. Electron-rich amino heterocycles for regiospecific synthesis of trifluoro-methyl-containing fused pyridines. Synthesis 2003, 2003, 1531–1540. [Google Scholar] [CrossRef]
- Boltacheva, N.S.; Filyakova, V.I.; Charushin, V.N. Fluoroalkyl-containing lithium 1,3-diketonates in reactions with amines and ammonium salts. Russ. J. Org. Chem. 2005, 41, 1452–1457. [Google Scholar] [CrossRef]
- Mormino, M.G.; Fier, P.S.; Hartwig, J.F. Copper-Mediated Perfluoroalkylation of Heteroaryl Bromides with (phen)CuRF. Org. Lett. 2014, 16, 1744–1747. [Google Scholar] [CrossRef] [PubMed]
- Duda, B.; Tverdomed, S.N.; Bassil, B.S.; Roschenthaler, G.V. Synthesis of highly substituted quinolines via heterocyclization of fluorinated acetylenephosphonates with ortho-aminoaryl ketones. Tetrahedron 2014, 70, 8084–8096. [Google Scholar] [CrossRef]
- Aribi, F.; Schmitt, E.; Panossian, A.; Vors, J.-P.; Pazenok, S.; Leroux, F.R. A new approach toward the synthesis of 2,4-bis(fluoroalkyl)-substituted quinoline derivatives using fluoroalkyl amino reagent chemistry. Org. Chem. Front. 2016, 3, 1392–1415. [Google Scholar] [CrossRef]
- Ogura, K.; Ogu, K.-i.; Ayabe, T.; Sonehara, J.-I.; Akazome, M. Stereoselective formation of α-fluoro-α-trifluoromethyl-γ-lactones starting from γ-hydroxy-α,β-unsaturated sulfones and a hexafluoropropene-diethylamine adduct (PPDA). Tetrahedron Lett. 1997, 38, 5173–5176. [Google Scholar] [CrossRef]
- Ogu, K.-i.; Akazome, M.; Ogura, K. A novel reaction of allylic alcohols with hexafluoropropene-diethylamine adduct (PPDA) to form 2-fluoro-2-trifluoromethyl-4-alkenamide. J. Fluorine Chem. 2003, 124, 69–80. [Google Scholar] [CrossRef]
- Ogu, K.; Akazome, M.; Ogura, K. Diastereoselective formation of 2-fluoro-2-trifluoromethyl-3,4-alkadienamides from propargyl alcohols and hexafluoropropene–diethylamine adduct (PPDA). J. Fluorine Chem. 2004, 125, 429–438. [Google Scholar] [CrossRef]
- Ogu, K.; Matsumoto, S.; Akazome, M.; Ogura, K. Novel Synthesis of α-Trifluoromethylated α-Amino Acid Derivatives from γ-Hydroxy-α-fluoro-α-trifluoromethyl Carboxamides. Org. Lett. 2005, 7, 589–592. [Google Scholar] [CrossRef] [PubMed]
- Takaoka, A.; Ibrahim, M.K.; Kagaruki, S.R.F.; Ishikawa, N. Synthesis of Monofluoro Heterocycles Using Fluoroolefins as Starting Materials. Nippon Kagaku Kaishi 1985, 1985, 2169–2176. [Google Scholar] [CrossRef]










© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Commare, B.; Schmitt, E.; Aribi, F.; Panossian, A.; Vors, J.-P.; Pazenok, S.; Leroux, F.R. Fluoroalkyl Amino Reagents (FARs): A General Approach towards the Synthesis of Heterocyclic Compounds Bearing Emergent Fluorinated Substituents. Molecules 2017, 22, 977. https://doi.org/10.3390/molecules22060977
Commare B, Schmitt E, Aribi F, Panossian A, Vors J-P, Pazenok S, Leroux FR. Fluoroalkyl Amino Reagents (FARs): A General Approach towards the Synthesis of Heterocyclic Compounds Bearing Emergent Fluorinated Substituents. Molecules. 2017; 22(6):977. https://doi.org/10.3390/molecules22060977
Chicago/Turabian StyleCommare, Bruno, Etienne Schmitt, Fallia Aribi, Armen Panossian, Jean-Pierre Vors, Sergiy Pazenok, and Frédéric R. Leroux. 2017. "Fluoroalkyl Amino Reagents (FARs): A General Approach towards the Synthesis of Heterocyclic Compounds Bearing Emergent Fluorinated Substituents" Molecules 22, no. 6: 977. https://doi.org/10.3390/molecules22060977