
Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders

Abstract
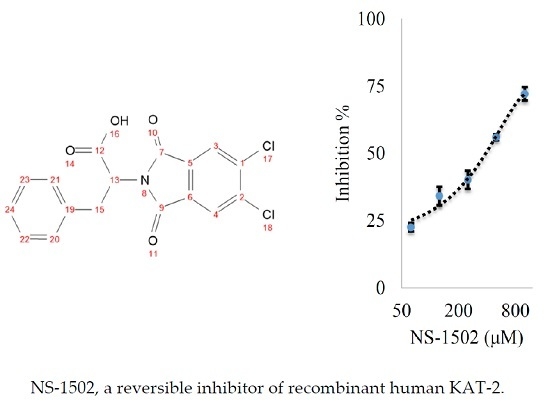
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
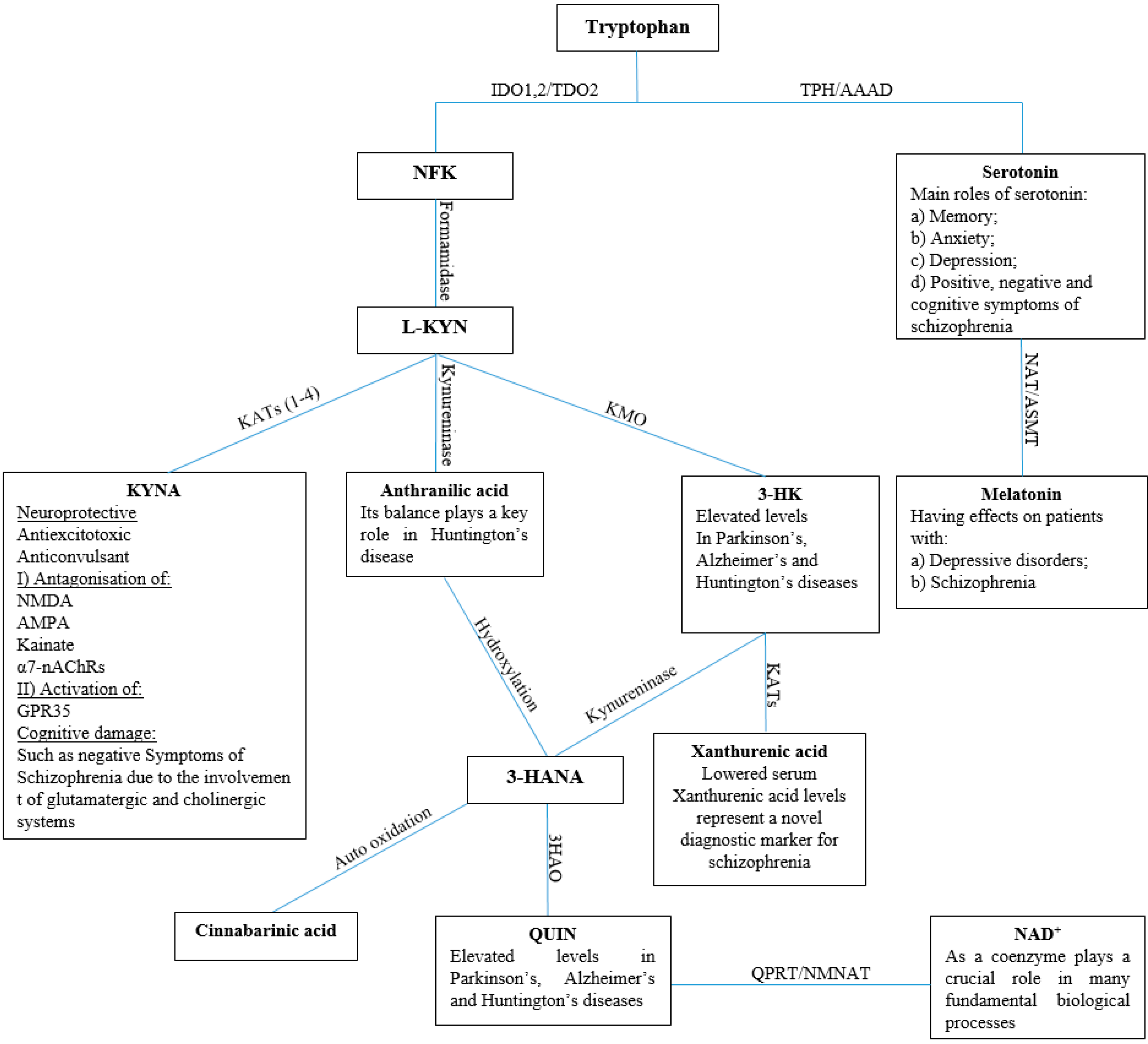
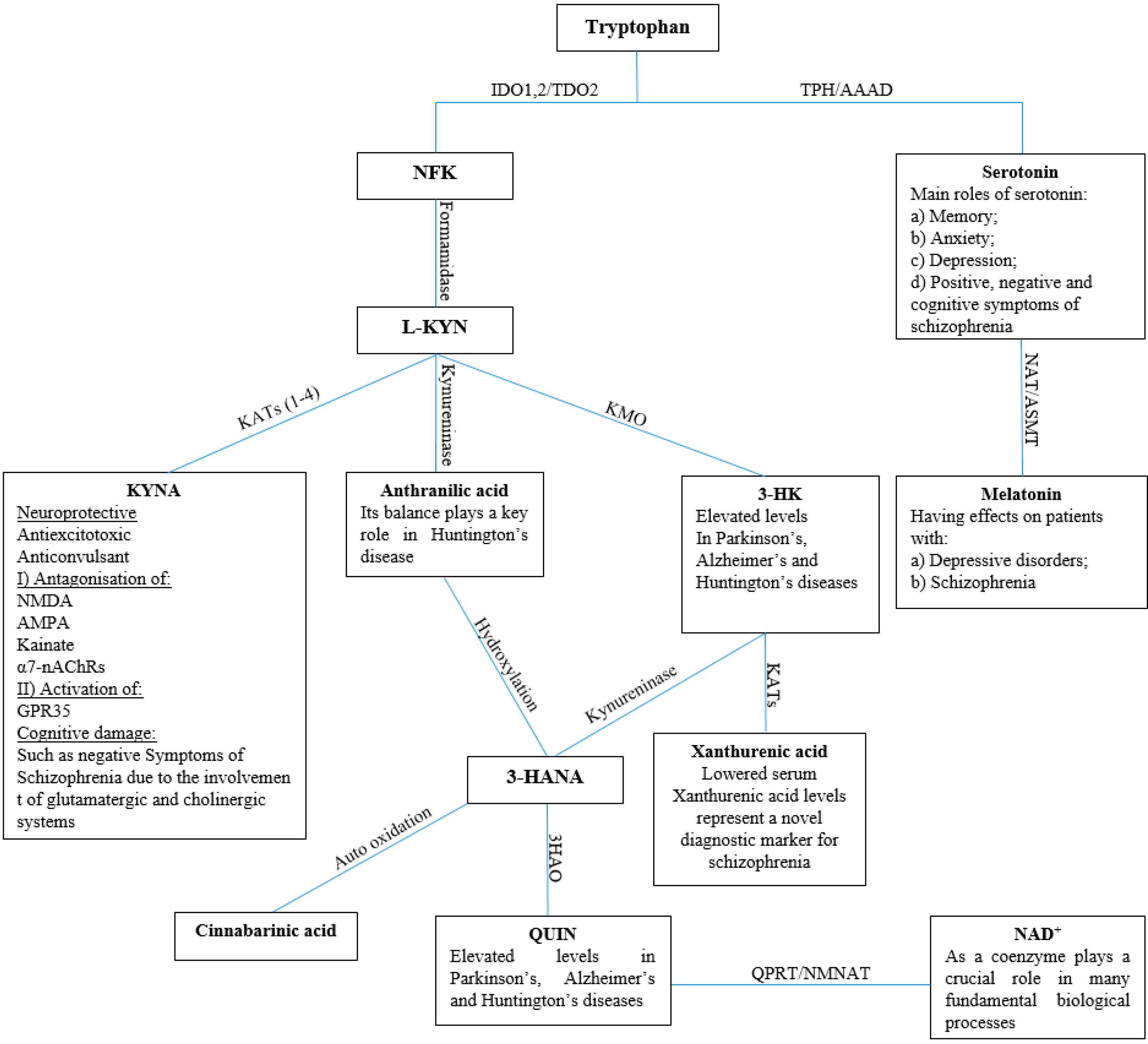
1. Introduction
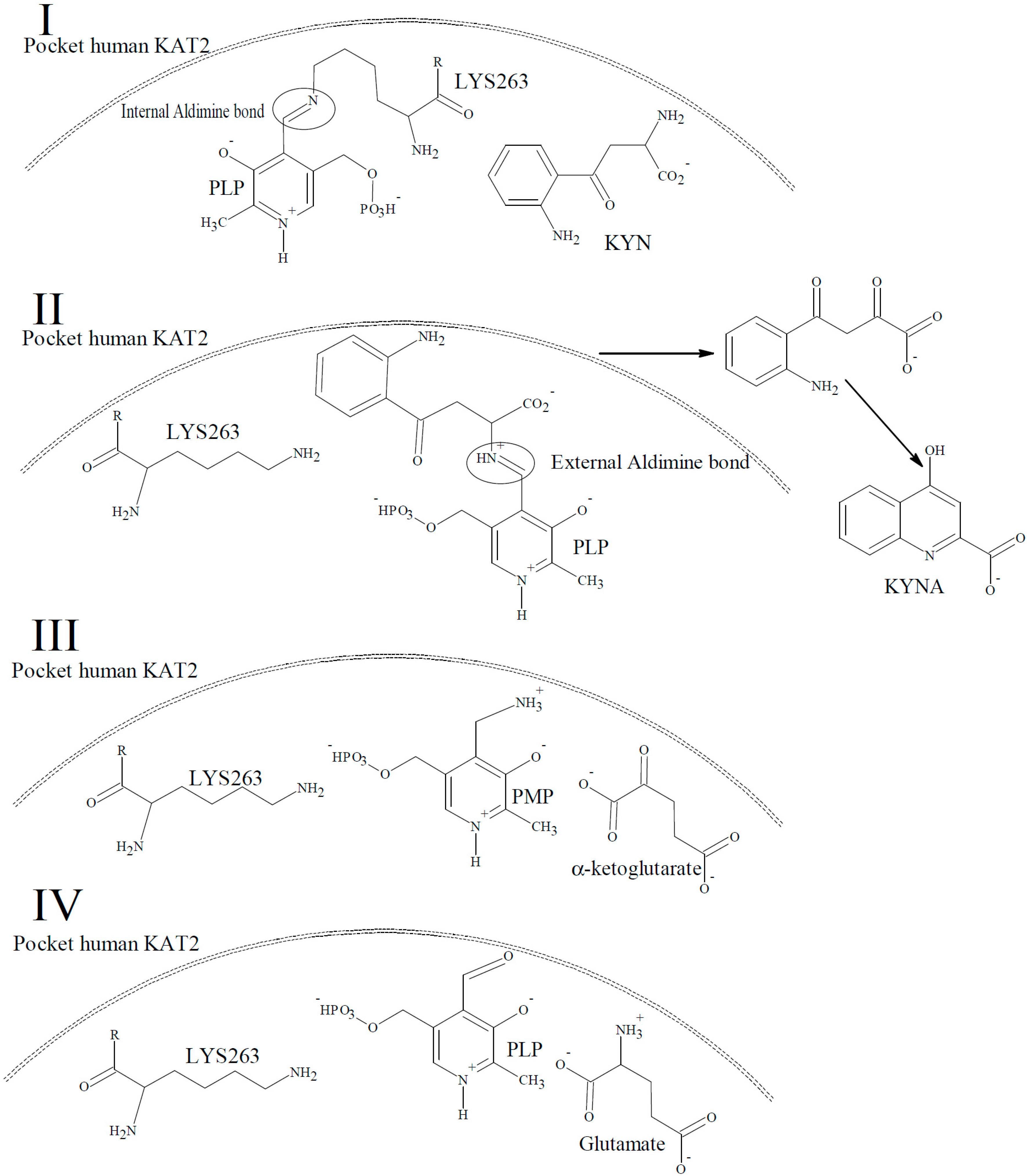
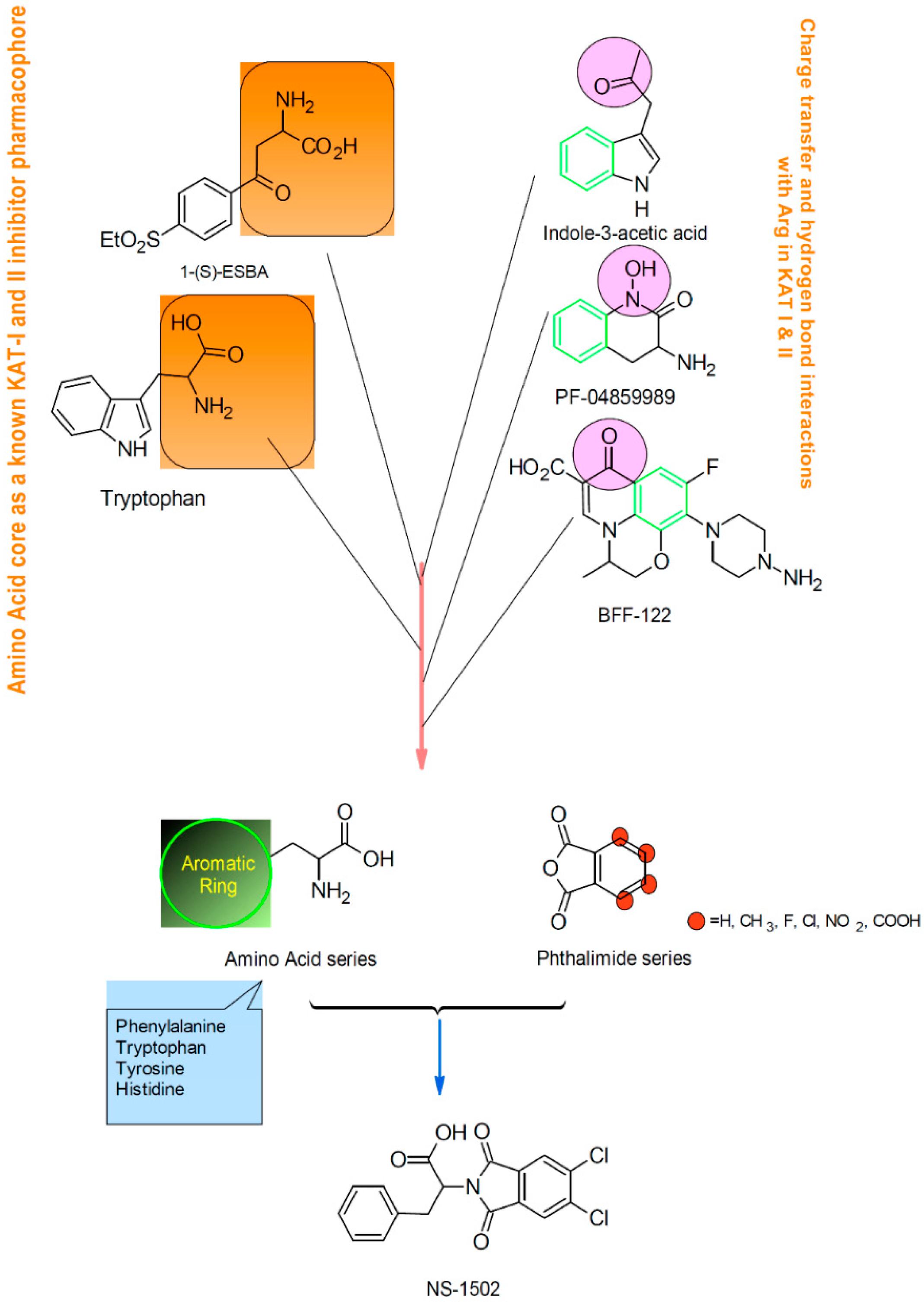
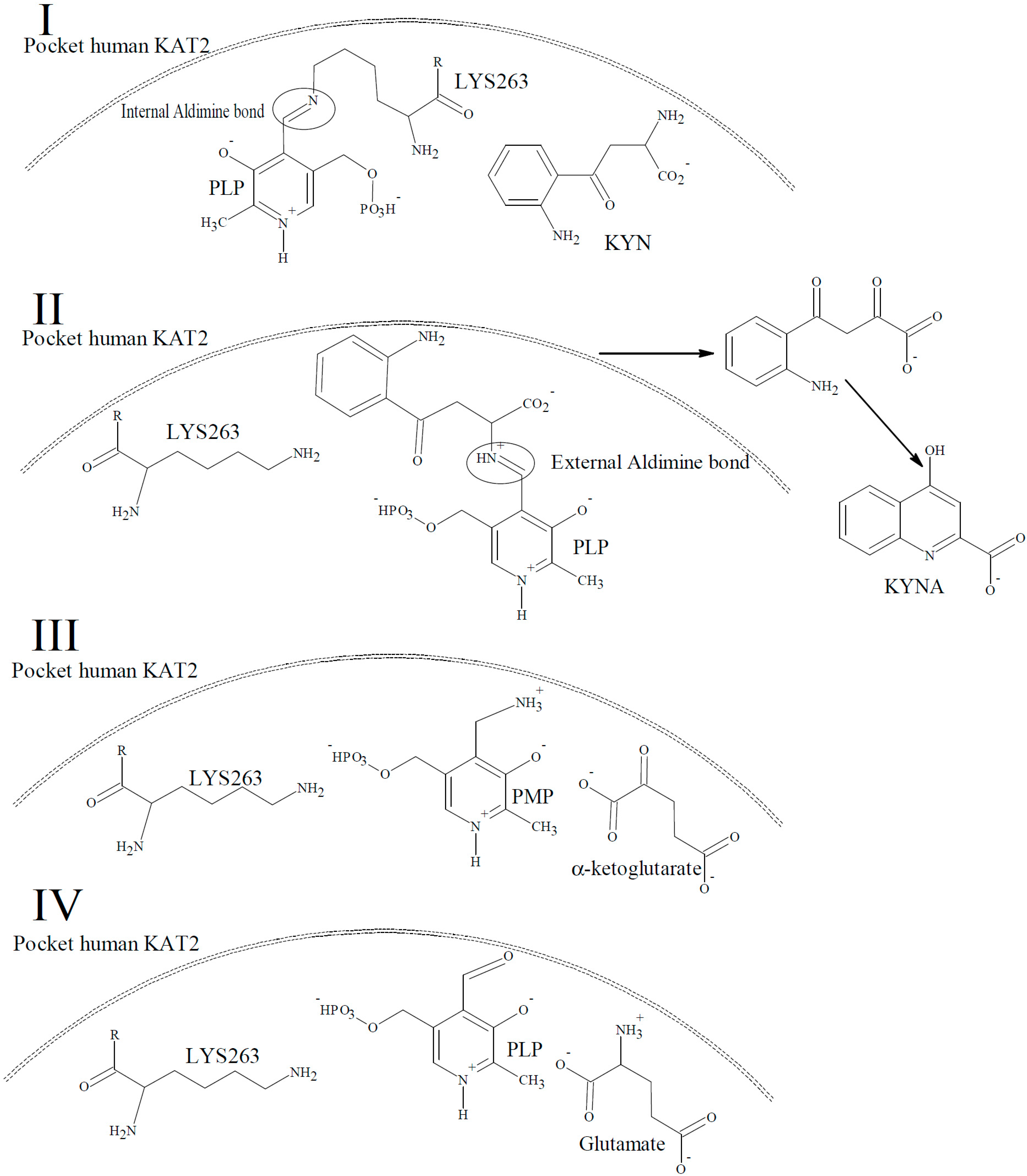
2. Results and Discussion
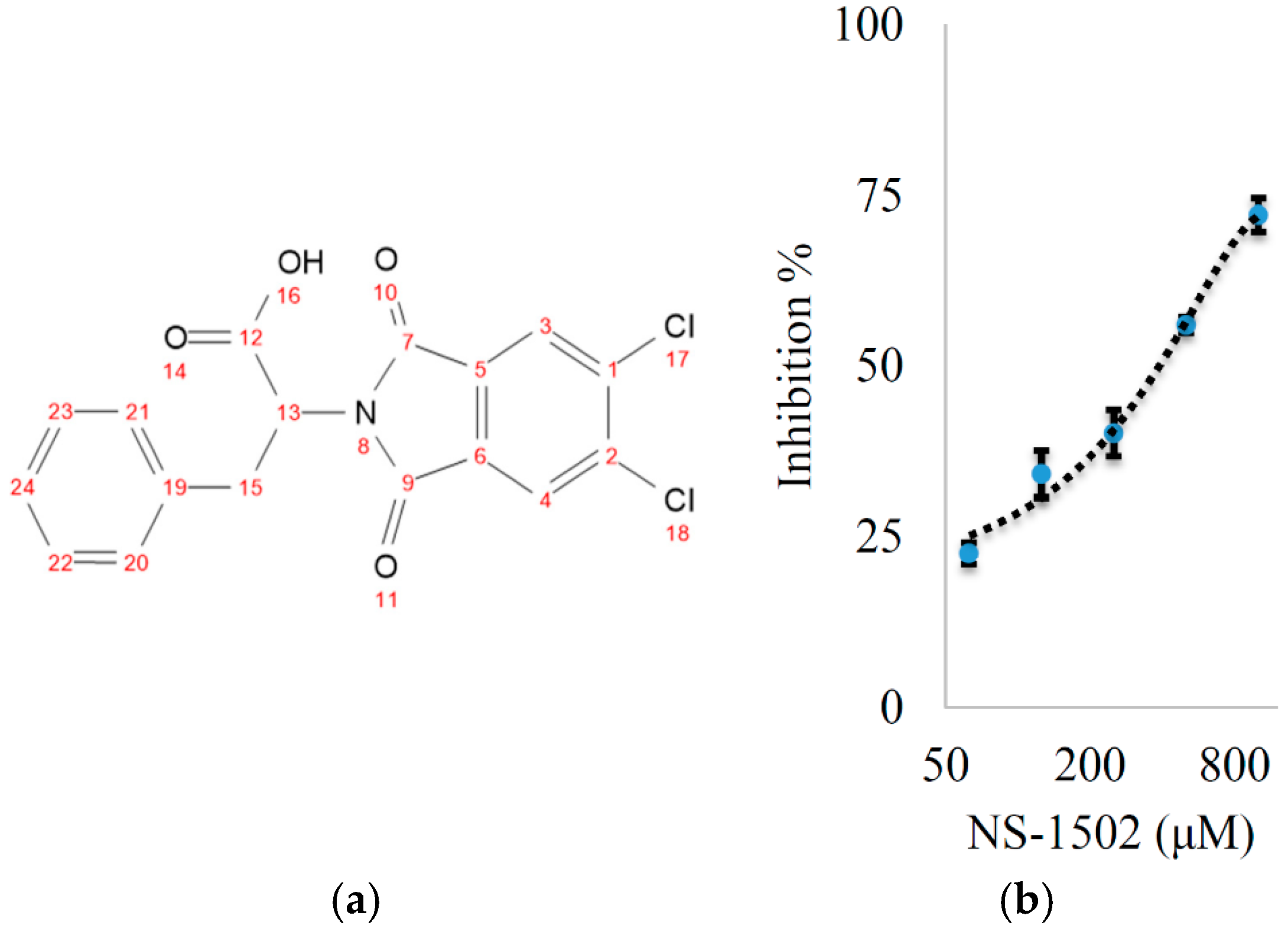
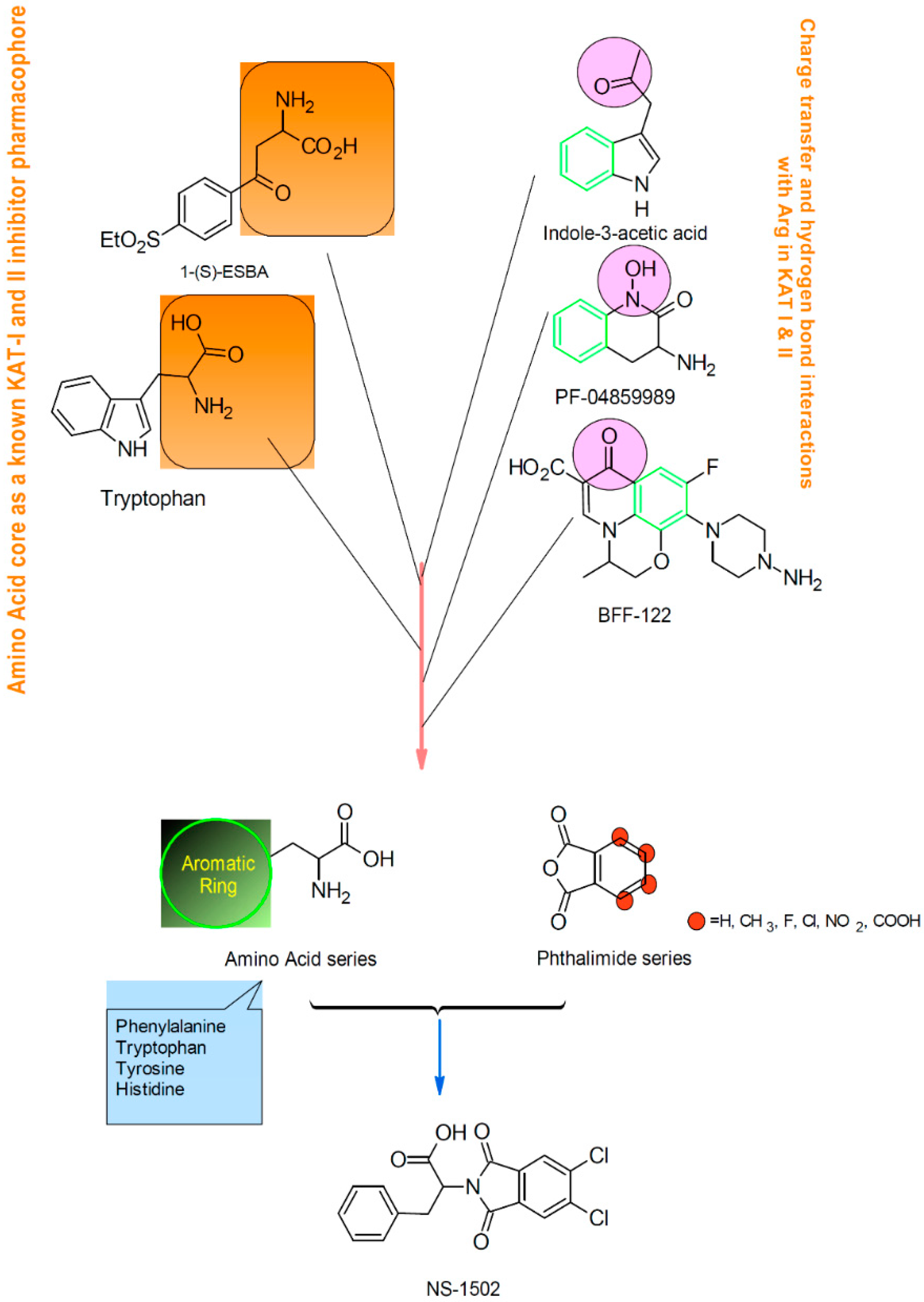
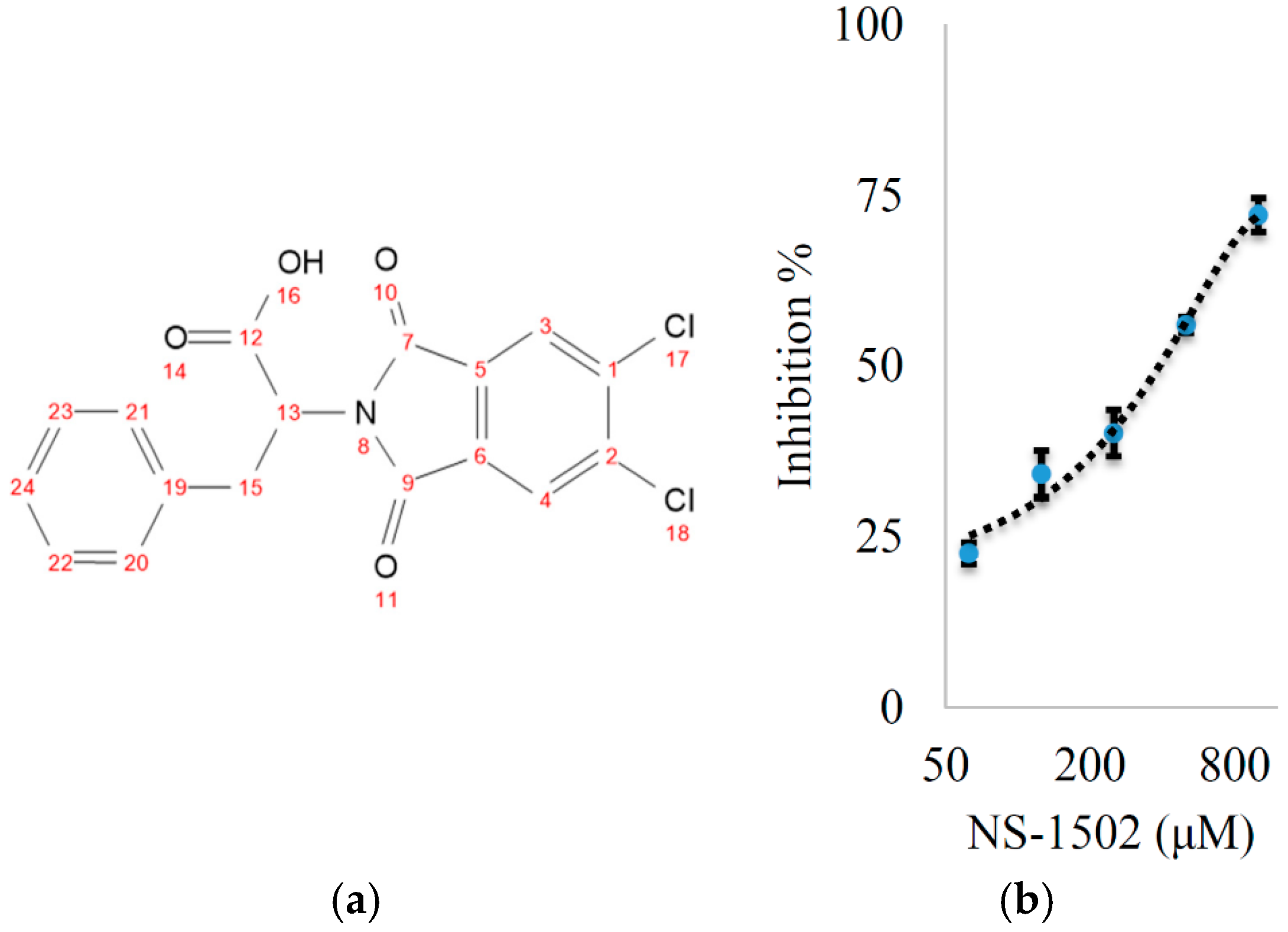
Characterization of the Inhibitory Activity and Binding Affinity of 2-(5,6-Dichloro-1,3-dioxo-1,3-dihydro-2H-isoindol-2-yl)-3-phenylpropanoic Acid (NS-1502) on hKAT2
3. Materials and Methods
3.1. General Procedures
3.2. Inhibitor Synthesis
3.3. Protein Preparation
3.4. Inhibition Studies Using Recombinant Human KAT2
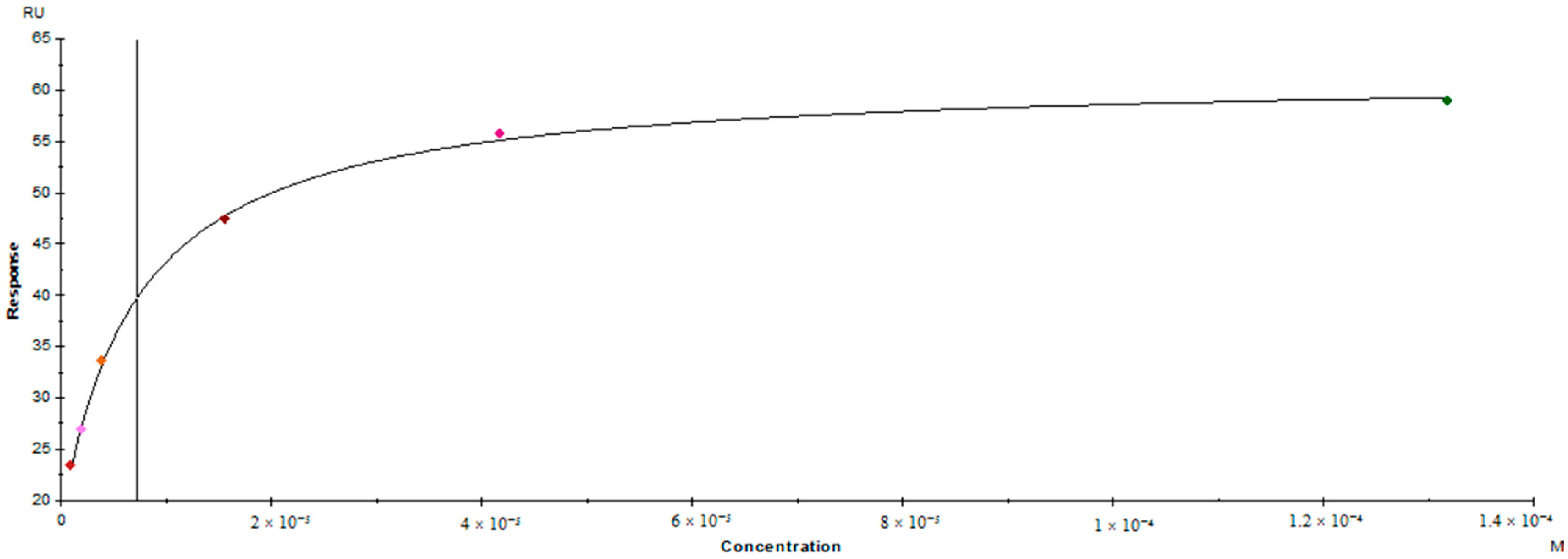
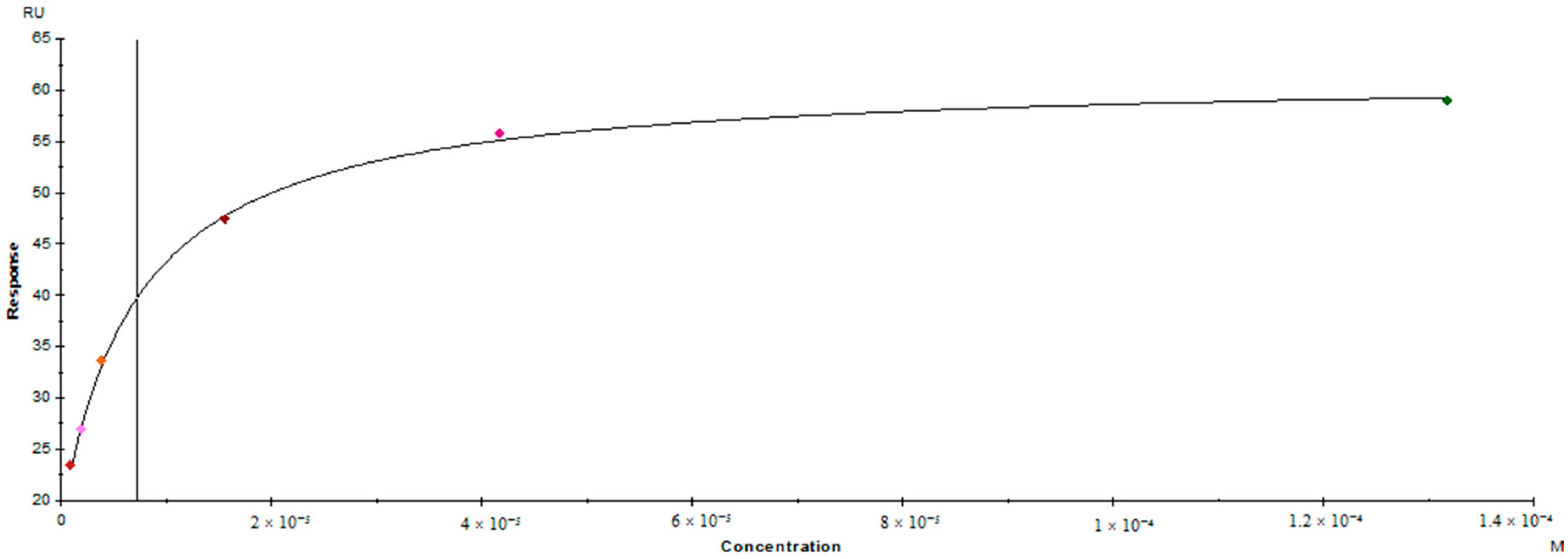
3.5. Surface Plasmon Resonance Binding Assay
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jones, S.P.; Guillemin, G.J.; Brew, B.J. The kynurenine pathway in stem cell biology. Int. J. Tryptophan Res. 2013, 6, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Vecsei, L.; Szalardy, L.; Fulop, F.; Toldi, J. Kynurenines in the CNS: Recent advances and new questions. Nat. Rev. Drug Discov. 2013, 12, 64–82. [Google Scholar] [CrossRef] [PubMed]
- Dalton, E.J.; Rotondi, D.; Levitan, R.D.; Kennedy, S.H.; Brown, G.M. Use of slow-release melatonin in treatment-resistant depression. J. Psychiatry Neurosci. 2000, 25, 48–52. [Google Scholar] [PubMed]
- Dolberg, O.T.; Hirschmann, S.; Grunhaus, L. Melatonin for the treatment of sleep disturbances in major depressive disorder. Am. J. Psychiatry 1998, 155, 1119–1121. [Google Scholar] [CrossRef] [PubMed]
- Guidetti, P.; Bates, G.P.; Graham, R.K.; Hayden, M.R.; Leavitt, B.R.; MacDonald, M.E.; Slow, E.J.; Wheeler, V.C.; Woodman, B.; Schwarcz, R. Elevated brain 3-hydroxykynurenine and quinolinate levels in Huntington disease mice. Neurobiol. Dis. 2006, 23, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Gulaj, E.; Pawlak, K.; Bien, B.; Pawlak, D. Kynurenine and its metabolites in Alzheimer’s disease patients. Adv. Med. Sci. 2010, 55, 204–211. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Matson, W.R.; Beal, M.F.; Myers, R.H.; Bird, E.D.; Milbury, P.; Saso, S. Kynurenine pathway abnormalities in Parkinson’s disease. Neurology 1992, 42, 1702–1706. [Google Scholar] [CrossRef] [PubMed]
- Schwarcz, R.; Bruno, J.P.; Muchowski, P.J.; Wu, H.Q. Kynurenines in the mammalian brain: When physiology meets pathology. Nat. Rev. Neurosci. 2012, 13, 465–477. [Google Scholar] [CrossRef] [PubMed]
- Shamir, E.; Laudon, M.; Barak, Y.; Anis, Y.; Rotenberg, V.; Elizur, A.; Zisapel, N. Melatonin improves sleep quality of patients with chronic schizophrenia. J. Clin. Psychiatry 2000, 61, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Szalardy, L.; Zadori, D.; Toldi, J.; Fulop, F.; Klivenyi, P.; Vecsei, L. Manipulating Kynurenic Acid Levels in the Brain—On the Edge Between Neuroprotection and Cognitive Dysfunction. Curr. Top. Med. Chem. 2012, 12, 1797–1806. [Google Scholar] [CrossRef] [PubMed]
- Fazio, F.; Lionetto, L.; Curto, M.; Iacovelli, L.; Cavallari, M.; Zappulla, C.; Ulivieri, M.; Napoletano, F.; Capi, M.; Corigliano, V. Xanthurenic Acid Activates mGlu2/3 Metabotropic Glutamate Receptors and is a Potential Trait Marker for Schizophrenia. Sci. Rep. 2015, 5, 17799. [Google Scholar] [CrossRef] [PubMed]
- Sun, G.; Nematollahi, A.; Nadvi, N.A.; Kwan, A.H.; Jeffries, C.M.; Church, W.B. Expression, purification and crystallization of human kynurenine aminotransferase 2 exploiting a highly optimized codon set. Protein Expr. Purif. 2016, 121, 41–45. [Google Scholar] [CrossRef] [PubMed]
- Jayawickrama, G.S.; Sadig, R.R.; Sun, G.; Nematollahi, A.; Nadvi, N.A.; Hanrahan, J.R.; Gorrell, M.D.; Church, W.B. Kynurenine Aminotransferases and the Prospects of Inhibitors for the Treatment of Schizophrenia. Curr. Med. Chem. 2015, 22, 2902–2918. [Google Scholar] [CrossRef] [PubMed]
- Pittaluga, A.; Vaccari, D.; Raiteri, M. The “kynurenate test”, a biochemical assay for putative cognition enhancers. J. Pharmacol. Exp. Ther. 1997, 283, 82–90. [Google Scholar] [PubMed]
- Nematollahi, A.; Church, W.B.; Nadvi, N.A.; Gorrell, M.D.; Sun, G. Homology modeling of human kynurenine aminotransferase III and observations on inhibitor binding using molecular docking. Cent. Nerv. Syst. Agents Med. Chem. 2014, 14, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Barcia, C.; Herrero, M.T.; Guillemin, G.J. The Involvement of Neuroinflammation and Kynurenine Pathway in Parkinson’s Disease. Parkinsons Dis. 2011, 2011, 716859. [Google Scholar] [CrossRef] [PubMed]
- Pellicciari, R.; Venturoni, F.; Bellocchi, D.; Carotti, A.; Marinozzi, M.; Macchiarulo, A.; Amori, L.; Schwarcz, R. Sequence variants in kynurenine aminotransferase II (KAT II) orthologs determine different potencies of the inhibitor S-ESBA. Chemmedchem 2008, 3, 1199–1202. [Google Scholar] [CrossRef] [PubMed]
- Henderson, J.L.; Sawant-Basak, A.; Tuttle, J.B.; Dounay, A.B.; McAllister, L.A.; Pandit, J.; Rong, S.B.; Hou, X.J.; Bechle, B.M.; Kim, J.Y.; et al. Discovery of hydroxamate bioisosteres as KAT II inhibitors with improved oral bioavailability and pharmacokinetics. Medchemcomm 2013, 4, 125–129. [Google Scholar] [CrossRef]
- Dounay, A.B.; Anderson, M.; Bechle, B.M.; Campbell, B.M.; Claffey, M.M.; Evdokimov, A.; Evrard, E.; Fonseca, K.R.; Gan, X.M.; Ghosh, S.; et al. Discovery of Brain-Penetrant, Irreversible Kynurenine Aminotransferase II Inhibitors for Schizophrenia. Acs. Med. Chem. Lett. 2012, 3, 187–192. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.Q.; Okuyama, M.; Kajii, Y.; Pocivavsek, A.; Bruno, J.P.; Schwarcz, R. Targeting Kynurenine Aminotransferase II in Psychiatric Diseases: Promising Effects of an Orally Active Enzyme Inhibitor. Schizophr. Bull. 2014, 40, S152–S158. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stein, A.; Cole, T. Parkinson’s disease: Carbidopa, nausea, and dyskinesia. Clin. Pharmacol. 2014, 6, 189–194. [Google Scholar] [PubMed]
- Wu, F.; Christen, P.; Gehring, H. A novel approach to inhibit intracellular vitamin B6-dependent enzymes: Proof of principle with human and plasmodium ornithine decarboxylase and human histidine decarboxylase. FASEB J. 2011, 25, 2109–2122. [Google Scholar] [CrossRef] [PubMed]
- Hinz, M.; Stein, A.; Cole, T. The Parkinson’s disease death rate: Carbidopa and vitamin B6. Clin. Pharmacol. 2014, 6, 161–169. [Google Scholar] [PubMed]
- Rossi, F.; Valentina, C.; Garavaglia, S.; Sathyasaikumar, K.V.; Schwarcz, R.; Kojima, S.; Okuwaki, K.; Ono, S.; Kajii, Y.; Rizzi, M. Crystal structure-based selective targeting of the pyridoxal 5′-phosphate dependent enzyme kynurenine aminotransferase II for cognitive enhancement. J. Med. Chem. 2010, 53, 5684–5689. [Google Scholar] [CrossRef] [PubMed]
- Mason, M.; Gullekson, E. Estrogen-enzyme inhibitions: Inhibition and protection of kynurenine transaminase by the sulfate esters of diethylstilbestrol, estradiol, and estrone. J. Biol. Chem. 1960, 235, 1312–1316. [Google Scholar] [PubMed]
- Mason, M.; Gullekson, E. Inhibition of pyridoxal phosphate-dependent enzymes by the sulfate esters of estradiol, estrone, and diethylstilbestrol. J. Am. Chem. Soc. 1959, 81, 1517. [Google Scholar] [CrossRef]
- Oliveira, E.; Santos, C.I.M.; Santos, H.M.; Fernandez-Lodeiro, A. From visible to far-red excitable chromophores derivatives of vitamin B6. Evaluation as pH-responsive probes and solvatochromic study. Dyes Pigments 2014, 110, 219–226. [Google Scholar] [CrossRef]
- Nematollahi, A.; Sun, G.; Harrop, S.J.; Hanrahan, J.R.; Church, W.B. Structure of the PLP-Form of the Human Kynurenine Aminotransferase II in a Novel Spacegroup at 1.83 A Resolution. Int. J. Mol. Sci. 2016, 17, 446. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Sample of the compound NS-1502 is available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nematollahi, A.; Sun, G.; Jayawickrama, G.S.; Hanrahan, J.R.; Church, W.B. Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders. Molecules 2016, 21, 856. https://doi.org/10.3390/molecules21070856
Nematollahi A, Sun G, Jayawickrama GS, Hanrahan JR, Church WB. Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders. Molecules. 2016; 21(7):856. https://doi.org/10.3390/molecules21070856
Chicago/Turabian StyleNematollahi, Alireza, Guanchen Sun, Gayan S. Jayawickrama, Jane R. Hanrahan, and W. Bret Church. 2016. "Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders" Molecules 21, no. 7: 856. https://doi.org/10.3390/molecules21070856
APA StyleNematollahi, A., Sun, G., Jayawickrama, G. S., Hanrahan, J. R., & Church, W. B. (2016). Study of the Activity and Possible Mechanism of Action of a Reversible Inhibitor of Recombinant Human KAT-2: A Promising Lead in Neurodegenerative and Cognitive Disorders. Molecules, 21(7), 856. https://doi.org/10.3390/molecules21070856