
Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells

 ,
, 
Abstract
:1. Introduction
2. Results
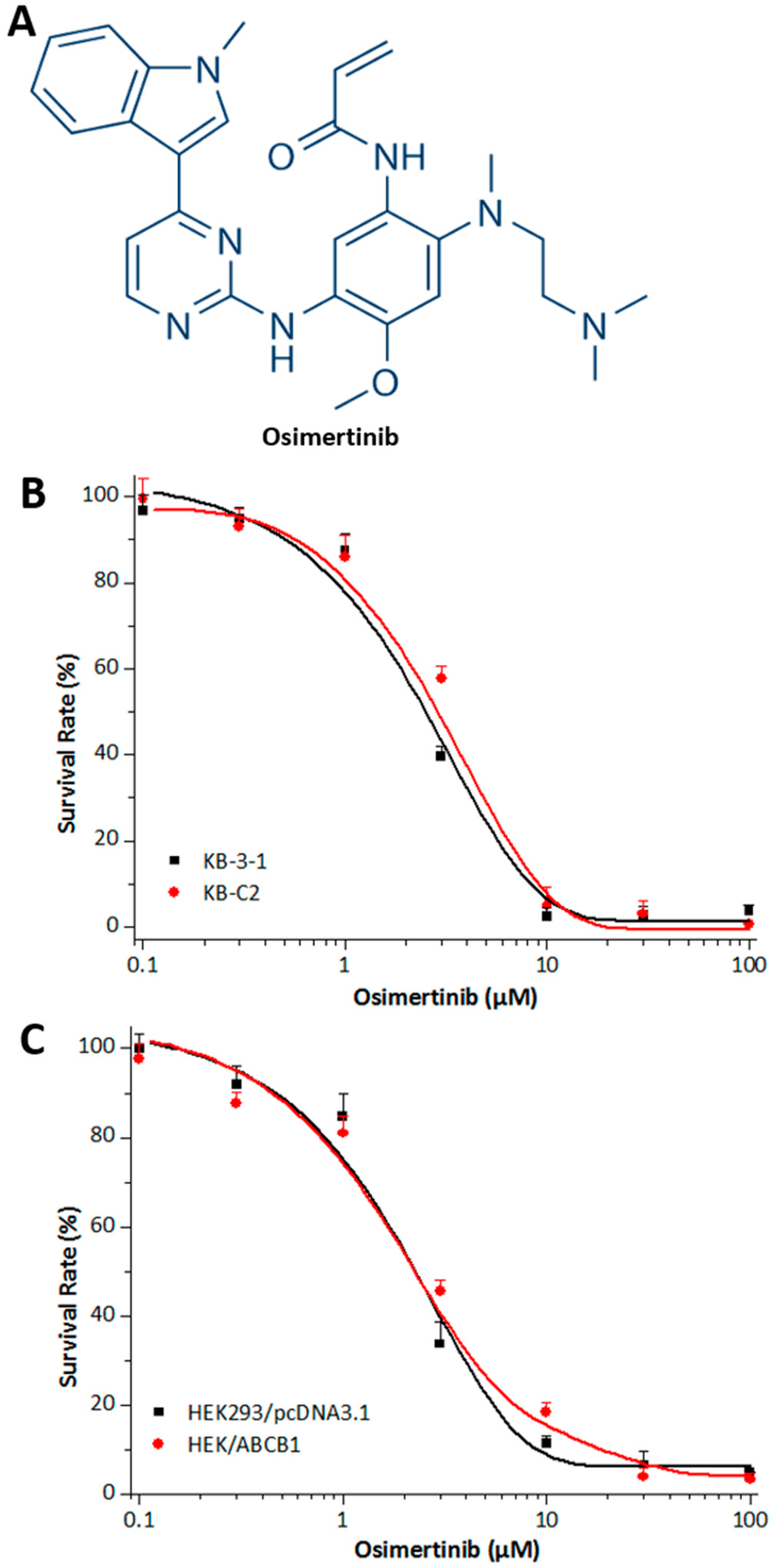
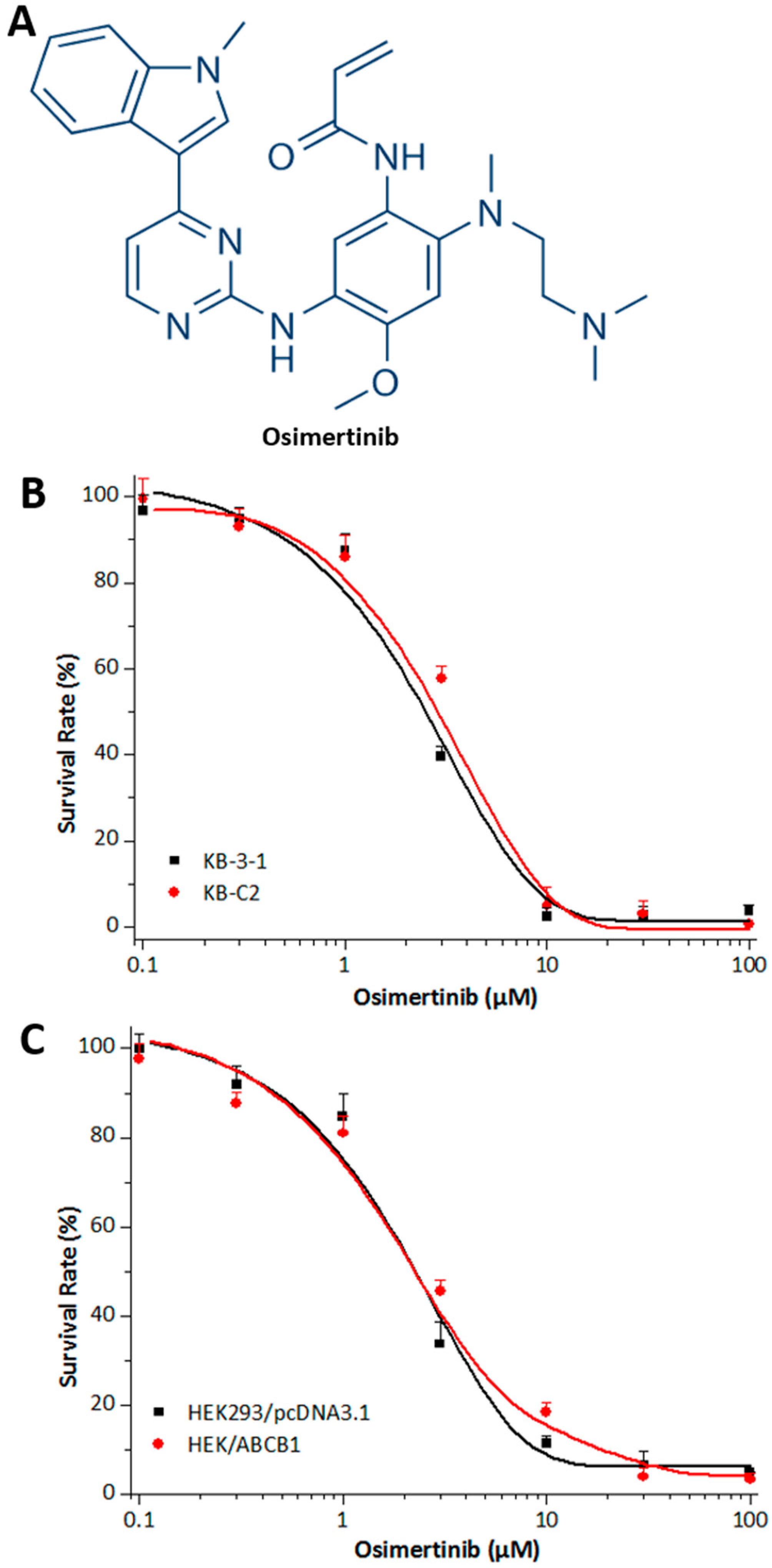
2.1. Effects of Osimertinib on ABCB1 Substrates in Cell Lines Overexpressing ABCB1
2.2. Effects of Osimertinib on Cell Lines Overexpressing ABCG2, ABCC1, or ABCC10
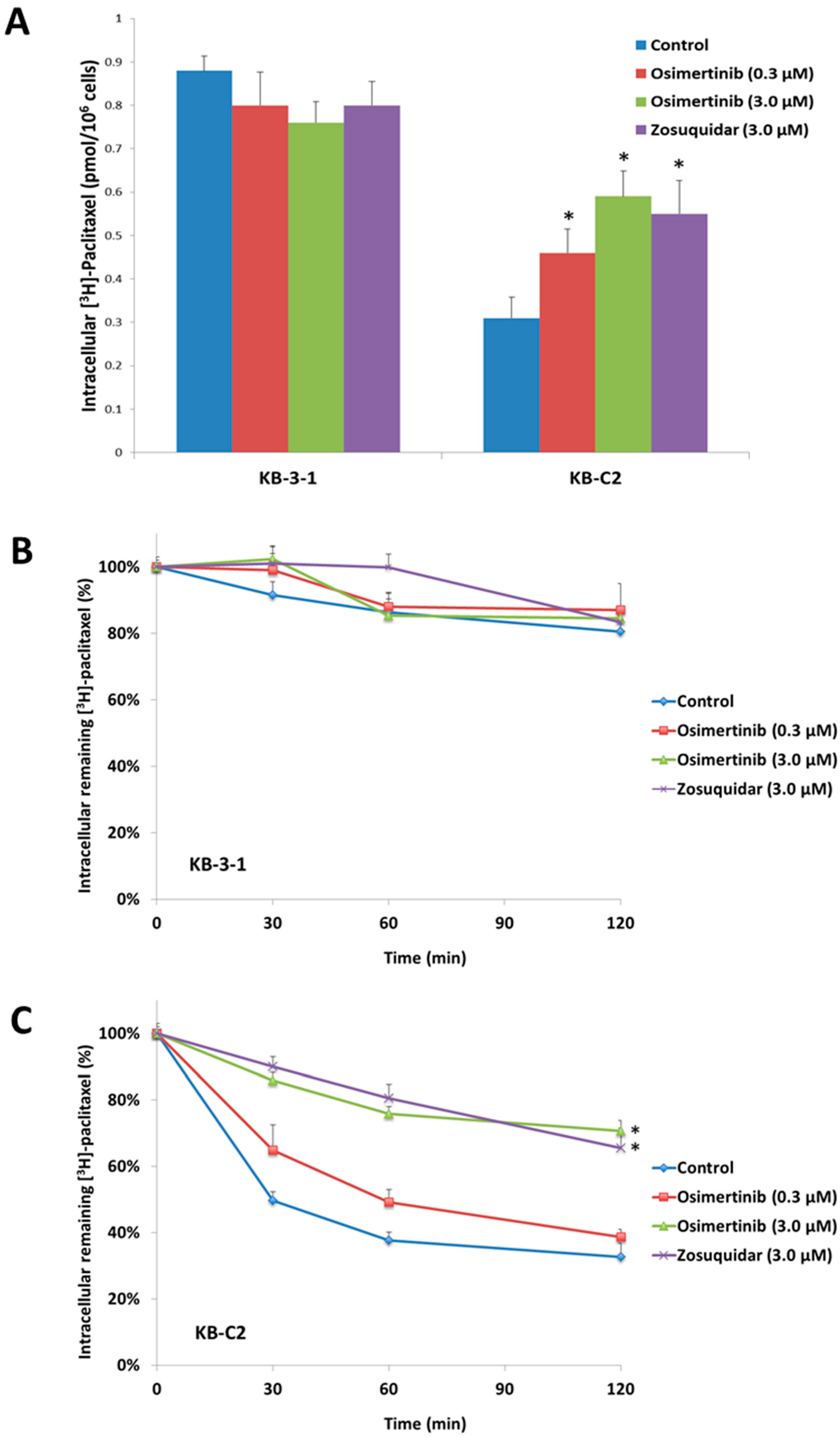
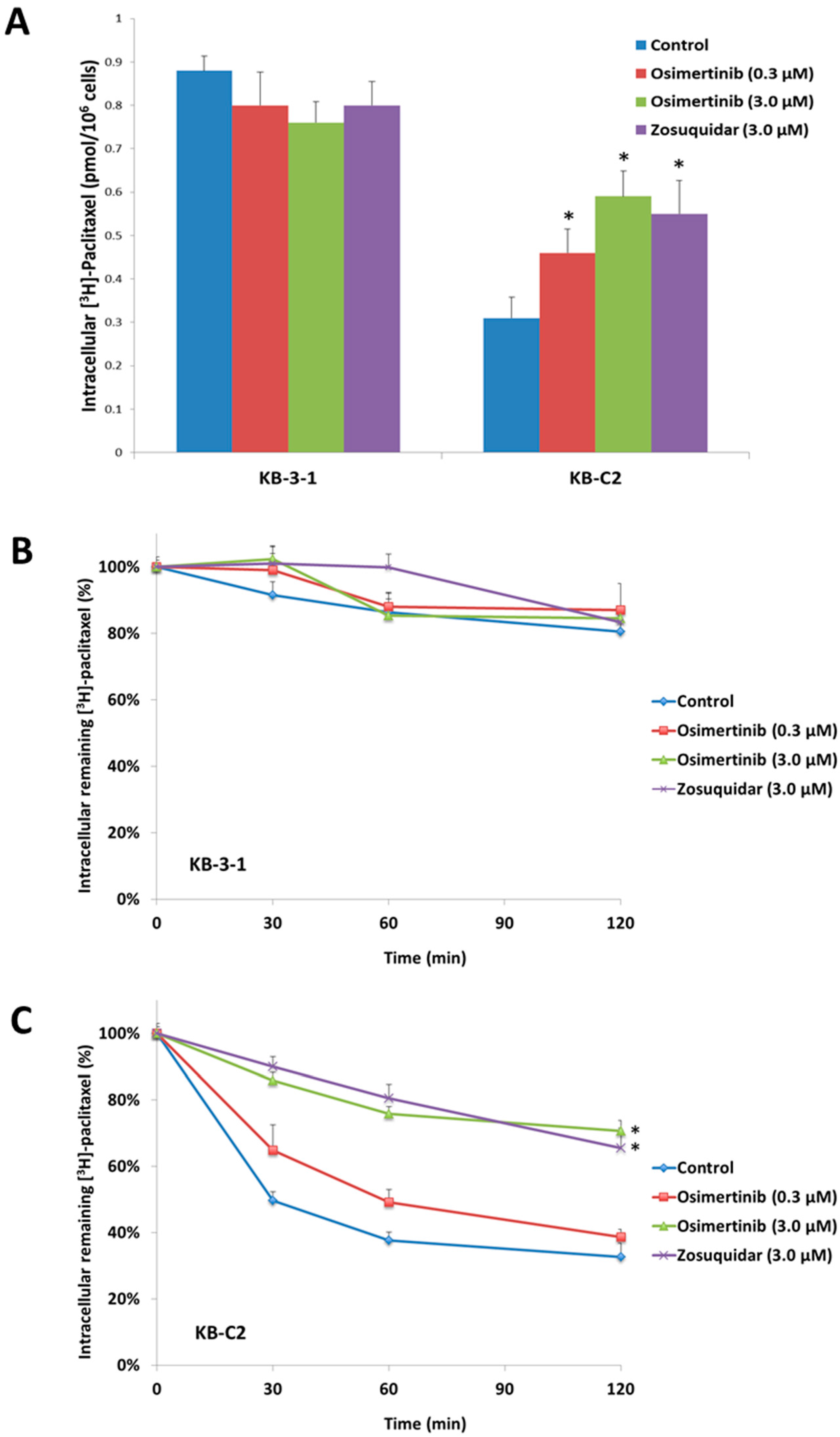
2.3. Effect of Osimertinib on the Intracellular Accumulation of [3H]-Paclitaxel
2.4. Effect of Osimertinib on the Efflux of [3H]-Paclitaxel
2.5. Effect of Osimertinib on the Protein Expression and Location of ABCB1
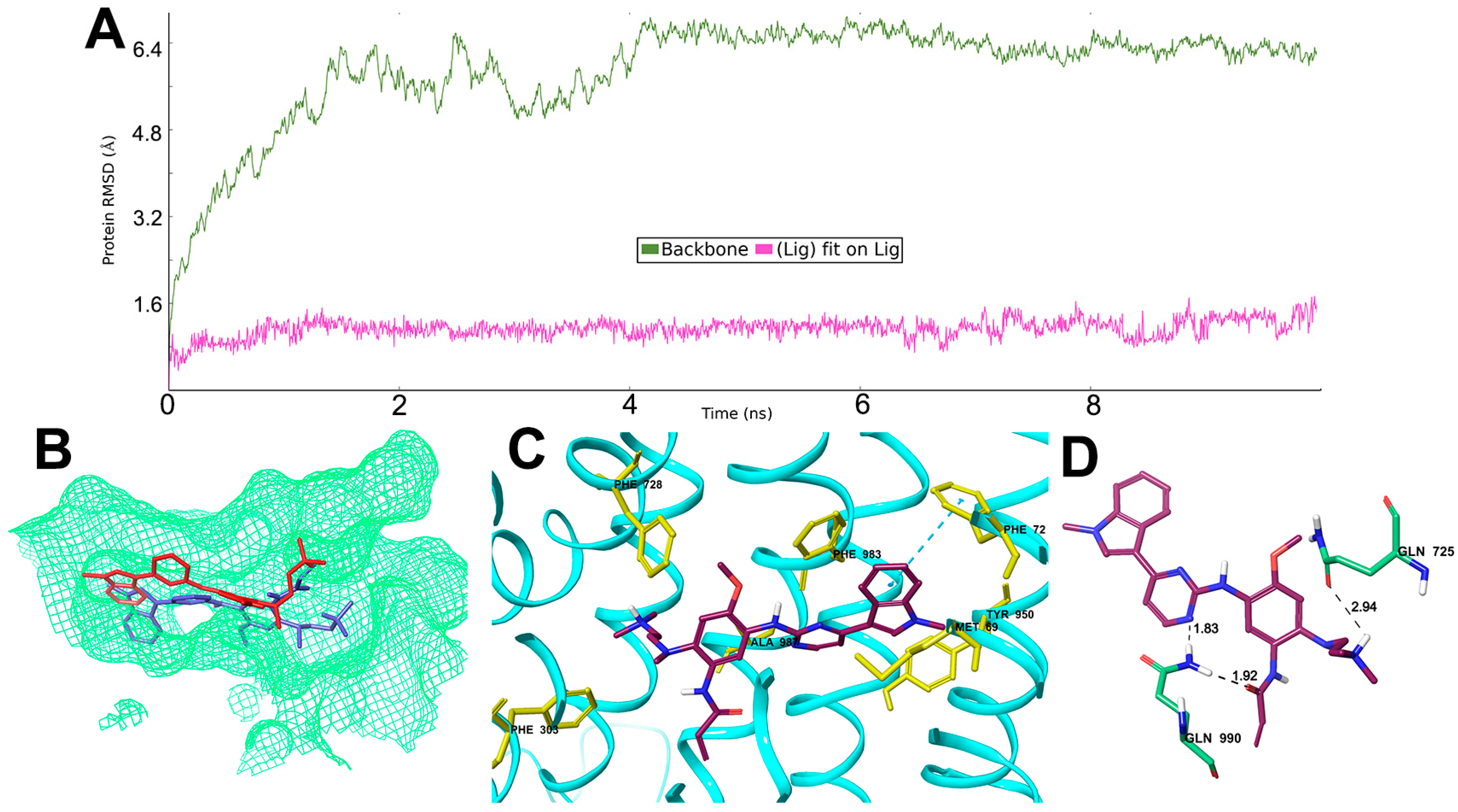
2.6. Interaction Analysis of MD-Stabilized Osimertinib-ABCB1 Complex
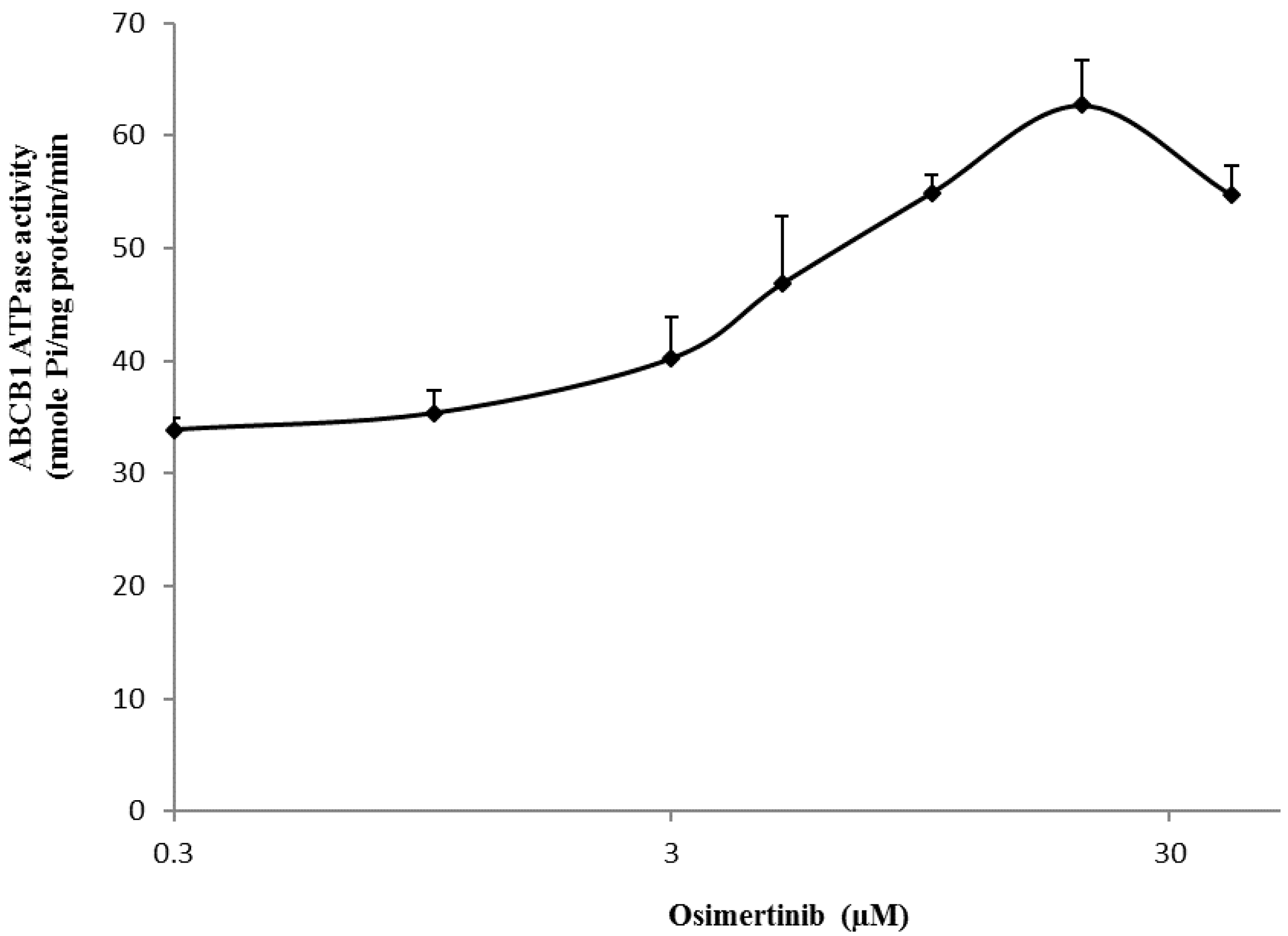
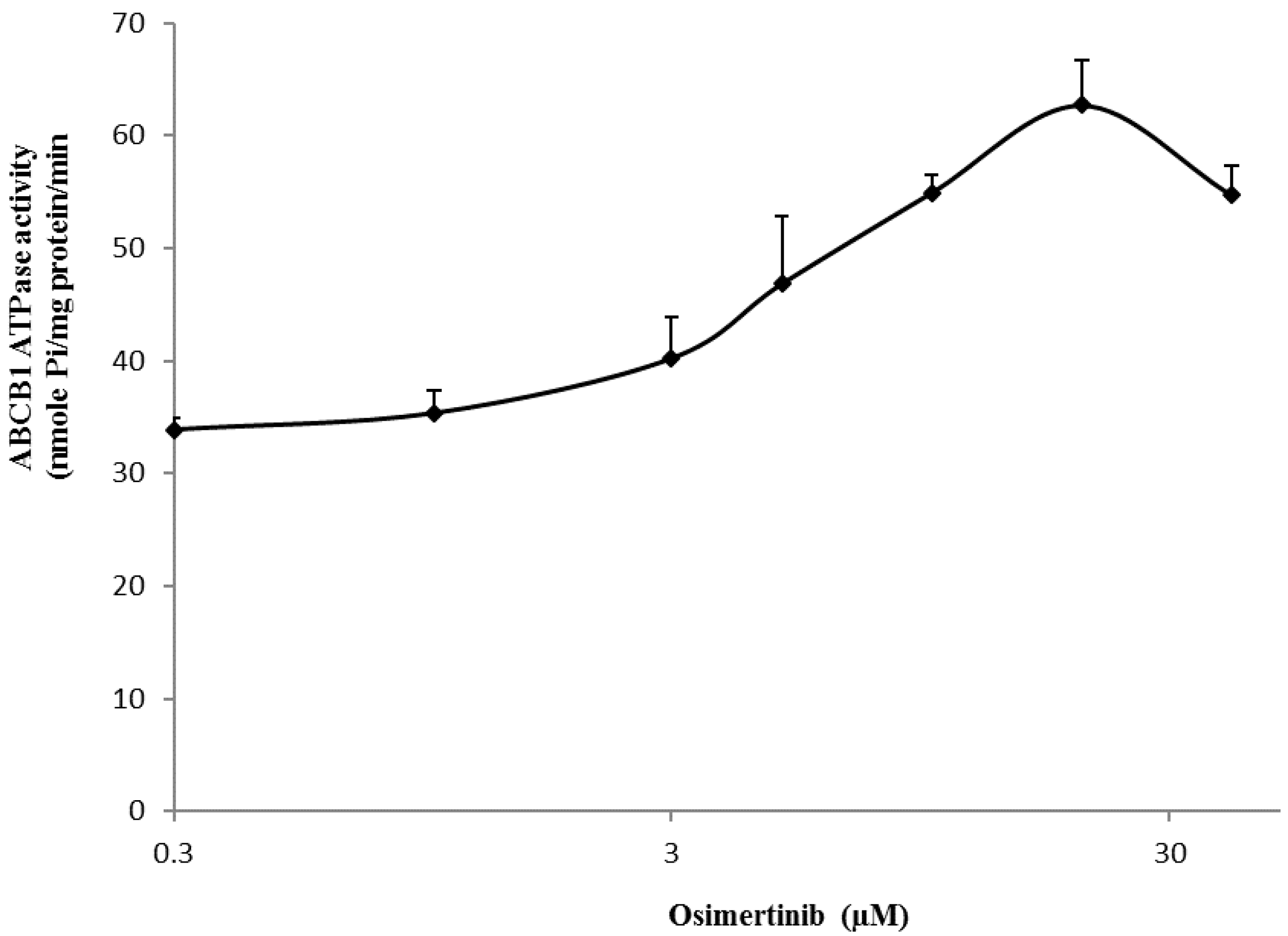
2.7. Effect of Osimertinib on the ABCB1 ATP Hydrolysis
3. Material and Methods
3.1. Chemicals
3.2. Cell Lines and Cell Culture
3.3. Cytotoxicity by MTT Assay
3.4. [3H]-Paclitaxel Accumulation Assay
3.5. [3H]-Paclitaxel Efflux Assay
3.6. Western Blot Analysis
3.7. Immunofluorescence
3.8. Induced-Fit Docking and Molecular Dynamics (MD) Simulation
3.9. ABCB1 ATPase Activity Assay
3.10. Statistical Analysis
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Gottesman, M.M.; Fojo, T.; Bates, S.E. Multidrug resistance in cancer: Role of ATP-dependent transporters. Nat. Rev. Cancer 2002, 2, 48–58. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Yang, Z.; Nie, Y.; Shi, Y.; Fan, D. Multi-drug resistance in cancer chemotherapeutics: Mechanisms and lab approaches. Cancer Lett. 2014, 347, 159–166. [Google Scholar] [CrossRef] [PubMed]
- Goldman, B. Multidrug resistance: Can new drugs help chemotherapy score against cancer? J. Natl. Cancer Inst. 2003, 95, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Fojo, A.; Hamilton, T.C.; Young, R.C.; Ozols, R.F. Multidrug resistance in ovarian cancer. Cancer 1987, 60 (Suppl. 8), 2075–2080. [Google Scholar] [CrossRef]
- Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Chen, Z.-S. Multidrug resistance proteins (MRPs) and cancer therapy. AAPS J. 2015, 17, 802–812. [Google Scholar] [CrossRef] [PubMed]
- Dean, M.; Annilo, T. Evolution of the ATP-binding cassette (ABC) transporter superfamily in vertebrates. Annu. Rev. Genom. Hum. Genet. 2005, 6, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Schinkel, A.H.; Mol, C.A.; Wagenaar, E.; van Deemter, L.; Smit, J.J.; Borst, P. Multidrug resistance and the role of P-glycoprotein knockout mice. Eur. J. Cancer 1995, 31a, 1295–1298. [Google Scholar] [CrossRef]
- Juliano, R.L.; Ling, V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim. Biophys. Acta BBA Biomembr. 1976, 455, 152–162. [Google Scholar] [CrossRef]
- Roskoski, R., Jr. STI-571: An anticancer protein-tyrosine kinase inhibitor. Biochem. Biophys. Res. Commun. 2003, 309, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Borst, P.; Elferink, R.O. Mammalian ABC transporters in health and disease. Annu. Rev. Biochem. 2002, 71, 537–592. [Google Scholar] [CrossRef] [PubMed]
- Longley, D.B.; Johnston, P.G. Molecular mechanisms of drug resistance. J. Pathol. 2005, 205, 275–292. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, J.; Zhang, L.; Zhao, L.; Fan, S.; Yang, Z.; Gao, F.; Kong, Y.; Xiao, G.G.; Wang, Q. Expression of P-gp, MRP, LRP, GST-Pi and TopoIIα and intrinsic resistance in human lung cancer cell lines. Oncol. Rep. 2011, 26, 1081–1089. [Google Scholar] [PubMed]
- Shukla, S.; Wu, C.P.; Ambudkar, S.V. Development of inhibitors of ATP-binding cassette drug transporters: Present status and challenges. Expert Opin. Drug Metab. Toxicol. 2008, 4, 205–223. [Google Scholar] [CrossRef] [PubMed]
- Shukla, S.; Chen, Z.-S.; Ambudkar, S.V. Tyrosine kinase inhibitors as modulators of ABC transporter-mediated drug resistance. Drug Resist. Updates 2012, 15, 70–80. [Google Scholar] [CrossRef] [PubMed]
- Burger, H.; van Tol, H.; Brok, M.; Wiemer, E.A.; de Bruijn, E.A.; Guetens, G.; de Boeck, G.; Sparreboom, A.; Verweij, J.; Nooter, K. Chronic imatinib mesylate exposure leads to reduced intracellular drug accumulation by induction of the ABCG2 (BCRP) and ABCB1 (MDR1) drug transport pumps. Cancer Biol. Ther. 2005, 4, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sodani, K.; Tiwari, A.K.; Singh, S.; Patel, A.; Xiao, Z.J.; Chen, J.J.; Sun, Y.L.; Talele, T.T.; Chen, Z.S. GW583340 and GW2974, human EGFR and HER-2 inhibitors, reverse ABCG2- and ABCB1-mediated drug resistance. Biochem. Pharmacol. 2012, 83, 1613–1622. [Google Scholar] [CrossRef] [PubMed]
- Hegedus, C.; Ozvegy-Laczka, C.; Apati, A.; Magocsi, M.; Nemet, K.; Orfi, L.; Keri, G.; Katona, M.; Takats, Z.; Varadi, A.; et al. Interaction of nilotinib, dasatinib and bosutinib with ABCB1 and ABCG2: Implications for altered anti-cancer effects and pharmacological properties. Br. J. Pharmacol. 2009, 158, 1153–1164. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Huang, C.J.; Yang, C.S.; Chu, Y.C.; Cheng, A.L.; Whang-Peng, J.; Yang, P.C. Gefitinib reverses chemotherapy resistance in gefitinib-insensitive multidrug resistant cancer cells expressing ATP-binding cassette family protein. Cancer Res. 2005, 65, 6943–6949. [Google Scholar] [CrossRef] [PubMed]
- Shi, Z.; Peng, X.X.; Kim, I.W.; Shukla, S.; Si, Q.S.; Robey, R.W.; Bates, S.E.; Shen, T.; Ashby, C.R., Jr.; Fu, L.W.; et al. Erlotinib (Tarceva, OSI-774) antagonizes ATP-binding cassette subfamily B member 1 and ATP-binding cassette subfamily G member 2-mediated drug resistance. Cancer Res. 2007, 67, 11012–11020. [Google Scholar] [CrossRef] [PubMed]
- Cataldo, V.D.; Gibbons, D.L.; Perez-Soler, R.; Quintas-Cardama, A. Treatment of non-small-cell lung cancer with erlotinib or gefitinib. N. Engl. J. Med. 2011, 364, 947–955. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.K.; Wu, Y.L.; Ding, P.N.; Lord, S.J.; Inoue, A.; Zhou, C.; Mitsudomi, T.; Rosell, R.; Pavlakis, N.; Links, M.; et al. Impact of Specific Epidermal Growth Factor Receptor (EGFR) Mutations and Clinical Characteristics on Outcomes After Treatment With EGFR Tyrosine Kinase Inhibitors Versus Chemotherapy in EGFR-Mutant Lung Cancer: A Meta-Analysis. J. Clin. Oncol. 2015, 33, 1958–1965. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and histological evolution of lung cancers acquiring resistance to EGFR inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed]
- Walter, A.O.; Sjin, R.T.; Haringsma, H.J.; Ohashi, K.; Sun, J.; Lee, K.; Dubrovskiy, A.; Labenski, M.; Zhu, Z.; Wang, Z.; et al. Discovery of a mutant-selective covalent inhibitor of EGFR that overcomes T790M-mediated resistance in NSCLC. Cancer Discov. 2013, 3, 1404–1415. [Google Scholar] [CrossRef] [PubMed]
- Park, K.; Han, J.Y.; Kim, D.W.; Bazhenova, L.A.; Ou, S.H.; Pang, Y.K.; Hin, H.S.; Juan, O.; Son, J.; Janne, P. 190TiP: ELUXA 1: Phase II study of BI 1482694 (HM61713) in patients (pts) with T790M-positive non-small cell lung cancer (NSCLC) after treatment with an epidermal growth factor receptor tyrosine kinase inhibitor (EGFR TKI). J. Thorac. Oncol. 2016, 11 (Suppl. 4), S139. [Google Scholar] [CrossRef]
- Wang, S.; Cang, S.; Liu, D. Third-generation inhibitors targeting EGFR T790M mutation in advanced non-small cell lung cancer. J. Hematol. Oncol. 2016, 9, 34. [Google Scholar] [CrossRef] [PubMed]
- Fung, K.L.; Pan, J.; Ohnuma, S.; Lund, P.E.; Pixley, J.N.; Kimchi-Sarfaty, C.; Ambudkar, S.V.; Gottesman, M.M. MDR1 synonymous polymorphisms alter transporter specificity and protein stability in a stable epithelial monolayer. Cancer Res. 2014, 74, 598–608. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Kathawala, R.J.; Zhang, Y.-K.; Patel, A.; Kumar, P.; Shukla, S.; Fung, K.L.; Ambudkar, S.V.; Talele, T.T.; Chen, Z.-S. Motesanib (AMG706), a potent multikinase inhibitor, antagonizes multidrug resistance by inhibiting the efflux activity of the ABCB1. Biochem. Pharmacol. 2014, 90, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.-J.; Huang, Y.; Anreddy, N.; Zhang, G.-N.; Zhang, Y.-K.; Xie, M.; Lin, D.; Yang, D.-H.; Zhang, M.; Chen, Z.-S. Tea nanoparticle, a safe and biocompatible nanocarrier, greatly potentiates the anticancer activity of doxorubicin. Oncotarget 2016, 7, 5877–5891. [Google Scholar] [PubMed]
- Zhang, Y.-K.; Zhang, G.-N.; Wang, Y.-J.; Patel, B.A.; Talele, T.T.; Yang, D.-H.; Chen, Z.-S. Bafetinib (INNO-406) reverses multidrug resistance by inhibiting the efflux function of ABCB1 and ABCG2 transporters. Sci. Rep. 2016, 6, 25694. [Google Scholar] [CrossRef] [PubMed]
- Kathawala, R.J.; Wei, L.; Anreddy, N.; Chen, K.; Patel, A.; Alqahtani, S.; Zhang, Y.-K.; Wang, Y.-J.; Sodani, K.; Kaddoumi, A.; et al. The small molecule tyrosine kinase inhibitor NVP-BHG712 antagonizes ABCC10-mediated paclitaxel resistance: A preclinical and pharmacokinetic study. Oncotarget 2015, 6, 510–521. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, Y.; Huang, H.; Wang, J.; Zhao, D.; Ji, L.; Chao, H. Cyclometalated Ruthenium (II) Anthraquinone Complexes Exhibit Strong Anticancer Activity in Hypoxic Tumor Cells. Chemistry 2015, 21, 15308–15319. [Google Scholar] [CrossRef] [PubMed]
- Zeng, L.; Chen, Y.; Liu, J.; Huang, H.; Guan, R.; Ji, L.; Chao, H. Ruthenium (II) Complexes with 2-Phenylimidazo [4, 5-f][1,10] phenanthroline Derivatives that Strongly Combat Cisplatin-Resistant Tumor Cells. Sci. Rep. 2016, 6, 19449. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.L.; Kathawala, R.J.; Singh, S.; Zheng, K.; Talele, T.T.; Jiang, W.Q.; Chen, Z.S. Zafirlukast antagonizes ATP-binding cassette subfamily G member 2-mediated multidrug resistance. Anticancer Drugs 2012, 23, 865–873. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.-S.; Patel, A.; Shukla, S.; Zhang, Y.-K.; Wang, Y.-J.; Kathawala, R.J.; Robey, R.W.; Zhang, L.; Yang, D.-H.; Talele, T.T.; et al. Icotinib antagonizes ABCG2-mediated multidrug resistance, but not the pemetrexed resistance mediated by thymidylate synthase and ABCG2. Oncotarget 2014, 5, 4529–4542. [Google Scholar] [CrossRef] [PubMed]
- Deng, W.; Dai, C.L.; Chen, J.J.; Kathawala, R.J.; Sun, Y.L.; Chen, H.F.; Fu, L.W.; Chen, Z.S. Tandutinib (MLN518) reverses multidrug resistance by inhibiting the efflux activity of the multidrug resistance protein 7 (ABCC10). Oncol. Rep. 2013, 29, 2479–2485. [Google Scholar] [PubMed]
- Zhang, Y.-K.; Zhang, H.; Zhang, G.-N.; Wang, Y.-J.; Kathawala, R.J.; Si, R.; Patel, B.A.; Xu, J.; Chen, Z.-S. Semi-synthetic ocotillol analogues as selective ABCB1-mediated drug resistance reversal agents. Oncotarget 2015, 6, 24277–24290. [Google Scholar] [CrossRef] [PubMed]
- Sherman, W.; Day, T.; Jacobson, M.P.; Friesner, R.A.; Farid, R. Novel procedure for modeling ligand/receptor induced fit effects. J. Med. Chem. 2006, 49, 534–553. [Google Scholar] [CrossRef] [PubMed]
- Shivakumar, D.; Williams, J.; Wu, Y.; Damm, W.; Shelley, J.; Sherman, W. Prediction of Absolute Solvation Free Energies using Molecular Dynamics Free Energy Perturbation and the OPLS Force Field. J. Chem. Theory Comput. 2010, 6, 1509–1519. [Google Scholar] [CrossRef] [PubMed]
- Szakács, G.; Paterson, J.K.; Ludwig, J.A.; Booth-Genthe, C.; Gottesman, M.M. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov. 2006, 5, 219–234. [Google Scholar] [CrossRef] [PubMed]
- Leonard, G.D.; Fojo, T.; Bates, S.E. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar] [CrossRef] [PubMed]
- Roe, M.; Folkes, A.; Ashworth, P.; Brumwell, J.; Chima, L.; Hunjan, S.; Pretswell, I.; Dangerfield, W.; Ryder, H.; Charlton, P. Reversal of P-glycoprotein mediated multidrug resistance by novel anthranilamide derivatives. Bioorg. Med. Chem. Lett. 1999, 9, 595–600. [Google Scholar] [CrossRef]
- Chen, K.-C.; Chen, H.-Y.; Chen, C.Y.-C. Potential protein phosphatase 2A agents from traditional Chinese medicine against cancer. Evid. Based Complement. Altern. Med. 2014, 2014, 436863. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: The compound osimertinib was purchase from Chemietek and the other test reagents were puchased from Sigma Aldrich, Thermo Fisher Scientific and others. Please refer to the Materials and Methods section for Vendor details.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatment | KB-3-1 | KB-C2 | ||
|---|---|---|---|---|
| IC50 ± SD a (nM) | RF b | IC50 ± SD (µM) | RF | |
| Paclitaxel | 3.58 ± 0.35 | [1.0] | 1.075 ± 0.299 | [307.1] |
| + Osimertinib (0.3 µM) | 3.63 ± 0.49 | [1.0] | 0.603 ± 0.045 * | [172.3] |
| + Osimertinib (0.5 µM) | 3.57 ± 0.41 | [1.0] | 0.092 ± 0.008 * | [26.3] |
| + Osimertinib (1.0 µM) | 3.12 ± 0.47 | [0.9] | 0.017 ± 0.005 * | [4.9] |
| + Verapamil (3.0 µM) | 3.24 ± 0.44 | [0.9] | 0.056 ± 0.011 * | [16.0] |
| + Zosuquidar (0.3 µM) | 2.81 ± 0.36 | [0.8] | 0.013 ± 0.003 * | [3.7] |
| Colchicine | 59.82 ± 8.57 | [1.0] | 15.528 ± 4.565 | [259.6] |
| + Osimertinib (0.3 µM) | 56.60 ± 7.13 | [0.9] | 5.120 ± 0.982 * | [85.6] |
| + Osimertinib (0.5 µM) | 58.66 ± 5.20 | [1.0] | 1.279 ± 0.606 * | [21.4] |
| + Osimertinib (1.0 µM) | 52.53 ± 7.39 | [0.9] | 0.637 ± 0.071 * | [10.6] |
| + Verapamil (3.0 µM) | 58.87 ± 9.65 | [1.0] | 0.279 ± 0.015 * | [4.7] |
| + Zosuquidar (0.3 µM) | 52.54 ± 9.12 | [0.9] | 0.078 ± 0.020 * | [3.7] |
| Vincristine | 5.05 ± 0.89 | [1.0] | 0.797 ± 0.019 | [157.8] |
| + Osimertinib (0.3 µM) | 5.49 ± 0.54 | [1.1] | 0.789 ± 0.027 | [156.3] |
| + Osimertinib (0.5 µM) | 5.26 ± 0.71 | [1.0] | 0.228 ± 0.037 * | [45.1] |
| + Osimertinib (1.0 µM) | 5.56 ± 0.96 | [1.1] | 0.027 ± 0.006 * | [5.2] |
| + Verapamil (3.0 µM) | 4.61 ± 0.86 | [0.9] | 0.062 ± 0.008 * | [12.3] |
| + Zosuquidar (0.3 µM) | 4.58 ± 0.65 | [0.9] | 0.013 ± 0.003 * | [2.6] |
| IC50 ± SD (µM) | RF | IC50 ± SD (µM) | RF | |
| Cisplatin | 2.90 ± 0.27 | [1.0] | 3.12 ± 0.20 | [1.1] |
| + Osimertinib (1.0 µM) | 2.78 ± 0.17 | [1.0] | 2.98 ± 0.22 | [1.0] |
| + Verapamil (3.0 µM) | 2.61 ± 0.20 | [0.9] | 2.66 ± 0.31 | [0.9] |
| Treatment | HEK293/pcDNA3.1 | HEK/ABCB1 | ||
|---|---|---|---|---|
| IC50 ± SD a (nM) | RF b | IC50 ± SD (nM) | RF | |
| Paclitaxel | 26.88 ± 2.74 | [1.0] | 1920.76± 150.32 | [71.5] |
| + Osimertinib (0.3 µM) | 27.53 ± 1.96 | [1.0] | 1497.33 ± 149.65 | [55.7] |
| + Osimertinib (0.5 µM) | 28.46 ± 3.35 | [1.1] | 906.75± 58.96 * | [33.7] |
| + Osimertinib (1.0 µM) | 30.45 ± 2.80 | [1.1] | 200.31 ± 22.04 * | [7.5] |
| + Verapamil (3.0 µM) | 23.30 ± 3.11 | [0.9] | 85.79 ± 5.86 * | [3.2] |
| + Zosuquidar (0.3 µM) | 29.50 ± 2.87 | [1.1] | 35.54 ± 5.77 * | [1.3] |
| Vincristine | 15.98 ± 2.88 | [1.0] | 566.19 ± 52.31 | [35.4] |
| + Osimertinib (0.3 µM) | 16.98 ± 2.34 | [1.1] | 434.10 ± 50.89 | [27.2] |
| + Osimertinib (0.5 µM) | 16.99 ± 1.96 | [1.1] | 269.55 ± 46.04 * | [16.9] |
| + Osimertinib (1.0 µM) | 15.22 ± 1.44 | [1.0] | 99.38 ± 38.92 * | [6.2] |
| + Verapamil (3.0 µM) | 14.84 ± 1.60 | [0.9] | 75.25 ± 27.55 * | [4.7] |
| + Zosuquidar (0.3 µM) | 13.26 ± 1.21 | [0.8] | 50.66 ± 20.54 * | [3.2] |
| Cisplatin | 1092.52 ± 100.26 | [1.0] | 1193.56 ± 111.2 | [1.0] |
| + Osimertinib (1.0 µM) | 985.27 ± 103.67 | [0.9] | 1011.04 ± 86.29 | [1.1] |
| + Verapamil (3.0 µM) | 900.79 ± 83.54 | [0.8] | 1121.36 ± 114.73 | [1.0] |
| Treatment | NCI-H460 | NCI-H460/MX20 | ||
|---|---|---|---|---|
| IC50 ± SD a (µM) | RF b | IC50 ± SD (µM) | RF | |
| Mitoxantrone | 0.13 ± 0.05 | [1.0] | 24.97 ± 5.46 | [193.86] |
| + Osimertinib (0.3 µM) | 0.15 ± 0.05 | [1.2] | 10.44 ± 5.92 * | [80.31] |
| + FTC (3 µM) | 0.12 ± 0.07 | [0.9] | 0.57 ± 0.11 * | [4.36] |
| Treatment | HEK293/pcDNA3.1 | HEK/ABCC1 | ||
| IC50 ± SD (nM) | RF | IC50 ± SD (nM) | RF | |
| Vincristine | 9.12 ± 1.12 | [1.0] | 91.74 ± 5.23 | [10.1] |
| + Osimertinib (0.3 µM) | 8.59 ± 1.57 | [0.9] | 72.96 ± 9.65 | [8.0] |
| + MK571 (25 µM) | 8.95 ± 1.48 | [1.0] | 9.86 ± 2.02 * | [1.1] |
| Treatment | HEK293/pcDNA3.1 | HEK/ABCC10 | ||
| IC50 ± SD (nM) | RF | IC50 ± SD (nM) | RF | |
| Paclitaxel | 13.45 ± 1.03 | [1.0] | 172.61± 8.99 | [12.8] |
| + Osimertinib (0.3 µM) | 14.48 ± 1.38 | [1.1] | 146.26 ± 14.21 | [10.9] |
| + Cepharanthine (3 µM) | 11.96 ± 1.26 | [0.9] | 21.15 ± 5.87 * | [1.6] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, X.-Y.; Zhang, Y.-K.; Wang, Y.-J.; Gupta, P.; Zeng, L.; Xu, M.; Wang, X.-Q.; Yang, D.-H.; Chen, Z.-S. Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells. Molecules 2016, 21, 1236. https://doi.org/10.3390/molecules21091236
Zhang X-Y, Zhang Y-K, Wang Y-J, Gupta P, Zeng L, Xu M, Wang X-Q, Yang D-H, Chen Z-S. Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells. Molecules. 2016; 21(9):1236. https://doi.org/10.3390/molecules21091236
Chicago/Turabian StyleZhang, Xiao-Yu, Yun-Kai Zhang, Yi-Jun Wang, Pranav Gupta, Leli Zeng, Megan Xu, Xiu-Qi Wang, Dong-Hua Yang, and Zhe-Sheng Chen. 2016. "Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells" Molecules 21, no. 9: 1236. https://doi.org/10.3390/molecules21091236
APA StyleZhang, X.-Y., Zhang, Y.-K., Wang, Y.-J., Gupta, P., Zeng, L., Xu, M., Wang, X.-Q., Yang, D.-H., & Chen, Z.-S. (2016). Osimertinib (AZD9291), a Mutant-Selective EGFR Inhibitor, Reverses ABCB1-Mediated Drug Resistance in Cancer Cells. Molecules, 21(9), 1236. https://doi.org/10.3390/molecules21091236