
Synthesis and Evaluation of Novel α-Aminoamides Containing an Indole Moiety for the Treatment of Neuropathic Pain

Abstract
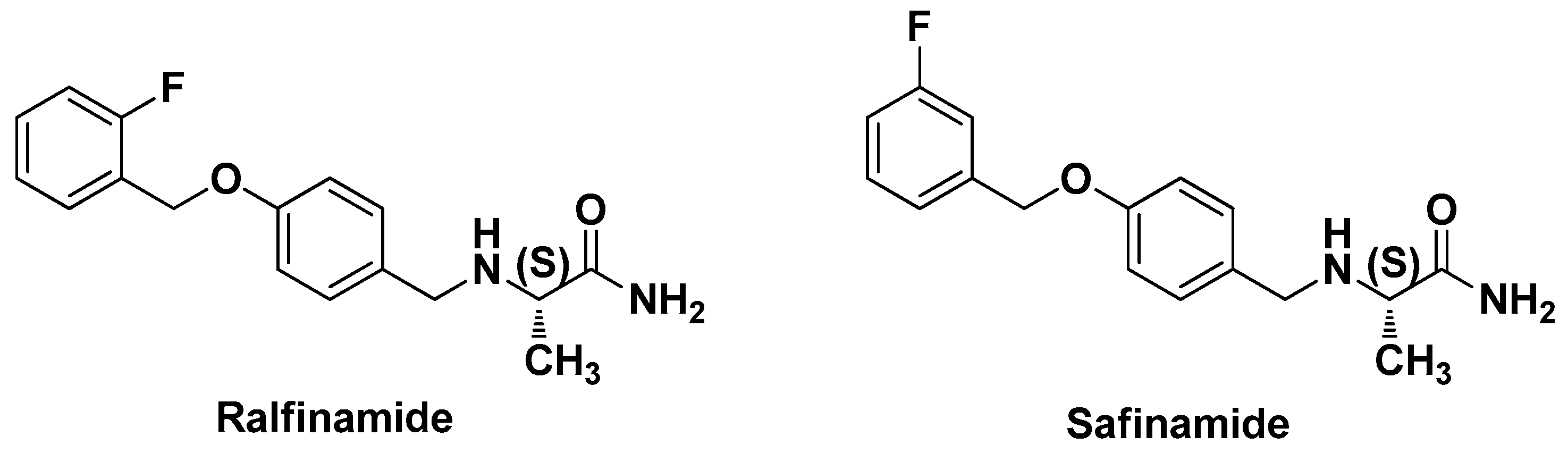
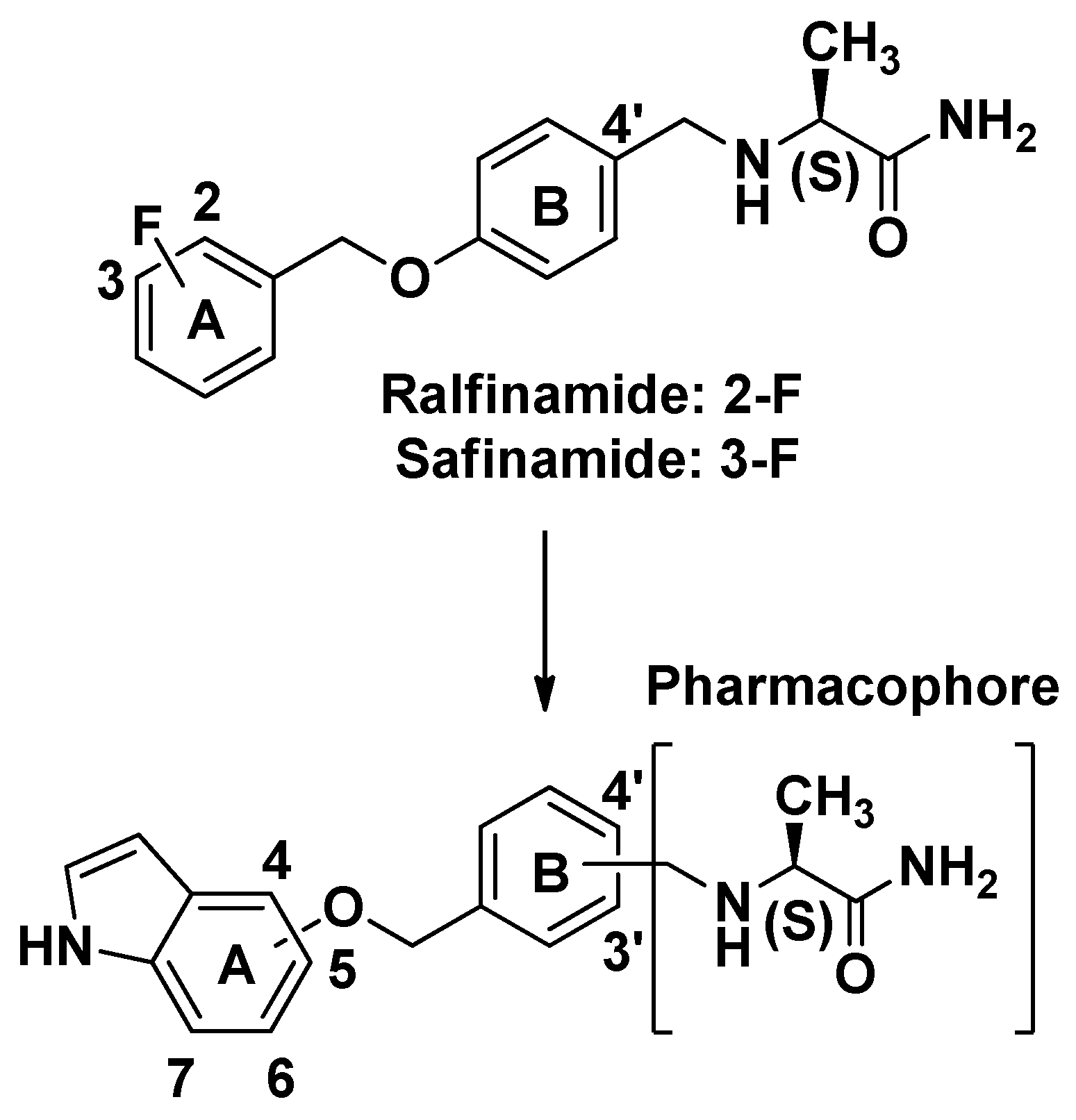
:1. Introduction
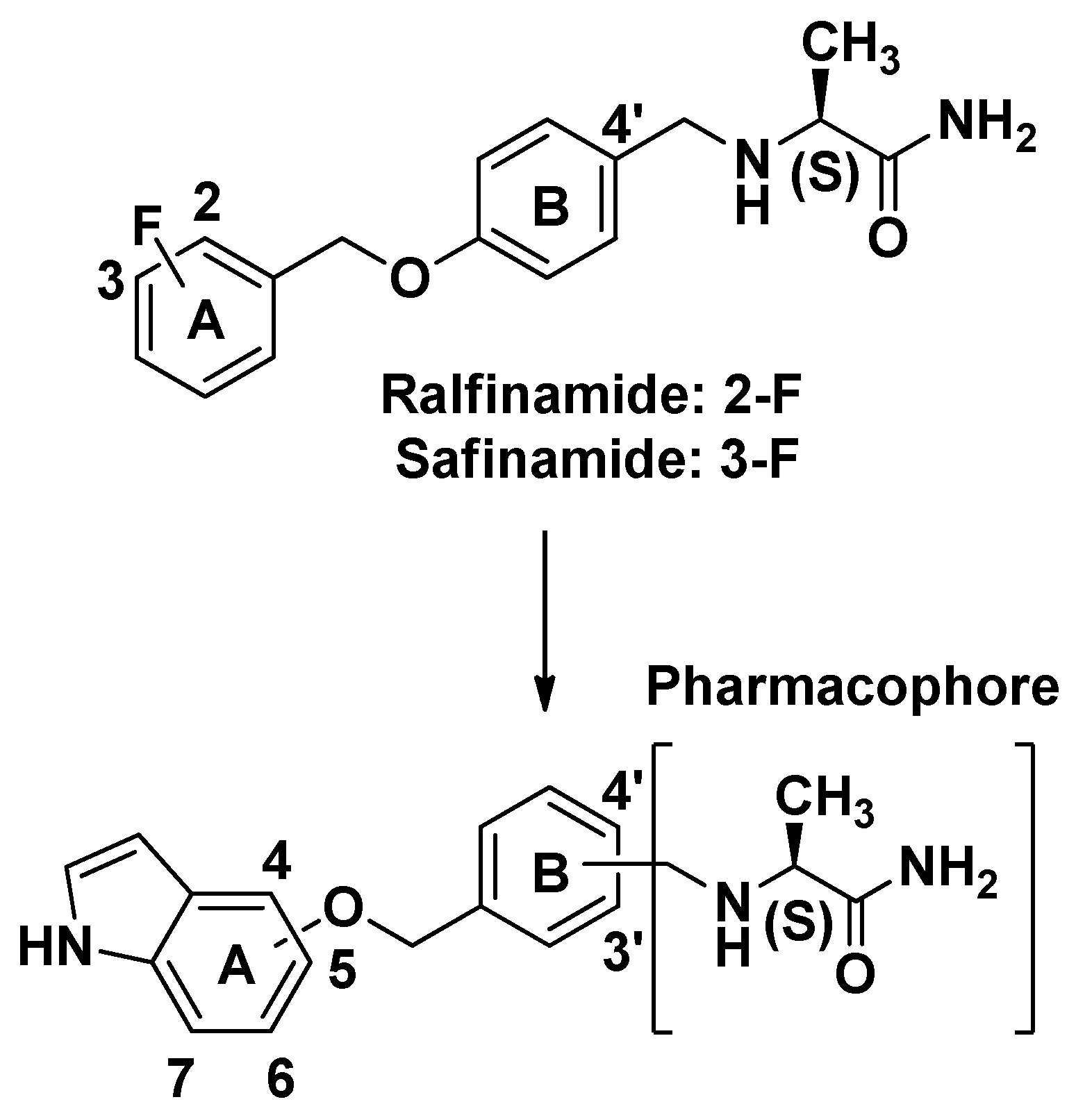
2. Results and Discussion
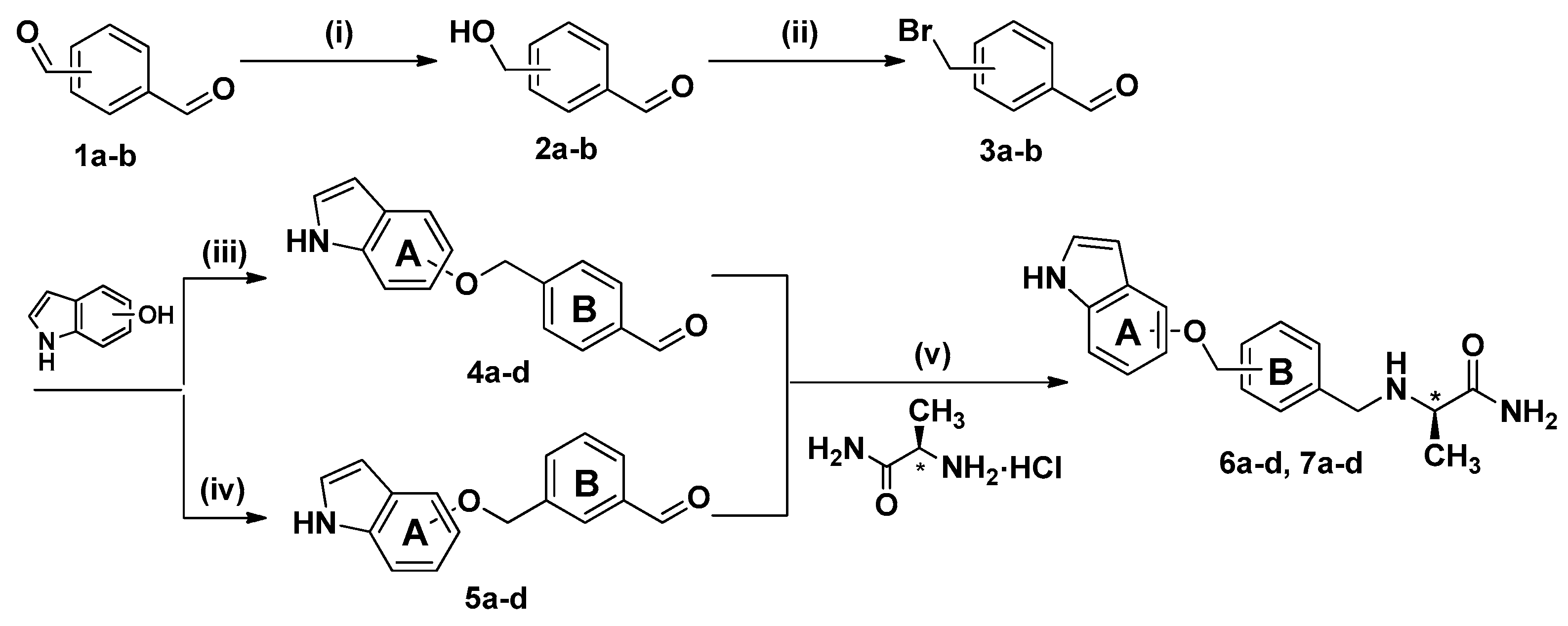
2.1. Synthesis
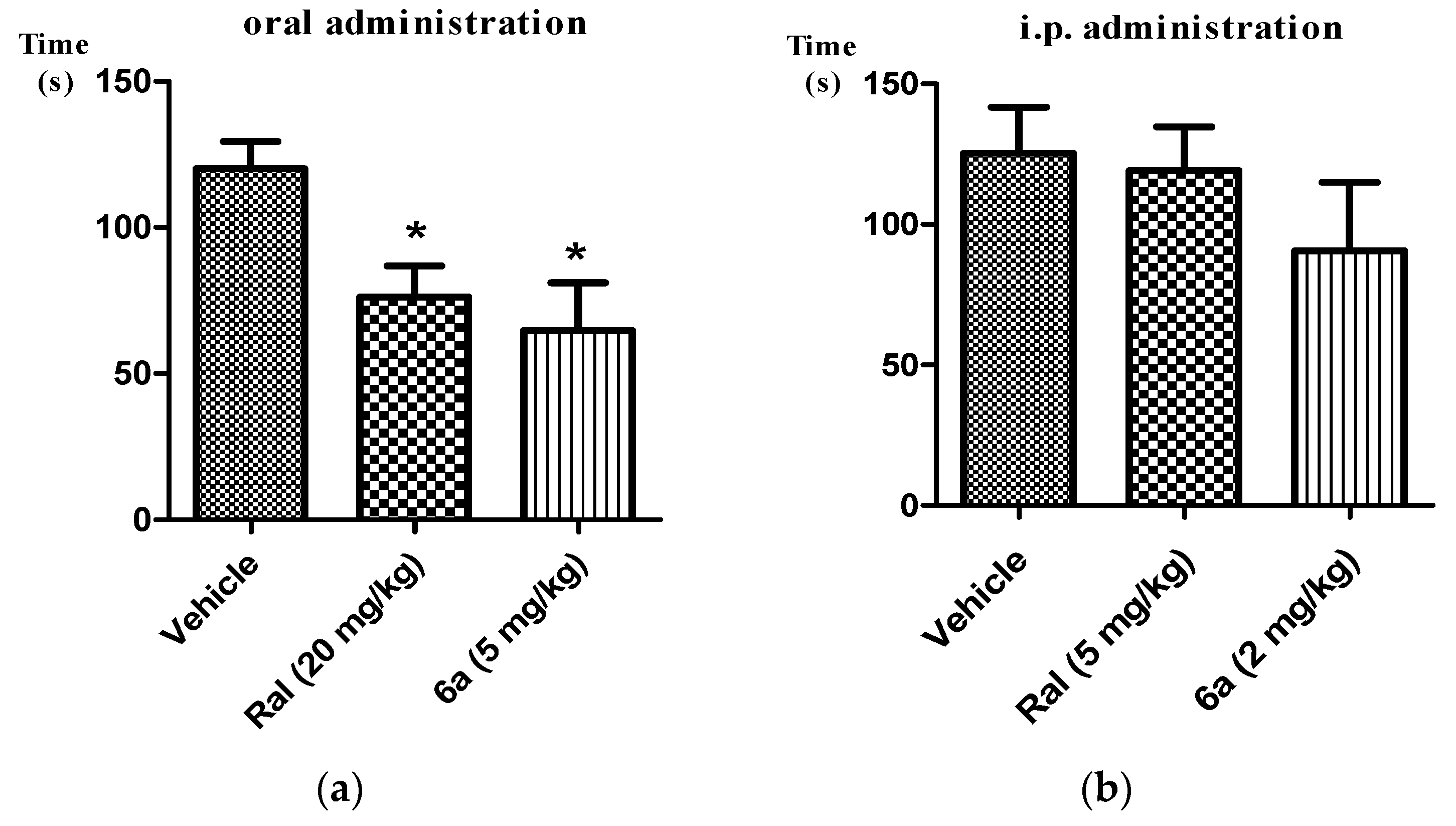
2.2. Analgesic Activity of Target Compounds in Formalin Test
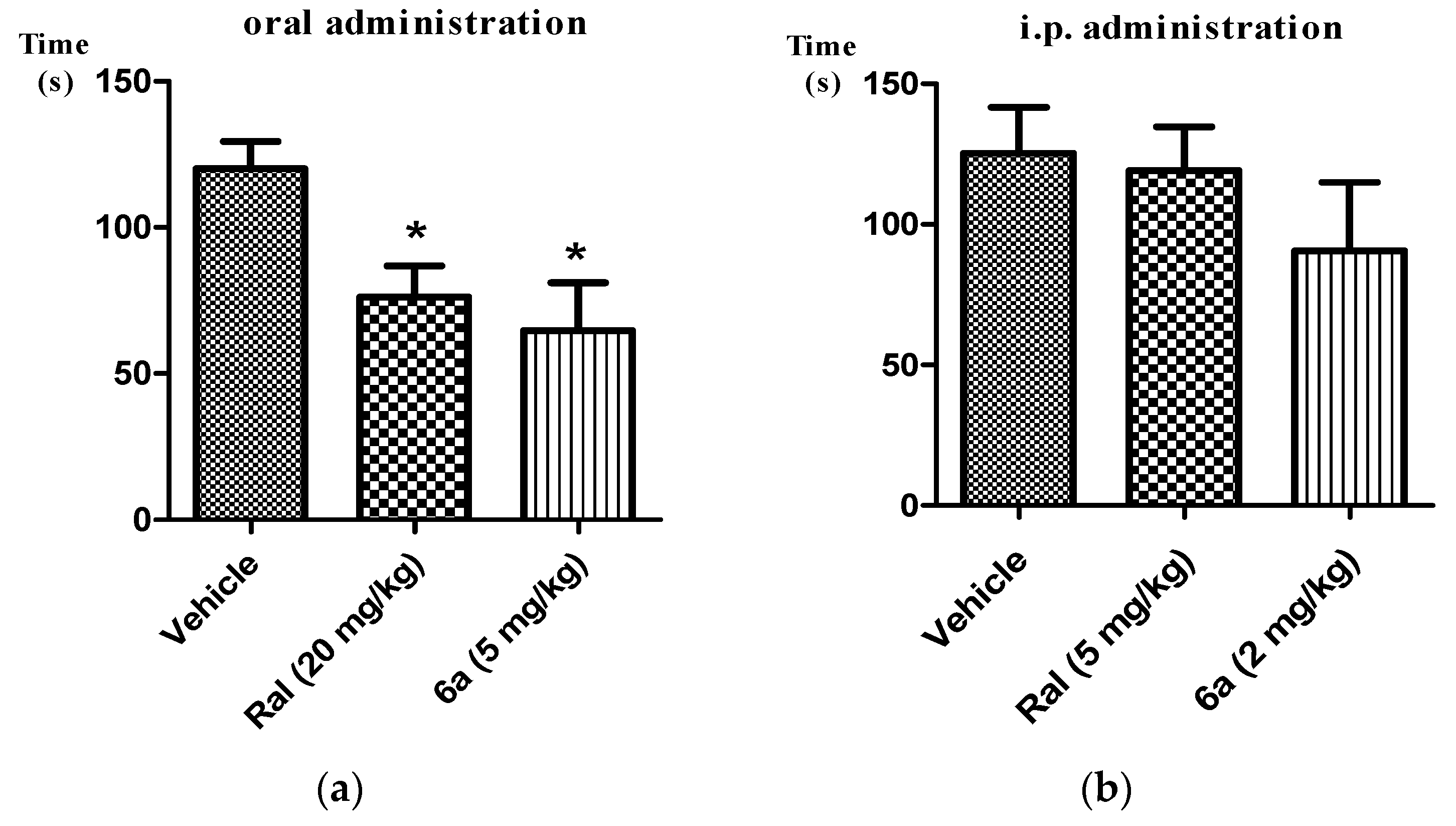
2.3. Inhibitory Activities of the Tested Compounds on the Tetrodoxin-Sensitive and Inactivated Human-Nav1.7 (hNav1.7)
3. Experimental Section
3.1. General Information
3.2. Chemistry
3.2.1. Synthesis of 4-(Hydroxymethyl)benzaldehyde (2a) and 3-(Hydroxymethyl)benzaldehyde (2b)
3.2.2. Synthesis of 4-(Bromomethyl)benzaldehyde (3a) and 3-(Bromomethyl)benzaldehyde (3b)
3.2.3. Preparation of Intermediates 4a–d
3.2.4. Preparation of Intermediates 5a–d
3.2.5. Preparation of Target Compounds 6a–d
3.2.6. Preparation of Target Compounds 7a–d
3.3. Formalin Test
3.4. Electrophysiology
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Baron, R.; Binder, A.; Wasner, G. Neuropathic pain: Diagnosis, pathophysiological mechanisms, and treatment. Lancet Neurol. 2010, 9, 807–819. [Google Scholar] [CrossRef]
- Costigan, M.; Scholz, J.; Woolf, C.J. Neuropathic pain: A maladaptive response of the nervous system to damage. Annu. Rev. Neurosci. 2009, 32, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Yalcin, I.; Barthas, F.; Barrot, M. Emotional consequences of neuropathic pain: Insight from preclinical studies. Neurosci. Biobehav. Rev. 2014, 47, 154–164. [Google Scholar] [CrossRef] [PubMed]
- Gangadhar, M.; Mishra, R.K.; Sriram, D.; Yogeeswari, P. Future directions in the treatment of neuropathic pain: A review on various therapeutic targets. CNS Neurol. Disord. Drug Tagets 2014, 13, 63–68. [Google Scholar] [CrossRef]
- Priest, B.T.; Kaczorowski, G.J. Blocking sodium channels to treat neuropathic pain. Expert Opin. Ther. Targets 2007, 11, 291–306. [Google Scholar] [CrossRef] [PubMed]
- Veneroni, O.; Maj, R.; Calabresi, M.; Faravelli, L.; Fariello, R.G.; Salvati, P. Anti-allodynic effect of NW-1029, a novel Na+ channel blocker, in experimental animal models of inflammatory and neuropathic pain. Pain 2003, 102, 17–25. [Google Scholar] [CrossRef]
- Yamane, H.; de Groat, W.C.; Sculptoreanu, A. Effects of ralfinamide, a Na+ channel blocker, on firing properties of nociceptive dorsal root ganglion neurons of adult rats. Exp. Neurol. 2007, 208, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Flaminio, C. Ralfinamide. Newron Pharmaceuticals. Idrugs 2004, 7, 935–939. [Google Scholar]
- Bauer, M.; Bliesath, H.; Leuratti, C.; Lackner, E.; Dieterle, W.; Müller, M.; Brunner, M. Disposition and metabolism of ralfinamide, a novel Na-channel blocker, in healthy male volunteers. Pharmacology 2010, 86, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Wilson, S.; Brittain, J.; Ripsch, M.S.; Salomé, C.; Park, K.D.; White, F.A.; Khanna, R.; Kohn, H. Merging structural motifs of functionalized amino acids and α-aminoamides results in novel anticonvulsant compounds with significant effects on slow and fast inactivation of voltage-gated sodium channels and in the treatment of neuropathic pain. ACS Chem. Neurosci. 2011, 2, 317–332. [Google Scholar] [CrossRef] [PubMed]
- Salom, C.; Salome-Grosjean, E.; Stables, J.; Kohn, H. Merging the structural motifs of functionalized amino acids and α-aminoamides: Compounds with significant anticonvulsant activities. J. Med. Chem. 2010, 53, 3756–3771. [Google Scholar] [CrossRef] [PubMed]
- Macsari, I.; Besidski, Y.; Csjernyik, G.; Nilsson, L.I.; Sandberg, L.; Yngve, U.; Ahlin, K.; Bueters, T.; Eriksson, A.B.; Lund, P.E. 3-oxoisoindoline-1-carboxamides: Potent, state-dependent blockers of voltage-gated sodium channel Nav1.7 with efficacy in rat pain models. J. Med. Chem. 2012, 55, 6866–6880. [Google Scholar] [CrossRef] [PubMed]
- Bregman, H.; Berry, L.; Buchanan, J.; Chen, A.; Du, B.; Feric, E.; Hierl, M.; Huang, L.; Immke, D.; Janosky, B. Identification of a potent, state-dependent inhibitor of Nav1.7 with oral efficacy in the formalin model of persistent pain. J. Med. Chem. 2011, 54, 4427–4445. [Google Scholar] [CrossRef] [PubMed]
- Deeks, E. Safinamide: First global approval. Drugs 2015, 75, 705–711. [Google Scholar] [CrossRef] [PubMed]
- Cattaneo, C.; Ferla, R.; Bonizzoni, E.; Sardina, M. Long-term effects of safinamide on dyskinesia in mid- to late-stage Parkinson’s disease: A post-hoc analysis. J. Parkinson’s Dis. 2015, 5, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, M.; Rosa, M.; Abreu, D.; Ferreira, J. Clinical pharmacology review of safinamide for the treatment of Parkinson’s disease. Neurodegener. Dis. Manag. 2015, 5, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Zuliani, V.; Rapalli, A.; Patel, M.K.; Rivara, M. Sodium channel blockers: A patent review (2010–2014). Expert Opin. Ther. Pat. 2015, 25, 279–290. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 6a–d and 7a–d are available from the authors.


| Compounds | Substituted Position of the Oxygen Atom in the A Ring | Substituted Position of –CH2– in the B Ring | % Analgesia a (10 mg/kg, i.p.) |
|---|---|---|---|
| 6a | 4 | 4’ | 76.3 ± 3.9 |
| 6b | 5 | 4’ | 62.1 ± 2.8 |
| 6c | 6 | 4’ | 71.0 ± 1.0 |
| 6d | 7 | 4’ | 68.6 ± 1.8 |
| 7a | 4 | 3’ | 64.9 ± 4.7 |
| 7b | 5 | 3’ | 67.2 ± 6.3 |
| 7c | 6 | 3’ | 68.8 ± 4.9 |
| 7d | 7 | 3’ | 58.5 ± 5.9 |
| Ralfinamide | - | - | 47.5 ± 2.9 |

| Compound | Substituted Position of the Oxygen Atom in the A Ring | Substituted Position of –CH2– in the B Ring | hNav1.7 (% Inhibition at 10 μM) |
|---|---|---|---|
| 6a | 4 | 4’ | 4 ± 2 |
| 7a | 4 | 3’ | 59 ± 7 |
| 7b | 5 | 3’ | 29 ± 2 |
| Ralfinamide | - | - | 66 ± 5 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, H.; Fan, S.; Cheng, J.; Zhang, P.; Zhong, B.; Shi, W. Synthesis and Evaluation of Novel α-Aminoamides Containing an Indole Moiety for the Treatment of Neuropathic Pain. Molecules 2016, 21, 793. https://doi.org/10.3390/molecules21070793
Li H, Fan S, Cheng J, Zhang P, Zhong B, Shi W. Synthesis and Evaluation of Novel α-Aminoamides Containing an Indole Moiety for the Treatment of Neuropathic Pain. Molecules. 2016; 21(7):793. https://doi.org/10.3390/molecules21070793
Chicago/Turabian StyleLi, Haotian, Shiyong Fan, Jingchao Cheng, Ping Zhang, Bohua Zhong, and Weiguo Shi. 2016. "Synthesis and Evaluation of Novel α-Aminoamides Containing an Indole Moiety for the Treatment of Neuropathic Pain" Molecules 21, no. 7: 793. https://doi.org/10.3390/molecules21070793
APA StyleLi, H., Fan, S., Cheng, J., Zhang, P., Zhong, B., & Shi, W. (2016). Synthesis and Evaluation of Novel α-Aminoamides Containing an Indole Moiety for the Treatment of Neuropathic Pain. Molecules, 21(7), 793. https://doi.org/10.3390/molecules21070793