A Systematic Review of Computational Drug Discovery, Development, and Repurposing for Ebola Virus Disease Treatment


Abstract
:1. Introduction
1.1. Rationale
1.2. Objectives
2. Results
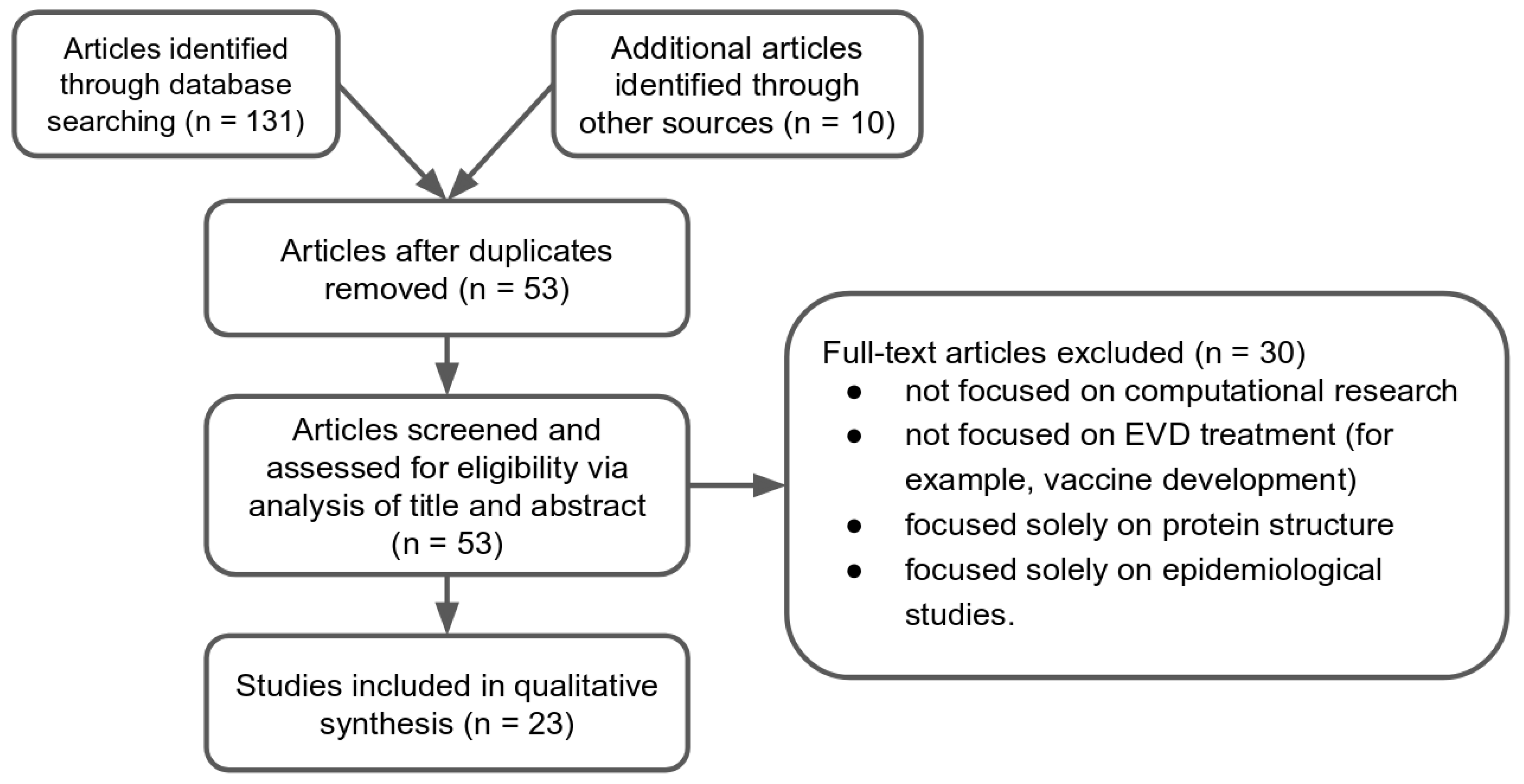
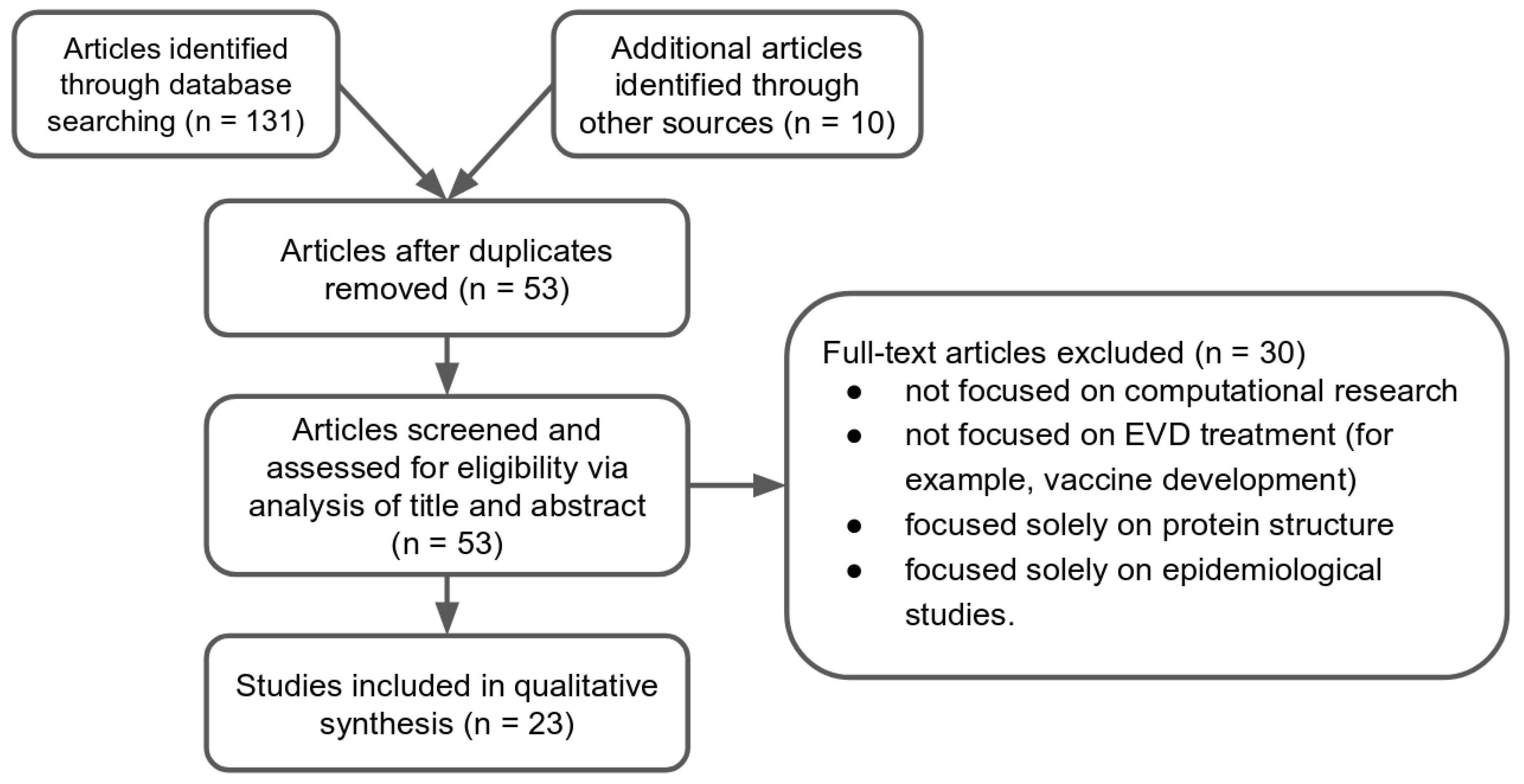
2.1. Study Selection
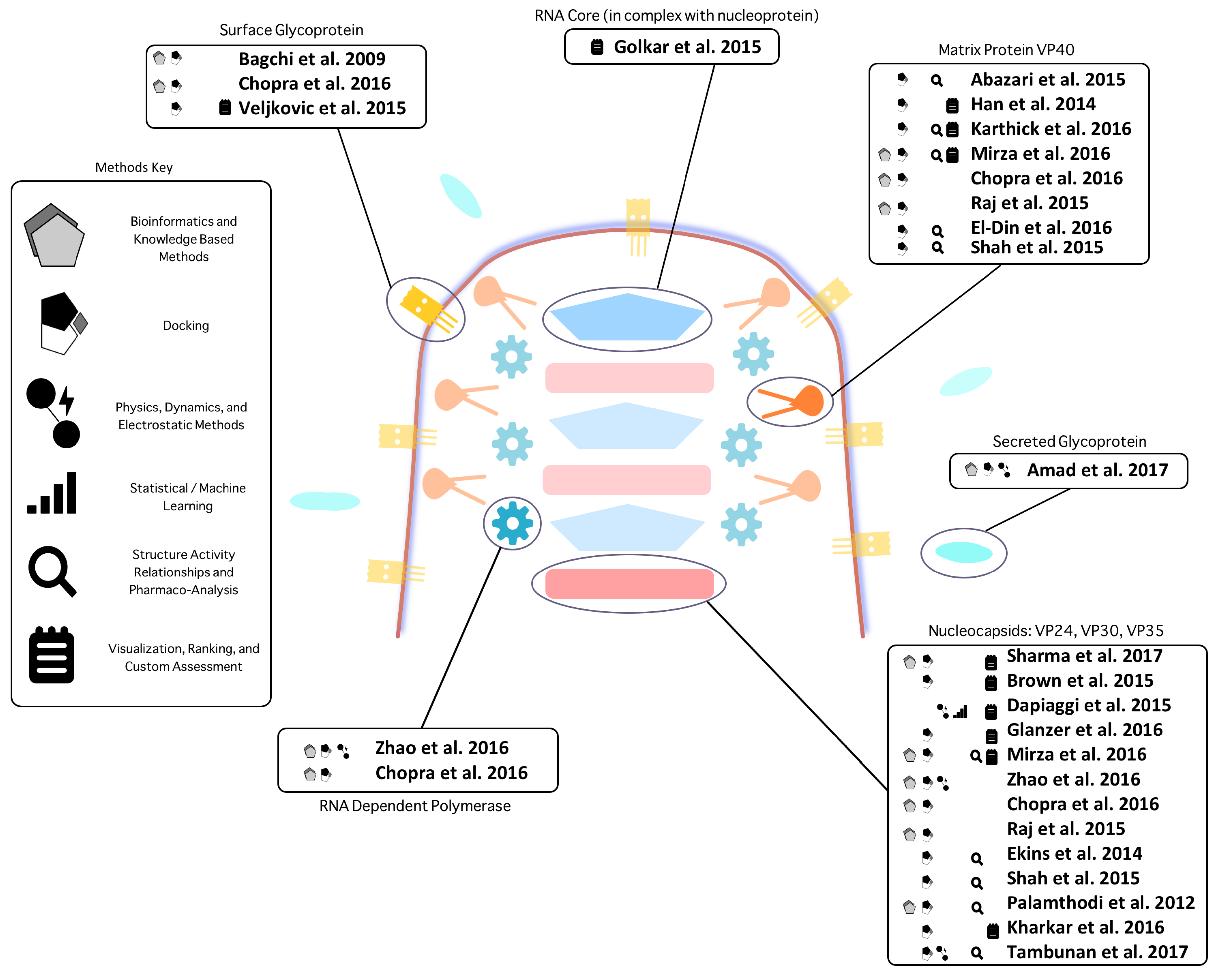
2.2. Study Characteristics
- Physics, dynamics, and electrostatic methods.
- Structure activity relationships and pharmacoanalysis.
- Bioinformatics and knowledge based methods.
- Statistical and machine learning methods.
- Visualization, ranking, and custom assessment.
3. Discussion
3.1. Synthesis of Results
3.2. Limitations
4. Materials and Methods
4.1. Protocol and Registration
4.2. Eligibility Criteria
4.3. Outcomes
4.4. Study Information Sources
4.5. Search Terms
4.6. Study Selection
4.7. Data Collection Process
4.8. Data Items
4.9. Bias in Individual Studies
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| ADME | absorption distribution metabolism excretion |
| ADMET | absorption distribution metabolism excretion toxicity |
| BSL-4 | biosafety level four |
| EBOV | Ebola virus |
| EVD | Ebola virus disease |
| NMR | Nuclear magnetic resonance |
| PDB | Protein Data Bank |
| QSAR | Quantitative structure-activity relationship |
| RAM | Random access memory |
| RG | ResearchGate |
| RMSD | Root mean square deviation |
| SVM | Support vector machine |
References
- 2014 Ebola Outbreak in West Africa—Case Counts. Available online: https://www.cdc.gov/vhf/ebola/outbreaks/2014-west-africa/case-counts.html (accessed on 25 July 2017).
- 2017 Democratic Republic of the Congo, Bas Uélé District. Available online: https://www.cdc.gov/vhf/ebola/outbreaks/drc/2017-may.html (accessed on 5 August 2017).
- World Health Organization; Regional Office for Africa, Health Emergencies Programme. Ebola Virus Disease Democratic Republic of Congo: External Situation Report; Technical Report 26; World Health Organization, Regional Office for Africa: Brazzaville, Republic of the Congo, 2017. [Google Scholar]
- Group, T.P.I.W. A randomized, controlled trial of ZMapp for Ebola virus infection. N. Engl. J. Med. 2016, 375, 1448–1456. [Google Scholar] [CrossRef] [PubMed]
- Van Griensven, J.; De Weiggheleire, A.; Delamou, A.; Smith, P.G.; Edwards, T.; Vandekerckhove, P.; Bah, E.I.; Colebunders, R.; Herve, I.; Lazaygues, C.; et al. The use of Ebola convalescent plasma to treat Ebola virus disease in resource-constrained settings: A perspective from the field. Clin. Infect. Dis. 2016, 62, 69–74. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A. New drug development in the United States from 1963 to 1999. Clin. Pharmacol. Ther. 2001, 69, 286–296. [Google Scholar] [CrossRef] [PubMed]
- DiMasi, J.A.; Hansen, R.W.; Grabowski, H.G. The price of innovation: New estimates of drug development costs. J. Health Econom. 2003, 22, 151–185. [Google Scholar] [CrossRef]
- Recognizing the Biosafety Levels. Available online: https://www.cdc.gov/training/quicklearns/biosafety/ (accessed on 25 July 2017).
- Drug Repurposing at NCATS. Available online: https://ncats.nih.gov/preclinical/repurpose (accessed on 25 July 2017).
- Langedijk, J.; Mantel-Teeuwisse, A.K.; Slijkerman, D.S.; Schutjens, M.H.D. Drug repositioning and repurposing: Terminology and definitions in literature. Drug Discov. Today 2015, 20, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.S.; Patel, C.J. A review of validation strategies for computational drug repositioning. Brief. Bioinform. 2016. [Google Scholar] [CrossRef]
- Zhao, Z.; Martin, C.; Fan, R.; Bourne, P.E.; Xie, L. Drug repurposing to target Ebola virus replication and virulence using structural systems pharmacology. BMC Bioinform. 2016, 17, 90. [Google Scholar] [CrossRef] [PubMed]
- Sliwoski, G.; Kothiwale, S.; Meiler, J.; Lowe, E.W. Computational methods in drug discovery. Pharmacol. Rev. 2014, 66, 334–395. [Google Scholar] [CrossRef] [PubMed]
- Ou-Yang, S.s.; Lu, J.y.; Kong, X.q.; Liang, Z.j.; Luo, C.; Jiang, H. Computational drug discovery. Acta Pharmacol. Sin. 2012, 33, 1131. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Zheng, S.; Chen, B.; Butte, A.J.; Swamidass, S.J.; Lu, Z. A survey of current trends in computational drug repositioning. Brief. Bioinform. 2015, 17, 2–12. [Google Scholar] [CrossRef] [PubMed]
- Kapetanovic, I. Computer-aided drug discovery and development (CADDD): In silico-chemico-biological approach. Chem. Biol. Inter. 2008, 171, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Clark, D.E.; Pickett, S.D. Computational methods for the prediction of ‘drug-likeness’. Drug Discov. Today 2000, 5, 49–58. [Google Scholar] [CrossRef]
- Dopazo, J. Genomics and transcriptomics in drug discovery. Drug Discov. today 2014, 19, 126–132. [Google Scholar] [CrossRef] [PubMed]
- March-Vila, E.; Pinzi, L.; Sturm, N.; Tinivella, A.; Engkvist, O.; Chen, H.; Rastelli, G. On the integration of in silico drug design methods for drug repurposing. Front. Pharmacol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Shoichet, B.K.; Walters, W.P.; Jiang, H.; Bajorath, J. Advances in computational medicinal chemistry: A reflection on the evolution of the field and perspective going forward. J. Med. Chem. 2016, 59, 4033–4034. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, A.L.; Trincão, J.; Romão, M.J. X-ray crystallography in drug discovery. In Ligand-Macromolecular Interactions in Drug Discovery: Methods and Protocols; Roque, A.C.A., Ed.; Humana Press: New York, NY, USA, 2010; pp. 31–56. [Google Scholar]
- Pellecchia, M.; Bertini, I.; Cowburn, D.; Dalvit, C.; Giralt, E.; Jahnke, W.; James, T.L.; Homans, S.W.; Kessler, H.; Luchinat, C.; et al. Perspectives on NMR in drug discovery: A technique comes of age. Nat. Rev. Drug Discov. 2008, 7, 738. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-Pdb Viewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Kucukural, A.; Zhang, Y. I-TASSER: A unified platform for automated protein structure and function prediction. Nat. Protocols 2010, 5, 725. [Google Scholar] [CrossRef] [PubMed]
- Laurie, A.T.; Jackson, R.M. Q-SiteFinder: An energy-based method for the prediction of protein–ligand binding sites. Bioinformatics 2005, 21, 1908–1916. [Google Scholar] [CrossRef] [PubMed]
- Willett, P. Similarity-based virtual screening using 2D fingerprints. Drug Discov. Today 2006, 11, 1046–1053. [Google Scholar] [CrossRef] [PubMed]
- Katara, P. Role of bioinformatics and pharmacogenomics in drug discovery and development process. Netw. Model. Anal. Health Inform. Bioinform. 2013, 2, 225–230. [Google Scholar] [CrossRef]
- Lamb, J.; Crawford, E.D.; Peck, D.; Modell, J.W.; Blat, I.C.; Wrobel, M.J.; Lerner, J.; Brunet, J.P.; Subramanian, A.; Ross, K.N.; et al. The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 2006, 313, 1929–1935. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y. Pharmacophore modeling and applications in drug discovery: Challenges and recent advances. Drug Discov. Today 2010, 15, 444–450. [Google Scholar] [CrossRef] [PubMed]
- Khedkar, S.A.; Malde, A.K.; Coutinho, E.C.; Srivastava, S. Pharmacophore modeling in drug discovery and development: An overview. Med. Chem. 2007, 3, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Cherkasov, A.; Muratov, E.N.; Fourches, D.; Varnek, A.; Baskin, I.I.; Cronin, M.; Dearden, J.; Gramatica, P.; Martin, Y.C.; Todeschini, R.; et al. QSAR modeling: Where have you been? Where are you going to? J. Med. Chem. 2014, 57, 4977–5010. [Google Scholar] [CrossRef] [PubMed]
- Perkins, R.; Fang, H.; Tong, W.; Welsh, W.J. Quantitative structure-activity relationship methods: Perspectives on drug discovery and toxicology. Environ. Toxicol. Chem. 2003, 22, 1666–1679. [Google Scholar] [CrossRef] [PubMed]
- Kitchen, D.B.; Decornez, H.; Furr, J.R.; Bajorath, J. Docking and scoring in virtual screening for drug discovery: Methods and applications. Nat. Rev. Drug Discov. 2004, 3, 935. [Google Scholar] [CrossRef] [PubMed]
- Hert, J.; Willett, P.; Wilton, D.J.; Acklin, P.; Azzaoui, K.; Jacoby, E.; Schuffenhauer, A. New methods for ligand-based virtual screening: Use of data fusion and machine learning to enhance the effectiveness of similarity searching. J. Chem. Inf. Model. 2006, 46, 462–470. [Google Scholar] [CrossRef] [PubMed]
- Medina-Franco, J.L.; Martínez-Mayorga, K.; Giulianotti, M.A.; Houghten, R.A.; Pinilla, C. Visualization of the chemical space in drug discovery. Curr. Comput. Aided Drug Des. 2008, 4, 322–333. [Google Scholar] [CrossRef]
- Lavecchia, A. Machine-learning approaches in drug discovery: Methods and applications. Drug Discov. Today 2015, 20, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balmith, M.; Faya, M.; Soliman, M.E. Ebola virus: A gap in drug design and discovery-experimental and computational perspective. Chem. Biol. Drug Des. 2016. [Google Scholar] [CrossRef] [PubMed]
- Suvannang, N.; Nantasenamat, C.; Isarankura-Na-Ayudhya, C.; Prachayasittikul, V. Molecular docking of aromatase inhibitors. Molecules 2011, 16, 3597–3617. [Google Scholar] [CrossRef]
- Jenwitheesuk, E.; Horst, J.A.; Rivas, K.L.; Van Voorhis, W.C.; Samudrala, R. Novel paradigms for drug discovery: Computational multitarget screening. Trends Pharmacol. Sci. 2008, 29, 62–71. [Google Scholar] [CrossRef] [PubMed]
- Méndez-Lucio, O.; Naveja, J.J.; Vite-Caritino, H.; Prieto-Martínez, F.D.; Medina-Franco, J.L. One drug for multiple targets: A computational perspective. J. Mex. Chem. Soc. 2016, 60. [Google Scholar]
- Overington, J.P.; Al-Lazikani, B.; Hopkins, A.L. How many drug targets are there? Nat. Rev. Drug Discov. 2006, 5, 993–996. [Google Scholar] [CrossRef] [PubMed]
- Imming, P.; Sinning, C.; Meyer, A. Drugs, their targets and the nature and number of drug targets. Nat. Rev. Drug Discov. 2006, 5, 821–834. [Google Scholar] [CrossRef] [PubMed]
- Campillos, M.; Kuhn, M.; Gavin, A.C.; Jensen, L.J.; Bork, P. Drug target identification using side-effect similarity. Science 2008, 321, 263–266. [Google Scholar] [CrossRef] [PubMed]
- Whitebread, S.; Hamon, J.; Bojanic, D.; Urban, L. Keynote review: In vitro safety pharmacology profiling: An essential tool for successful drug development. Drug Discov. Today 2005, 10, 1421–1433. [Google Scholar] [CrossRef]
- Liebler, D.C.; Guengerich, F.P. Elucidating mechanisms of drug-induced toxicity. Nat. Rev. Drug Discov. 2005, 4, 410. [Google Scholar] [CrossRef] [PubMed]
- Hodos, R.A.; Kidd, B.A.; Shameer, K.; Readhead, B.P.; Dudley, J.T. In silico methods for drug repurposing and pharmacology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2016, 8, 186–210. [Google Scholar] [CrossRef] [PubMed]
- What are the Possible Side Effects of a Drug and Where Can I Find the Most Current Information About My Drug? Available online: https://www.fda.gov/aboutfda/transparency/basics/ucm194959.htm (accessed on 25 July 2017).
- Morphy, R.; Rankovic, Z. Designed multiple ligands. An emerging drug discovery paradigm. J. Med. Chem. 2005, 48, 6523–6543. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, A.L. Network pharmacology: The next paradigm in drug discovery. Nat. Chem. Biol. 2008, 4, 682–690. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, G.R.; Lehar, J.; Keith, C.T. Multi-target therapeutics: When the whole is greater than the sum of the parts. Drug Discov. Today 2007, 12, 34–42. [Google Scholar] [CrossRef] [PubMed]
- Korcsmáros, T.; Szalay, M.S.; Böde, C.; Kovács, I.A.; Csermely, P. How to design multi-target drugs: Target search options in cellular networks. Expert Opin. Drug Discov. 2007, 2, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Lounkine, E.; Keiser, M.J.; Whitebread, S.; Mikhailov, D.; Hamon, J.; Jenkins, J.; Lavan, P.; Weber, E.; Doak, A.K.; Côté, S.; et al. Large scale prediction and testing of drug activity on side-effect targets. Nature 2012, 486, 361. [Google Scholar] [CrossRef] [PubMed]
- Roth, B.L.; Sheffler, D.J.; Kroeze, W.K. Magic shotguns versus magic bullets: Selectively non-selective drugs for mood disorders and schizophrenia. Nat. Rev. Drug Discov. 2004, 3, 353–359. [Google Scholar] [CrossRef] [PubMed]
- Rix, U.; Hantschel, O.; Dürnberger, G.; Rix, L.L.R.; Planyavsky, M.; Fernbach, N.V.; Kaupe, I.; Bennett, K.L.; Valent, P.; Colinge, J.; et al. Chemical proteomic profiles of the BCR-ABL inhibitors imatinib, nilotinib, and dasatinib reveal novel kinase and nonkinase targets. Blood 2007, 110, 4055–4063. [Google Scholar] [CrossRef] [PubMed]
- Keiser, M.J.; Setola, V.; Irwin, J.J.; Laggner, C.; Abbas, A.; Hufeisen, S.J.; Jensen, N.H.; Kuijer, M.B.; Matos, R.C.; Tran, T.B.; et al. Predicting new molecular targets for known drugs. Nature 2009, 462, 175. [Google Scholar] [CrossRef] [PubMed]
- De Lera, A.R.; Ganesan, A. Epigenetic polypharmacology: From combination therapy to multitargeted drugs. Clin. Epigenet. 2016, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Horst, J.A.; Laurenzi, A.; Bernard, B.; Samudrala, R. Computational multitarget drug discovery. In Polypharmacology in Drug Discovery; Peters, J.U., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2012; pp. 263–301. [Google Scholar]
- Jadhav, A.; Bansode, B.; Phule, D.; Shelar, A.; Patil, R.; Gade, W.; Kharat, K.; Karuppayil, S.M. The antibacterial agent, moxifloxacin inhibits virulence factors of Candida albicans through multitargeting. World J. Microbiol. Biotechnol. 2017, 33, 96. [Google Scholar] [CrossRef] [PubMed]
- Melisi, D.; Piro, G.; Tamburrino, A.; Carbone, C.; Tortora, G. Rationale and clinical use of multitargeting anticancer agents. Curr. Opin. Pharmacol. 2013, 13, 536–542. [Google Scholar] [CrossRef] [PubMed]
- Silver, L.L. Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 2007, 6, 41. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Schurig-Briccio, L.A.; Feng, X.; Upadhyay, A.; Pujari, V.; Lechartier, B.; Fontes, F.L.; Yang, H.; Rao, G.; Zhu, W.; et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014, 57, 3126–3139. [Google Scholar] [CrossRef] [PubMed]
- Giordano, S.; Petrelli, A. From single-to multi-target drugs in cancer therapy: When aspecificity becomes an advantage. Curr. Med. Chem. 2008, 15, 422–432. [Google Scholar] [CrossRef]
- Petrelli, A.; Valabrega, G. Multitarget drugs: The present and the future of cancer therapy. Expert Opin. Pharmacother. 2009, 10, 589–600. [Google Scholar] [CrossRef] [PubMed]
- Jenwitheesuk, E.; Samudrala, R. Identification of potential multitarget antimalarial drugs. JAMA 2005, 294, 1487–1491. [Google Scholar]
- Bugatti, A.; Urbinati, C.; Ravelli, C.; De Clercq, E.; Liekens, S.; Rusnati, M. Heparin-mimicking sulfonic acid polymers as multitarget inhibitors of human immunodeficiency virus type 1 Tat and gp120 proteins. Antimicrob. Agents Chemother. 2007, 51, 2337–2345. [Google Scholar] [CrossRef] [PubMed]
- Balzarini, J.; Andrei, G.; Balestra, E.; Huskens, D.; Vanpouille, C.; Introini, A.; Zicari, S.; Liekens, S.; Snoeck, R.; Holỳ, A.; et al. A multi-targeted drug candidate with dual anti-HIV and anti-HSV activity. PLoS Pathog. 2013, 9, e1003456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, L.; Draizen, E.J.; Bourne, P.E. Harnessing big data for systems pharmacology. Ann. Rev. Pharmacol. Toxicol. 2017, 57, 245–262. [Google Scholar] [CrossRef] [PubMed]
- Forli, S. Charting a path to success in virtual screening. Molecules 2015, 20, 18732–18758. [Google Scholar] [CrossRef] [PubMed]
- Cavasotto, C.N.; Phatak, S.S. Homology modeling in drug discovery: Current trends and applications. Drug Discov. Today 2009, 14, 676–683. [Google Scholar] [CrossRef] [PubMed]
- Klebe, G. Virtual ligand screening: Strategies, perspectives and limitations. Drug Discov. Today 2006, 11, 580–594. [Google Scholar] [CrossRef] [PubMed]
- Sweiti, H.; Ekwunife, O.; Jaschinski, T.; Lhachimi, S.K. Repurposed therapeutic agents targeting the Ebola virus: A systematic review. Curr. Ther. Res. 2017. [Google Scholar] [CrossRef] [PubMed]
- Lai, K.Y.; Ng, W.Y.G.; Cheng, F.F. Human Ebola virus infection in West Africa: A review of available therapeutic agents that target different steps of the life cycle of Ebola virus. Infect. Dis. Poverty 2014, 3, 43. [Google Scholar] [CrossRef] [PubMed]
- Arp, R.; Smith, B.; Spear, A.D. Building Ontologies with Basic Formal Ontology; Mit Press: Cambridge, MA, USA, 2015. [Google Scholar]
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Brooks, B.R.; Brooks, C.L.; MacKerell, A.D.; Nilsson, L.; Petrella, R.J.; Roux, B.; Won, Y.; Archontis, G.; Bartels, C.; Boresch, S.; et al. CHARMM: The biomolecular simulation program. J. Comput. Chem. 2009, 30, 1545–1614. [Google Scholar] [CrossRef] [PubMed]
- Biovia, D.S. Dassault Systemes; Discovery Studio Modeling Environment: San Diego, CA, USA, 2015. [Google Scholar]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Bekker, H.; Berendsen, H.; Dijkstra, E.; Achterop, S.; Van Drunen, R.; Van der Spoel, D.; Sijbers, A.; Keegstra, H.; Reitsma, B.; Renardus, M. Gromacs: A parallel computer for molecular dynamics simulations. Phys. Comput. 1993, 92, 252–256. [Google Scholar]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.Y.; Sali, A. Protein structure modeling with MODELLER. In Structural Proteomics: High-Throughput Methods; Humana Press: New York, NY, USA, 2008; pp. 145–159. [Google Scholar]
- Molsoft Software. Available online: https://www.molsoft.com/ (accessed on 25 July 2017).
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [PubMed]
- Lovell, S.C.; Davis, I.W.; Arendall, W.B.; de Bakker, P.I.; Word, J.M.; Prisant, M.G.; Richardson, J.S.; Richardson, D.C. Structure validation by Cα geometry: ϕ, ψ and Cβ deviation. Proteins Struct. Funct. Bioinform. 2003, 50, 437–450. [Google Scholar] [CrossRef] [PubMed]
- NCBI, R.C. Database Resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2017, 45, D12–D17. [Google Scholar]
- Bernstein, F.C.; Koetzle, T.F.; Williams, G.J.; Meyer, E.F.; Brice, M.D.; Rodgers, J.R.; Kennard, O.; Shimanouchi, T.; Tasumi, M. The protein data bank. FEBS J. 1977, 80, 319–324. [Google Scholar]
- Bairoch, A.; Apweiler, R.; Wu, C.H.; Barker, W.C.; Boeckmann, B.; Ferro, S.; Gasteiger, E.; Huang, H.; Lopez, R.; Magrane, M.; et al. The universal protein resource (UniProt). Nucleic Acids Res. 2005, 33, D154–D159. [Google Scholar] [CrossRef] [PubMed]
- Tong, Y.G.; Shi, W.F.; Liu, D.; Qian, J.; Liang, L.; Bo, X.C.; Liu, J.; Ren, H.G.; Fan, H.; Ni, M.; et al. Genetic diversity and evolutionary dynamics of Ebola virus in Sierra Leone. Nature 2015, 524, 93–96. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. ZINC- a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Wishart, D.S.; Knox, C.; Guo, A.C.; Cheng, D.; Shrivastava, S.; Tzur, D.; Gautam, B.; Hassanali, M. DrugBank: A knowledgebase for drugs, drug actions and drug targets. Nucleic Acids Res. 2007, 36, D901–D906. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2015, 44, D1202–D1213. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.Y.C. TCM Database@ Taiwan: The world’s largest traditional Chinese medicine database for drug screening in silico. PLoS ONE 2011, 6, e15939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kozomara, A.; Griffiths-Jones, S. miRBase: Annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2013, 42, D68–D73. [Google Scholar] [CrossRef] [PubMed]
- Thelwall, M.; Kousha, K. ResearchGate: Disseminating, communicating, and measuring Scholarship? J. Assoc. Inf. Sci. Technol. 2015, 66, 876–889. [Google Scholar] [CrossRef]
- Han, Z.; Lu, J.; Liu, Y.; Davis, B.; Lee, M.S.; Olson, M.A.; Ruthel, G.; Freedman, B.D.; Schnell, M.J.; Wrobel, J.E.; et al. Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses. J. Virol. 2014, 88, 7294–7306. [Google Scholar] [CrossRef] [PubMed]
- Brown, C.S.; Lee, M.S.; Leung, D.W.; Wang, T.; Xu, W.; Luthra, P.; Anantpadma, M.; Shabman, R.S.; Melito, L.M.; MacMillan, K.S.; et al. In silico derived small molecules bind the filovirus VP35 protein and inhibit its polymerase cofactor activity. J. Mol Biol. 2014, 426, 2045–2058. [Google Scholar] [CrossRef] [PubMed]
- Mirza, M.U.; Ikram, N. Integrated computational approach for virtual hit identification against ebola viral proteins VP35 and VP40. Int. J. Mol. Sci. 2016, 17, 1748. [Google Scholar] [CrossRef] [PubMed]
- Kharkar, P.S.; Ramasami, P.; Choong, Y.S.; Rhyman, L.; Warrier, S. Discovery of anti-Ebola drugs: A computational drug repositioning case study. RSC Adv. 2016, 6, 26329–26340. [Google Scholar] [CrossRef]
- Karthick, V.; Nagasundaram, N.; Doss, C.G.P.; Chakraborty, C.; Siva, R.; Lu, A.; Zhang, G.; Zhu, H. Virtual screening of the inhibitors targeting at the viral protein 40 of Ebola virus. Infect. Dis. Poverty 2016, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Chopra, G.; Kaushik, S.; Elkin, P.L.; Samudrala, R. Combating ebola with repurposed therapeutics using the CANDO platform. Molecules 2016, 21, 1537. [Google Scholar] [CrossRef] [PubMed]
- Dapiaggi, F.; Pieraccini, S.; Sironi, M. In silico study of VP35 inhibitors: From computational alanine scanning to essential dynamics. Mol. BioSyst. 2015, 11, 2152–2157. [Google Scholar] [CrossRef] [PubMed]
- Glanzer, J.G.; Byrne, B.M.; McCoy, A.M.; James, B.J.; Frank, J.D.; Oakley, G.G. In silico and in vitro methods to identify ebola virus VP35-dsRNA inhibitors. Bioorg. Med. Chem. 2016, 24, 5388–5392. [Google Scholar] [CrossRef] [PubMed]
- Golkar, Z.; Battaria, R.; Pace, D.G.; Bagasra, O. Inhibition of Ebola virus by anti-Ebola miRNAs in silico. J. Infect. Dev. Ctries 2016, 10, 626–634. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, N.; Farman, A.; Badshah, S.L.; Rahman, A.U.; ur Rashid, H.; Khan, K. Molecular modeling, simulation and docking study of ebola virus glycoprotein. J. Mol. Gr. Model. 2017, 72, 266–271. [Google Scholar] [CrossRef] [PubMed]
- Abazari, D.; Moghtadaei, M.; Behvarmanesh, A.; Ghannadi, B.; Aghaei, M.; Behruznia, M.; Rigi, G. Molecular docking based screening of predicted potential inhibitors for VP40 from Ebola virus. Bioinformation 2015, 11, 243. [Google Scholar] [CrossRef] [PubMed]
- El-Din, H.M.A.; Loutfy, S.A.; Fathy, N.; Elberry, M.H.; Mayla, A.M.; Kassem, S.; Naqvi, A. Molecular docking based screening of compounds against VP40 from Ebola virus. Bioinformation 2016, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Raj, U.; Varadwaj, P.K. Flavonoids as multi-target inhibitors for proteins associated with Ebola virus: In silico discovery using virtual screening and molecular docking studies. Interdiscip. Sci. Comput. Life Sci. 2016, 8, 132–141. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, P.; Mahesh, M.; Somashekhar, R. Pharmacoinformatics: Homology modelling of the target protein (GP1, 2) for Ebola hemorrhagic fever and predicting an Ayurvedic remediation of the disease. J. Proteom. Bioinform. 2009, 2, 287–294. [Google Scholar] [CrossRef]
- Tambunan, U.; Nasution, M. Identification of novel Ebola virus (EBOV) VP24 inhibitor from Indonesian natural products through in silico drug design approach. In AIP Conference Proceedings; AIP Publishing: Melville, NY, USA, 2017; Volume 1862, p. 030091. [Google Scholar]
- Shah, R.; Panda, P.K.; Patel, P.; Panchal, H. Pharmacophore based virtual screening and molecular docking studies of inherited compounds against Ebola virus receptor proteins. World J. Pharm. Pharm. Sci. 2015, 4, 1268–1282. [Google Scholar]
- Ekins, S.; Freundlich, J.S.; Clark, A.M.; Anantpadma, M.; Davey, R.A.; Madrid, P. Machine learning models identify molecules active against the Ebola virus in vitro. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Tracz, V. The five deadly sins of science publishing. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Freundlich, J.S.; Coffee, M. A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus. F1000Research 2014, 3. [Google Scholar] [CrossRef]
- Sharma, D.; Sharma, N.; Pathak, M.; Sharma, R.; Tyagi, P.; Chawla, R.; Basu, M.; Ojha, H. Homology modeling and docking studies of VP24 protein of Ebola virus with an antiviral drug and its derivatives. Chem. Biol. Lett. 2017, 4, 27–32. [Google Scholar]
- Veljkovic, V.; Loiseau, P.M.; Figadere, B.; Glisic, S.; Veljkovic, N.; Perovic, V.R.; Cavanaugh, D.P.; Branch, D.R. Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection. F1000Research 2015, 4. [Google Scholar] [CrossRef]
- Veljkovic, V.; Goeijenbier, M.; Glisic, S.; Veljkovic, N.; Perovic, V.R.; Sencanski, M.; Branch, D.R.; Paessler, S. In silico analysis suggests repurposing of ibuprofen for prevention and treatment of EBOLA virus disease. F1000Research 2015, 4. [Google Scholar] [CrossRef] [PubMed]
- Palamthodi, S.; Patil, D.; Sankpal, A.; Zarekar, S.; Patil, Y. Identification of drug lead molecules against Ebola virus: An in silico approach. J. Comput. Methods Mol. Des. 2012, 2, 76–84. [Google Scholar]
- Gupta, R. Rethinking the development of Ebola treatments. Lancet Glob. Health 2014, 2, e563–e564. [Google Scholar] [CrossRef]
- Ban, T.A. The role of serendipity in drug discovery. Dialogues Clin. Neurosci. 2006, 8, 335. [Google Scholar] [PubMed]
- Hu, Y.; Bajorath, J. Learning from ‘big data’: Compounds and targets. Drug Discov. Today 2014. [Google Scholar] [CrossRef] [PubMed]
- Bajorath, J. On data sharing in computational drug discovery and the need for data notes. F1000Research 2014, 3. [Google Scholar] [CrossRef] [PubMed]
- Halgren, T.A. Identifying and characterizing binding sites and assessing druggability. J. Chem. Inf. Model. 2009, 49, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Van Tongeren, S.A.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G.; et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014, 508, 402. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, J.; Sun, W.; Martínez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti–Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. [Google Scholar] [CrossRef] [PubMed]
- Box, G.E. Science and statistics. J. Am. Stat. Assoc. 1976, 71, 791–799. [Google Scholar] [CrossRef]
- Sun, Z.; Chaichana, T. A systematic review of computational fluid dynamics in type B aortic dissection. Int. J. Cardiol. 2016, 210, 28–31. [Google Scholar] [CrossRef] [PubMed]
- Das, D.; Mukherjee, R.; Chakraborty, C. Computational microscopic imaging for malaria parasite detection: A systematic review. J. Microsc. 2015, 260, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Moher, D.; Shamseer, L.; Clarke, M.; Ghersi, D.; Liberati, A.; Petticrew, M.; Shekelle, P.; Stewart, L.A. Preferred reporting items for systematic review and meta-analysis protocols (PRISMA-P) 2015 statement. Syst. Rev. 2015, 4, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meerpohl, J.J.; Herrle, F.; Antes, G.; von Elm, E. Scientific value of systematic reviews: Survey of editors of core clinical journals. PloS ONE 2012, 7, e35732. [Google Scholar] [CrossRef]
- Scannell, J.W.; Blanckley, A.; Boldon, H.; Warrington, B. Diagnosing the decline in pharmaceutical R&D efficiency. Nat. Rev. Drug Discov. 2012, 11, 191. [Google Scholar] [PubMed]
- Cleves, A.E.; Jain, A.N. Effects of inductive bias on computational evaluations of ligand-based modeling and on drug discovery. J. Comput. Aided Mol. Des. 2008, 22, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Hert, J.; Irwin, J.J.; Laggner, C.; Keiser, M.J.; Shoichet, B.K. Quantifying biogenic bias in screening libraries. Nat. Chem. Biol. 2009, 5, 479–483. [Google Scholar] [CrossRef] [PubMed]
- Huang, N.; Shoichet, B.K.; Irwin, J.J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Title | Author, Reference | Journal | Publisher | Impact Factor |
|---|---|---|---|---|
| Small-molecule probes targeting the viral PPxY-host Nedd4 interface block egress of a broad range of RNA viruses | Han et al., 2014 [96] | Journal of Virology | American Society for Microbiology | 5.076 (self reported), 4.69 (RG) |
| In silico derived small molecules bind the filovirus VP35 protein and inhibit its polymerase co-factor activity | Brown et al., 2014 [97] | Journal of Molecular Biology | Elsevier | 4.632 (self reported), 4.68 (RG) |
| Integrated computational approach for virtual hit identification against Ebola viral proteins VP35 and VP40 | Mirza et al., 2016 [98] | International Journal of Molecular Sciences | MDPI | 3.226 (self reported), 4.01 (RG) |
| Discovery of anti-Ebola drugs: a computational drug repositioning case study | Kharkar et al., 2016 [99] | RSC Advances | Royal Society of Chemistry | 3.108 (self reported), 3.06 (RG) |
| Virtual screening of inhibitors targeting at the viral protein 40 of Ebola virus | Karthick et al., 2016 [100] | Infectious Diseases of Poverty | BioMed Central | 3.181 (self reported), 2.97 (RG) |
| Combating Ebola with Repurposed Therapeutics using the CANDO platform | Chopra et al., 2016 [101] | Molecules | MDPI | 2.861 (self reported), 2.80 (RG) |
| In silico study of VP35 inhibitors: from computational alanine scanning to essential dynamics | Dapiaggi et al., 2015 [102] | Molecular BioSystems | Royal Society of Chemistry | 2.781 (self reported), 2.92 (RG) |
| Drug repurposing to target Ebola virus replication and virulence using structural systems pharmacology | Zhao et al., 2016 [12] | BMC Bioinformatics | BioMed Central | 2.448 (self reported), 2.97 (RG) |
| In silico and in vitro methods to identify ebola virus VP35-dsRNA inhibitors | Glanzer et al., 2016 [103] | Bioorganic & Medicinal Chemistry | Elsevier | 2.930 (self reported), 2.29 (RG) |
| Inhibition of Ebola Virus by anti-Ebola miRNAs in silico | Golkar et al., 2015 [104] | The Journal of Infection in Developing Countries | Open Learning on Enteric Pathogens | 1.67 (RG) |
| Molecular modeling, simulation and docking study of Ebola Virus glycoprotein | Ahmad et al., 2017 [105] | Journal of Molecular Graphics and Modelling | Molecular Graphics and Modelling Society; American Chemical Society | 1.12 (RG) |
| Molecular docking based screening of predicted potential inhibitors for VP40 from Ebola virus | Abazari et al., 2015 [106] | Bioinformation | Biomedical Informatics Publishing Group | 0.80 (RG) |
| Molecular docking based screening of compounds against VP40 from Ebola virus | El-Din et al., 2016 [107] | Bioinformation | Biomedical Informatics Publishing Group | 0.80 (RG) |
| Flavonoids as multi-target inhibitors for proteins associated with Ebola virus: in silico discovery using virtual screening and molecular docking studies | Raj et al., 2015 [108] | Interdisciplinary Sciences: Computational Life Sciences | Springer | 0.753 (self reported), 0.64 (RG) |
| Pharmaco-Informatics: Homology Modeling of the Target Protein (GP1,2) for Ebola Hemorrhagic Fever and Predicting an Ayurvedic Remediation of the Disease | Bagchi et al., 2009 [109] | Journal of Proteomics & Bioinformatics | OMICS International | 1.57 (self reported), 0.41 (RG) |
| Identification of novel Ebola virus (EBOV) VP24 inhibitor from Indonesian natural products through in silico drug design approach | Tambunan et al., 2017 [110] | AIP Conference Proceedings 10 July 2017 | American Institute of Physics | 0.22 (RG) |
| Pharmacophore based virtual screening and molecular docking studies of inherited compounds against Ebola virus receptor proteins | Shah et al., 2015 [111] | World Journal of Pharmacy and Pharmaceutical Sciences | WJPPS | 6.647 (self reported), 0.19 (RG) |
| Machine learning models identify molecules active against the Ebola virus in vitro | Ekins et al., 2015 [112] | F1000Research | Faculty of 1000 | NA [113] |
| A common feature pharmacophore for FDA-approved drugs inhibiting the Ebola virus | Ekins et al., 2014 [114] | F1000Research | Faculty of 1000 | NA [113] |
| Homology modeling and docking studies of VP24 protein of Ebola virus with Oseltamivir and its derivatives | Sharma et al., 2017 [115] | Chemical Biology Letters | Integrated Science | NA |
| Virtual screen for repurposing approved and experimental drugs for candidate inhibitors of EBOLA virus infection | Veljkovic et al., 2015 [116] | F1000Research | Faculty of 1000 | NA [113] |
| In silico analysis suggests repurposing of ibuprofen for prevention and treatment of EBOLA virus disease | Veljkovic et al., 2015 [117] | F1000Research | Faculty of 1000 | NA [113] |
| Identification of Drug Lead Molecules against Ebola Virus: an In Silico Approach | Palamthodi et al., 2012 [118] | Journal of Computational Methods in Molecular Design | Scholars Research Library | NF |
| Author, Reference | Method/Protocol/Pipeline | In Vitro Integration | Multitargeting |
|---|---|---|---|
| Han et al., 2014 [96] | in vitro methods, docking, energy minimization, ranking, substructure similarity searching, statistical analysis (analysis of variance), testing in vitro | Yes | No |
| Brown et al., 2014 [97] | docking, energy minimization, ranking and interaction fingerprint comparison, medicinal chemistry: crystallography, compound synthesis, NMR spectroscopy, structural study, pulldown assay, mini genome study, EBOV assays | Yes | No |
| Mirza et al., 2016 [98] | binding site prediction, drug similarity analysis, pharmacokinetics, pharmacodynamics, energy minimization, metabolic site prediction, docking, validation | No | Yes |
| Kharkar et al., 2016 [99] | ligand-based virtual screening, molecular docking | No | No |
| Sharma et al., 2017 [115] | template-based modeling, structure prediction, energy minimization, validation, docking | No | No |
| Karthick et al., 2016 [100] | energy minimization, virtual screening, docking, intermolecular interaction analysis, dynamics, absorption-distribution-metabolism-excretion analysis (ADME), drug likeness analysis, toxicity prediction | No | No |
| Chopra et al., 2016 [101] | binding site prediction, docking, interaction signature ranking similarity | No | Yes |
| Dapiaggi et al., 2015 [102] | molecular dynamics, computational alanine scanning, RMSD fluctuations, bootstrap/principal component analysis | No | No |
| Zhao et al., 2016 [12] | binding site prediction, proteome wide binding site comparison, template-based modeling, docking, molecular dynamics | No | Yes |
| Glanzer et al., 2016 [103] | docking, alignment, in vitro testing, compound property analysis, residue analysis | Yes | No |
| Golkar et al., 2015 [104] | sequence alignment, other algorithms to predict miRNA-EBOV RNA inhibitory activity/post-transcriptional silencing | No | No |
| Ahmad et al., 2017 [105] | template-based modeling, structure prediction, validation, molecular dynamics, docking | No | No |
| Abazari et al., 2015 [106] | dynamics, docking, pharmacokinetic analysis | No | No |
| El-Din et al., 2016 [107] | fingerprint comparison, compound modeling, energy minimization, docking, pharmacokenetics | No | No |
| Raj et al., 2015 [108] | energy minimization, binding site prediction, docking, active site residue interaction analysis, absorption-distribution-metabolism-excretion-toxicity (ADMET) analysis | No | Yes |
| Bagchi et al., 2009 [109] | template-based modeling, structure prediction, validation, docking | No | No |
| Tambunan et al., 2017 [110] | dynamics/energy minimization, ADMET screening, molecular docking | No | No |
| Shah et al., 2015 [111] | pharmacophore modeling, docking | No | Yes |
| Ekins et al., 2014 [114] | pharmacophore modeling, docking | No | No |
| Ekins et al., 2015 [112] | viral pseudotype entry assay and EBOV replication assay, machine learning models (Bayesian, SVM, Recursive Partitioning forest, single tree), validation, virtual screening | Yes | No |
| Veljkovic et al., 2015 [116] | library curation/data mining, compound virtual screening (electron-ion interaction potential/average quasi valence number) | Yes | N/A |
| Veljkovic et al., 2015 [117] | compound virtual screening (electron-ion interaction potential/average quasi valence number), ligand optimization, molecular docking | No | No |
| Palamthodi et al., 2012 [118] | screening of lead molecules, docking, pharmacoanalysis | No | No |
| Author, Year, Country | Reported Candidate Compounds and Biologics |
|---|---|
| Han et al., 2014, USA [96] | compounds ’4’ (Amb123203) and ’5’ (Amb21795397) |
| Brown et al., 2014, USA [97] | GA-017, GA-246, VPL-42, VPL-57, VPL-60, VPL-51, VPL-58, VPL-15, VPL-48, VPL-29 |
| Mirza et al., 2016, Pakistan, Belgium [98] | Timtec-ST45161107, Otava_7118230235, Timtec-ST50912611, Timtec-ST50616170, Analyticon-NP-010155, Otava-0115540195, Analyticon-NP-019744 (kihadarnin A), Analyticon-NP-0005474, PubChem CID 17597017, Analyticon-NP-000375 (lactupicrin), Analyticon_NP-014205 (parfumine), Analyticon-NP-014522, Analyticon-003228 (isorutarin) |
| Kharkar et al., 2016, India [99] | sitaxentan, alitretinoin, ceftriaxone, acitretin, cidofovir, telmisartan, nateglinide, ceftizoxime, treprostinil, tenoxicam |
| Sharma et al., 2017, India [115] | ZINC_77287098 (an oseltamivir derivative) |
| Karthick et al., 2016, (Hong Kong) China [100] | Top results: emodin-8-beta- d-glucoside, tonkinochromane_G. Other results: neoglucobrassicin; glisoflavanone; rosmarinic_acid_ethyl_ester; 2-[(6Z.9Z_12Z)-heptadeca-6_9_12-trienyI]-6-hydroxybenzoic_acid; chrysophanol-8-beta-d-glucoside; 3_4-dihydro-3-methoxypaederoside; Melittoside; beta-methoxylforsythoside; glucobrassicin; manninotriose; d-mannitol_monohexadecanoate; 4__O-methyl_myricetin_3-O-(6-O-alpha-l-rhamnopyranosyl)-beta-d-glucopyranoside; (-)-epicatechin-3-O-gallate |
| Chopra et al., 2016, USA [101] | niclosamide, sertraline, clomifene, mebendazole, deslanoside, digoxin, raloxifene, clemastine, tamoxifen |
| Dapiaggi et al., 2015, Italy [102] | GA-017, GA-246, VPL-27, VPL-29, VPL-42, VPL-48, VPL-57, VPL-58, VPL-60 |
| Zhao et al., 2016, USA [12] | Top results: indinavir, sinefungin. Other results: maraviroc, abacavir, telbivudine, cidofovir, montelukast, iloprost, salmeterol xinafoate, travoprost, latanoprost, remikiren, vitamin K1, mitoxantrone, labetalol hydrochloride, tafluprost, misoprostol, carboprost, fosinopril, Benzylpenicilloyl Polylysine, Bimatoprost, Nebivolol, valrubicin, Tamsulosin, Mycophenolate Mofetil, SAM, aza-Sadenosyl-Lmethionine, A9145C, Maraviroc, Telbivudine, Cidofovir |
| Glanzer et al., 2016, USA [103] | ZINC_05328460 |
| Golkar et al., 2015, Denmark, USA [104] | hsa-miR-607, hsa-miR-5699-5p, hsa-miR-4682, hsa-miR-4692, hsa-miR-548az, hsa-miR-4526, hsa-miR-3065-5p, hsa-miR-145-3p, hsa-miR-491-3p, hsa-miR-4633-3p, hsa-miR-491-3p, hsa-miR-548-3p |
| Ahmad et al., 2017, Pakistan [105] | dronedarone 1D, amiodarone 2A, and other dronedarone and amiodarone derivatives |
| Abazari et al., 2015, Iran [106] | 10 unlabeled, 4 selected as top candidates |
| El-Din et al., 2016, Egypt [107] | PubChem CIDs: 416,724, 374,108, 3,851,453, 256,623, 44,149,862, 254,616, 3183 |
| Raj et al., 2015, India [108] | Gossypetin and Toxifolin (top 2). Other relevant results: ST50903219, ST50940361, ST101866, ST078351. |
| Bagchi et al., 2009, India [109] | andrographolide |
| Tambunan et al., 2017, Indonesia [110] | cycloartocarpin, letestuianin B, lissoclin A, varamine A, lissoclibadin 4, cystodytin J, (−)- N-methylcoclaurine, (−)-matairesinol, cardamonine, reticuline |
| Shah, et al., 2015, India [111] | deslanoside, digoxin, vincristine, vinorelbine, and several unnamed ZINC compounds and investigational compounds |
| Ekins et al., 2014, USA [114] | selective estrogen receptor modulators (SERMs) and anti-malarials |
| Ekins et al., 2015, USA [112] | quinacrine, pyronaridine, tilorone |
| Veljkovic et al., 2015, Serbia, France, USA, Canada [117] | 267 approved and 382 experimental drugs. Notable classes: antimalarials and antibiotics (macrolides, pleuromutilins , aminoglycosides). |
| Veljkovic et al., 2015, Serbia, The Netherlands, Canada, USA [116] | ibuprofen |
| Palamthodi et al., 2012, India [118] | VP35 compounds: 2-(2,3-diamino-3-oxopropyl)sulfynyl acetic acid; 5-cyclohexypyridine 2-caboxylic acid; Copper carboxymethoxyananide dihydrate; 2, 3-dihydroxy-3-[(4-methylphenyl)carbamoyl]propanoic acid. VP40 compounds: 2-(1,3-benzothiazol-2-ylsulfanyl)acetate; 2-(1,8-dihydroxy-9-oxo-10h-anthracen-2yl)acetic acid; 1-[(2 s,4s,5r)-4-hydroxy-5-methyloxolan-2-yl]-5-methylpyrimidine,2,4 dione; 1-[(2r, 4s, 5s)-5-(hydroxymethyl)-4-methyloxolan-2-yl]-1,2,4-triazole-3-carboxamide. |
| Author, Year, Country | Selected Software, Algorithms, and Version Numbers |
|---|---|
| Han et al., 2014, USA [96] | Autodock 4.0, CHARMM (MMFF), Accelrys LigScore2 |
| Brown et al., 2014, USA [97] | Autodock 4.0 (DOVIS PIPELINE), CHARMM35b2 (MMFF), LigScore2, REFMACS, Phenix, PRODRG2, Coot, MolProbity, GraphPad Prism |
| Mirza et al., 2016, Pakistan, Belgium [98] | OpenBabel, Discovery Studio, UCSF Chimera (AMBER ff12SB force field for protein energy minimization), CASTP, MUSCLE, Jalview 2.7, BLAST (PSI-BLAST), ALIGN2D, MODELLER 9.12, CHARMM22, Autodock Vina, Mcule-pipeline, PyMol, DrugScore eXtended, DUD-e, Daylight, NSCC.11, Molinspiration, OSIRIS, ADMET prediction suite (ACD/LABS), Molsoft, AdmetSAR, Aggregator Advisor, MetaPrint2D, PAINS, clustalX, ligPlots |
| Kharkar et al., 2016, India [99] | ROCS OpenEye Scientific Software Suite, AutoDock |
| Sharma et al., 2017, India [115] | Phyre2, EasyModeller4, RAMPAGE, BLASTP, YASARA, Schrodinger Suite, GLIDE, Autodock, Discovery Studio Visualizer |
| Karthick et al., 2016, (Hong Kong) China [100] | GROMACS (GROMOS43a1), iScreen (PLANTS), Autodock 4.2.6, Autodock Tools, AutoGrid, PEARLS, PDBsum, UCSF Chimera, GROMACS (GROMOS43a1, SPC water model), PRODRG (EWALD Algorithm, Lincs Algorithm), GROMACS_UTIL (g_rms, g_hbond), Molsoft, OSIRIS Property Explorer, Protox Web Server, PROCHECK |
| Chopra et al., 2016, USA [101] | COFACTOR, CANDOCK, ITASSER (HHBLITS, LOMETS, SPICKER, ModRefiner, KobaMin, et al.), ChemAxon MarvinBeans molconverter v.5.11.3, Xemistry Cactvs Chemoinformatics Toolkit, CANDO |
| Dapiaggi et al., 2015, Italy [102] | GROMACS (AMBER99SB-ILDN, Generalized Amber Force Field, TIP3P, LINCS algorithm, velocity rescale algorithm, Berendsen barostat, Particles Mesh Ewald algorithm), (MM/PBSA, APBS), GROMACS_utility (g_covar, g_anaeig), VMD, Naccess |
| Zhao et al., 2016, USA [12] | Verify3D, PROCHECK, SMAP, Modeller v9.14, I-TASSER, Autodock4, Autodock Vina, PLANTS, Surflex, AutoDockTools 4, CASTp, Xleap, ACEMD (AMBER99SB, SHAKE algorithm), Pymol, Ligplot |
| Glanzer et al., 2016, USA [103] | Molegro Docking, Molinspiration Property Calculation Service, Molegro Structure Protein Alignment, UCSF CHIMERA |
| Golkar et al., 2015, Denmark, USA [104] | Software described in a cited publication |
| Ahmad et al., 2017, Pakistan [105] | PREDATOR, PHD, GOR4, DPM, HNN, DSC, SIMPA96, SOPM, RONN, GLOBPLOT, DISSEMBLE, MOE, RAMPAGE, ERRATE, Expasy-ProtoParam, MUSCLE server, PSI-BLAST |
| Abazari et al., 2015, Iran [106] | GROMACS 4.5.4, PyRx / AutoDock Vina, FAFDrugs3, admetSAR, PROTOX |
| El-Din et al., 2016., Egypt [107] | Chem Sketch, Swiss PDB Viewer, Autodock4 , Auto Grid, Auto Dock hydrogen module, UCSF Chimera, PROTOX, Molsoft |
| Raj et al., 2015, India [108] | Protein Preparation Wizard, SiteMap, GLIDE (Receptor Grid Generation Panel), QikProp v3.9 |
| Bagchi et al., 2009, India [109] | MODELLER, Swiss-PDBViewer, ACD/ChemSketch, RAMPAGE, ArgusLab 4.0.1, HEX_SERVER, HHpred, BLAST |
| Tambunan et al., 2017, Indonesia [110] | Molecular Operating Environment (MOE) 2014.09, UCSF Chimera 1.9, Vega ZZ 3.0.5, OSIRIS DataWarrior 4.2.2 |
| Shah, et al., 2015, India [111] | Discovery Studio Visualizer 4.0, Swiss PDB viewer, Chimera, CastP server, AutoDock Vina 4.2, GEMDOCK, Molinspiration |
| Ekins et al., 2014, USA [114] | Discovery Studio 4.1 (CAESAR, FAST conformer generation, LibDock), CHARMM |
| Ekins et al., 2015, USA [112] | Discovery studio, Mobile Molecular Data Sheet, R (programming language), other software described in citation |
| Veljkovic et al., 2015, Serbia, France, USA, Canada [116] | Software undisclosed, showed equations for AQVN and EIIP and explained high level process, secondary goal of this publication was to establish a web server for this type of study, “ebola screen” web server ( http://www.biomedconsulting.info/ebolascreen.php) |
| Veljkovic et al., 2015, Serbia, The Netherlands, Canada, USA [117] | Custom software for calculating AQVN and EIIP, VEGA ZZ, MOPAC 2009, Autodock Vina |
| Palamthodi et al., 2012, India [118] | PYMOL, Chimera, Arguslab, AutoDock |
| Software | Count | Method/Technique |
|---|---|---|
| Autodock | 7 | molecular docking |
| UCSF CHIMERA | 7 | visualization and analysis suite |
| Discovery Studio | 5 | affinity, ranking, modeling, workflow tooling |
| Autodock Vina | 5 | molecular docking |
| BLAST Suite | 4 | sequence alignment |
| CHARMM | 4 | molecular dynamics, minimization, analysis |
| Autodock Tools | 3 | structure preparation utilities, workflow tooling |
| GROMACS | 3 | molecular dynamics |
| Modeller | 3 | template-based modeling |
| Molsoft | 3 | suite of bioinformatic and cheminformatic tools |
| PROTOX | 3 | toxicity prediction |
| RAMPAGE | 3 | Ramachandran plot analysis |
| CASTP | 3 | binding and active site prediction/analysis |
| Molinspiration | 3 | cheminformatic software suite |
| Swiss PDB Viewer | 3 | visualization and analysis |
| PyMol | 3 | visualization |
| OSIRIS | 3 | chemical property analysis |
| ACD ChemSketch | 2 | chemical structure modeling, property analysis, logp |
| admetSAR | 2 | cheminformatics/ADMET analysis |
| AutoGrid | 2 | support tooling for AutoDock |
| GLIDE | 2 | docking |
| GROMACSutils | 2 | support tooling for GROMACS simulations |
| I-TASSER | 2 | protein structure and function prediction |
| LigScore2 | 2 | binding affinity prediction |
| MUSCLE | 2 | multiple alignment |
| PROCHECK | 2 | protein structure steriochemical quality analysis |
| PRODRG | 2 | small molecule topology generation for simulation use, energy minimization |
| ArgusLab 4.0.1 | 2 | molecular modeling |
| MOE | 2 | suite of protein modeling, assessment, and analysis software |
| VEGA ZZ | 2 | molecular modeling suite |
| ADMET prediction suite (ACD/LABS) | 1 | ADMET analysis |
| Aggregator Advisor | 1 | molecular aggregation prediction for biochemical assays |
| ALIGN2D | 1 | sequence structure alignment |
| biomedconsulting Ebola screen server | 1 | AQVN and EIIP screening server for Ebola research |
| CANDO | 1 | drug discovery platform (docking, dynamics, multitargeting, and drug repurposing) |
| CANDOCK | 1 | fragment based docking with dynamics |
| ChemAxon MarvinBeans | 1 | computational chemistry suite |
| clustalX | 1 | multiple alignment |
| COFACTOR | 1 | protein functional annotation |
| Coot | 1 | visualization, modeling, analysis, validation |
| Daylight | 1 | cheminformatics, fingerprinting |
| DisEMBL | 1 | Intrinsic disorder prediction |
| DOVIS PIPELINE | 1 | virtual screening pipeline |
| DPM | 1 | promoter structure of co-regulated gene modeling |
| DrugScore eXtended | 1 | knowledge based protein-ligand complex scoring |
| DSC | 1 | protein secondary structure prediction |
| DUD-e | 1 | docking benchmarking, virtual screening support |
| EasyModeller4 | 1 | interface for MODELLER with integrated analysis tooling |
| ERRAT | 1 | verifying protein structures determined by crystallography |
| Expasy-ProtoParam | 1 | protein physical and chemical analysis |
| FAFDrugs3 | 1 | ADMET screening, virtual screening filtering |
| GLOBPLOT | 1 | intrinstic protein disorder, domain, and globularity prediction |
| GOR4 | 1 | secondary structure prediction |
| GraphPad Prism | 1 | graphing and statistics |
| HEXSERVER | 1 | docking |
| Hhpred | 1 | protein structure and function prediction |
| HNN | 1 | secondary structure prediction |
| iScreen | 1 | docking platform |
| Jalview | 1 | multiple sequence alignment editing, visualization and analysis |
| Ligplot+ | 1 | schematic diagrams of protein-ligand interactions |
| Mcule-pipeline | 1 | molecular modeling and cheminformatic screening interface for mcule database |
| MetaPrint2D | 1 | xenobiotic metabolism prediction, phase I metabolic site prediction |
| Mobile Molecular Data Sheet | 1 | mobile cheminformatics modeling and analysis |
| molconverter | 1 | utility for file format conversion |
| Molegro Docking | 1 | docking |
| Molegro Structure Protein Alignment | 1 | alignment |
| MolProbity | 1 | protein structure validation |
| MOPAC | 1 | semiempirical quantum chemistry program |
| Naccess | 1 | atomic solvent accessible area prediction |
| NSCC 11 | 1 | statistical software |
| OpenBabel | 1 | computational chemistry software suite |
| PDBsum | 1 | protein visualization and analysis |
| PEARLS | 1 | energetic analysis of receptor-ligand systems |
| PHD | 1 | secondary structure prediction |
| Phenix | 1 | structure determination software for X-ray crystallography and other methods |
| Phyre2 | 1 | protein fold recognition |
| PLANTS | 1 | docking |
| PREDATOR | 1 | secondary structure prediction |
| protein preparation wizard | 1 | tools for protein structure preparation for simulation |
| PyRx | 1 | virtual screening pipeline |
| QikProp | 1 | ADME screening, prediction |
| R | 1 | statistical programming language/platform |
| REFMACS | 1 | maximum likelihood refinement analysis for protein structure data |
| ROCS OpenEye | 1 | virtual screening by shape comparison tool |
| RONN | 1 | protein disorder prediction |
| Schrodinger Suite | 1 | suite of protein modeling, assessment, and analysis software |
| SIMPA96 | 1 | secondary structure prediction |
| SiteMap | 1 | binding site prediction |
| SMAP | 1 | protein-ligand interaction analysis |
| SOPM | 1 | secondary structure prediction |
| Surflex | 1 | docking |
| Verify3D | 1 | structure-sequence compatibility assessment |
| VMD | 1 | visualization, analysis |
| Xemistry Cactvs Cheminformatics Toolkit | 1 | cheminformatics software suite |
| Xleap | 1 | tool that interfaces with LEAP/AMBER |
| YASARA | 1 | visualization, modeling, analysis |
| ACEMD | 1 | dynamics |
| Gemdock | 1 | docking |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schuler, J.; Hudson, M.L.; Schwartz, D.; Samudrala, R. A Systematic Review of Computational Drug Discovery, Development, and Repurposing for Ebola Virus Disease Treatment. Molecules 2017, 22, 1777. https://doi.org/10.3390/molecules22101777
Schuler J, Hudson ML, Schwartz D, Samudrala R. A Systematic Review of Computational Drug Discovery, Development, and Repurposing for Ebola Virus Disease Treatment. Molecules. 2017; 22(10):1777. https://doi.org/10.3390/molecules22101777
Chicago/Turabian StyleSchuler, James, Matthew L. Hudson, Diane Schwartz, and Ram Samudrala. 2017. "A Systematic Review of Computational Drug Discovery, Development, and Repurposing for Ebola Virus Disease Treatment" Molecules 22, no. 10: 1777. https://doi.org/10.3390/molecules22101777