
Prenylated Chalcone 2 Acts as an Antimitotic Agent and Enhances the Chemosensitivity of Tumor Cells to Paclitaxel


 , , , and
, , , and
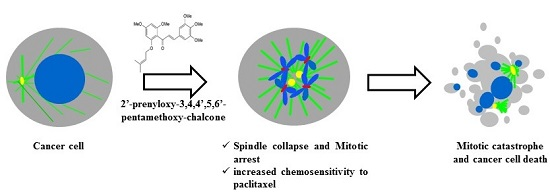
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
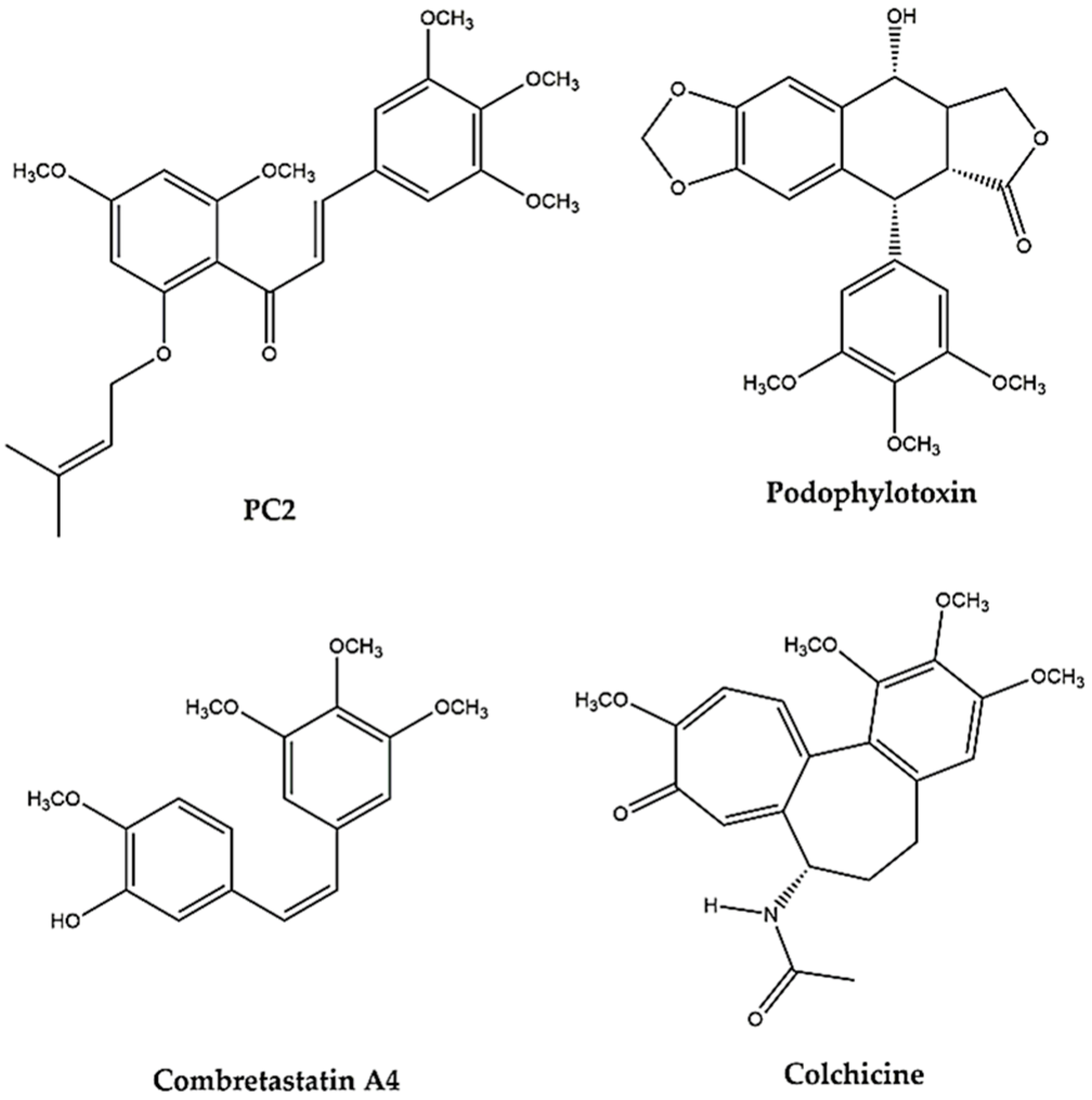
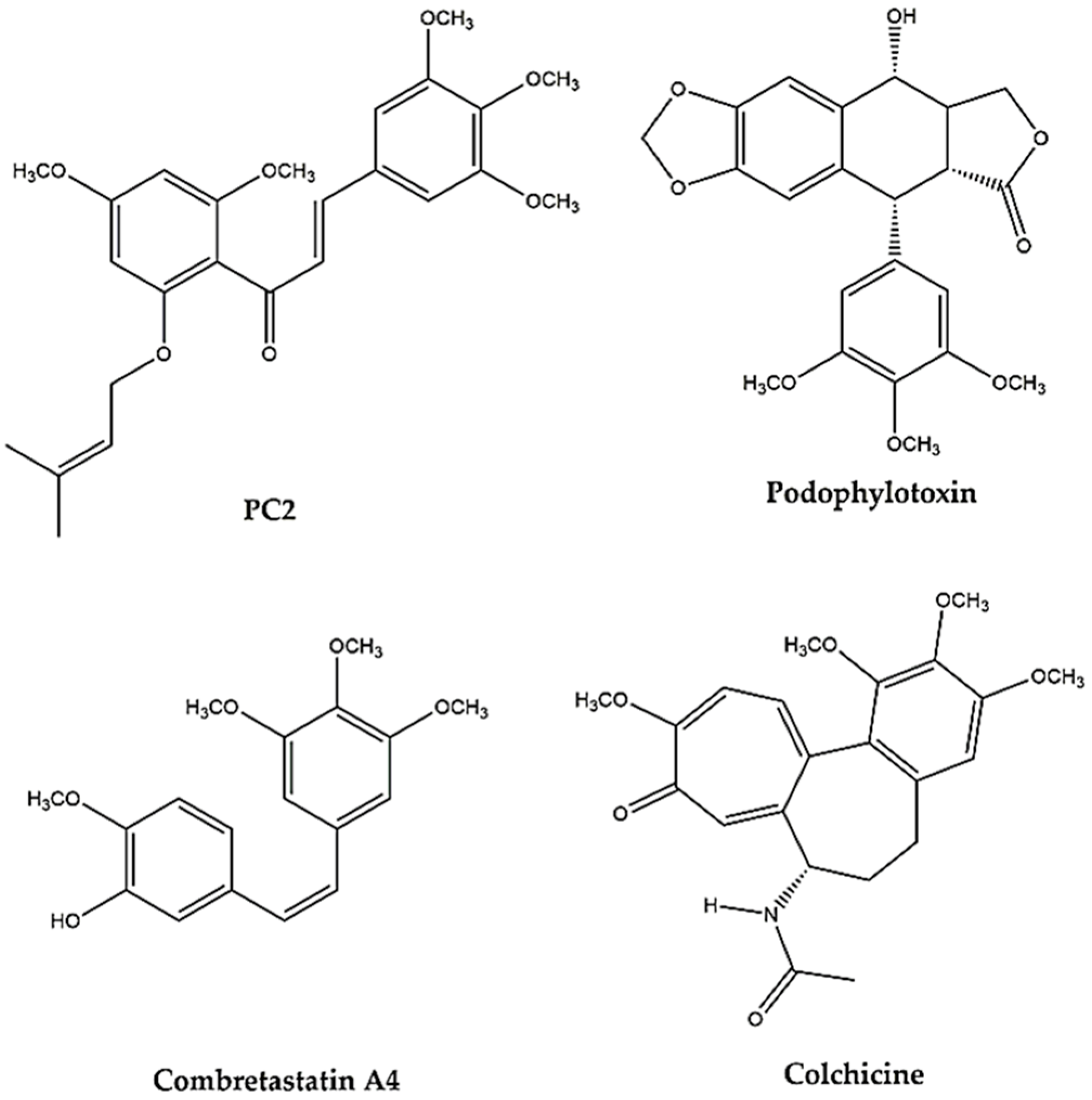
1. Introduction
2. Results
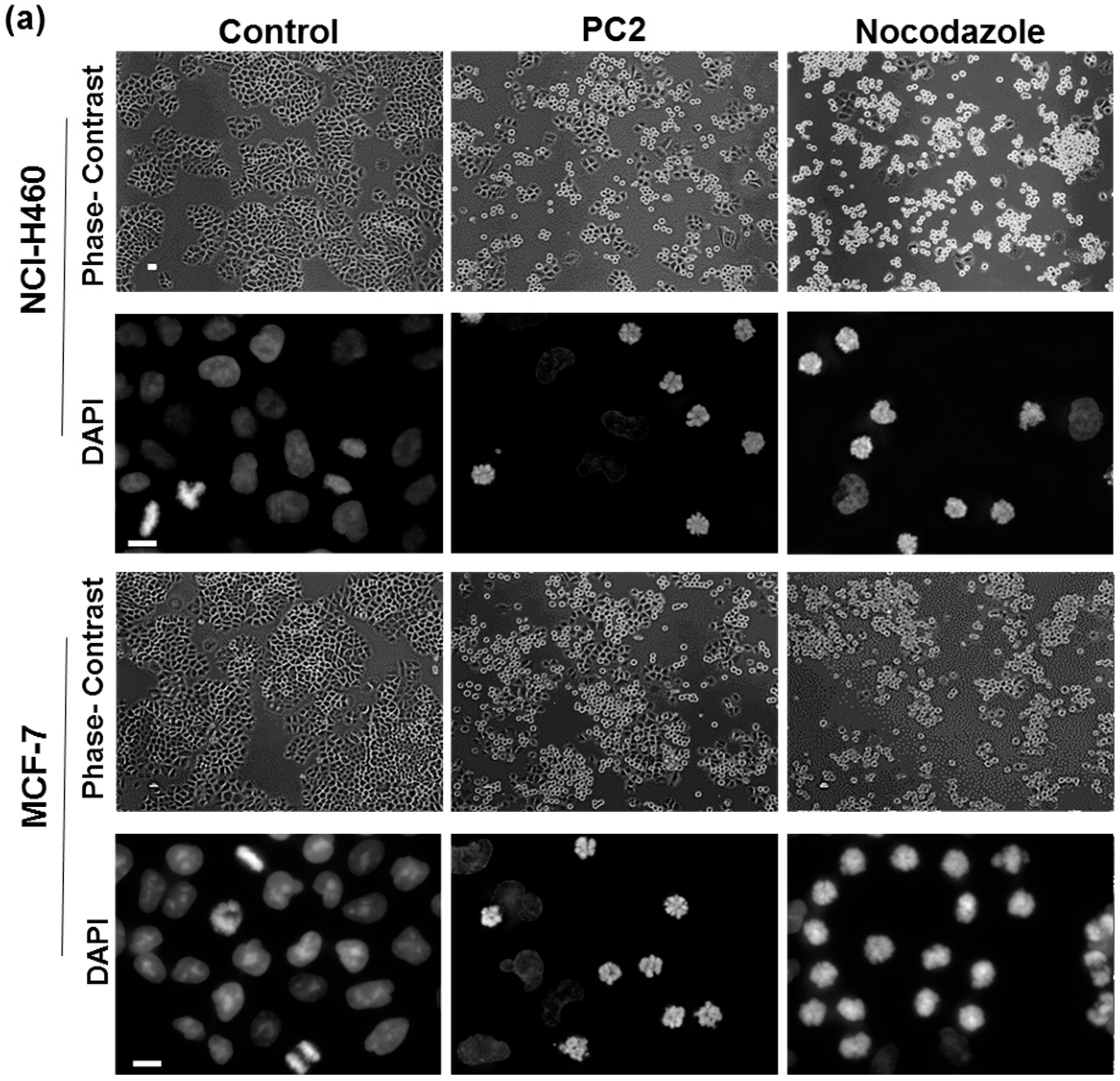
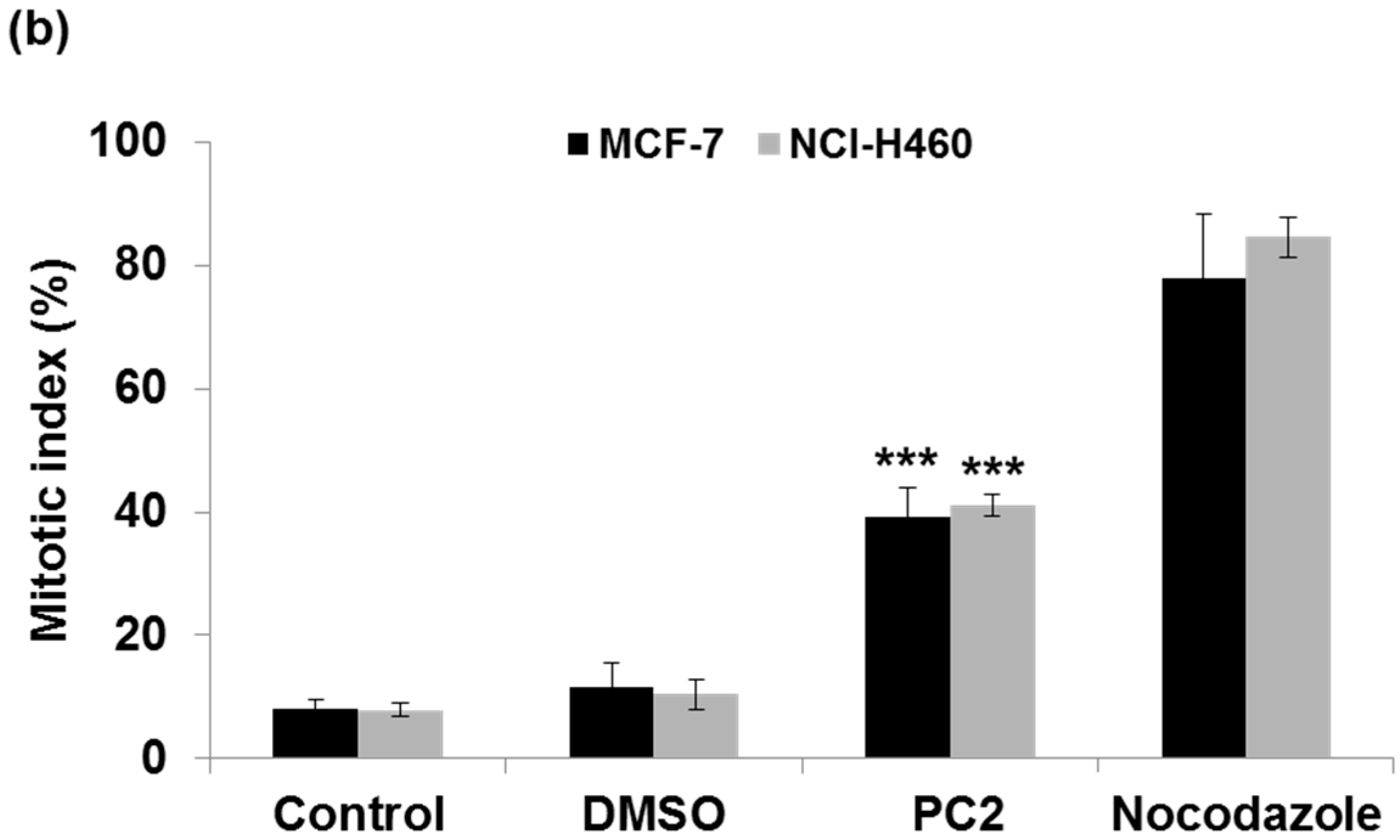
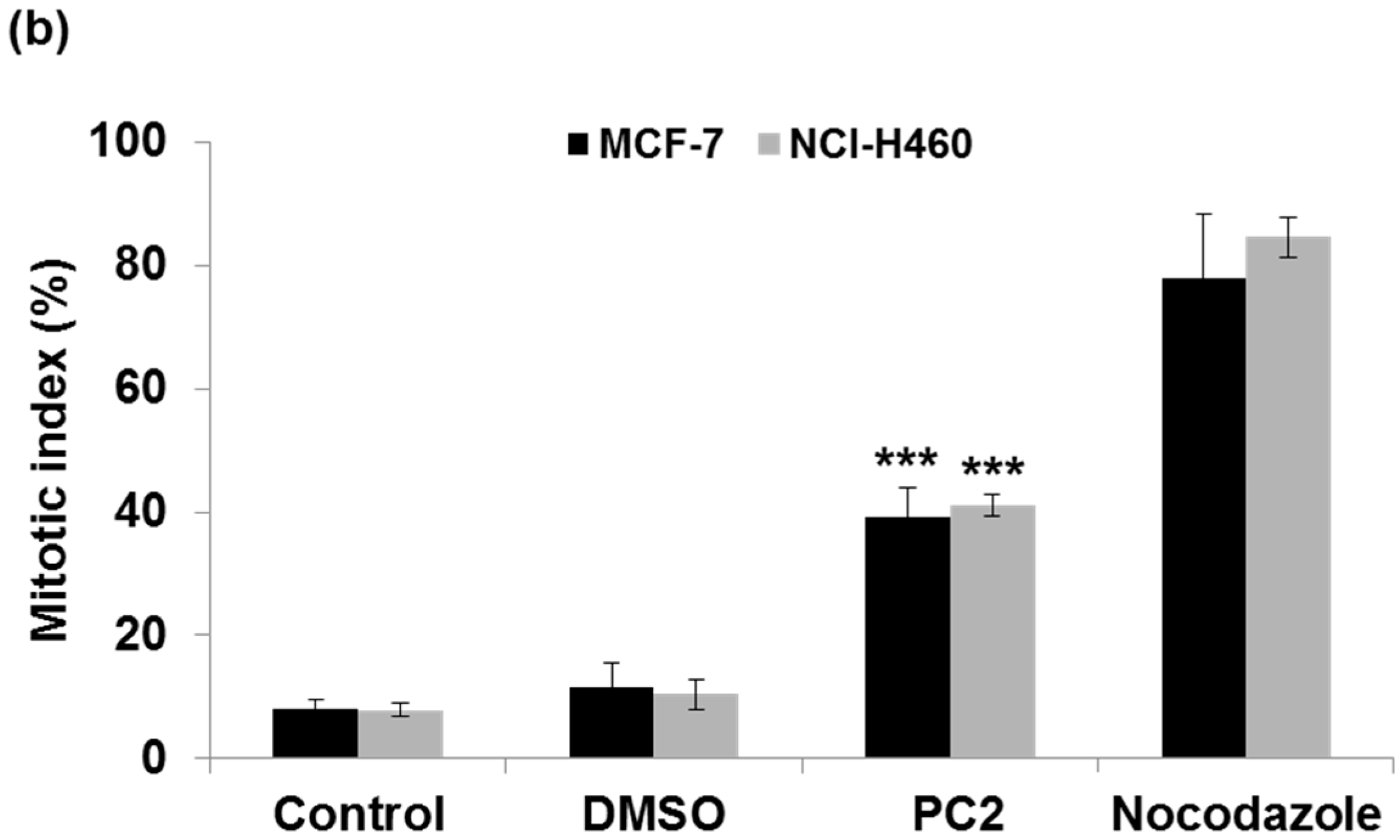
2.1. Treatment with PC2 Arrests MCF-7 and NCI-H460 Cells in Mitosis
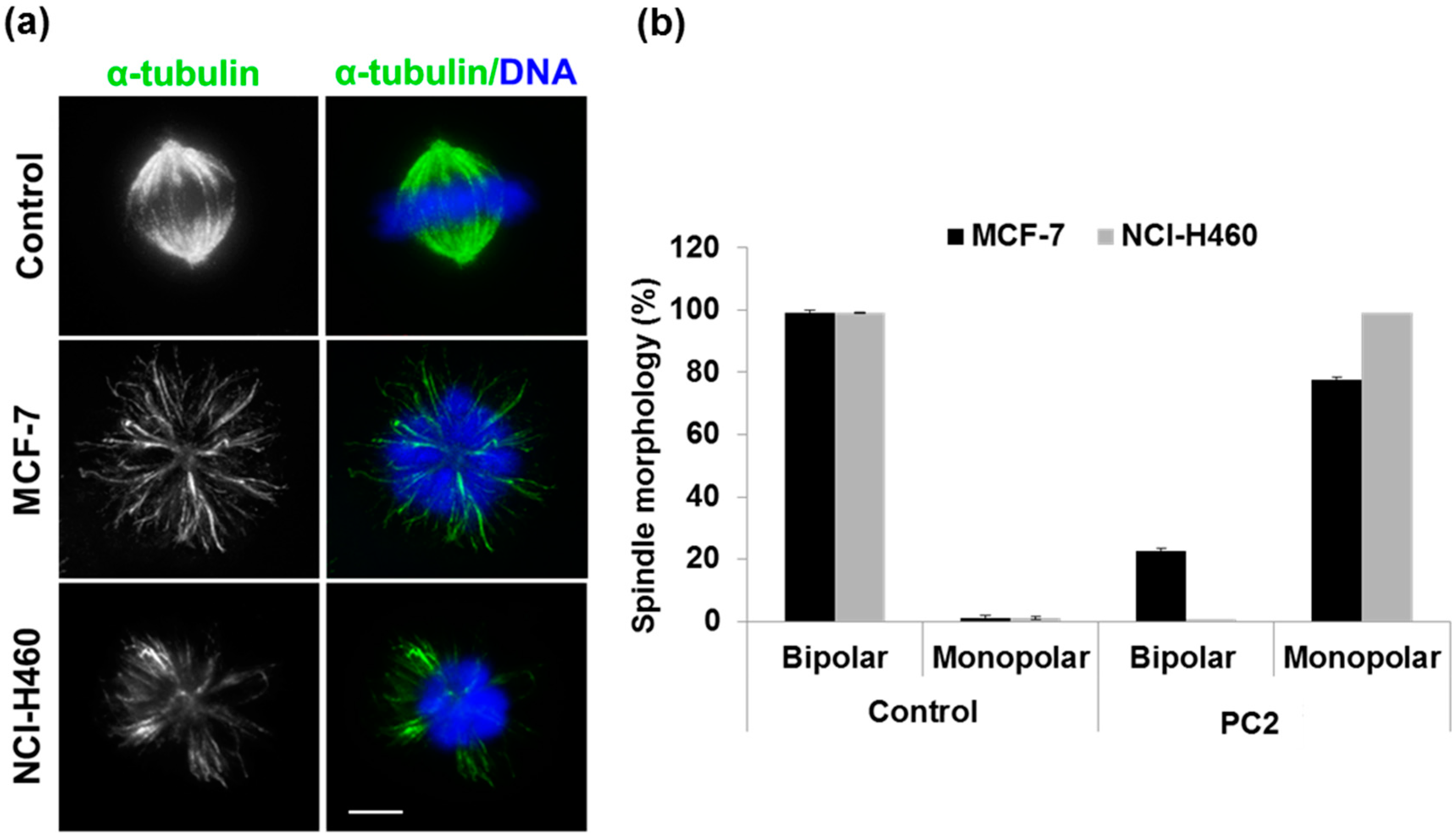
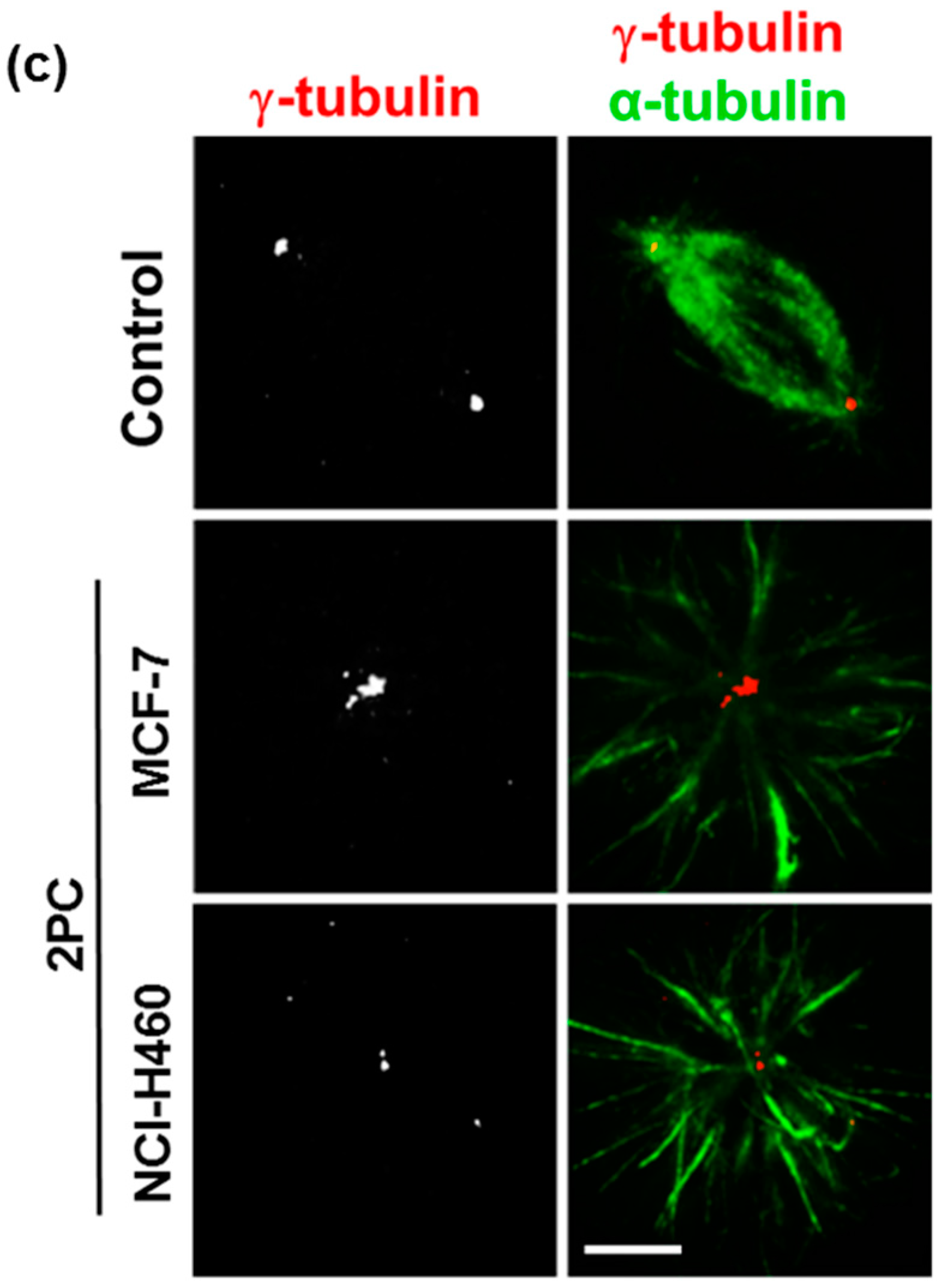
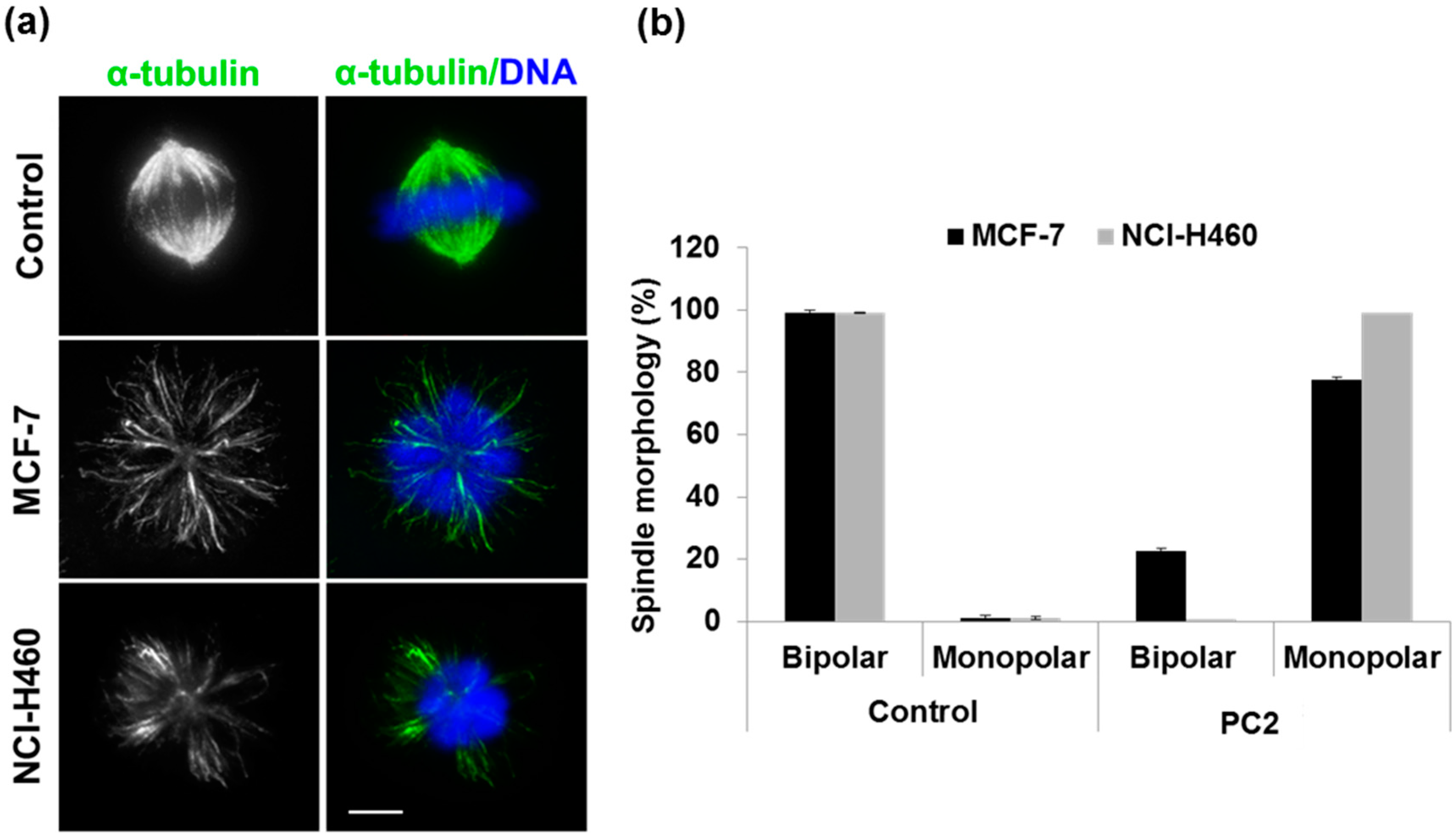
2.2. Treatment with PC2 Induces Mitotic Spindle Collapse
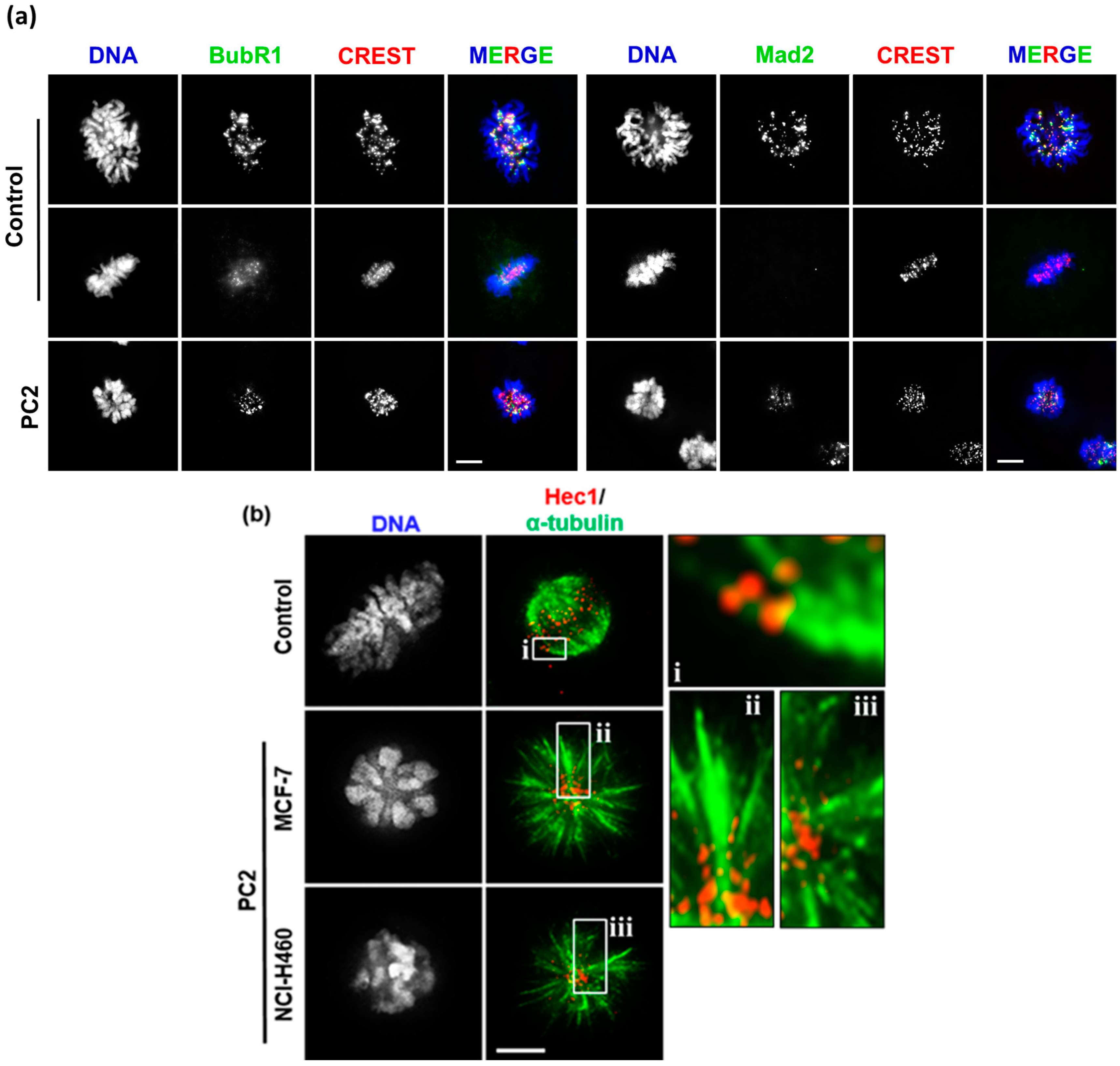
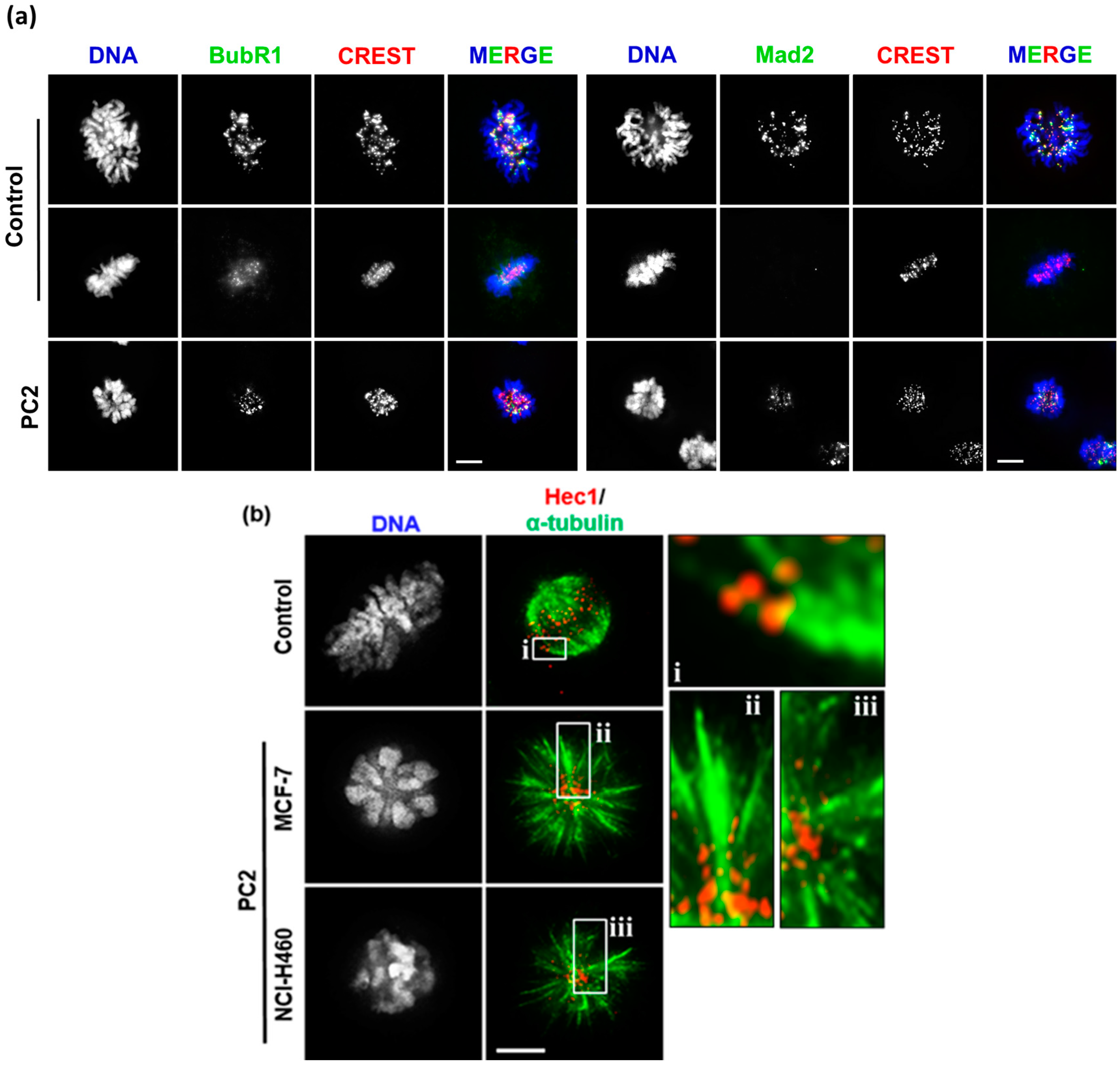
2.3. Treatment with PC2 Induces Activation of the Spindle Assembly Checkpoint
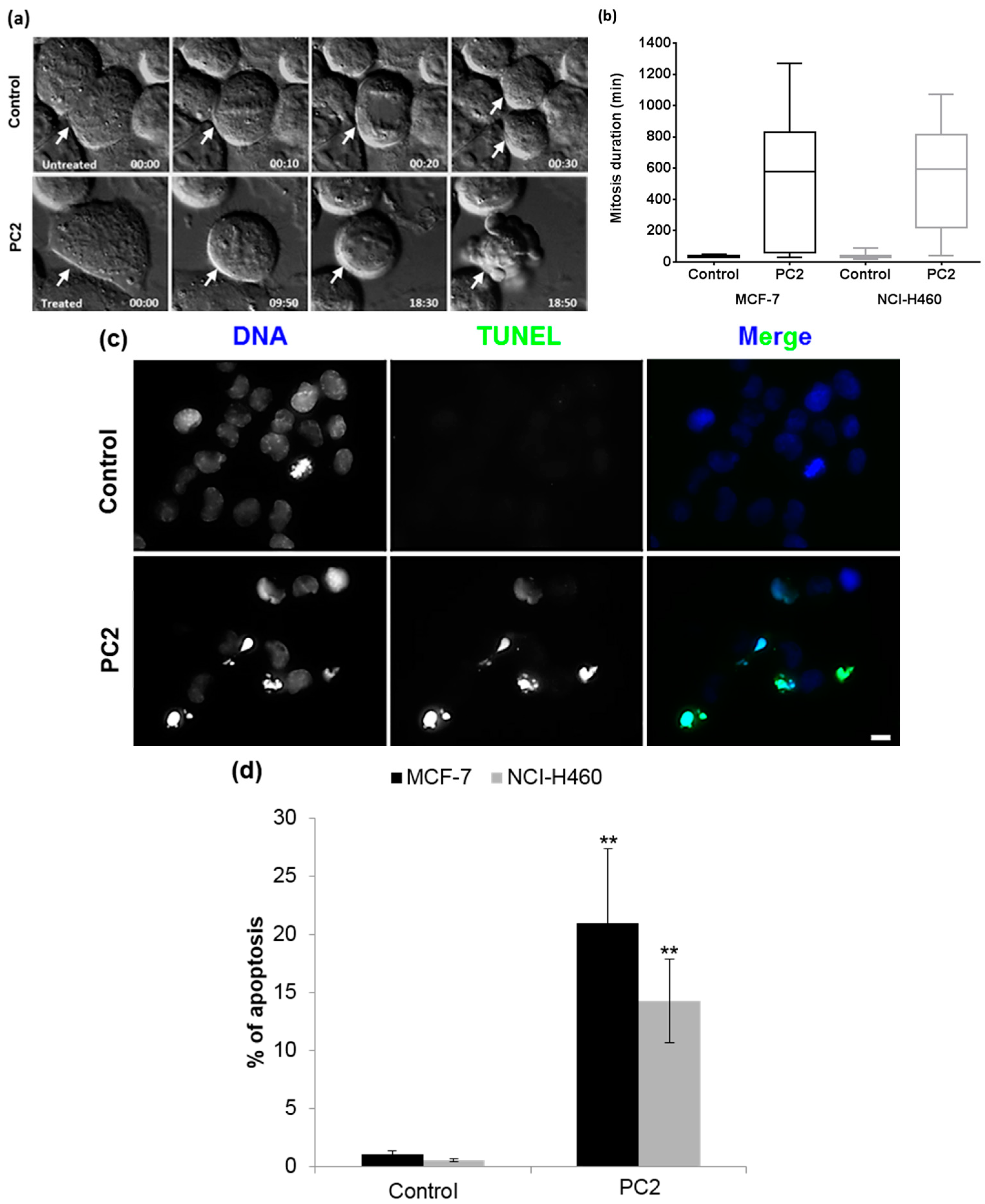
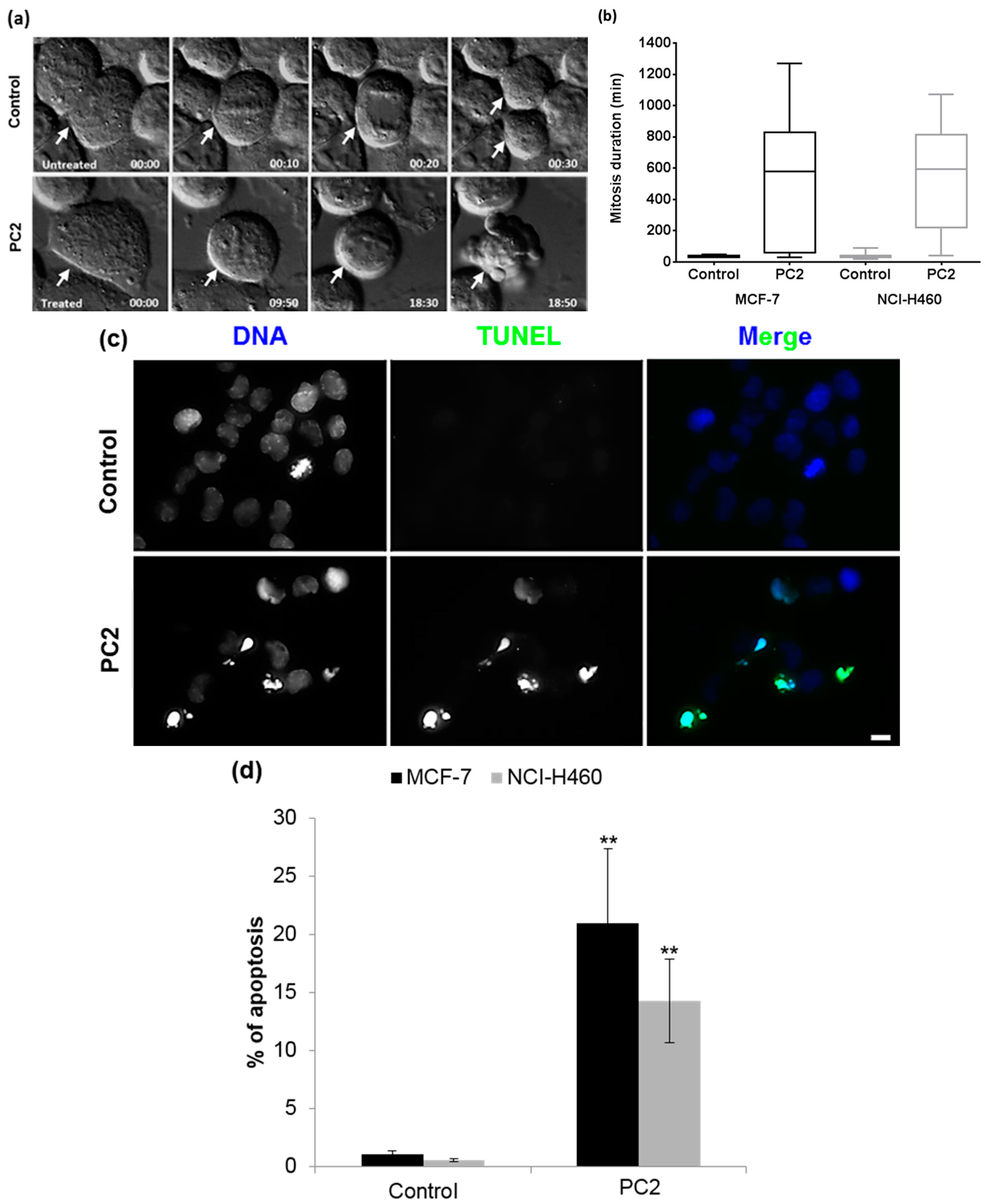
2.4. Treatment with PC2 Causes a Long Mitotic Delay which Triggers Mitotic Catastrophe Accompanied by Apoptosis
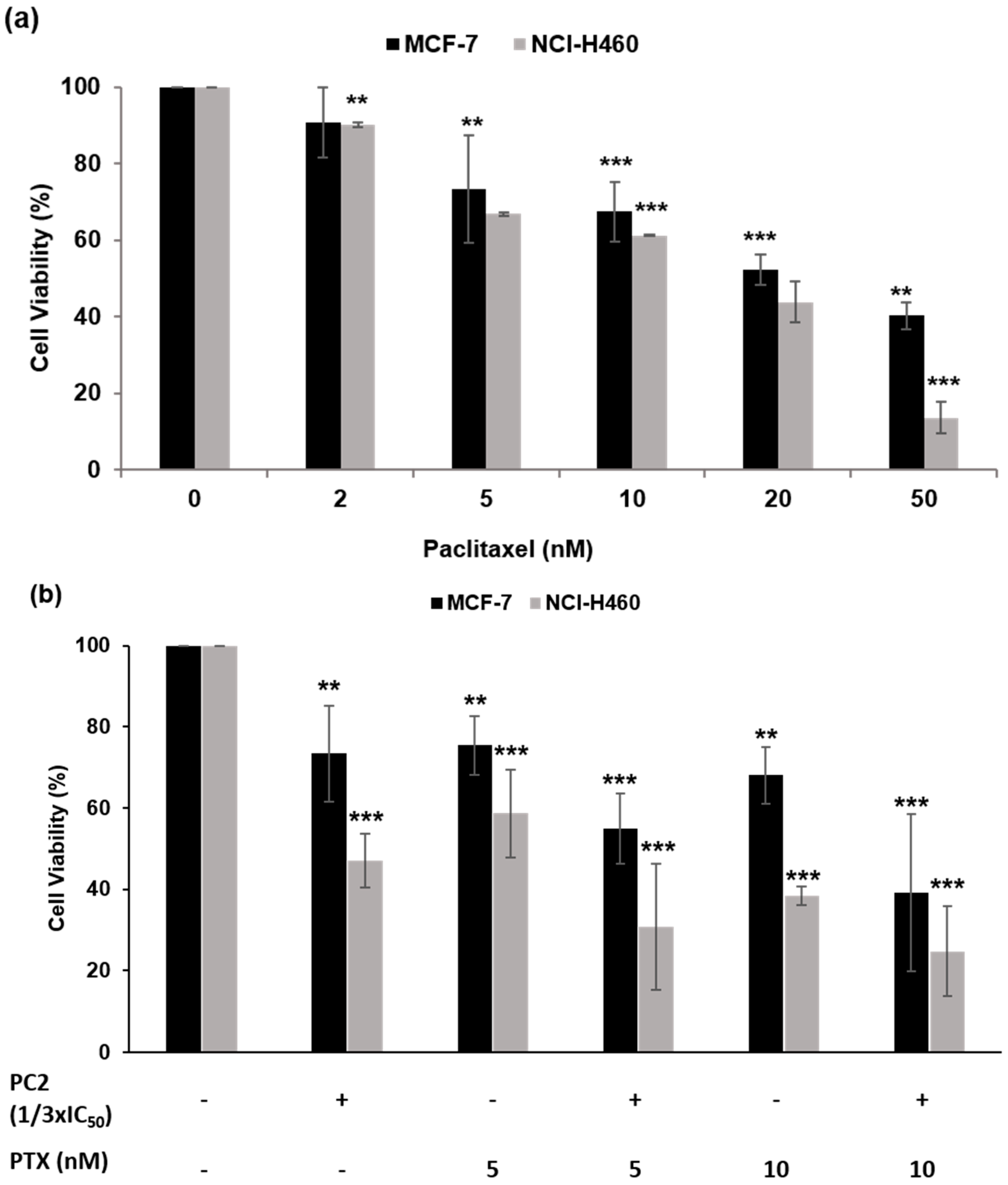
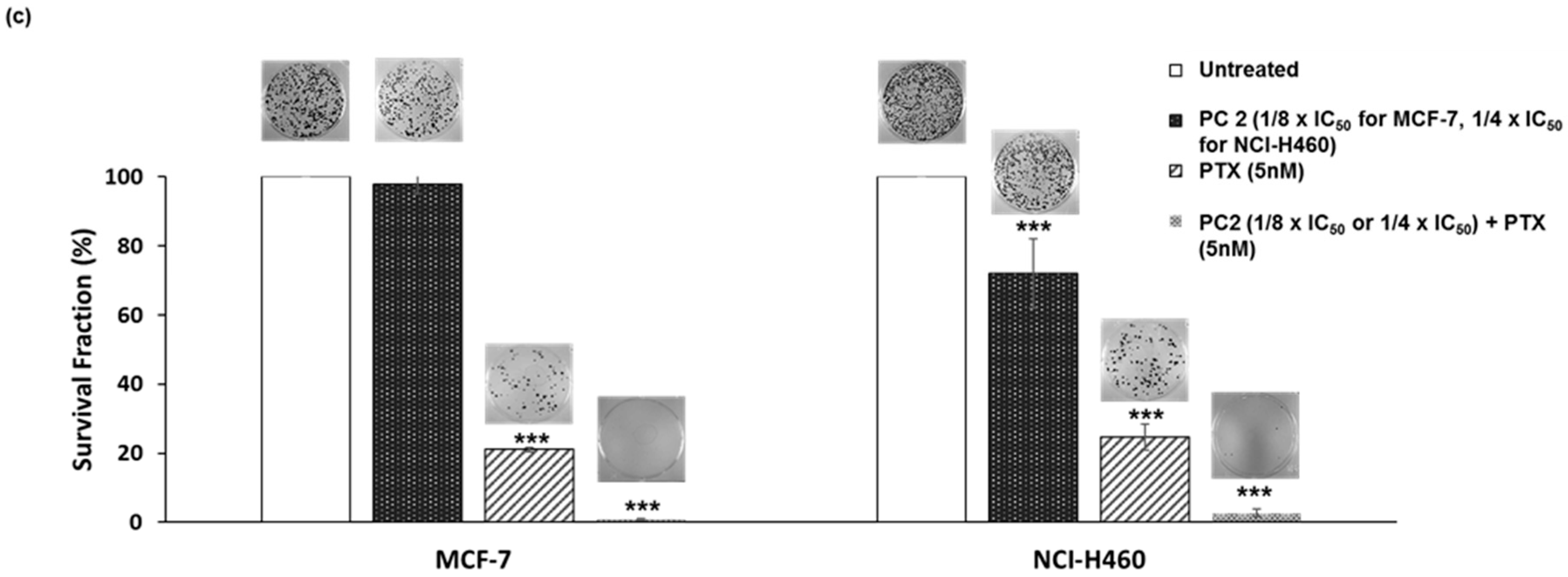
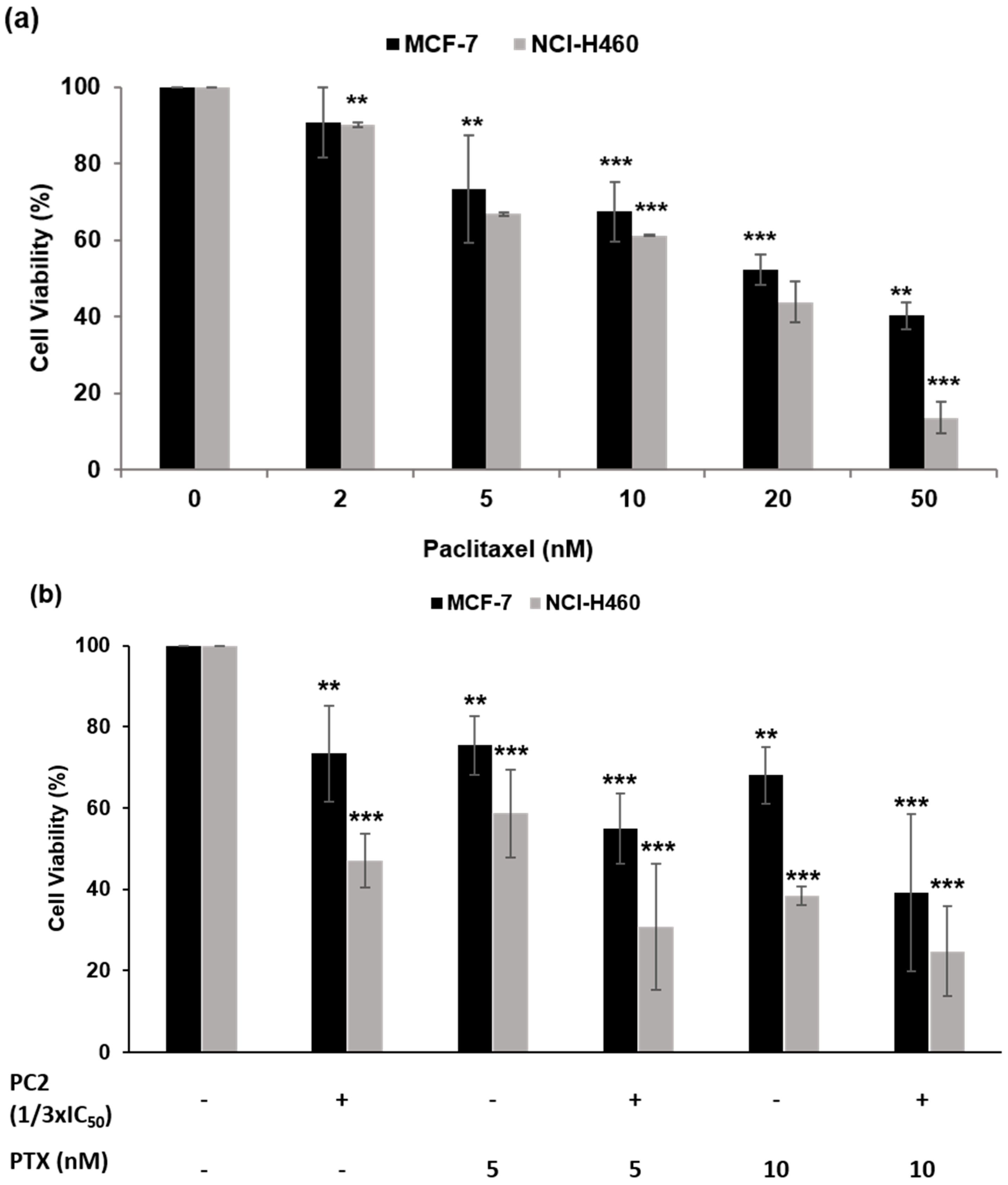
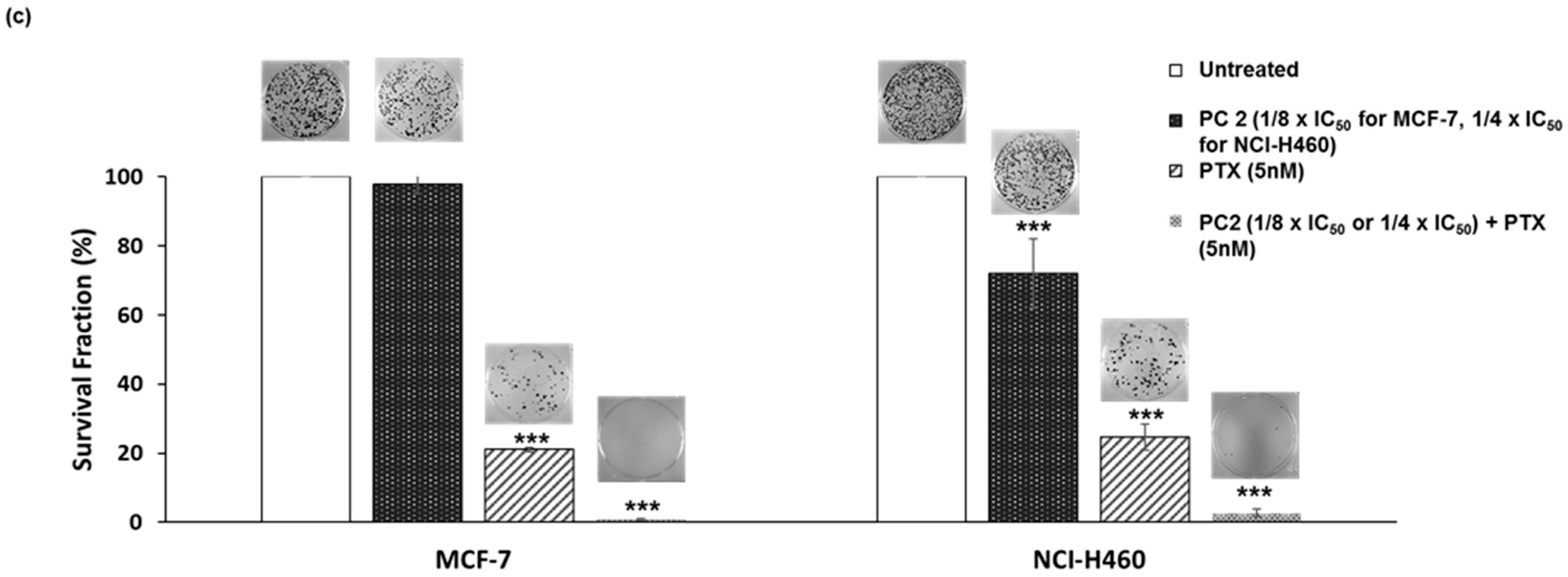
2.5. Treatment with PC2 Enhances Tumor Cell Sensitivity to Paclitaxel
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Culture
4.3. Viability Assay
4.4. Clonogenic Assay
4.5. Determination of Mitotic Index
4.6. Immunofluorescence
4.7. TUNEL Assay
4.8. Time-Lapse Video Microscopy
4.9. Image Acquisition and Processing
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Van der Vaart, B.; Akhmanova, A.; Straube, A. Regulation of microtubule dynamic instability. Biochem. Soc. Trans. 2009, 37, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Perez, E.A. Microtubule inhibitors: Differentiating tubulin-inhibiting agents based on mechanisms of action, clinical activity, and resistance. Mol. Cancer Ther. 2009, 8, 2086–2095. [Google Scholar] [CrossRef] [PubMed]
- Fojo, T.; Menefee, M. Mechanisms of multidrug resistance: The potential role of microtubule-stabilizing agents. Ann. Oncol. 2007, 18 (Suppl. S5), v3–v8. [Google Scholar] [CrossRef] [PubMed]
- Kavallaris, M. Microtubules and resistance to tubulin-binding agents. Nat. Rev. Cancer 2010, 10, 194–204. [Google Scholar] [CrossRef] [PubMed]
- Mukhtar, E.; Adhami, V.M.; Mukhtar, H. Targeting microtubules by natural agents for cancer therapy. Mol. Cancer Ther. 2014, 13, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Negi, A.S.; Gautam, Y.; Alam, S.; Chanda, D.; Luqman, S.; Sarkar, J.; Khan, F.; Konwar, R. Natural antitubulin agents: Importance of 3,4,5-trimethoxyphenyl fragment. Bioorg. Med. Chem. 2015, 23, 373–389. [Google Scholar] [CrossRef] [PubMed]
- Luduena, R.F.; Roach, M.C. Tubulin sulfhydryl groups as probes and targets for antimitotic and antimicrotubule agents. Pharmacol. Ther. 1991, 49, 133–152. [Google Scholar] [CrossRef]
- Boumendjel, A.; Ronot, X.; Boutonnat, J. Chalcones derivatives acting as cell cycle blockers: Potential anti cancer drugs? Curr. Drug Targets 2009, 10, 363–371. [Google Scholar] [PubMed]
- Lopez-Lazaro, M. Flavonoids as anticancer agents: Structure-activity relationship study. Curr. Med. Chem. Anticancer Agents 2002, 2, 691–714. [Google Scholar] [CrossRef] [PubMed]
- Sahu, N.K.; Balbhadra, S.S.; Choudhary, J.; Kohli, D.V. Exploring pharmacological significance of chalcone scaffold: A review. Curr. Med. Chem. 2012, 19, 209–225. [Google Scholar] [CrossRef] [PubMed]
- Botta, B.; Vitali, A.; Menendez, P.; Misiti, D.; Delle Monache, G. Prenylated flavonoids: Pharmacology and biotechnology. Curr Med. Chem. 2005, 12, 717–739. [Google Scholar] [CrossRef] [PubMed]
- Ducki, S. Antimitotic chalcones and related compounds as inhibitors of tubulin assembly. Anti-Cancer Agents Med. Chem. 2009, 9, 336–347. [Google Scholar] [CrossRef]
- Ducki, S.; Mackenzie, G.; Greedy, B.; Armitage, S.; Chabert, J.F.; Bennett, E.; Nettles, J.; Snyder, J.P.; Lawrence, N.J. Combretastatin-like chalcones as inhibitors of microtubule polymerisation. Part 2: Structure-based discovery of alpha-aryl chalcones. Bioorg. Med. Chem. 2009, 17, 7711–7722. [Google Scholar] [CrossRef] [PubMed]
- Edwards, M.L.; Stemerick, D.M.; Sunkara, P.S. Chalcones: A new class of antimitotic agents. J. Med. Chem. 1990, 33, 1948–1954. [Google Scholar] [CrossRef] [PubMed]
- Peyrot, V.; Leynadier, D.; Sarrazin, M.; Briand, C.; Menendez, M.; Laynez, J.; Andreu, J.M. Mechanism of binding of the new antimitotic drug mdl 27048 to the colchicine site of tubulin: Equilibrium studies. Biochemistry 1992, 31, 11125–11132. [Google Scholar] [CrossRef] [PubMed]
- Peyrot, V.; Leynadier, D.; Sarrazin, M.; Briand, C.; Rodriquez, A.; Nieto, J.M.; Andreu, J.M. Interaction of tubulin and cellular microtubules with the new antitumor drug mdl 27048. A powerful and reversible microtubule inhibitor. J. Biol. Chem. 1989, 264, 21296–21301. [Google Scholar] [PubMed]
- Wang, G.; Peng, F.; Cao, D.; Yang, Z.; Han, X.; Liu, J.; Wu, W.; He, L.; Ma, L.; Chen, J.; et al. Design, synthesis and biological evaluation of millepachine derivatives as a new class of tubulin polymerization inhibitors. Bioorg. Med. Chem. 2013, 21, 6844–6854. [Google Scholar] [CrossRef] [PubMed]
- Neves, M.P.; Lima, R.T.; Choosang, K.; Pakkong, P.; de Sao Jose Nascimento, M.; Vasconcelos, M.H.; Pinto, M.; Silva, A.M.; Cidade, H. Synthesis of a natural chalcone and its prenyl analogs—Evaluation of tumor cell growth-inhibitory activities, and effects on cell cycle and apoptosis. Chem. Biodivers. 2012, 9, 1133–1143. [Google Scholar] [CrossRef] [PubMed]
- Leao, M.; Soares, J.; Gomes, S.; Raimundo, L.; Ramos, H.; Bessa, C.; Queiroz, G.; Domingos, S.; Pinto, M.; Inga, A.; et al. Enhanced cytotoxicity of prenylated chalcone against tumour cells via disruption of the p53-mdm2 interaction. Life Sci. 2015, 142, 60–65. [Google Scholar] [CrossRef] [PubMed]
- Silva, P.; Barbosa, J.; Nascimento, A.V.; Faria, J.; Reis, R.; Bousbaa, H. Monitoring the fidelity of mitotic chromosome segregation by the spindle assembly checkpoint. Cell Prolif. 2011, 44, 391–400. [Google Scholar] [CrossRef] [PubMed]
- Logarinho, E.; Bousbaa, H. Kinetochore-microtubule interactions “in check” by bub1, bub3 and bubr1: The dual task of attaching and signalling. Cell Cycle 2008, 7, 1763–1768. [Google Scholar] [CrossRef] [PubMed]
- Stanton, R.A.; Gernert, K.M.; Nettles, J.H.; Aneja, R. Drugs that target dynamic microtubules: A new molecular perspective. Med. Res. Rev. 2011, 31, 443–481. [Google Scholar] [CrossRef] [PubMed]
- Galan-Malo, P.; Vela, L.; Gonzalo, O.; Calvo-Sanjuan, R.; Gracia-Fleta, L.; Naval, J.; Marzo, I. Cell fate after mitotic arrest in different tumor cells is determined by the balance between slippage and apoptotic threshold. Toxicol. Appl. Pharmacol. 2012, 258, 384–393. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. How do anti-mitotic drugs kill cancer cells? J. Cell Sci. 2009, 122, 2579–2585. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Vitale, I.; Abrams, J.M.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; Dawson, T.M.; Dawson, V.L.; El-Deiry, W.S.; Fulda, S.; et al. Molecular definitions of cell death subroutines: Recommendations of the nomenclature committee on cell death 2012. Cell Death Differ. 2012, 19, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Kroemer, G.; Galluzzi, L.; Vandenabeele, P.; Abrams, J.; Alnemri, E.S.; Baehrecke, E.H.; Blagosklonny, M.V.; El-Deiry, W.S.; Golstein, P.; Green, D.R.; et al. Classification of cell death: Recommendations of the nomenclature committee on cell death 2009. Cell Death Differ. 2009, 16, 3–11. [Google Scholar] [CrossRef] [PubMed]
- Vitale, I.; Galluzzi, L.; Castedo, M.; Kroemer, G. Mitotic catastrophe: A mechanism for avoiding genomic instability. Nat. Rev. Mol. Cell Biol. 2011, 12, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Gascoigne, K.E.; Taylor, S.S. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 2008, 14, 111–122. [Google Scholar] [CrossRef] [PubMed]
- Colin, D.J.; Hain, K.O.; Allan, L.A.; Clarke, P.R. Cellular responses to a prolonged delay in mitosis are determined by a DNA damage response controlled by bcl-2 family proteins. Open Biol. 2015, 5, 140156. [Google Scholar] [CrossRef] [PubMed]
- Orth, J.D.; Loewer, A.; Lahav, G.; Mitchison, T.J. Prolonged mitotic arrest triggers partial activation of apoptosis, resulting in DNA damage and p53 induction. Mol. Biol. Cell 2012, 23, 567–576. [Google Scholar] [CrossRef] [PubMed]
- Weaver, B.A. How taxol/paclitaxel kills cancer cells. Mol. Biol. Cell 2014, 25, 2677–2681. [Google Scholar] [CrossRef] [PubMed]
- Zasadil, L.M.; Andersen, K.A.; Yeum, D.; Rocque, G.B.; Wilke, L.G.; Tevaarwerk, A.J.; Raines, R.T.; Burkard, M.E.; Weaver, B.A. Cytotoxicity of paclitaxel in breast cancer is due to chromosome missegregation on multipolar spindles. Sci. Trans. Med. 2014, 6, 229ra243. [Google Scholar] [CrossRef] [PubMed]
- Masawang, K.; Pedro, M.; Cidade, H.; Reis, R.M.; Neves, M.P.; Correa, A.G.; Sudprasert, W.; Bousbaa, H.; Pinto, M.M. Evaluation of 2′,4′-dihydroxy-3,4,5-trimethoxychalcone as antimitotic agent that induces mitotic catastrophe in mcf-7 breast cancer cells. Toxicol. Lett. 2014, 229, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not Available.


© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fonseca, J.; Marques, S.; Silva, P.M.A.; Brandão, P.; Cidade, H.; Pinto, M.M.; Bousbaa, H. Prenylated Chalcone 2 Acts as an Antimitotic Agent and Enhances the Chemosensitivity of Tumor Cells to Paclitaxel. Molecules 2016, 21, 982. https://doi.org/10.3390/molecules21080982
Fonseca J, Marques S, Silva PMA, Brandão P, Cidade H, Pinto MM, Bousbaa H. Prenylated Chalcone 2 Acts as an Antimitotic Agent and Enhances the Chemosensitivity of Tumor Cells to Paclitaxel. Molecules. 2016; 21(8):982. https://doi.org/10.3390/molecules21080982
Chicago/Turabian StyleFonseca, Joana, Sandra Marques, Patrícia M. A. Silva, Pedro Brandão, Honorina Cidade, Madalena M. Pinto, and Hassan Bousbaa. 2016. "Prenylated Chalcone 2 Acts as an Antimitotic Agent and Enhances the Chemosensitivity of Tumor Cells to Paclitaxel" Molecules 21, no. 8: 982. https://doi.org/10.3390/molecules21080982