
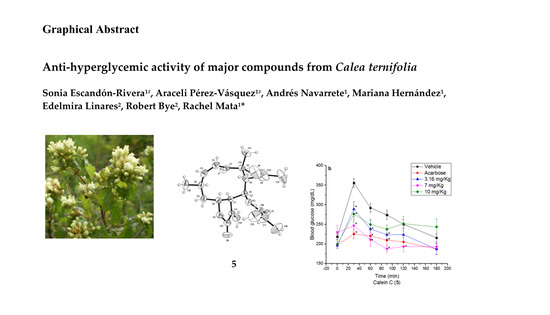
Anti-Hyperglycemic Activity of Major Compounds from Calea ternifolia

 , ,
, , 
Abstract
:
1. Introduction
2. Results and Discussion
2.1. Acute Toxicity Study in Mice
2.2. Isolation of Compounds
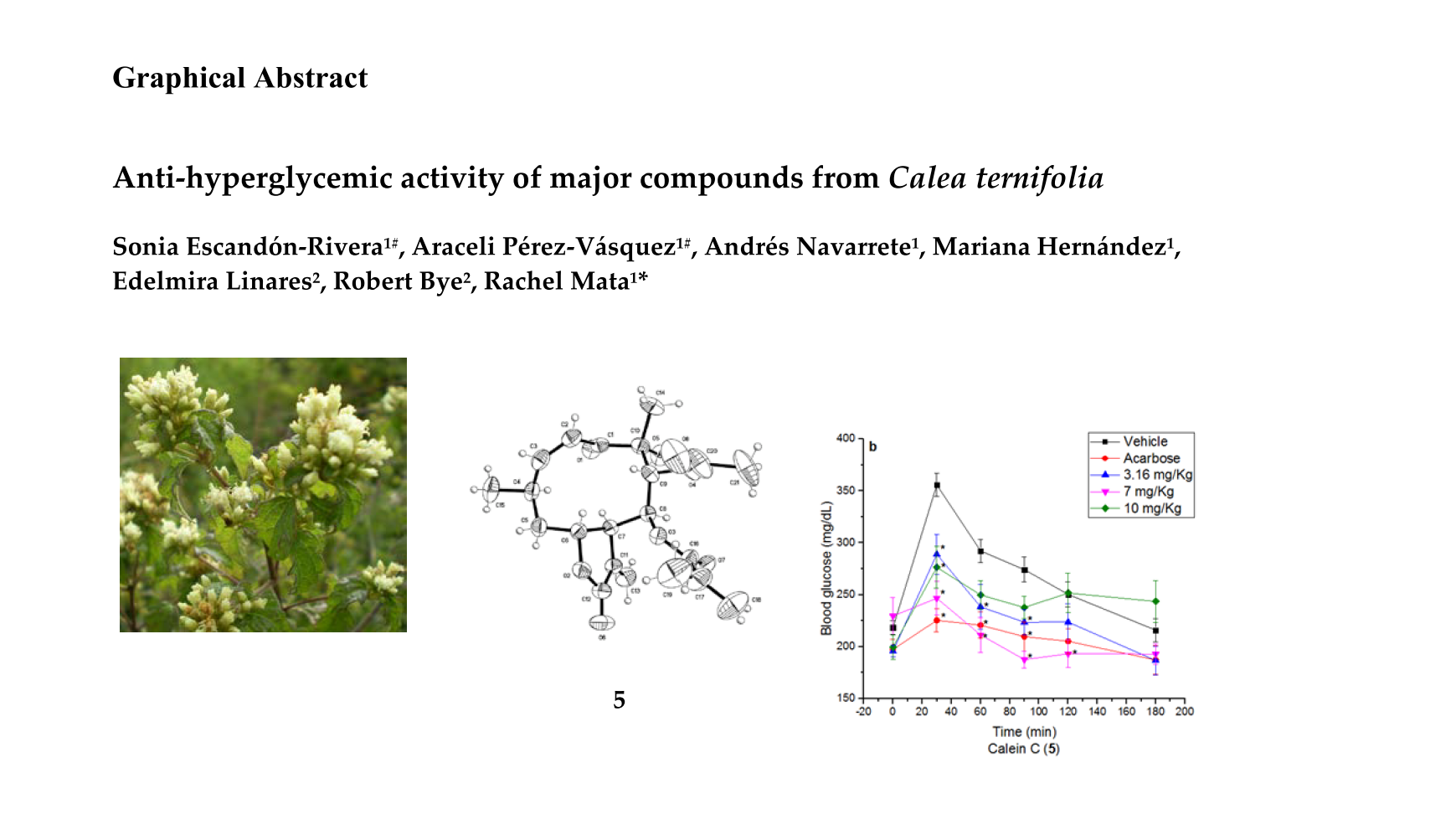
2.3. Oral Sucrose Tolerance Test of Compounds 1, 4, 5 and Essential Oil
2.4. Essential Oil Composition
2.5. HS-SPME Analysis
2.6. Quantification of the Active Markers 1 and 2
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. General Experimental Procedures
3.3. Plant Material
3.4. Preparation of the Extracts and Isolation of the Metabolites
3.5. Preparation of the Chromene-Rich Fraction
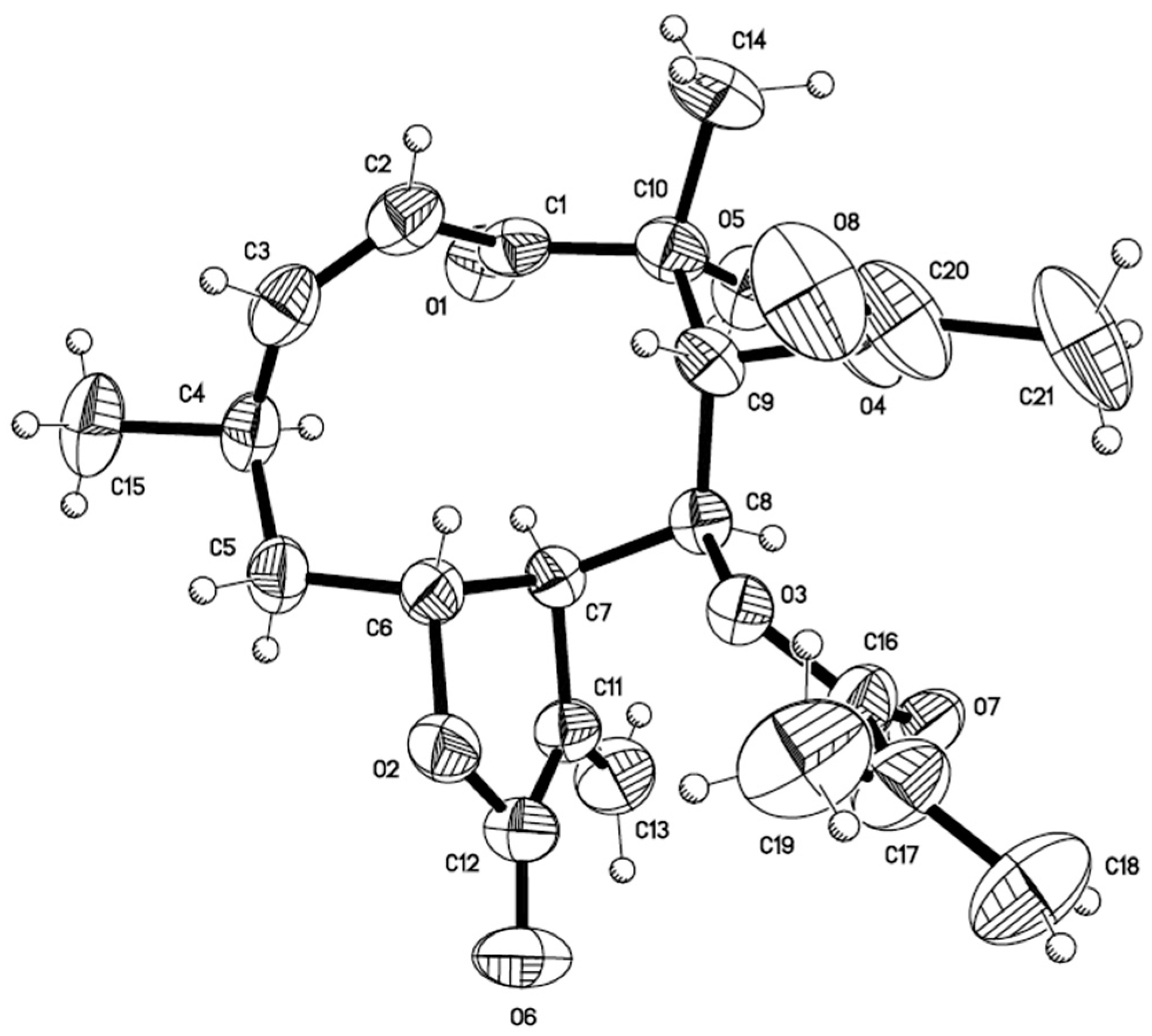
3.6. X-ray Crystallographic Data of Calein C (5)
3.7. Volatile Composition
3.7.1. Essential Oil
3.7.2. Headspace Solid-Phase Microextraction
3.7.3. Gas Chromatography-Mass Spectrometry Analysis
3.8. HPLC Analysis
3.9. HPLC Method Validation
3.10. In Vivo Assays
3.10.1. Experimental Animals
3.10.2. Nicotinamide-Streptozotocin (NA-STZ) Experimental Induced Hyperglicemia in Mice
3.10.3. Oral Sucrose Tolerance Test
3.10.4. Statistical Analysis
3.10.5. Acute Toxicity Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Andrade-Cetto, A.; Heinrich, M. Mexican plants with hypoglycaemic effect used in treatment of diabetes. J. Ethnopharmacol. 2005, 99, 325–348. [Google Scholar] [CrossRef] [PubMed]
- Leonti, M.; Sticher, O.; Heinrich, M. Antiquity of medicinal plant usage in two macro-mayan ethnic groups (México). J. Ethnopharmacol. 2003, 88, 119–124. [Google Scholar] [CrossRef]
- Ramos, R.; Alarcón-Aguilar, F.; Lara-Lemus, A.; Flores-Saenz, J.L. Hypoglycemic effect of plants used in Mexico as antidiabetics. Arch. Med. Res. 1992, 23, 59–64. [Google Scholar]
- Bork, P.M.; Schmitz, M.L.; Kuhnt, M.; Escher, C.; Heinrich, M. Sesquiterpene lactone containing Mexican Indian medicinal plants and pure sesquiterpene lactones as potent inhibitors of transcription factor NF-κB. FEBS Lett. 1997, 402, 85–90. [Google Scholar] [CrossRef]
- Venegas-Flores, H.; Segura-Cobos, D.; Vázquez-Cruz, B. Antiinflammatory activity of the aqueous extract of Calea zacatechichi. Proc. West Pharmacol. Soc. 2002, 45, 110–111. [Google Scholar] [PubMed]
- Köhler, I.; Jenett-Siems, K.; Siems, K.; Hernández, M.A.; Ibarra, R.A.; Berendsohn, W.G.; Bienzle, U.; Eich, E. In vitro Antiplasmodial investigation of medicinal plants from el Salvador. Z. Naturforsch. 2002, 57c, 277–281. [Google Scholar] [CrossRef]
- Wu, H.; Fronczek, F.R.; Burandt, C.L.; Zjawiony, J.K. Antileishmanial germacranolides from Calea zacatechichi. Planta. Med. 2011, 77, 749–753. [Google Scholar] [CrossRef] [PubMed]
- Salaga, M.; Kowalczuk, A.; Zielinska, M.; Blazewicz, A.; Fichna, J. Calea zacatechichi dichloromethane extract exhibits antidiarrheal and antinociceptive effects in mouse models mimicking irritable bowel syndrome. Naunyn-Schmiedeberg Planta Med. 2015, 388, 1069–1077. [Google Scholar] [CrossRef] [PubMed]
- Salaga, M.; Fichna, J.; Socala, K.; Nieoczym, D.; Pieróg, M.; Zielinska, M.; Kowalczuk, A.; Wlaz, P. Neuropharmacological characterization of the oneirogenic Mexican plant Calea zacatechichi aqueous extract in mice. Metab. Brain Dis. 2016, 31, 631–641. [Google Scholar] [CrossRef] [PubMed]
- Mossoba, M.E.; Flynn, T.J.; Vohra, S.; Wiesenfeld, P.; Sprando, R.L. Evaluation of (Dream Herb,) Calea zacatechichi, for nephrotoxicity using human kidney proximal tubule cells. J. Toxicol. 2016, 9794570. [Google Scholar] [CrossRef] [PubMed]
- Escandón-Rivera, S.; Gonzalez-Andrade, M.; Bye, R.; Linares, E.; Navarrete, A.; Mata, R. α-Glucosidase inhibitors from Brickellia cavanillesii. J. Nat. Prod. 2012, 75, 968–974. [Google Scholar] [CrossRef] [PubMed]
- Escandón-Rivera, S.; Gonzalez-Andrade, M.; Bye, R.; Linares, E.; Navarrete, A.; Mata, R. Correction to α-glucosidase inhibitors from Brickellia cavanillesii. J. Nat. Prod. 2017. [Google Scholar] [CrossRef] [PubMed]
- Mata, R.; Cristians, S.; Escandón-Rivera, S.; Juárez-Reyes, K.; Rivero-Cruz, I. Mexican antidiabetic herbs: Valuable sources of inhibitors of α-glucosidases. J. Nat. Prod. 2013, 76, 468–483. [Google Scholar] [CrossRef] [PubMed]
- Quijano, L.; Romo de Vivar, A.; Rios, T. Revision of the structures of calein A and B, germacranolide sesquiterpenes from Calea zacatechichi. Phytochemistry 1979, 18, 1745–1747. [Google Scholar] [CrossRef]
- Lee, I.Y.; Olivier, E.J.; Urbatsch, L.E.; Fischer, N.H. Two sesquiterpene lactones of Calea ternifolia Var. calyculata. Phytochemistry 1982, 21, 2313–2316. [Google Scholar]
- Martinez, M.; Esquivel, B.; Ortega, A. Two caleins from Calea zacatechichi. Phytochemistry 1987, 26, 2104–2106. [Google Scholar] [CrossRef]
- Herz, W.; Kumar, N. Sesquiterpene lactones of Calea zacatechichi and C. Urticifolia. Phytochemistry 1980, 19, 593–597. [Google Scholar] [CrossRef]
- Ober, A.G.; Urbatsch, L.E.; Fischer, N.H. Guaianolides and chromenes from Calea species. Phytochemistry 1985, 24, 795–799. [Google Scholar] [CrossRef]
- IDF Diabetes Atlas, 7th ed. International Diabetes Federation. Available online: http://www.diabetesatlas.org/ (accessed on 1 February 2017).
- WHO Expert Committee on Specifications for Pharmaceutical Preparations—WHO Technical Report Series, No. 863, Annex 11 (Guidelines for the Assessment of Herbal Medicines)—Thirty-fourth Report. Available online: http://apps.who.int/medicinedocs/en/d/Jh2984e/#Jh2984e (accessed on 7 February 2017).
- Lorke, D.A. New approach to practical acute toxicity testing. Arch. Toxicol. 1983, 54, 275–287. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, S.K.; Verma, S.; Singh, S.S.; Tripathi, A.K.; Khan, Z.K.; Kmar, S. Antifeedant and antifungal activity of chromene compounds isolated from Blepharispermum subsessile. J. Ethnopharmacol. 2000, 71, 231–234. [Google Scholar] [CrossRef]
- Kulkarni, M.M.; Nagasampagi, B.A.; Deshpande, S.G.; Sharma, R.N. Five chromenes from Blepharispermum subsessile. Phytochemistry 1987, 26, 2969–2971. [Google Scholar] [CrossRef]
- Elgamal, M.H.A.; Elewa, N.H.; Elkhrisy, A.M.; Duddeck, H. 13C NMR Chemical shifts and carbon-proton coupling constants of some furocoumarins and furochromones. Phytochemistry 1979, 18, 139–143. [Google Scholar] [CrossRef]
- Ma, C.H.; Ke, W.; Sun, Z.L.; Peng, J.Y.; Li, Z.X.; Zhou, X.; Fan, G.R.; Huang, C.G. Large-Scale isolation and purification of scoparone from herba Artemisiae scopariae by high-speed counter-current chromatography. Chromatographia 2006, 64, 83–87. [Google Scholar] [CrossRef]
- Bandoni, A.L.; Medina, J.E.; Rondina, R.V.D.; Conssio, J.D. Genus Baccharis L.I. Phytochemical analysis of a non polar fraction from B. crispa. Planta Med. 1978, 34, 328–331. [Google Scholar] [CrossRef]
- Suzuki, K.; Katsura, D.; Sagara, M.; Aoki, C.; Nishida, M.; Aso, Y. Postprandial reactive hypoglycemia treated with a low-dose alpha-glucosidase Inhibitor: Voglibose may suppress oxidative stress and prevent endothelial dysfunction. Intern. Med. 2016, 55, 949–953. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.X.; Chen, Y.S.; Zhang, W.R.; Chen, B.; Qiu, X.M.; He, L.H.; Mu, L.L.; Yang, C.H.; Chen, R. polysaccharide from Gynura divaricata modulates the activities of intestinal disaccharidases in streptozotocin-induced diabetic rats. Br. J. Nutr. 2011, 106, 1323–1329. [Google Scholar] [CrossRef] [PubMed]
- Dudeja, P.K.; Wali, R.K.; Klitzke, A.; Brasitus, A. Intestinal d-glucose transport and membrane fluidity along crypt-villus axis of streptozocin-induced diabetic rats. Am. J. Physiol. Gastrointest. Liver Physiol. 1990, 259, G571–G577. [Google Scholar]
- Liu, L.; Yu, Y.L.; Liu, C.; Wang, X.T.; Liu, X.D.; Xie, L. Insulin deficiency induces abnormal increase in intestinal disaccharidase activities and expression under diabetic states, evidences from in vivo and in vitro study. Biochem. Pharmacol. 2011, 82, 1963–1970. [Google Scholar] [CrossRef] [PubMed]
- Islam, M.S.; Loots du, T. Experimental rodent models of type 2 diabetes: A review. Methods Find. Exp. Clin. Pharmacol. 2009, 31, 249–261. [Google Scholar] [CrossRef] [PubMed]
- Bösenberg, L.H.; van Zyl, D.G. The mechanism of action of oral antidiabetic drugs: A review of recent literature. J. Endocrinol. Metab. Diabetes S. Afr. 2008, 13, 80–88. [Google Scholar] [CrossRef]
- ICH. Text on Validation of Analytical Procedures. Harmonized Tripartite Guideline [Q2(R1)]. In International Conference on Harmonization, Geneva, Switzerland, 1–13 November 2005; Available online: http://www.ich.org/fileadmin/Public_Web_Site/ICH_Products/Guidelines/Quality/Q2_R1/Step4/Q2_R1__Guideline.pdf (accessed on 15 May 2016).
- NOM-062-ZOO-1999. Especificaciones Técnicas Para la Producción, Cuidado y uso de los Animales de Laboratorio. Available online: http://www.economia-noms.gob.mx/normas/noms/2001/062zoo.pdf (accessed on 12 February 2015).
- Fröde, T.S.; Medeiros, Y.S. Animal models to test drugs with potential antidiabetic activity. J. Ethnopharmacol. 2008, 115, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Masiello, P.; Broca, C.; Gross, R.; Roye, M.; Manteghetti, M.; Hillaire-Buys, D.; Novelli, M.; Ribes, G. Experimental NIDDM: Development of a new model in adult rats administered streptozotocin and nicotinamide. Diabetes 1998, 47, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Wojcikowski, K.; Gobe, G. Animal studies on medicinal herbs: Predictability, dose conversion and potential value. Phytother. Res. 2014, 28, 22–27. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.
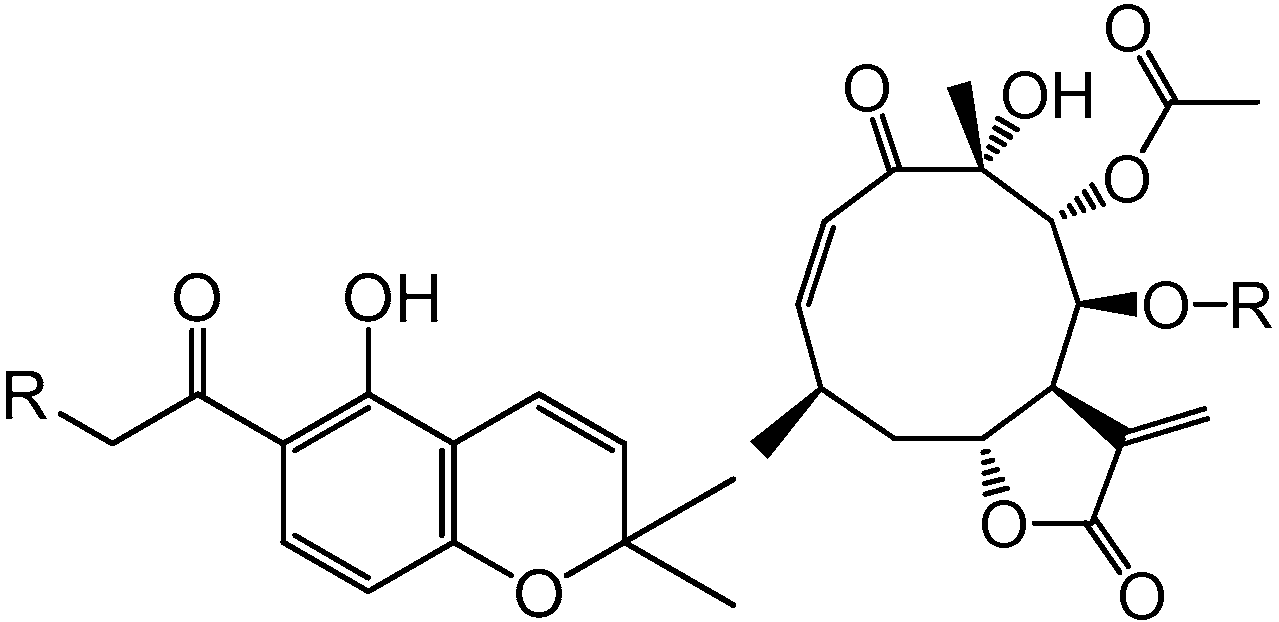
| R | R | |||
| 1 | H | 4 | CH2=C(CH3)CO | |
| 2 | OH | 5 | CH3CH=C(CH3)CO |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Compounds | IR | DVB/CAR/PDMS | PDMS | CAR/PDMS | PDMS/DVB |
|---|---|---|---|---|---|---|
| 12 | α-Pinene | 996 | 4.25 | 0.44 | 3.55 | 3.19 |
| 13 | Camphene | 1018 | 4.35 | 0.45 | 2.56 | 1.60 |
| 14 | β-Pinene | 1046 | 7.19 | 0.82 | 2.41 | 1.21 |
| 42 | o-Cymene | 1109 | 0.95 | - | - | 5.45 |
| 43 | d-Limonene | 1111 | 6.80 | - | - | 5.45 |
| 15 | Eucalyptol | 1113 | 1.91 | 0.36 | 2.00 | 5.45 |
| 16 | Camphor | 1242 | 4.06 | 1.79 | 9.89 | 5.10 |
| 17 | Pinocarvone | 1261 | 1.40 | - | 1.84 | 0.88 |
| 18 | 1,2,3-Trimethylcyclopentane | 1277 | - | 0.31 | 2.75 | 1.22 |
| 19 | S-Pinocamphone | 1278 | 1.16 | 0.29 | 2.75 | - |
| 44 | 4-Carvomenthol | 1288 | 0.32 | - | - | |
| 45 | Thymol | 1429 | 0.25 | - | - | - |
| 46 | α-Cubebene | 1526 | - | 3.83 | - | - |
| 20 | β-Caryophyllene | 1527 | 3.31 | 3.83 | 4.01 | 1.99 |
| 21 | β-Cubebene | 1539 | 1.17 | 1.24 | - | - |
| 22 | α-Ocimene | 1563 | 0.98 | 4.07 | 1.79 | |
| 23 | Germacrene D | 1580 | 1.33 | 0.41 | 4.07 | 1.55 |
| 24 | Curcumene | 1591 | 5.02 | 10.90 | 1.04 | 8.90 |
| 47 | α-Zingiberene | 1604 | 1.11 | 1.80 | - | - |
| 25 | α-Selinene | 1607 | 0.66 | 0.79 | 1.19 | - |
| 26 | Germacrene A | 1627 | 0.67 | 1.25 | 0.42 | - |
| 27 | δ-Cadinene | 1632 | 1.79 | - | 2.71 | 2.61 |
| 28 | Calamenene | 1639 | 0.33 | 0.81 | 1.42 | 0.88 |
| 29 | cis-α-Farnesene | 1641 | 1.37 | 1.83 | - | 0.50 |
| 30 | α-Calacorene | 1666 | 0.11 | 0.01 | - | 0.08 |
| 31 | Hedicariol | 1678 | 1.36 | 1.22 | 2.18 | - |
| 32 | (E)-Nerolidol | 1682 | 2.68 | 1.97 | 1.61 | 1.94 |
| 33 | Aromadendrene oxide 2 | 1698 | 0.22 | 0.13 | - | - |
| 34 | Spathulenol | 1714 | 4.40 | 7.58 | 3.83 | 4.12 |
| 35 | Caryophyllene oxide | 1716 | 3.77 | 7.69 | 3.54 | 5.02 |
| 48 | trans-Chrysanthemal | 1723 | - | 7.64 | 14.35 | 1.89 |
| 49 | Carotol | 1738 | 3.15 | 4.38 | 4.00 | 7.00 |
| 50 | Humulene epoxide II | 1748 | 0.87 | 0.93 | - | - |
| 51 | α-Muurolol | 1780 | - | 2.73 | - | - |
| 38 | τ-Cadinol | 1783 | 2.90 | - | - | 3.31 |
| 39 | β-Eudesmol | 1800 | 1.12 | 3.81 | 2.14 | 5.49 |
| 40 | α-Bisabolol | 1829 | 0.52 | - | - | - |
| 41 | Ledene oxide II | 1843 | 0.56 | - | - | - |
| 1 | Demethylisoencecalin | 1883 | 2.68 | 10.68 | - | 8.48 |
| Rt | Linear Range (mg/mL) | Calibration Equation | R2 a | LOD (mg/mL) | LOQ (mg/mL) | Precision | Recovery (%mean) | Stability (%RSD) | ||
|---|---|---|---|---|---|---|---|---|---|---|
| Intraday (%RSD) | Interday (%RSD) | |||||||||
| 1 | 18.8 | 20–65 | y = 206712x − 640270 | 0.9991 | 0.3 | 0.8 | 0.7 | 0.6 | 100.20 | 0.91 |
| 2 | 7.0 | 10–80 | y = 190874x − 138782 | 0.9997 | 0.1 | 0.3 | 0.9 | 0.9 | 100.03 | 0.20 |
| Batch | % CRF a | Content in mg/mg b | |
|---|---|---|---|
| 1 | 2 | ||
| I | 0.04 | 21.91 ± 1.07 | 22.74 ± 1.62 |
| II | 0.17 | 7.74 ± 0.89 | 28.99 ± 1.23 |
| III | 0.13 | 58.04 ± 1.62 | 40.90 ± 1.10 |
| IV | 0.14 | 8.46 ± 1.48 | 5.04 ± 1.28 |
| V | 0.33 | nd | nd |
| VI | 0.38 | nd | nd |
| VII | 0.32 | nd | nd |
| VIII | 0.24 | nd | nd |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Escandón-Rivera, S.; Pérez-Vásquez, A.; Navarrete, A.; Hernández, M.; Linares, E.; Bye, R.; Mata, R. Anti-Hyperglycemic Activity of Major Compounds from Calea ternifolia. Molecules 2017, 22, 289. https://doi.org/10.3390/molecules22020289
Escandón-Rivera S, Pérez-Vásquez A, Navarrete A, Hernández M, Linares E, Bye R, Mata R. Anti-Hyperglycemic Activity of Major Compounds from Calea ternifolia. Molecules. 2017; 22(2):289. https://doi.org/10.3390/molecules22020289
Chicago/Turabian StyleEscandón-Rivera, Sonia, Araceli Pérez-Vásquez, Andrés Navarrete, Mariana Hernández, Edelmira Linares, Robert Bye, and Rachel Mata. 2017. "Anti-Hyperglycemic Activity of Major Compounds from Calea ternifolia" Molecules 22, no. 2: 289. https://doi.org/10.3390/molecules22020289
APA StyleEscandón-Rivera, S., Pérez-Vásquez, A., Navarrete, A., Hernández, M., Linares, E., Bye, R., & Mata, R. (2017). Anti-Hyperglycemic Activity of Major Compounds from Calea ternifolia. Molecules, 22(2), 289. https://doi.org/10.3390/molecules22020289