
Spectroelectrochemical Study of Carbon Monoxide and Ethanol Oxidation on Pt/C, PtSn(3:1)/C and PtSn(1:1)/C Catalysts



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Physicochemical Characterization
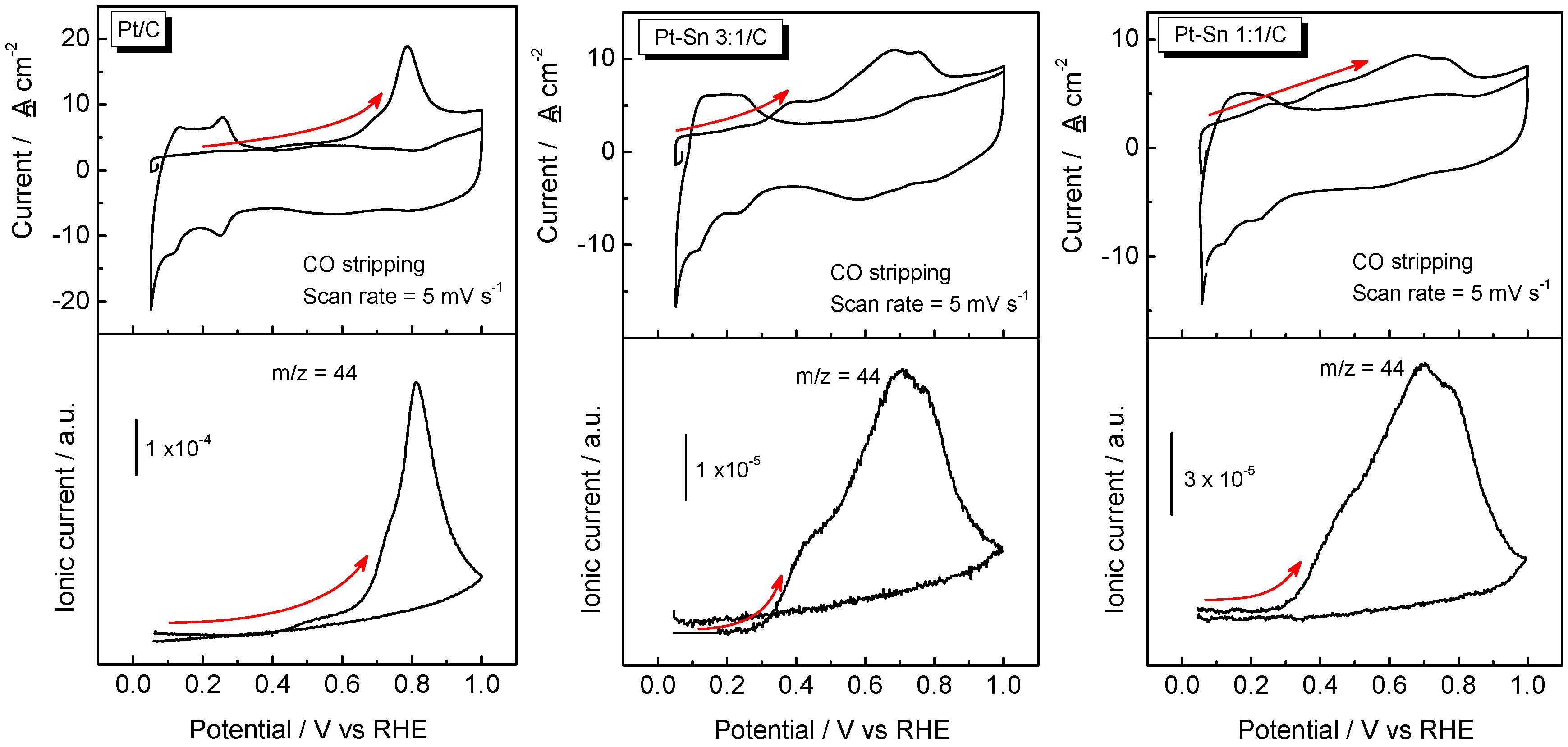
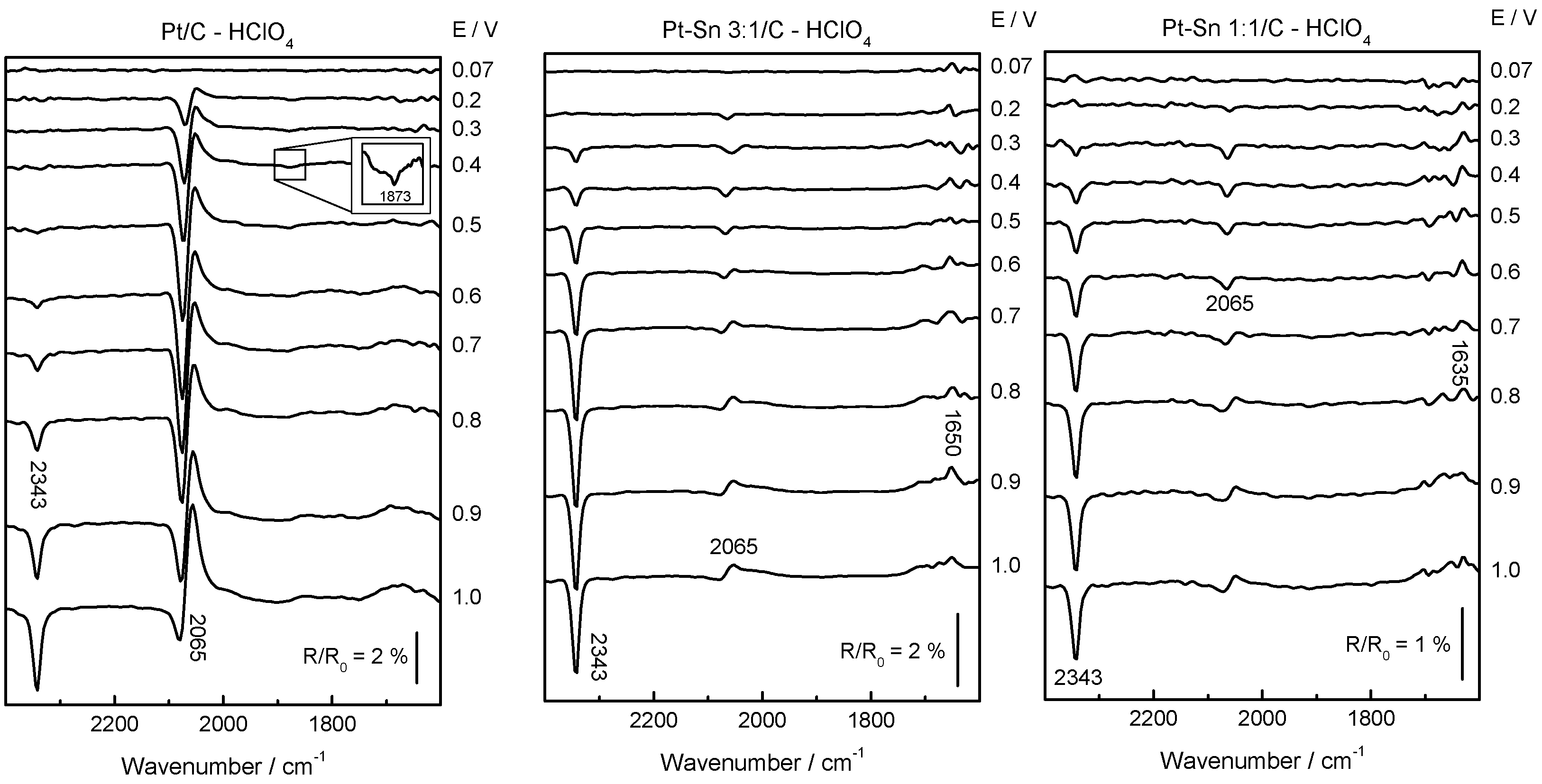
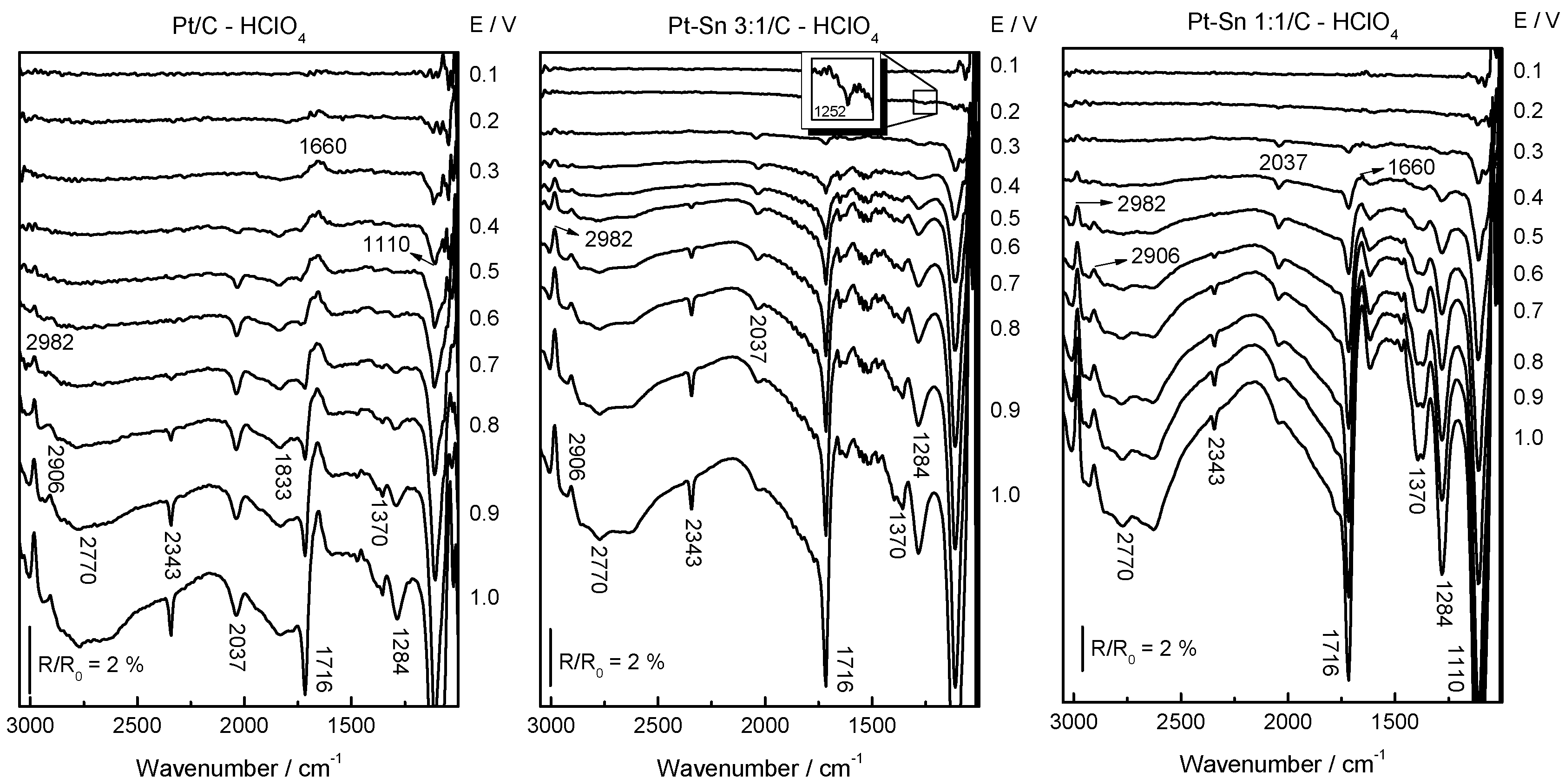
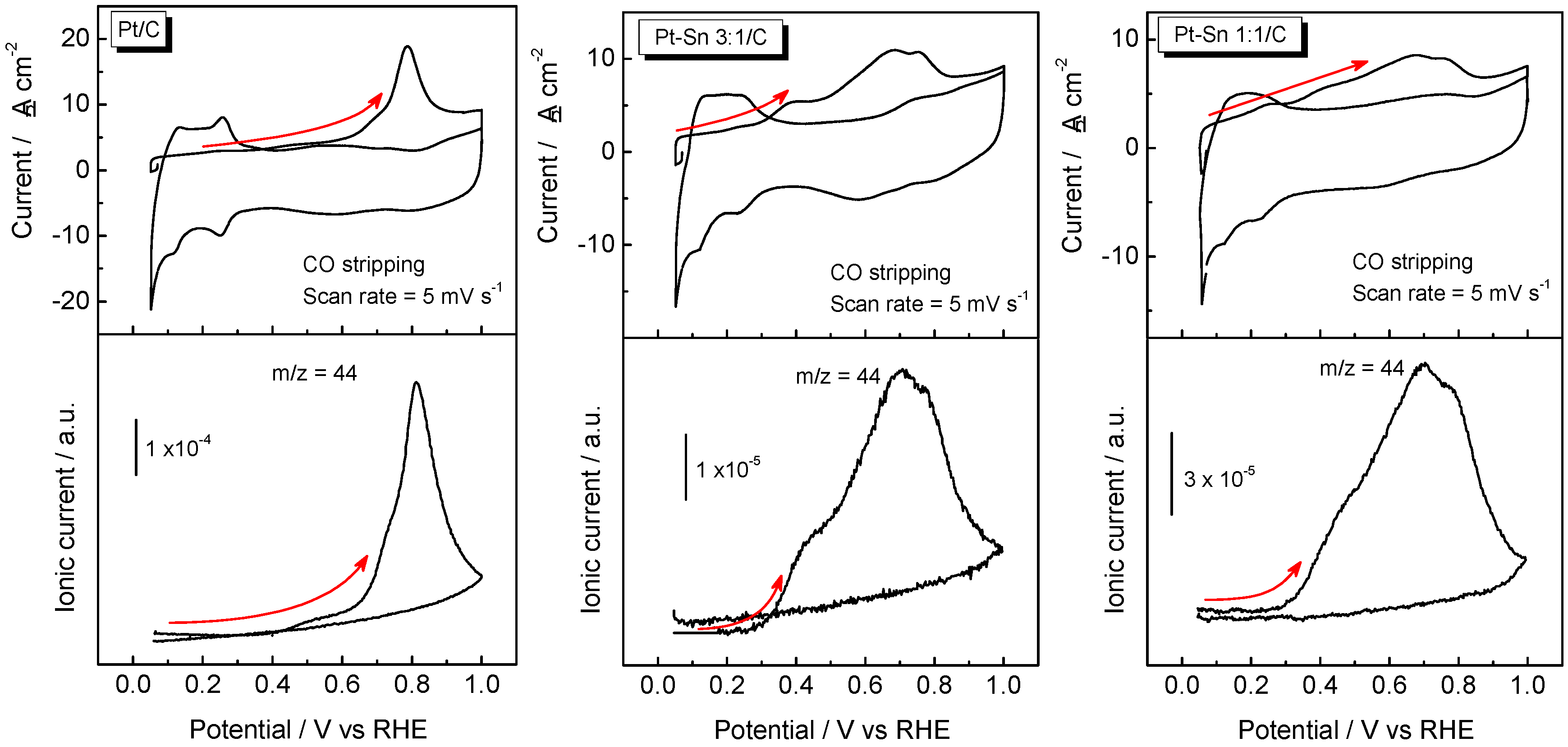
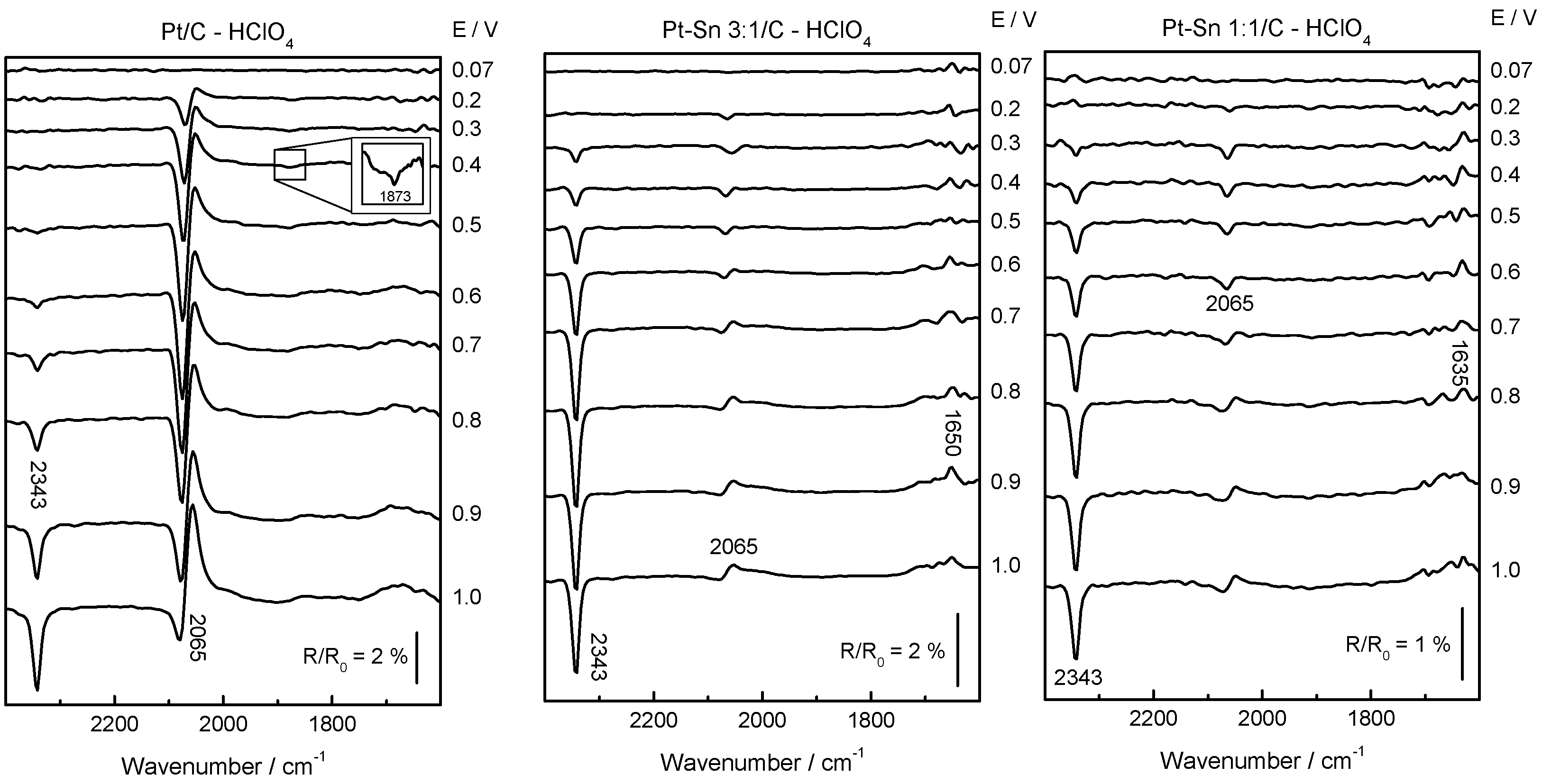
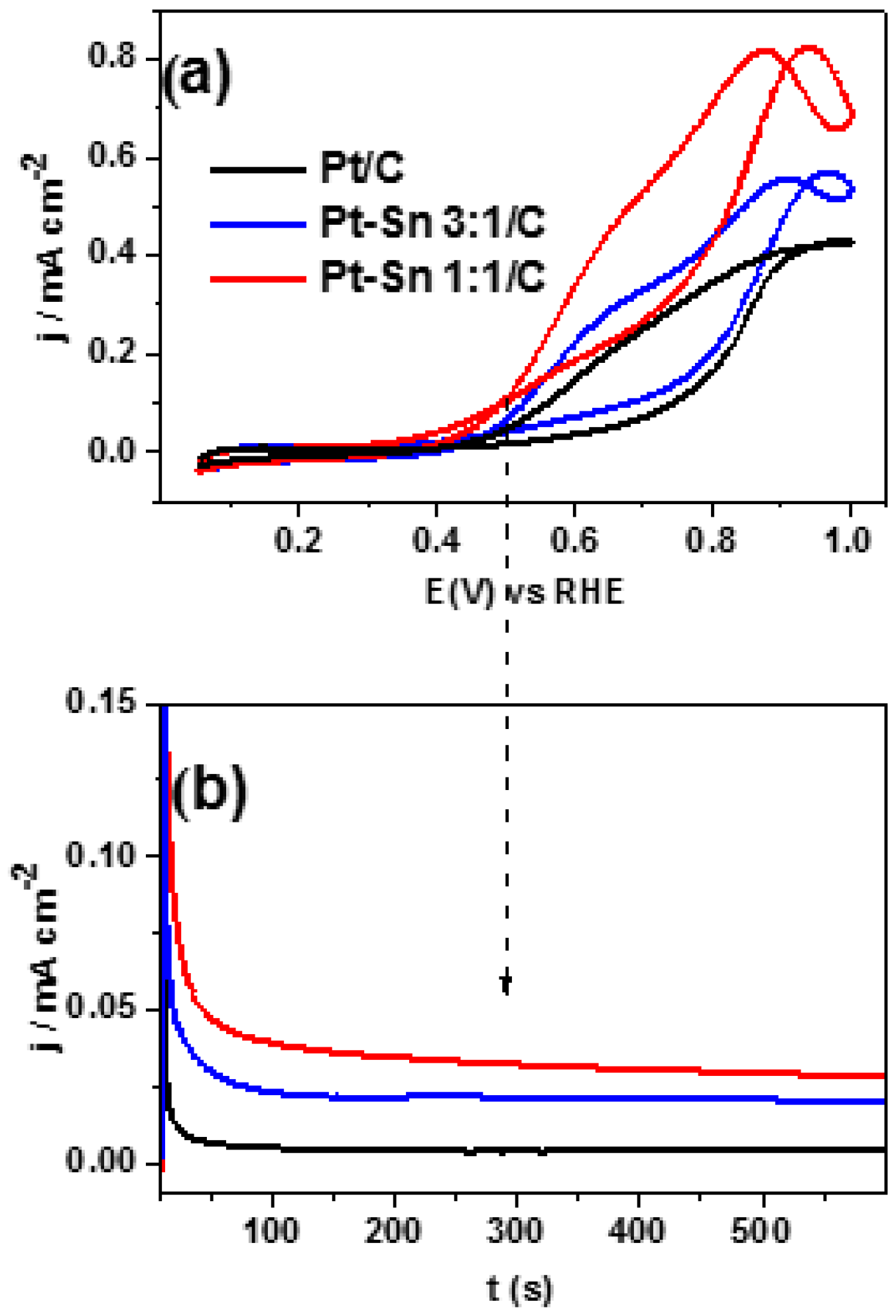
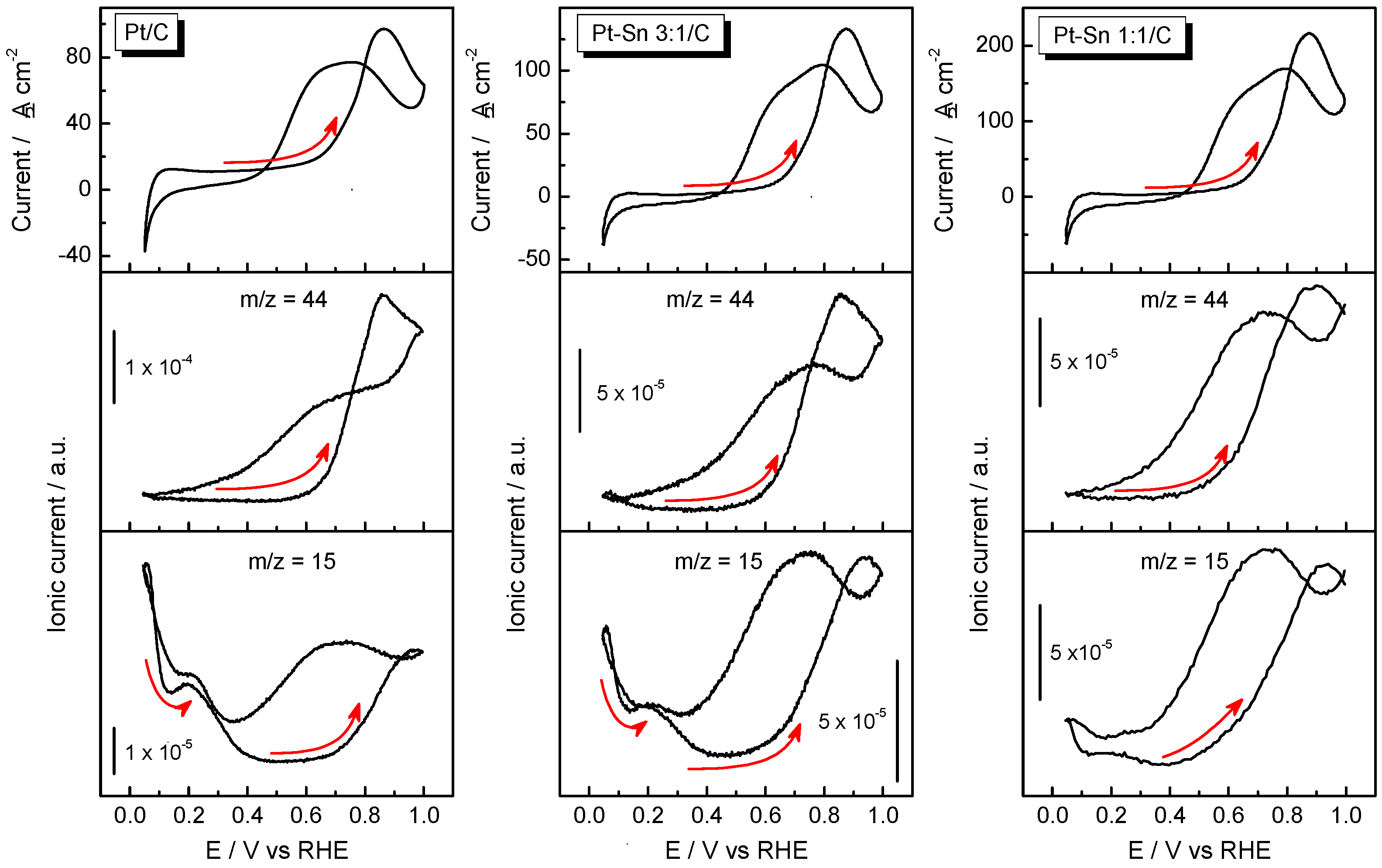
2.2. Adsorbed CO ELectrooxidation (CO Stripping)
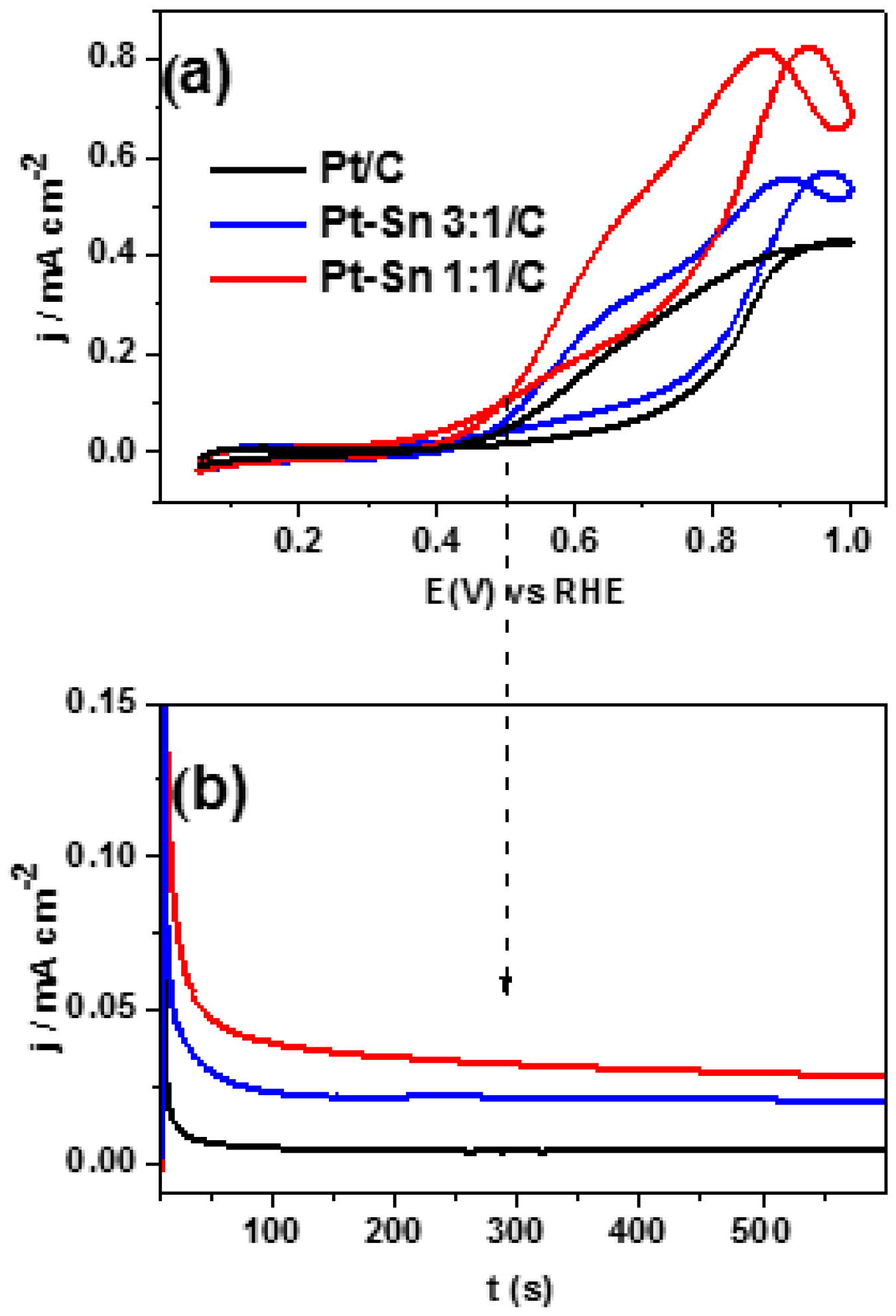
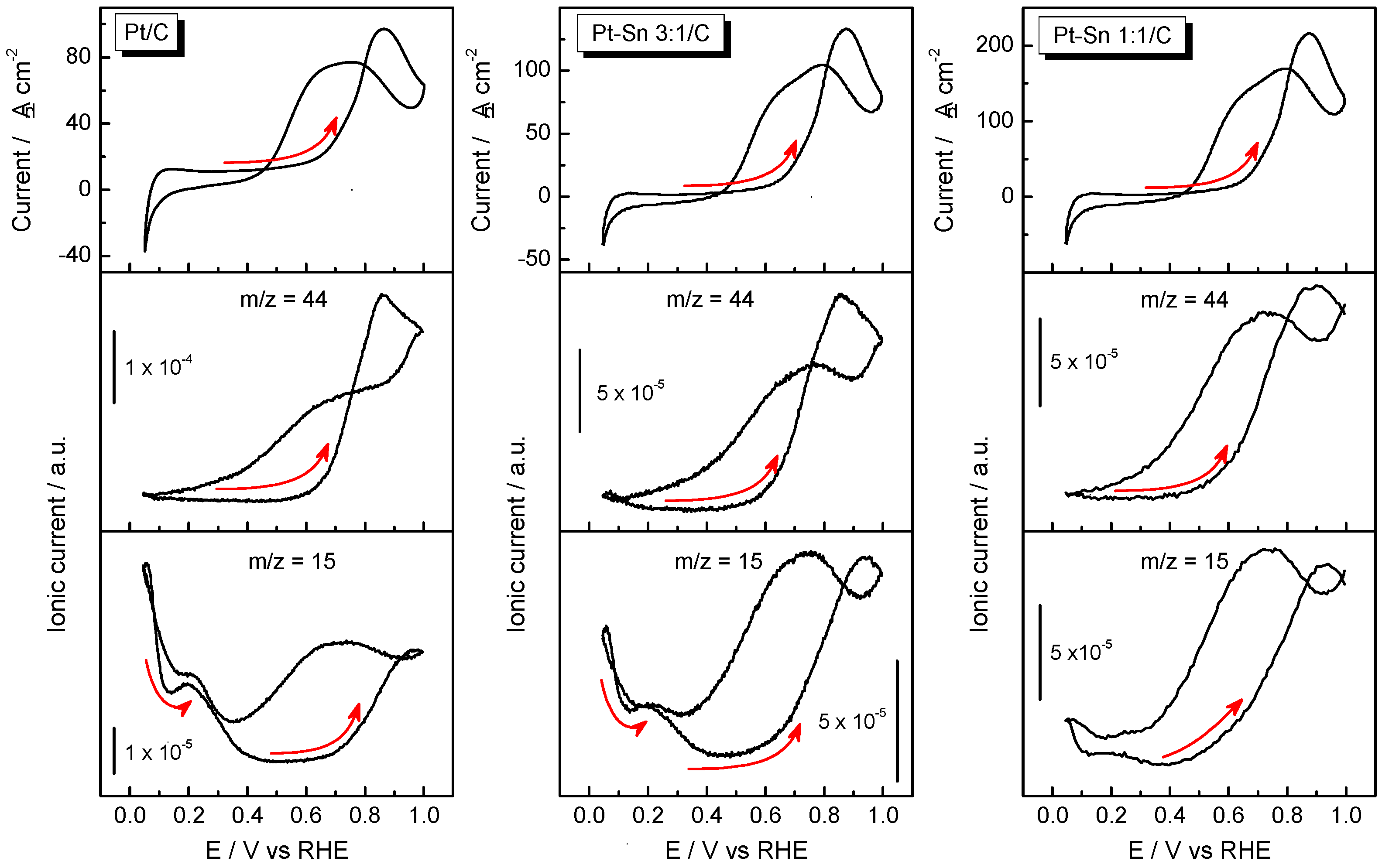
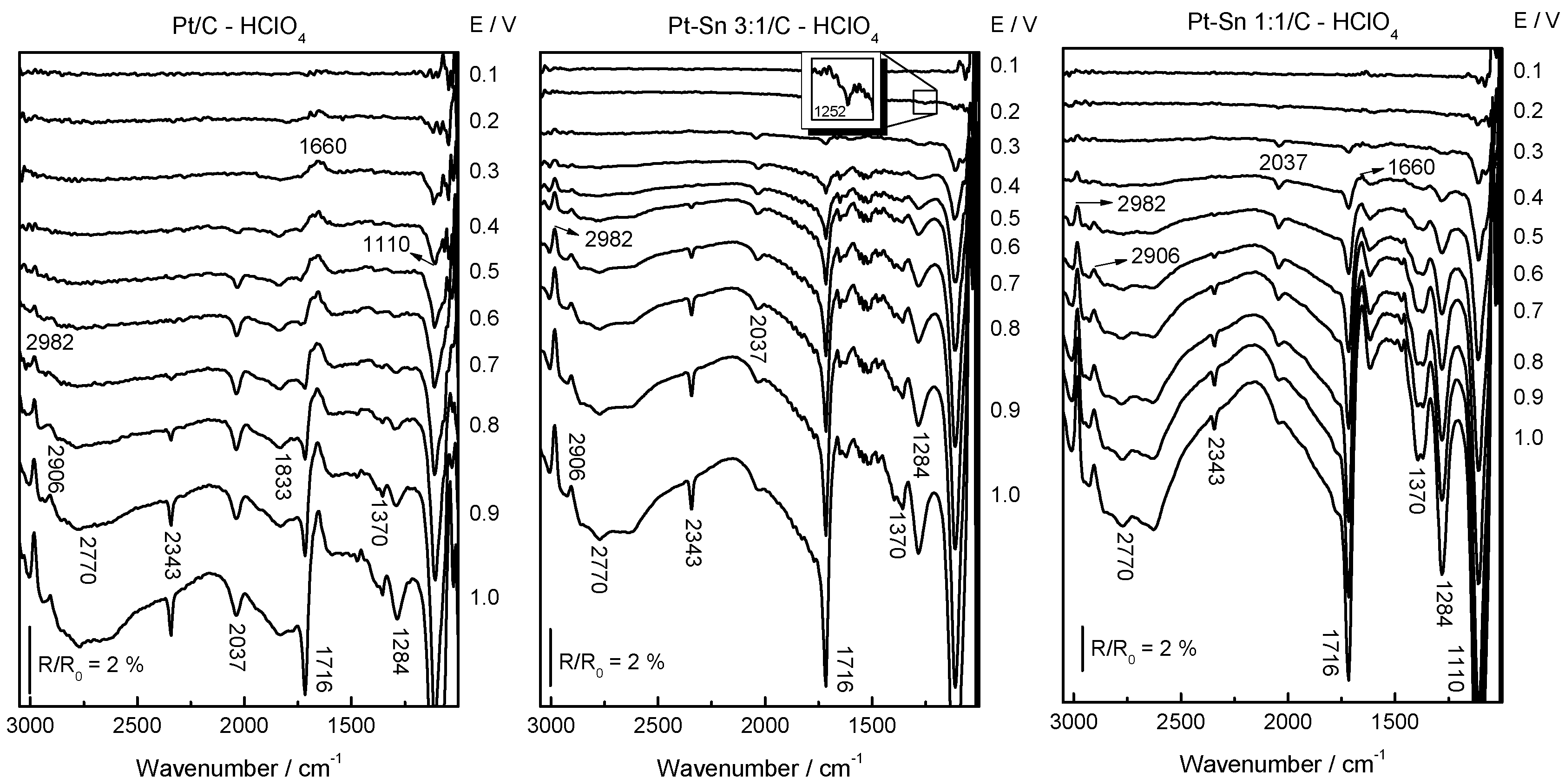
2.3. Ethanol Electrooxidation
3. Materials and Methods
3.1. Catalysts Preparation and Physicochemical Characterization
3.2. Electrochemical Characterization
3.3. EC-MS Set-Up
3.4. In-Situ FTIRS
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vielstich, W.; Paganin, V.A.; Alves, O.B.; Ciapina, E.G. Handbook of Fuel Cells—Fundamentals, Technology and Applications; Vielstich, W., Lamm, A., Gaseiger, H.A., Eds.; Wiley: New York, NY, USA, 2003; pp. 174–182. [Google Scholar]
- García, G.; Koper, M. Carbon monoxide oxidation on Pt single crystal electrodes: Understanding the catalysis for low temperature fuel cells. Chem. Phys. Chem. 2011, 12, 2064–2072. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Adzic, R.R. Low-Platinum-Content Electrocatalysts for Methanol and Ethanol Electrooxidation. In Electrocatalysis in Fuel Cells: A Non-Low-Platinum Approach; Shao, M., Ed.; Springer London: London, UK, 2013; pp. 1–25. [Google Scholar]
- Martínez-Huerta, M.V.; Tsiouvaras, N.; García, G.; Peña, M.A.; Pastor, E.; Rodriguez, J.L.; Fierro, J.L. Carbon-Supported PtRuMo Electrocatalysts for Direct Alcohol Fuel Cells. Catalysts 2013, 3, 811–838. [Google Scholar]
- Martínez Huerta, M.V.; García, G. Fabrication of electro-catalytic nano-particles and applications to proton exchange membrane fuel cells. In Micro and Nano-Engineering of Fuel Cells; Leung, D.Y.C., Xuan, J., Eds.; CRC Press: London, UK, 2015; pp. 95–129. [Google Scholar]
- Flórez-Montaño, J.; García, G.; Guillén-Villafuerte, O.; Rodríguez, J.L.; Planes, G.A.; Pastor, E. Mechanism of ethanol electrooxidation on mesoporous Pt electrode in acidic medium studied by a novel electrochemical mass spectrometry set-up. Electrochim. Acta 2016, 209, 121–131. [Google Scholar] [CrossRef]
- Guillén-Villafuerte, O.; García, G.; Arévalo, M.C.; Rodríguez, J.L.; Pastor, E. New insights on the electrochemical oxidation of ethanol on carbon-supported Pt electrode by a novel electrochemical mass spectrometry configuration. Electrochem. Comm. 2016, 63, 48–51. [Google Scholar] [CrossRef]
- Calvillo, L.; García, G.; Paduano, A.; Guillen-Villafuerte, O.; Valero-Vidal, C.; Vittadini, A.; Bellini, M.; Lavacchi, A.; Agnoli, S.; Martucci, A.; et al. Electrochemical Behavior of TiOxCy as Catalyst Support for Direct Ethanol Fuel Cells at Intermediate Temperature: From Planar Systems to Powders. ACS Appl. Mater. Interfaces 2016, 8, 716–725. [Google Scholar] [CrossRef] [PubMed]
- Roca-Ayats, M.; García, G.; Soler-Vicedo, M.; Pastor, E.; Lázaro, M.J.; Martínez-Huerta, M.V. The role of Sn, Ru and Ir. on the ethanol electrooxidation on Pt3M/TiCN electrocatalysts. Int. J. Hydrogen Energ. 2015, 40, 14519–14528. [Google Scholar] [CrossRef]
- Asgardi, J.; Calderón, J.C.; Alcaide, F.; Querejeta, A.; Calvillo, L.; Lázaro, M.J.; García, G.; Pastor, E. Carbon monoxide and ethanol oxidation on PtSn supported catalysts: Effect of the nature of the carbon support and Pt: Sn composition. Appl. Catal. B Environ. 2015, 168, 33–41. [Google Scholar] [CrossRef]
- Flórez-Montaño, J.; García, G.; Rodríguez, J.L.; Pastor, E.; Cappellari, P.; Planes, G.A. On the design of Pt based catalysts. Combining porous architecture with surface modification by Sn for electrocatalytic activity enhancement. J. Power Sources 2015, 282, 34–44. [Google Scholar] [CrossRef]
- Zhou, W.J.; Song, S.Q.; Li, W.Z.; Zhou, Z.H.; Sun, G.Q.; Xin, Q.; Douvartzides, S.; Tsiakaras, P. Direct ethanol fuel cells based on PtSn anodes: The effect of Sn content on the fuel cell performance. J. Power Sources 2005, 140, 50–58. [Google Scholar] [CrossRef]
- Tsiakaras, P.E. PtM/C (M = Sn, Ru, Pd, W) based anode direct ethanol–PEMFCs: Structural characteristics and cell performance. J. Power Sources 2007, 171, 107–112. [Google Scholar] [CrossRef]
- Kim, J.H.; Choi, S.M.; Nam, S.H.; Seo, M.H.; Choi, S.H.; Kim, W.B. Influence of Sn content on PtSn/C catalysts for electrooxidation of C1–C3 alcohols: Synthesis, characterization, and electrocatalytic activity. App. Catal. B Environ. 2008, 82, 89–102. [Google Scholar] [CrossRef]
- Antolini, E. Catalysts for direct ethanol fuel cells. J. Power Sources 2007, 170, 1–12. [Google Scholar] [CrossRef]
- García, G.; Silva-Chong, J.; Rodríguez, J.L.; Pastor, E. Spectroscopic elucidation of reaction pathways of acetaldehyde on platinum and palladium in acidic media. J. Solid State Electrochem. 2014, 18, 1205–1213. [Google Scholar] [CrossRef]
- García, G.; Tsiouvaras, N.; Pastor, E.; Peña, M.A.; Fierro, J.L.G.; Martínez-Huerta, M.V. Ethanol oxidation on PtRuMo/C catalysts: In situ FTIR spectroscopy and DEMS studies. Int. J. Hydrogen Energy 2012, 37, 7131–7140. [Google Scholar] [CrossRef]
- García, G.; Bruno, M.M.; Planes, G.A.; Rodriguez, J.L.; Barbero, C.A.; Pastor, E. Probe beam deflection studies of nanostructured catalyst materials for fuel cells. Phys. Chem. Chem. Phys. 2008, 10, 6677–6685. [Google Scholar] [CrossRef] [PubMed]
- Godoi, D.R.; Villullas, H.M.; Zhu, F.C.; Jiang, Y.X.; Sun, S.G.; Guo, J.; Chen, R. A comparative investigation of metal-support interactions on the catalytic activity of Pt nanoparticles for ethanol oxidation in alkaline medium. J. Power Sources 2016, 311, 81–90. [Google Scholar] [CrossRef]
- Busó-Rogero, C.; Brimaud, S.; Solla-Gullon, J.; Vidal-Iglesias, F.J.; Herrero, E.; Behm, R.J.; Feliu, J.M. Ethanol oxidation on shape-controlled platinum nanoparticles at different pHs: A combined in situ IR spectroscopy and online mass spectrometry study. J. Electroanal. Chem. 2016, 763, 116–124. [Google Scholar] [CrossRef]
- Busó-Rogero, C.; Solla-Gullón, J.; Vidal-Iglesias, F.J.; Herrero, E.; Feliu, J.M. Adatom modified shape-controlled platinum nanoparticles towards ethanol oxidation. Electrochim. Acta 2016, 196, 270–279. [Google Scholar] [CrossRef]
- De Souza, J.; Queiroz, S.L.; Bergamaski, K.; Gonzalez, E.R.; Nart, F.C. Electro-oxidation of ethanol on Pt, Rh, and PtRh electrodes. A study using DEMS and in-situ FTIR techniques. J. Phys. Chem. B 2002, 106, 9825–9830. [Google Scholar]
- Iwasita, T.; Pastor, E. D/H exchange of ethanol at platinum electrodes. Electrochim. Acta 1994, 39, 547–551. [Google Scholar]
- Rizo, R.; Sebastián, D.; Lázaro, M.J.; Pastor, E. On the design of Pt-Sn efficient catalyst for carbon monoxide and ethanol oxidation in acid and alkaline media. Appl. Catal. B 2017, 200, 246–254. [Google Scholar] [CrossRef]
- Akhairi, M.A.F.; Kamarudin, S.K. Catalysts in direct ethanol fuel cell (DEFC): An overview. Int. J. Hydrogen Energy 2016, 41, 4214–4228. [Google Scholar] [CrossRef]
- Soares, L.A.; Morais, C.; Napporn, T.W.; Kokoh, K.B.; Olivi, P. Beneficial effects of rhodium and tin oxide on carbon supported platinum catalysts for ethanol electrooxidation. J. Power Sources 2016, 315, 47–55. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, D.; Peng, M.; Yu, Z.; Wang, X.; Guo, G.; Sun, Y. Microfluidic Synthesis Enables Dense and Uniform Loading of Surfactant-Free PtSn Nanocrystals on Carbon Supports for Enhanced Ethanol Oxidation. Angew. Chem. Int. Ed. 2016, 55, 4952–4956. [Google Scholar] [CrossRef] [PubMed]
- Colmati, F.; Antolini, E.; Gonzalez, E.R. Effect of temperature on the mechanism of ethanol oxidation on carbon supported Pt, PtRu and Pt3Sn electrocatalysts. J. Power Sources 2006, 157, 98–103. [Google Scholar] [CrossRef]
- Wang, H.; Jusys, Z.; Behm, R.J. Ethanol electro-oxidation on carbon-supported Pt, PtRu and Pt3Sn catalysts: A quantitative DEMS study. J. Power Sources 2006, 154, 351–359. [Google Scholar] [CrossRef]
- Zhou, W.P.; Axnanda, S.; White, M.G.; Adzic, R.R.; Hrbek, J. Enhancement in ethanol electrooxidation by SnOx nanoislands grown on Pt (111): Effect of metal oxide–metal interface sites. J. Phys. Chem. C 2011, 115, 16467–16473. [Google Scholar] [CrossRef]
- Zhou, W.; Zhou, Z.; Song, S.; Li, W.; Sun, G.; Tsiakaras, P.; Xin, Q. Pt based anode catalysts for direct ethanol fuel cells. Appl. Catal. B 2003, 46, 273–285. [Google Scholar] [CrossRef]
- Antolini, E.; Gonzalez, E.R. Effect of synthesis method and structural characteristics of Pt–Sn fuel cell catalysts on the electro-oxidation of CH3OH and CH3CH2OH in acid medium. Catal. Today 2011, 160, 28–38. [Google Scholar] [CrossRef]
- Vigier, F.; Coutanceau, C.; Hahn, F.; Belgsir, E.M.; Lamy, C. On the mechanism of ethanol electro-oxidation on Pt and PtSn catalysts: electrochemical and in situ IR reflectance spectroscopy studies. J. Electroanal. Chem. 2004, 563, 81–94. [Google Scholar] [CrossRef]
- Garcia, G.; Silva-Chong, J.; Guillen-Villafuerte, O.; Rodriguez, J.L.; Gonzalez, E.; Pastor, E. CO tolerant catalysts for PEM fuel cells: Spectroelectrochemical studies. Catal. Today 2006, 116, 415–421. [Google Scholar] [CrossRef]
- Roca-Ayats, M.; García, G.; Galante, J.L.; Peña, M.A.; Martínez-Huerta, M.V. TiC, TiCN, and TiN supported Pt electrocatalysts for CO and methanol oxidation in acidic and alkaline media. J. Phys. Chem. C 2013, 117, 20769–20777. [Google Scholar] [CrossRef]
- Roca-Ayats, M.; García, G.; Peña, M.A.; Martinez-Huerta, M.V. Titanium carbide and carbonitride electrocatalyst supports: Modifying Pt–Ti interface properties by electrochemical potential cycling. J. Mater. Chem. A 2014, 2, 18786–18790. [Google Scholar] [CrossRef]
- Iwasita, T.; Rasch, B.; Cattaneo, E.; Vielstich, W. A SNIFTIRS study of ethanol oxidation on platinum. Electrochim. Acta 1989, 34, 1073–1079. [Google Scholar] [CrossRef]
- Wang, Q.; Sun, G.Q.; Jiang, L.H.; Xin, Q.; Sun, S.G.; Jiang, Y.X.; Chen, S.P.; Jusys, Z.; Behm, R.J. Adsorption and oxidation of ethanol on colloid-based Pt/C, PtRu/C and Pt3Sn/C catalysts: In situ FTIR spectroscopy and on-line DEMS studies. Phys. Chem. Chem. Phys. 2007, 9, 2686–2696. [Google Scholar] [CrossRef] [PubMed]
- Iwasita, T.; Nart, F.C. In situ infrared spectroscopy at electrochemical interfaces. Prog. Surf. Sci. 1997, 55, 271–340. [Google Scholar] [CrossRef]
- Iwasita, T.; Pastor, E. A DEMS and FTIR spectroscopic investigation of adsorbed ethanol on polycrystalline platinum. Electrochim. Acta 1994, 39, 531–537. [Google Scholar] [CrossRef]
- Liang, Z.; Chen, W.; Liu, J.; Wang, S.; Zhou, Z.; Li, W.; Sun, G.; Xin, Q. FT-IR study of the microstructure of Nafion® membrane. J. Membr. Sci. 2004, 233, 39–44. [Google Scholar] [CrossRef]
- Gonzalez, E.R.; Ticianelli, E.A.; Pinheiro, A.L.N.; Perez, J. Braz. Patent INPl-SP no. 0032l, 1997.
- García, G.; Rodríguez, J.L.; Lacconi, G.I.; Pastor, E. Adsorption and oxidation pathways of thiourea at polycrystalline platinum electrodes. J. Electroanal. Chem. 2006, 588, 169–178. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rizo, R.; Lázaro, M.J.; Pastor, E.; García, G. Spectroelectrochemical Study of Carbon Monoxide and Ethanol Oxidation on Pt/C, PtSn(3:1)/C and PtSn(1:1)/C Catalysts. Molecules 2016, 21, 1225. https://doi.org/10.3390/molecules21091225
Rizo R, Lázaro MJ, Pastor E, García G. Spectroelectrochemical Study of Carbon Monoxide and Ethanol Oxidation on Pt/C, PtSn(3:1)/C and PtSn(1:1)/C Catalysts. Molecules. 2016; 21(9):1225. https://doi.org/10.3390/molecules21091225
Chicago/Turabian StyleRizo, Rubén, María Jesús Lázaro, Elena Pastor, and Gonzalo García. 2016. "Spectroelectrochemical Study of Carbon Monoxide and Ethanol Oxidation on Pt/C, PtSn(3:1)/C and PtSn(1:1)/C Catalysts" Molecules 21, no. 9: 1225. https://doi.org/10.3390/molecules21091225