
Inhibitors of the Hydrolytic Enzyme Dimethylarginine Dimethylaminohydrolase (DDAH): Discovery, Synthesis and Development


Abstract
:1. Introduction
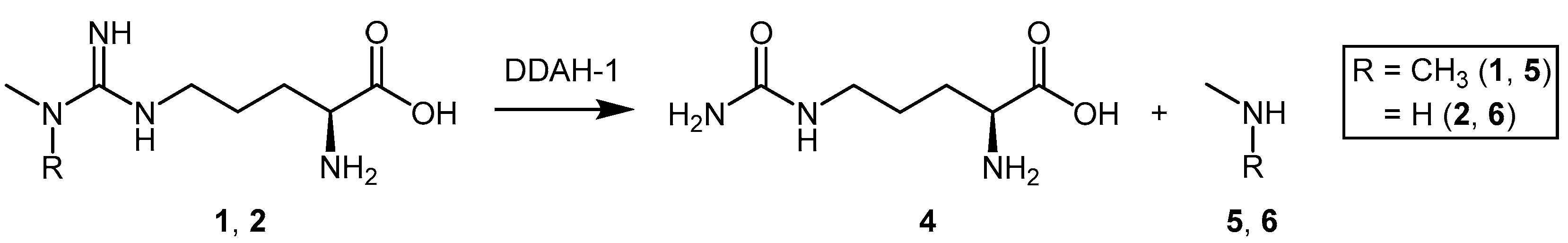
2. Endogenous Modulation of DDAH Activity
3. Pharmacological Induction of DDAH
4. Pharmacological Inhibition of DDAH
4.1. Substrate-Like Inhibitors
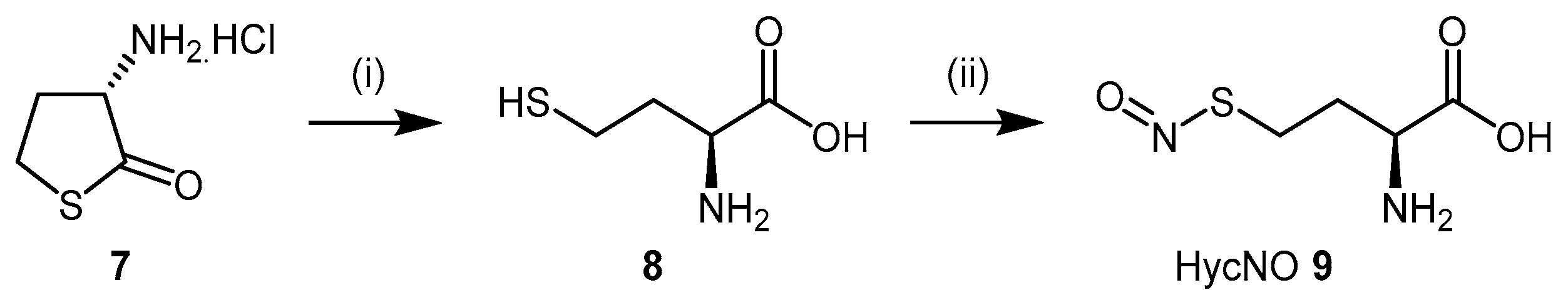
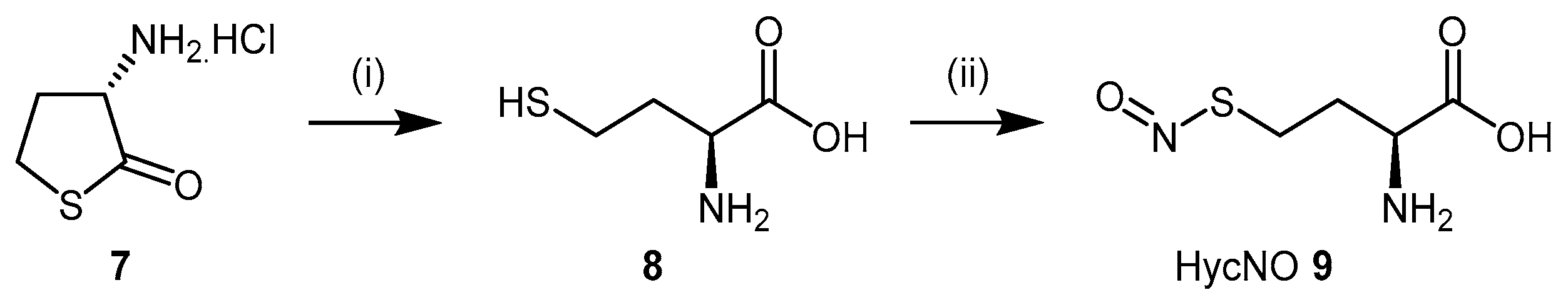
4.1.1. S-Nitroso-l-Homocysteine (HcyNO)
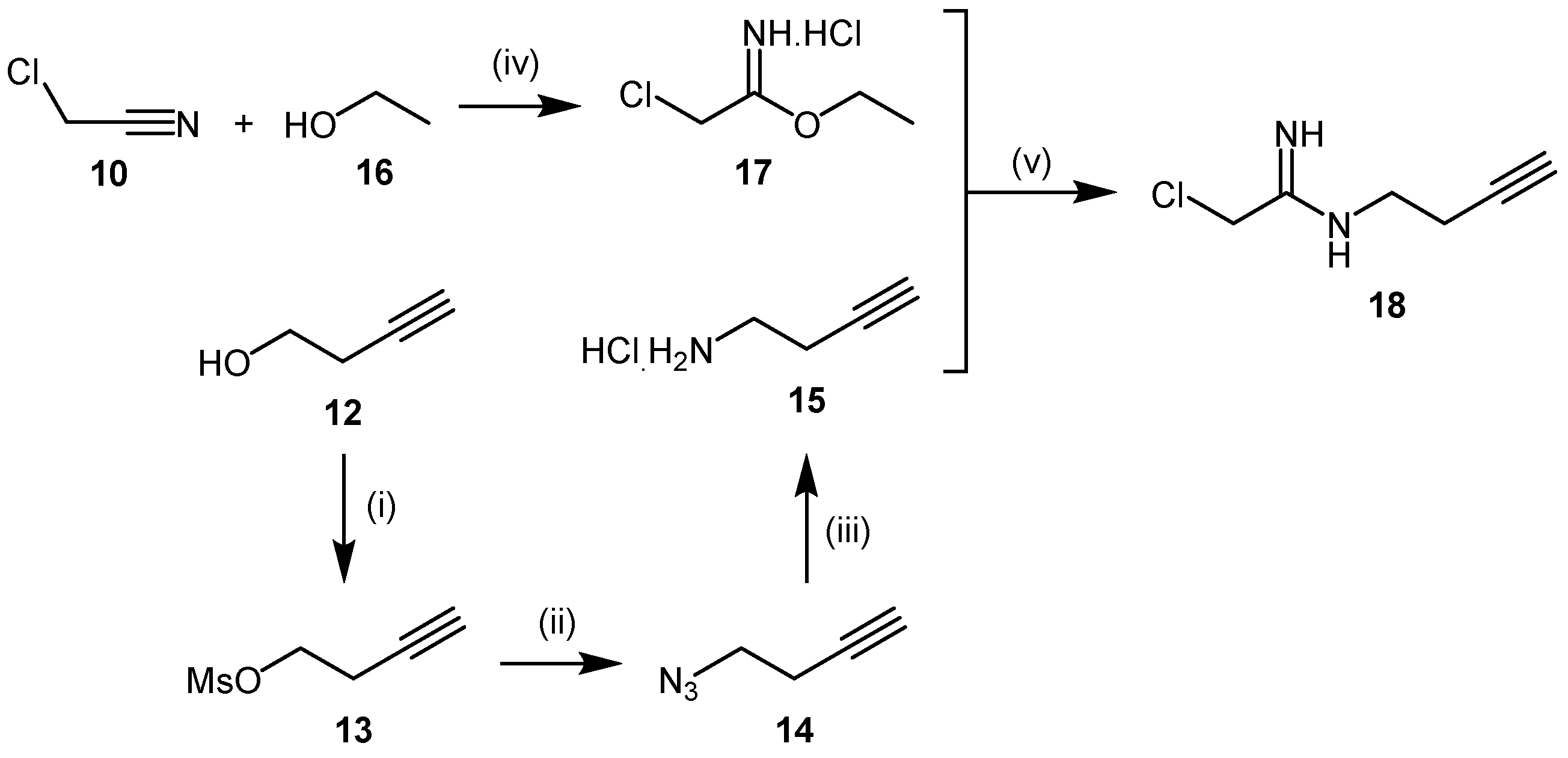
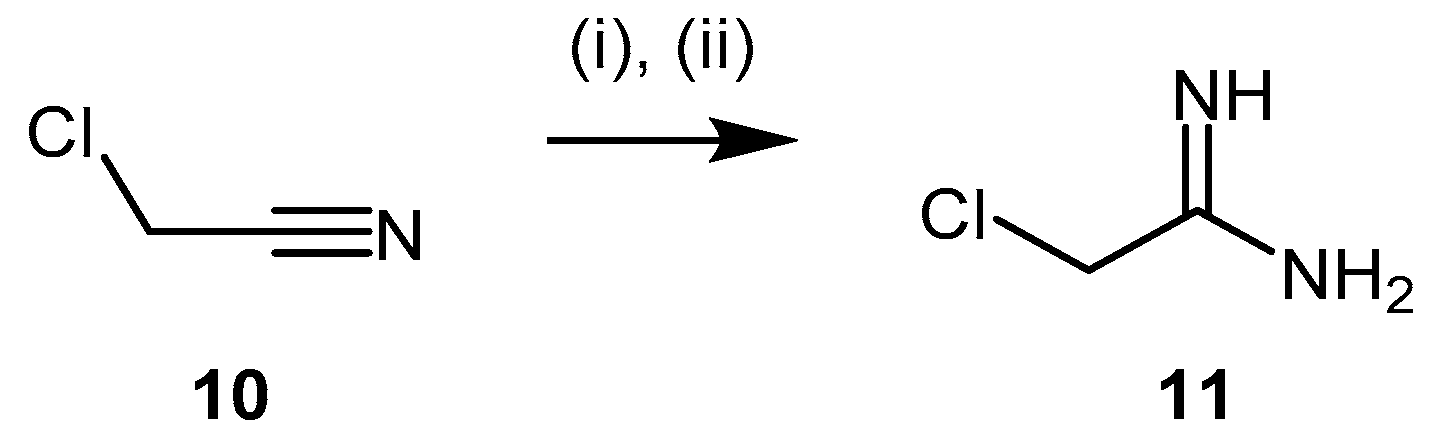
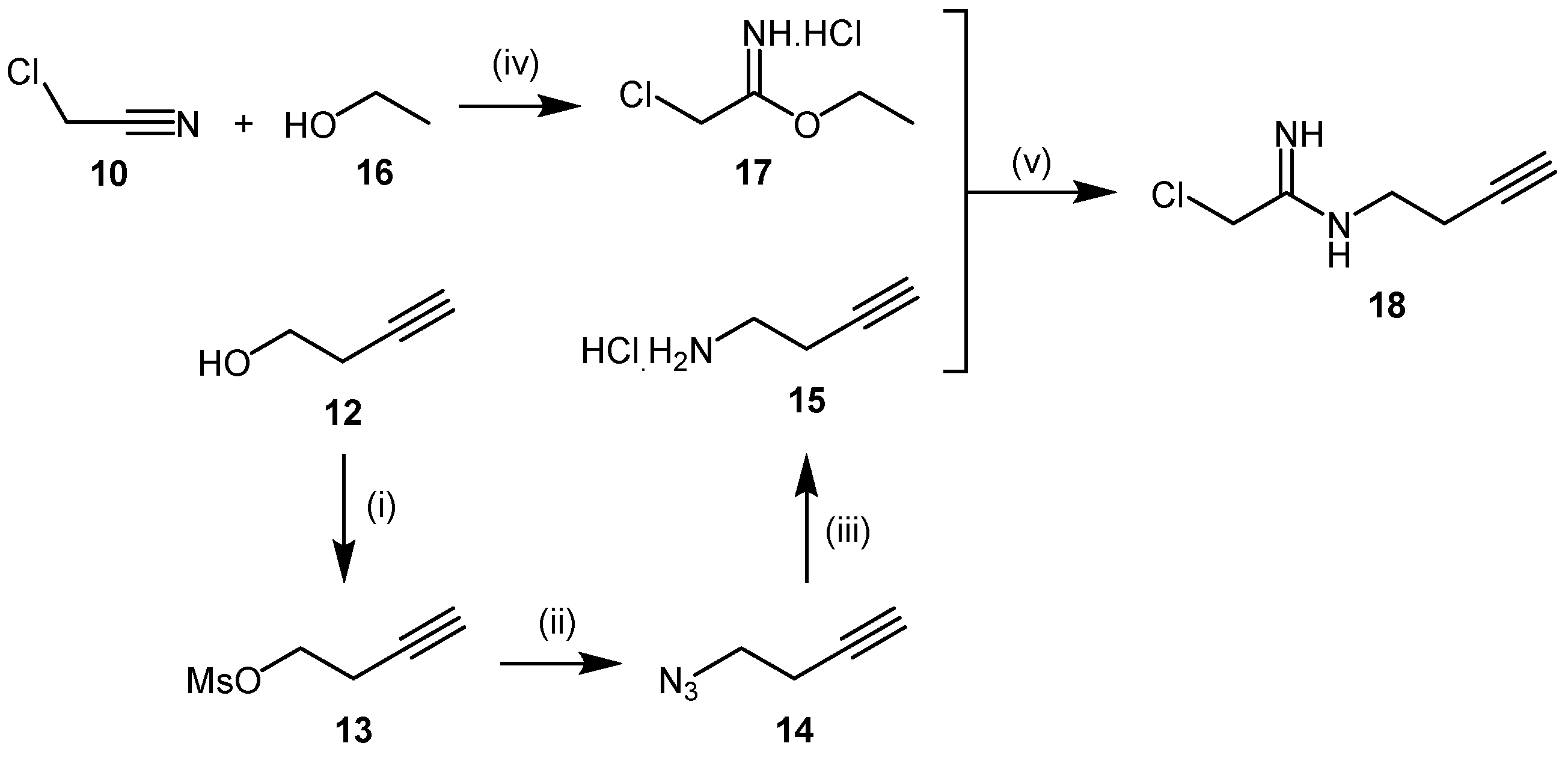
4.1.2. 2-Chloroacetamidine and N-But-3-ynyl-2-chloroacetamidine
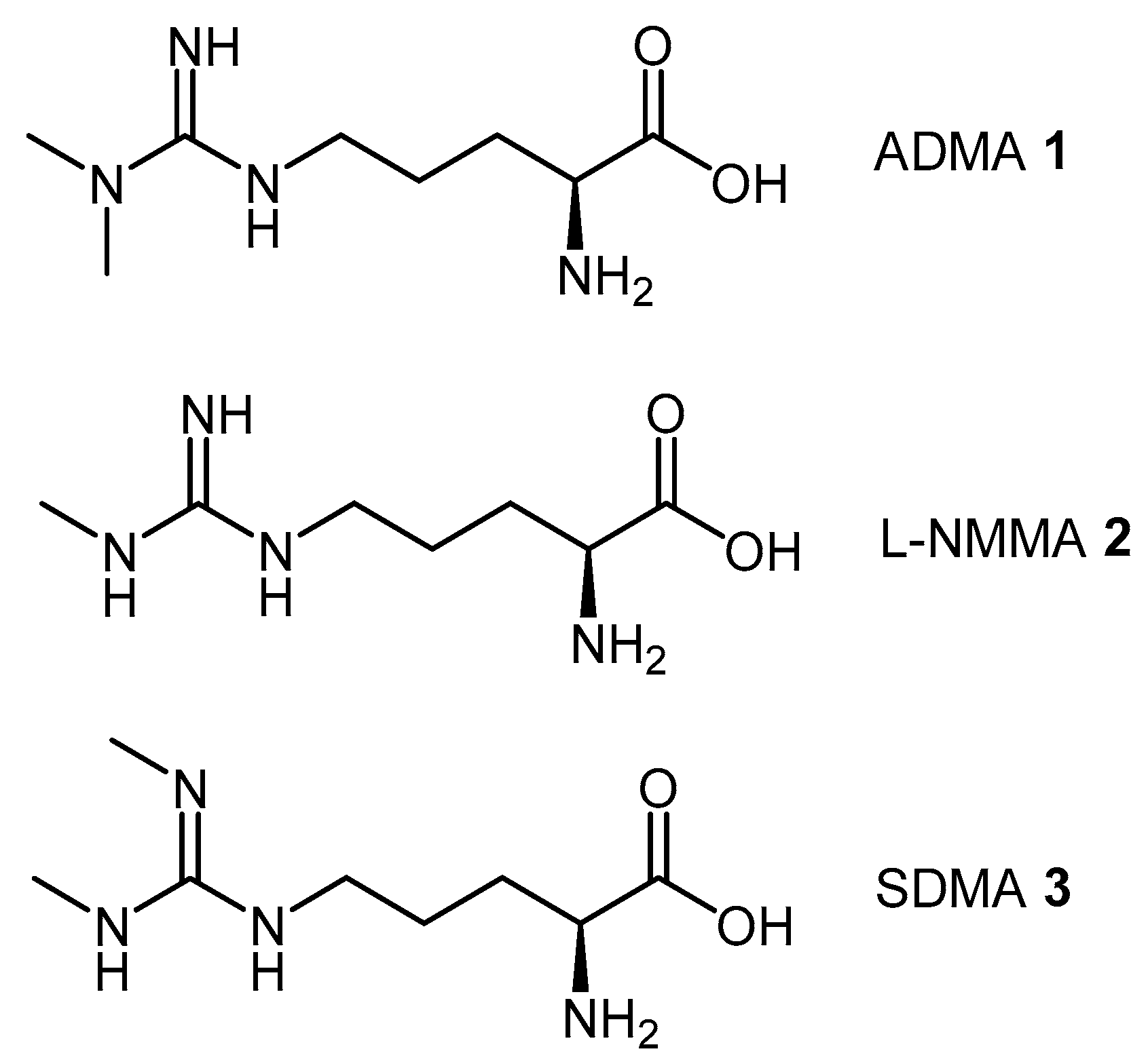
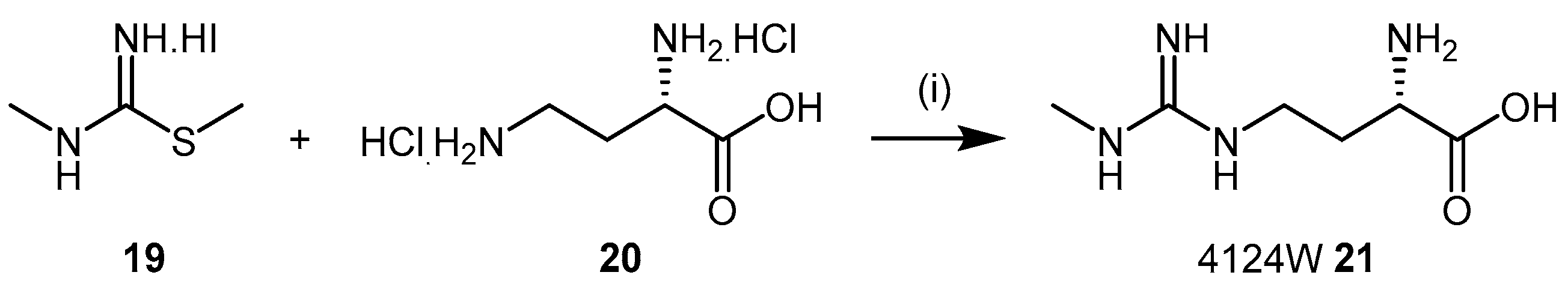
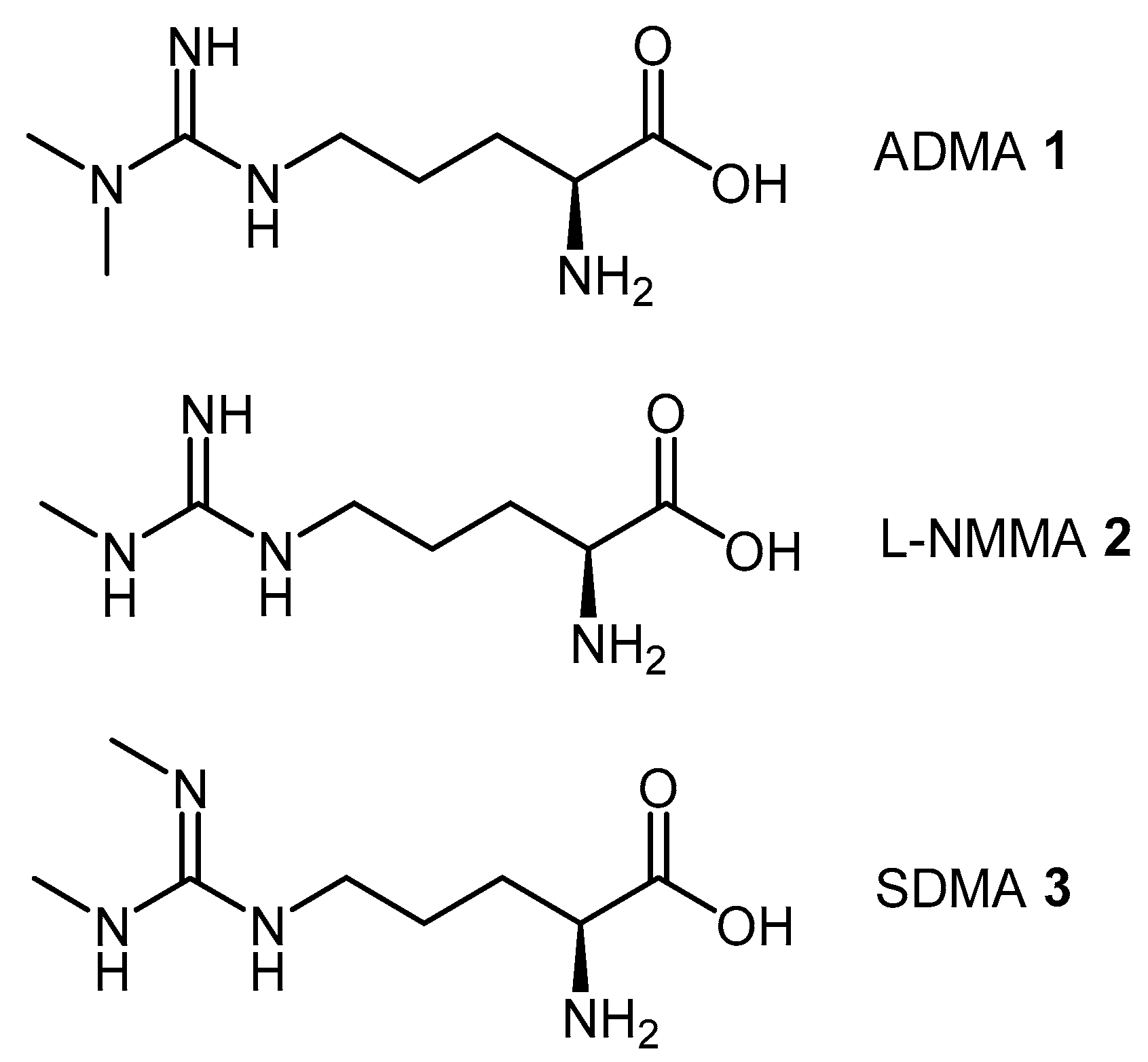
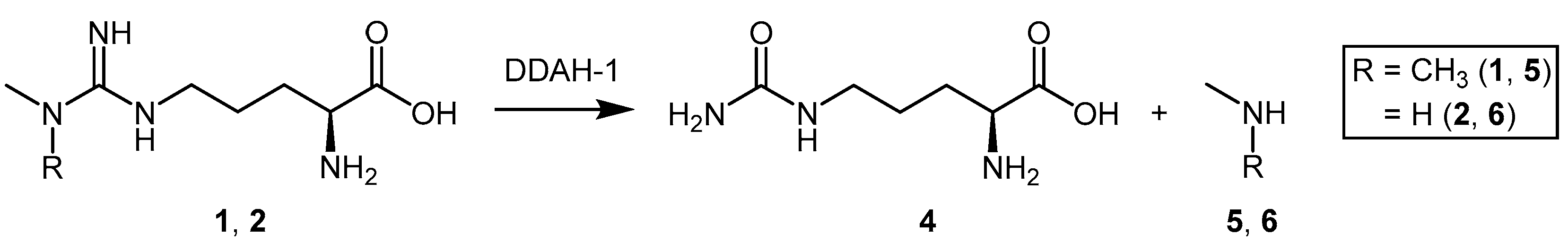
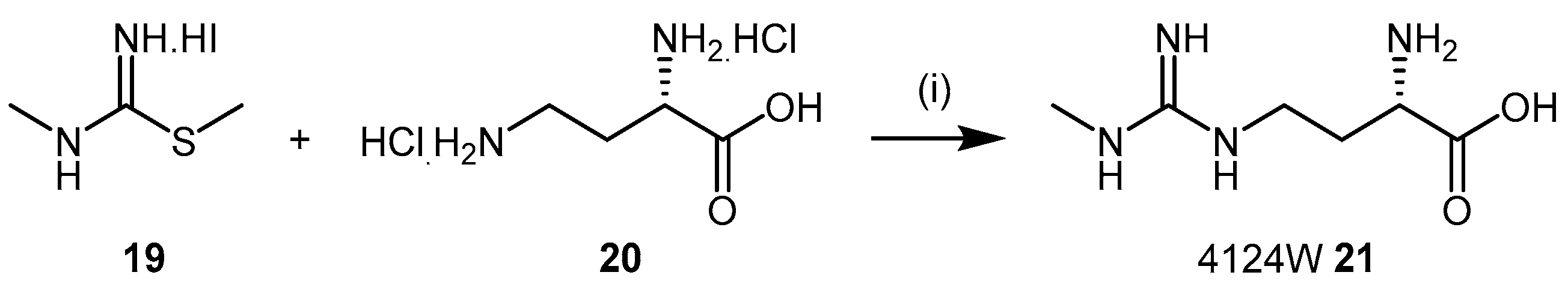
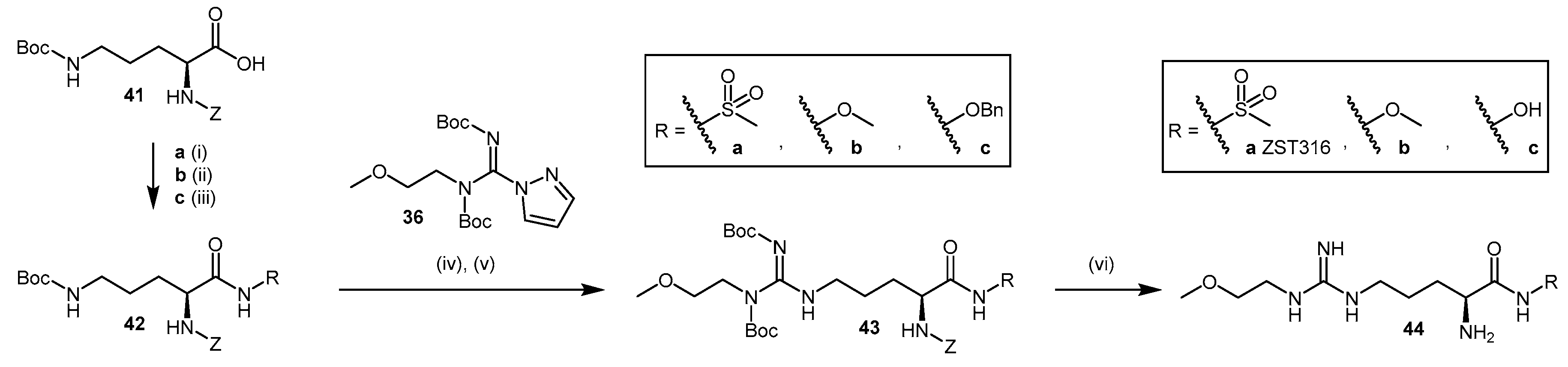
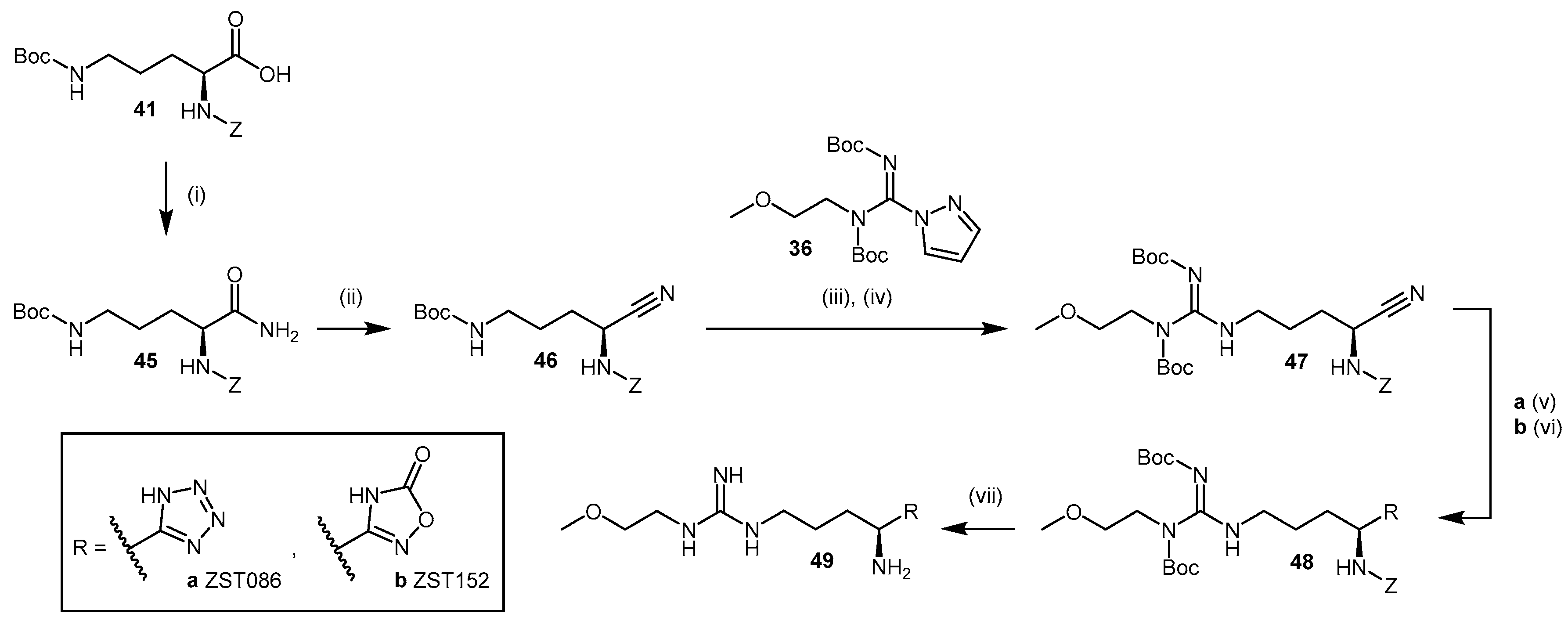
4.1.3. (S)-2-Amino-4-(3-Methylguanidino)butanoic Acid Analogues
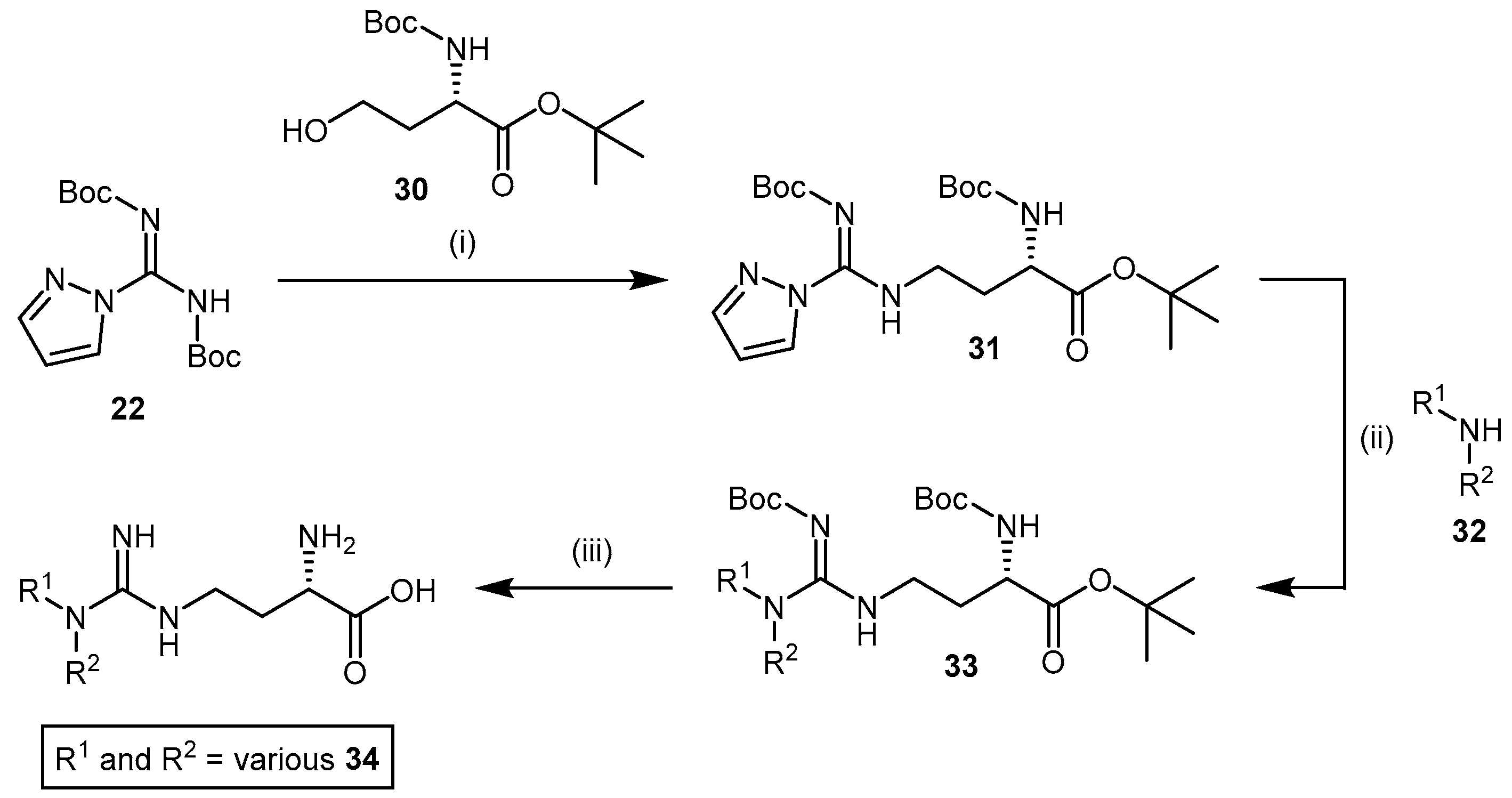
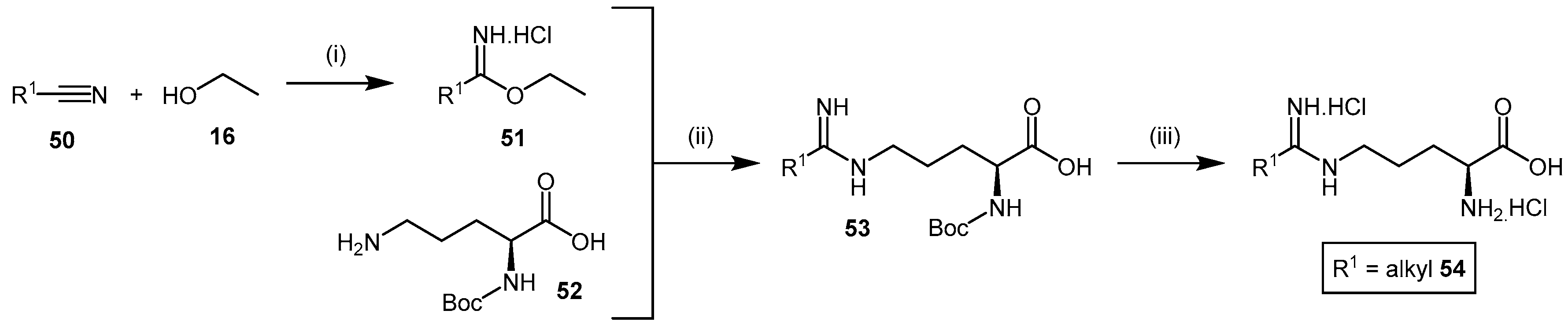
4.1.4. NG-Substituted-l-Arginine Analogues
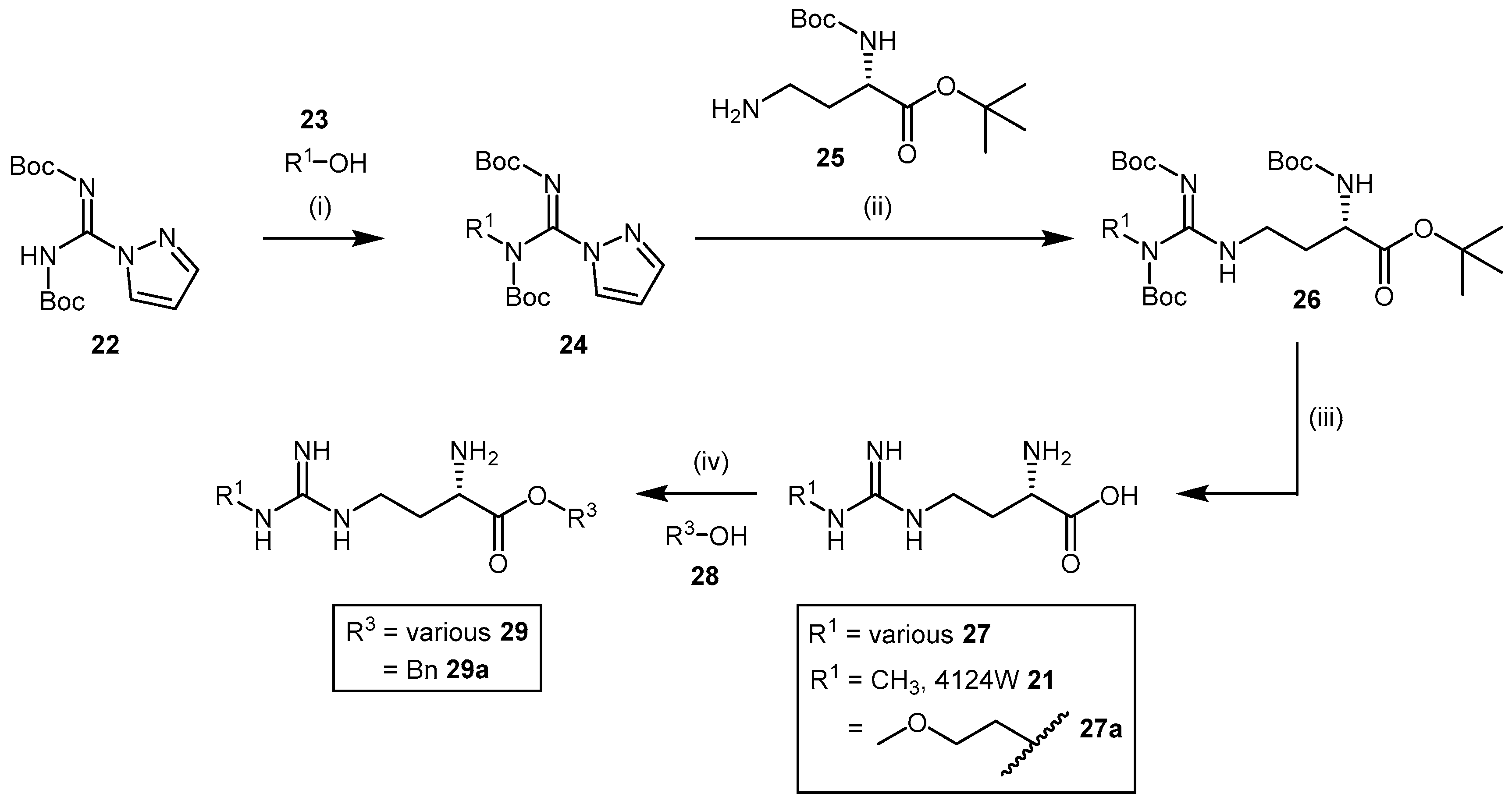
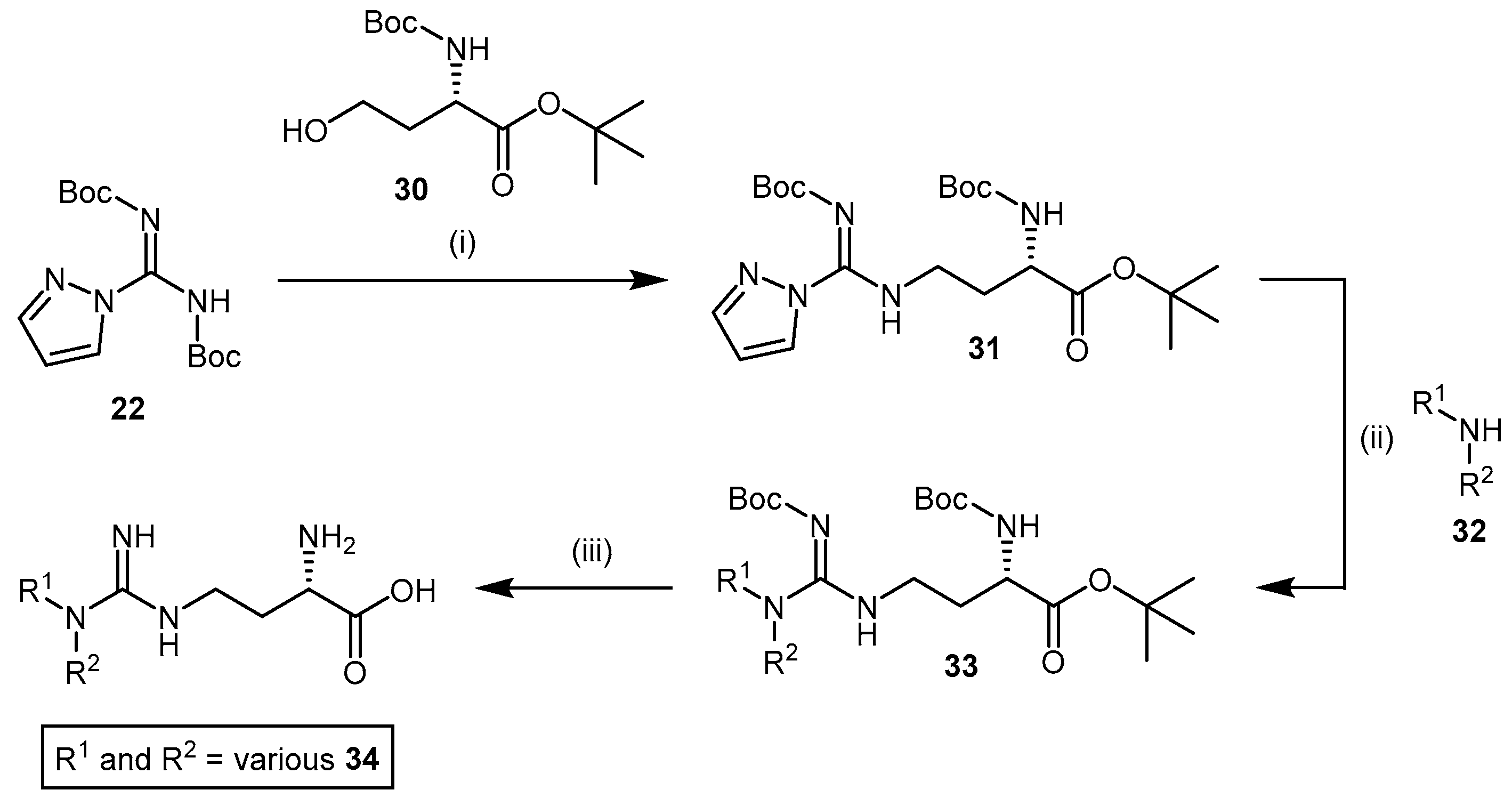
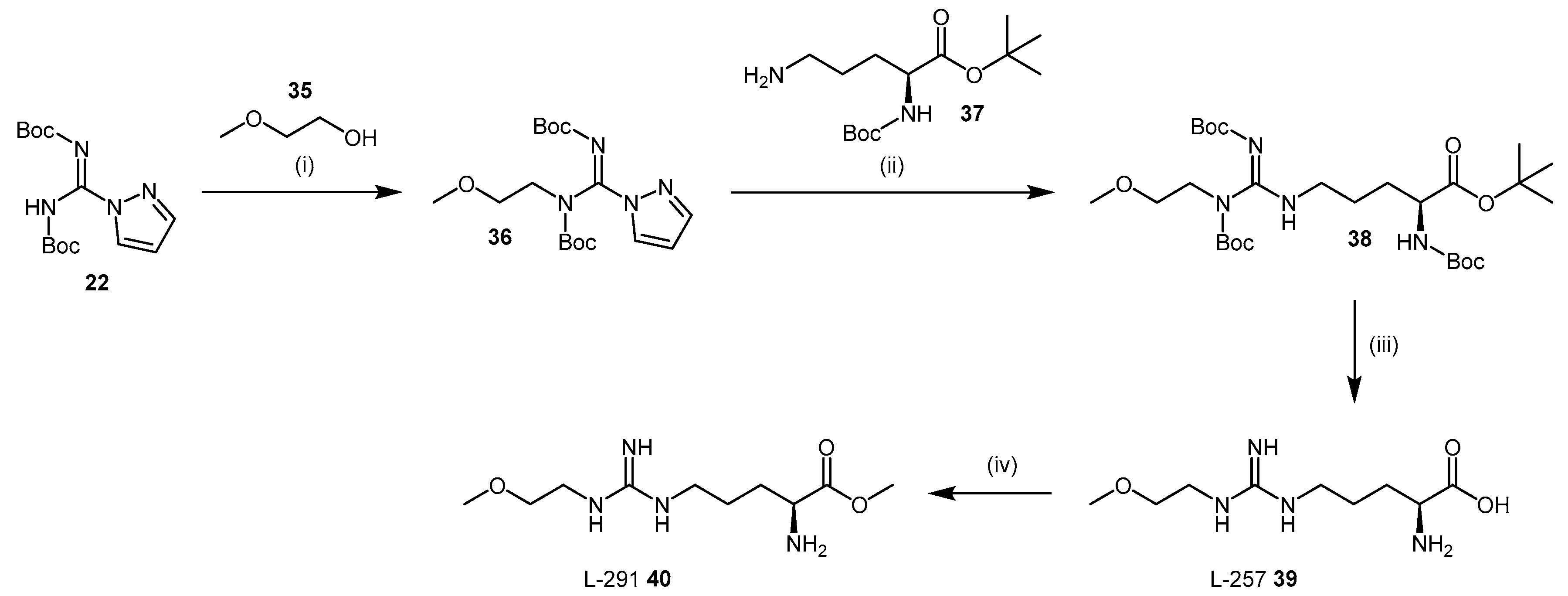
4.1.5. NG-(2-Methoxyethyl)-l-Arginine Carboxylate Bioisosteres
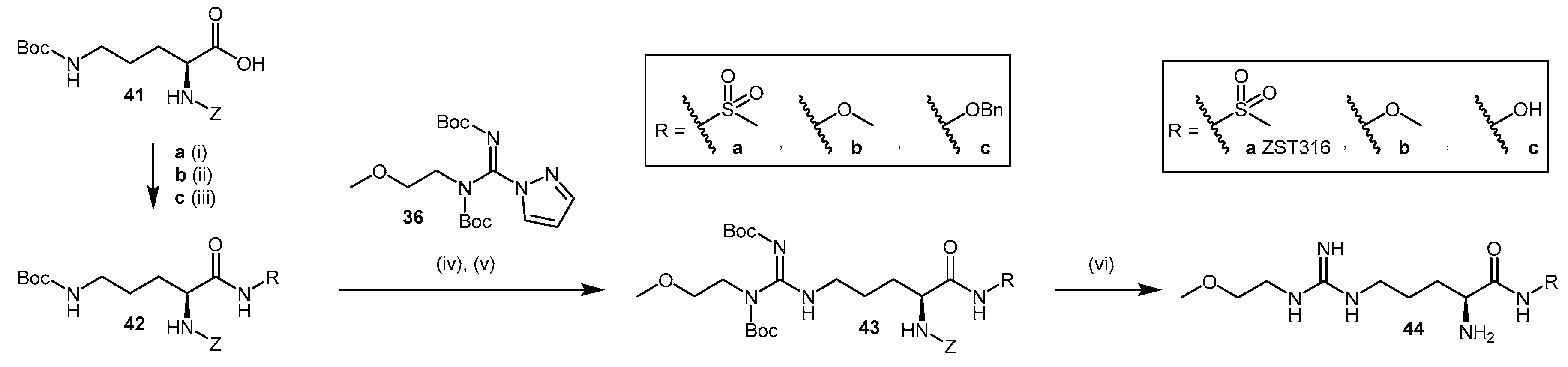
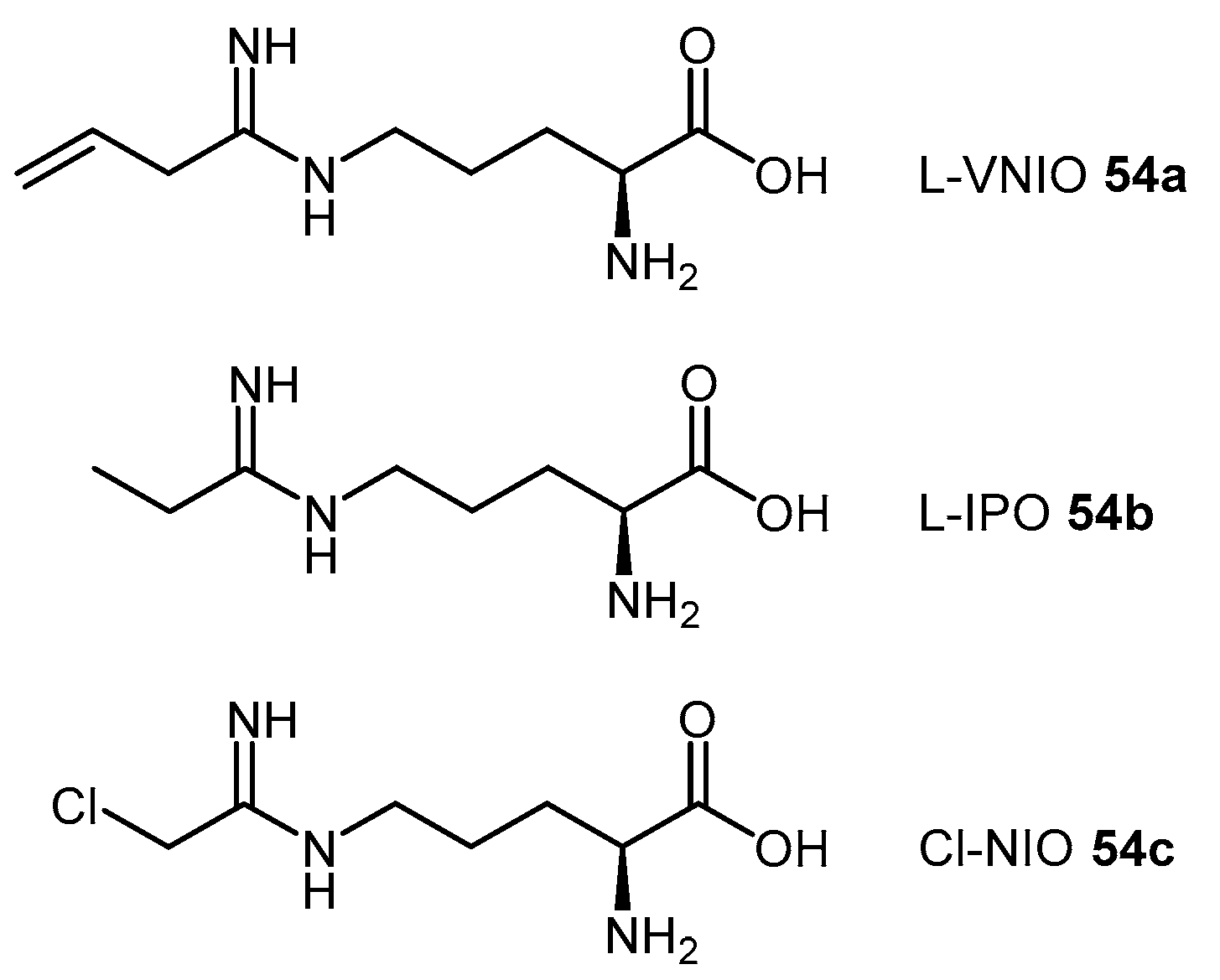
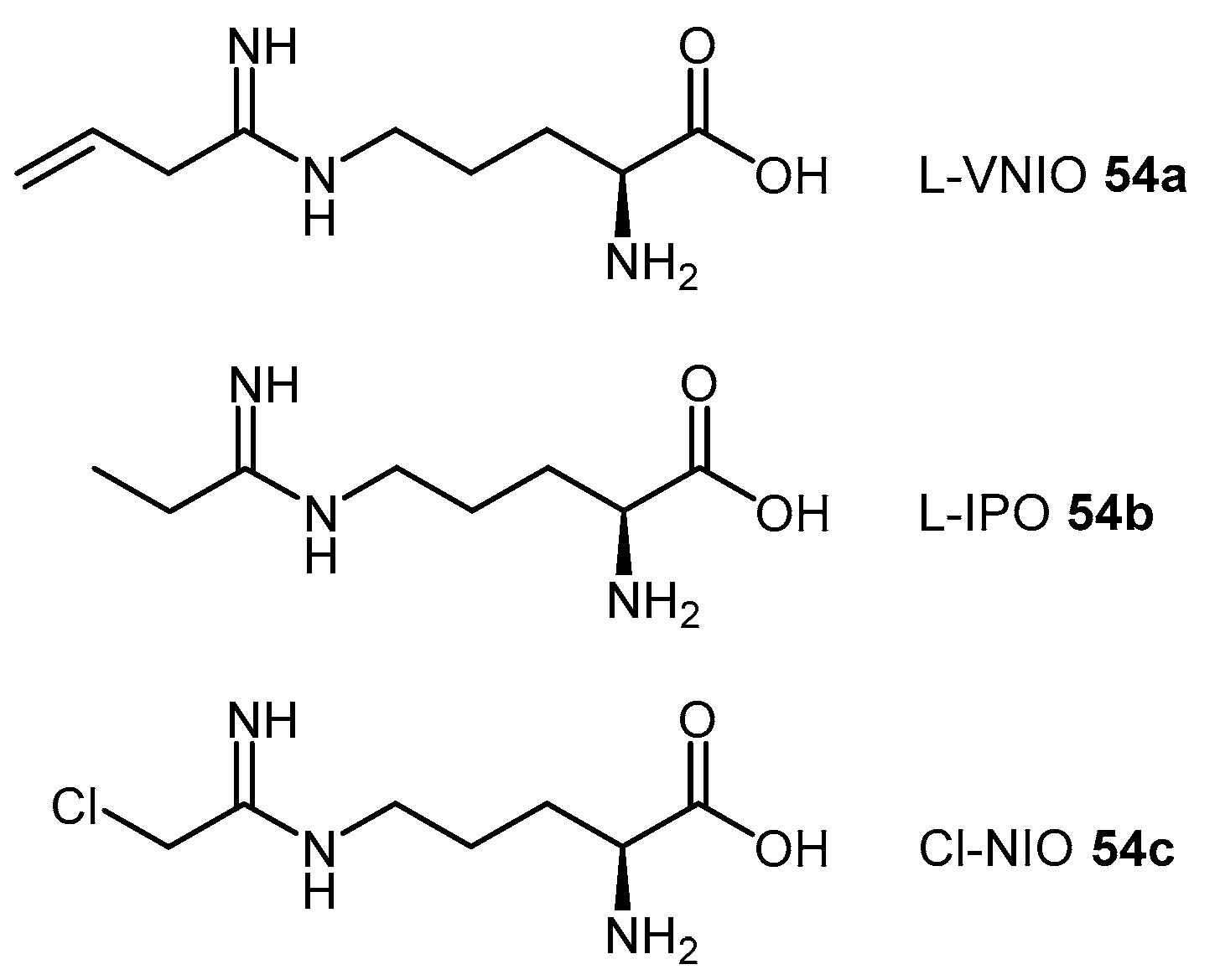
4.1.6. N5-(1-Iminoalk(en)yl)-l-Ornithine Derivatives
4.2. Non-Substrate-Like Inhibitors
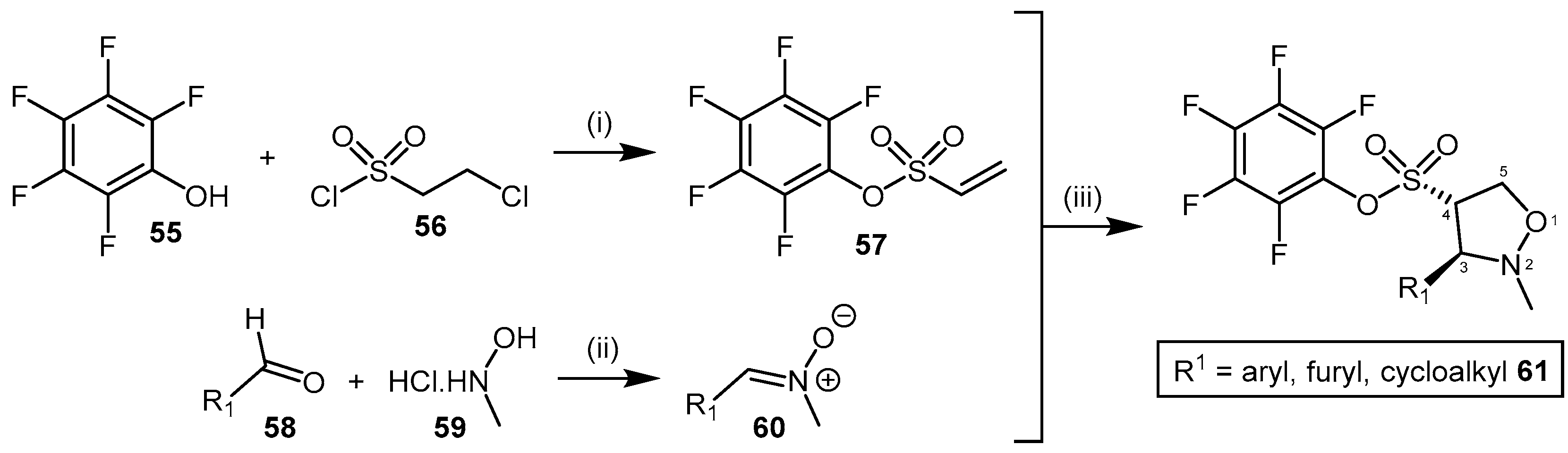
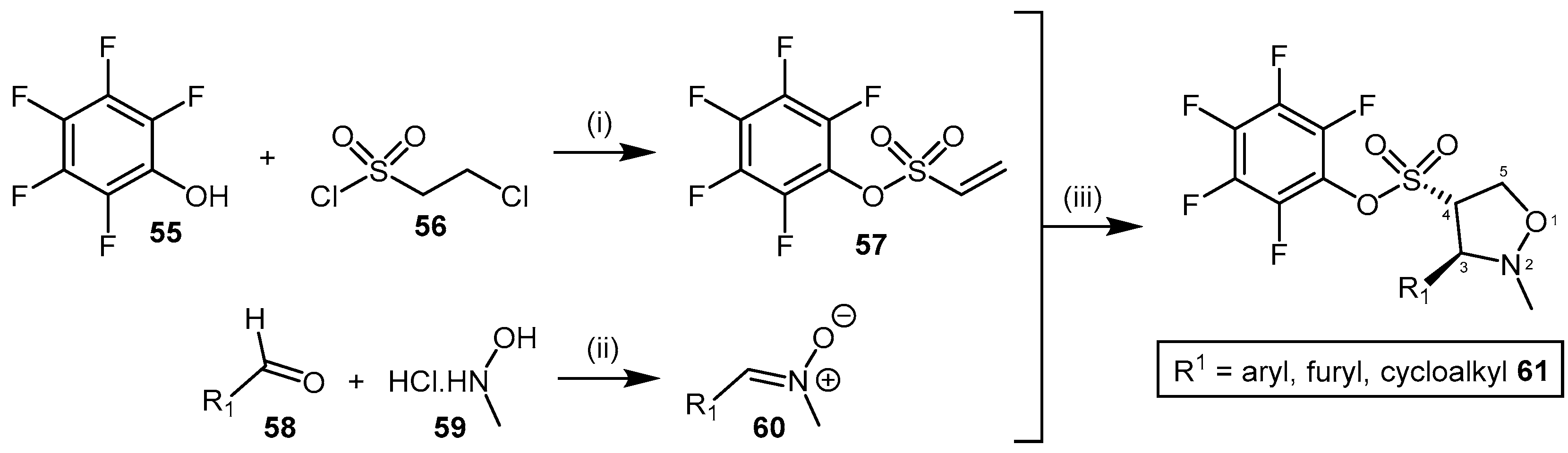
4.2.1. Pentafluorophenyl (PFP) Sulfonate Esters
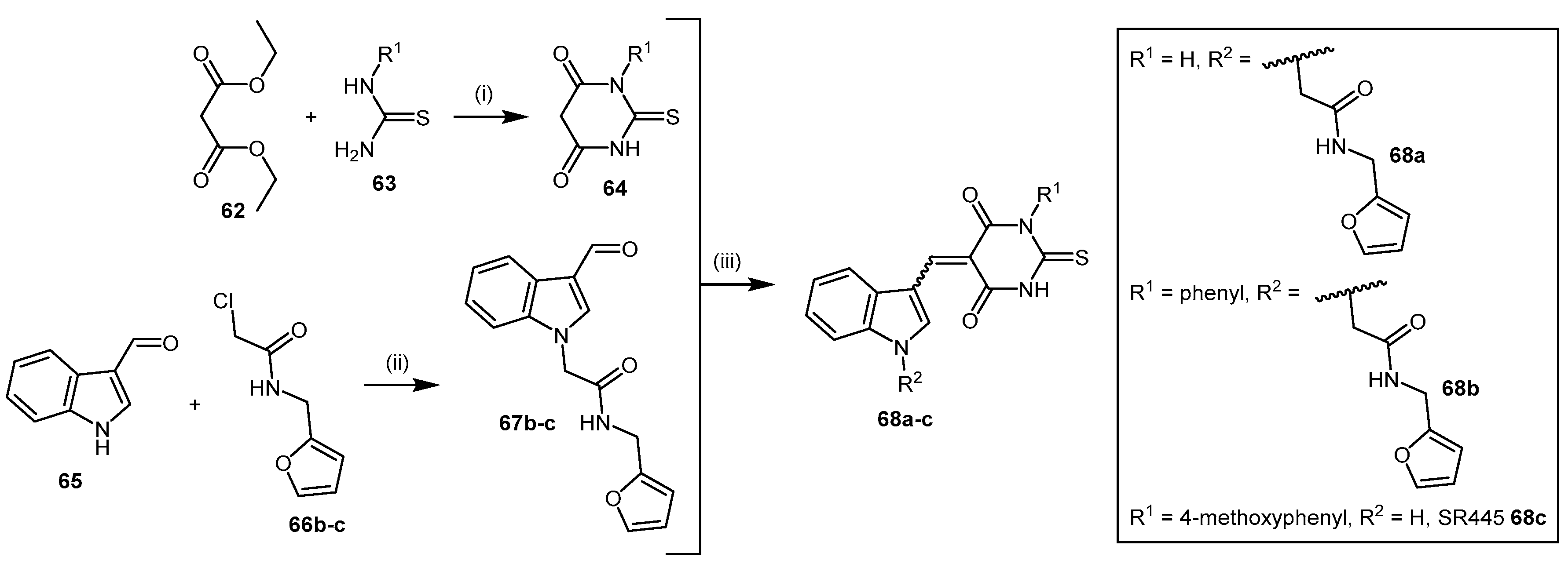
4.2.2. Indolylthiobarbituric Acid Derivatives
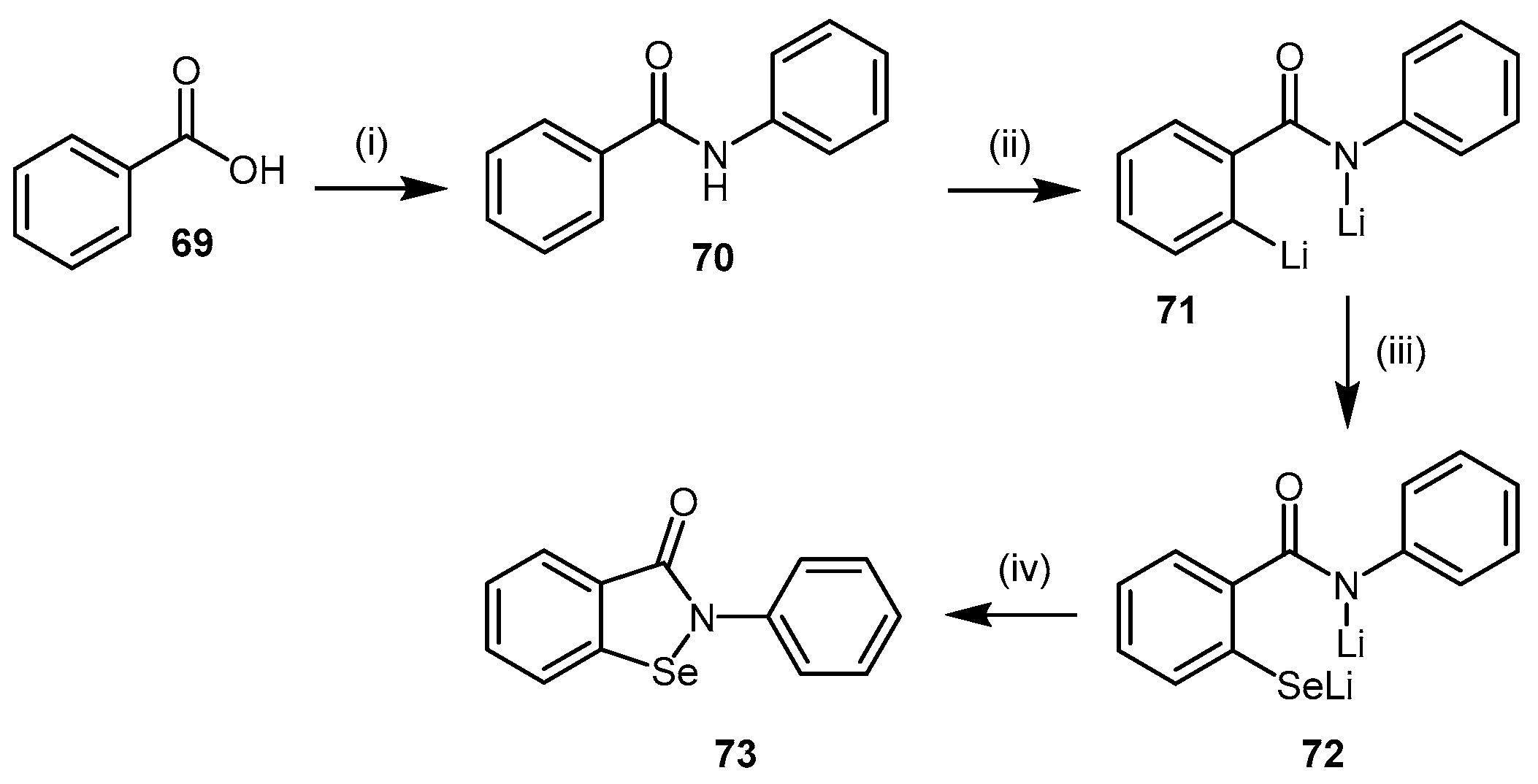
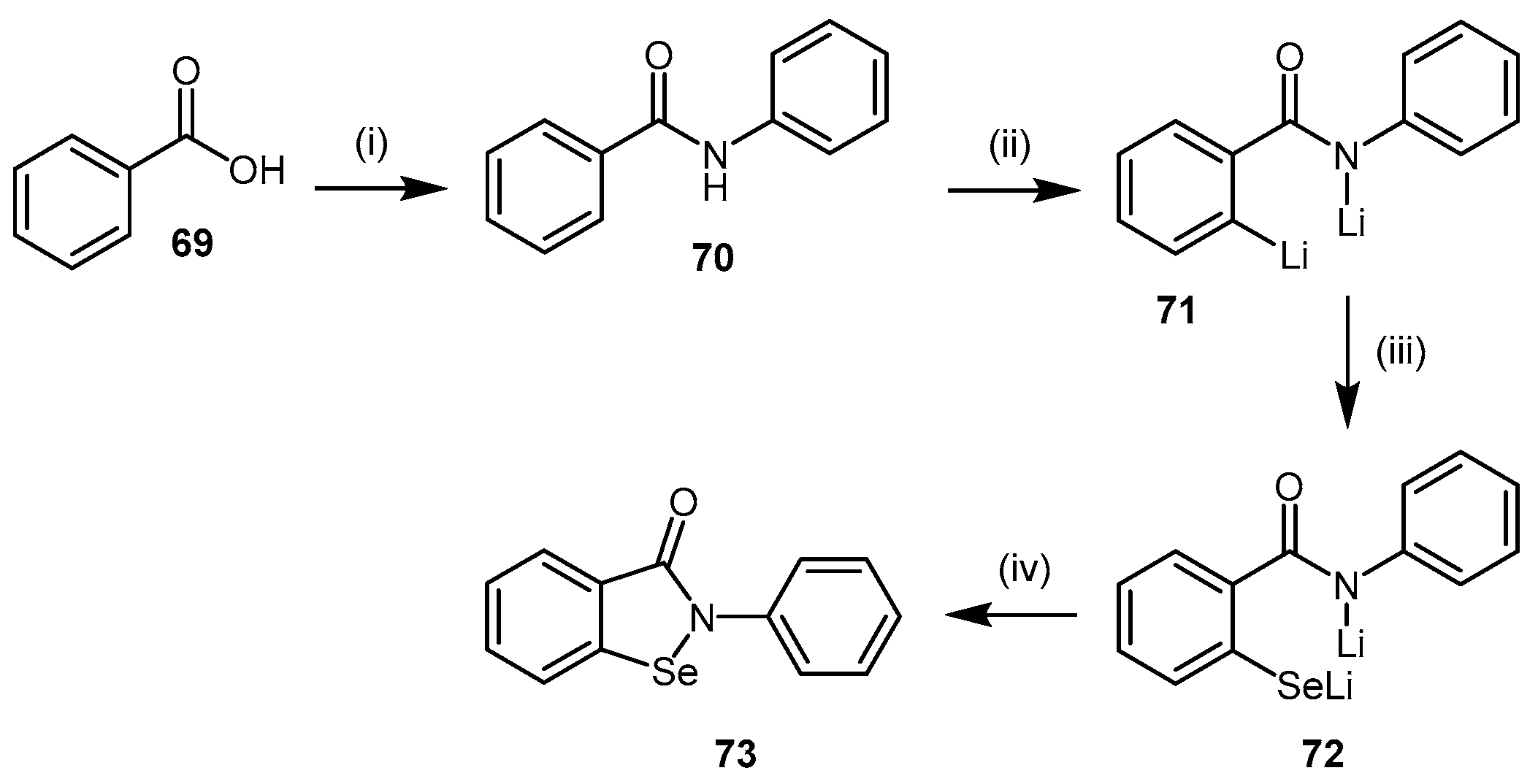
4.2.3. Ebselen
4.2.4. 4-Hydroxy-2-Nonenal
4.2.5. PD 404182
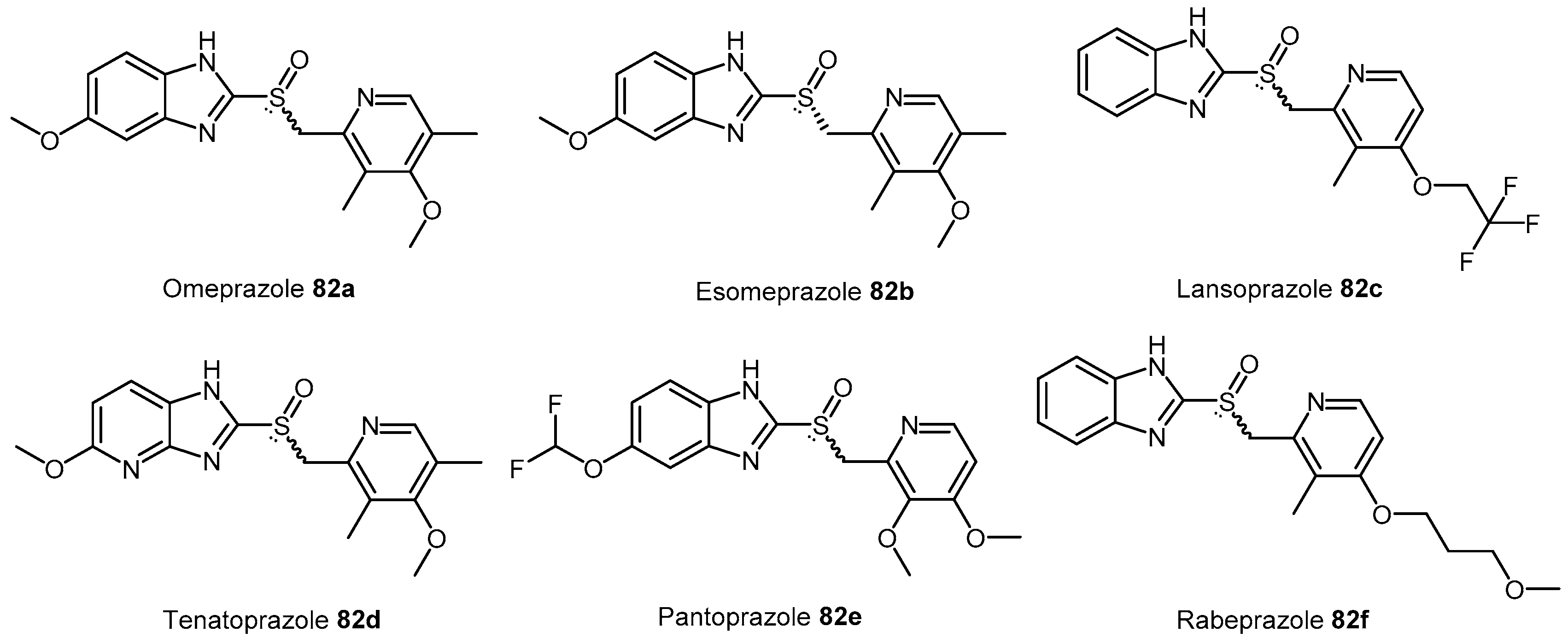
4.2.6. Proton Pump Inhibitors
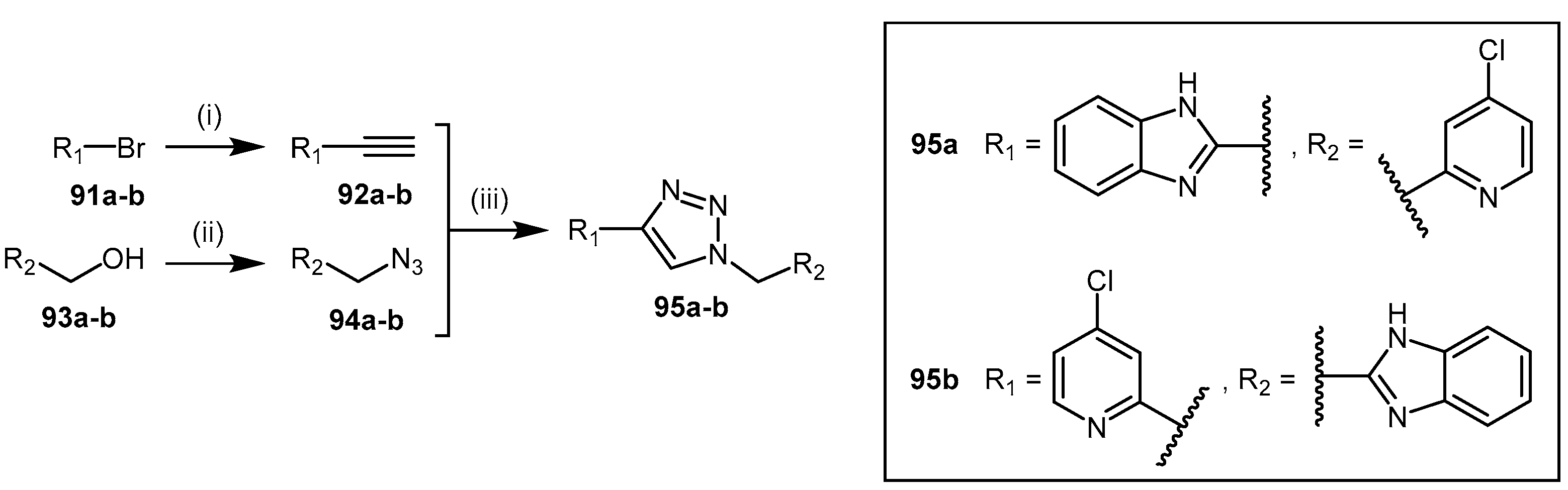
4.2.7. 1,2,3-Triazoles
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| DDAH | Dimethylarginine dimethylamino hydrolase |
| ADMA | Asymmetric dimethylarginine |
| L-NMMA | Monomethyl arginine |
| NOS | Nitric oxide synthase |
| NO | Nitric oxide |
| SDMA | Symmetric dimethylarginine |
| PRMTs | Protein arginine methyl transferases |
| CAT | Cationic amino acid transporter |
| AGXT2 | Alanine-glyoxylate amino transferase 2 |
| PKA | Protein kinase A |
| Akt | Protein kinase B |
| VEGF | Vascular endothelial growth factor |
| iNOS | Inducible nitric oxide synthase |
| TNF-α | Tumour necrosis factor α |
| LDL | Low density lipoprotein |
| IL-1β | Interleukin-1β |
| AT1-R | Type 1 angiotensin receptor |
| DNA | Deoxyribonucleic acid |
| FXR | Farnesoid X receptor |
| HcyNO | S-Nitroso-l-homocysteine |
| NaNO2 | Sodium nitrite |
| NaOH | Sodium hydroxide |
| HCl | Hydrochloric acid |
| Min | Minutes |
| Ki | Inhibition constant |
| µM | Micromolar |
| H2O | Water |
| Cys | Cysteine |
| His | Histidine |
| PaDDAH | Pseudomonas aeruginosa dimethylarginine dimethylamino hydrolase |
| PAD | Peptydilarginine deaminase |
| NaOMe | Sodium methoxide |
| MeOH | Methanol |
| AcOH | Acetic acid |
| NH4Cl | Ammonium chloride |
| PEO | Poly(ethylene oxide) |
| DCM | Dichloromethane |
| TEA | Triethylamine |
| DMAP | 4-Dimethylaminopyridine |
| MsCl | Methanesulfonyl chloride |
| Et2O | Diethyl ether |
| RT | Room temperature |
| NaN3 | Sodium azide |
| DMF | N,N-Dimethylformamide |
| Ph3P | Triphenylphosphine |
| 4124W | (S)-2-Amino-4-(3-methylguanidino)butanoic acid |
| Boc | tert-Butyloxycarbonyl |
| M | Molar |
| DEAD | Diethyl azodicarboxylate |
| THF | Tetrahydrofuran |
| DIPEA | N,N-Diisopropylethylamine |
| CH3CN | Acetonitrile |
| SOCl2 | Thionyl chloride |
| SAR | Structure activity relationship |
| IC50 | Inhibition constant |
| L-257 | NG-(2-Methoxyethyl)-l-arginine |
| L-291 | NG-(2-Methoxyethyl)-l-arginine methyl ester |
| Orn | Ornithine |
| Bu | Butyl |
| Z | Carboxybenzyl |
| TFMSA | Trifluoromethanesulfonic acid |
| TFA | Trifluoroacetic acid |
| CDI | 1,1′-Carbonyldiimidazole |
| DBU | 1,8-Diazabicyclo[5.4.0]undec-7-ene |
| h | Hours |
| HATU | 1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxide hexafluorophosphate |
| mM | Millimolar |
| DMSO | Dimethyl sulfoxide |
| i-BuCOCl | Isobutyl chloroformate |
| NH4OH | Ammonium hydroxide |
| TFAA | Trifluoroacetic anhydride |
| ZnBr2 | Zinc bromide |
| NH2OH.HCl | Hydroxylamine hydrochloride |
| NaHCO3 | Sodium bicarbonate |
| L-VNIO | N5-(1-Imino-3-butenyl)-l-ornithine |
| EtOH | Ethanol |
| EtOAc | Ethyl acetate |
| L-IPO | N5-(1-Iminopropyl)-l-ornithine |
| Cl-NIO | N5-(1-Imino-2-chloroethyl)-l-ornithine |
| PFP | Pentafluorophenyl |
| NaOAc | Sodium acetate |
| ADI | Arginine deaminase |
| HTS | High throughput screening |
| SMTC | S-methyl-l-thiocitrulline |
| ESI-MS | Electrospray ionization mass spectrometry |
| BuLi | Butyl lithium |
| Se | Selenium |
| CuBr2 | Copper(II) bromide |
| NADPH | Nicotinamide adenine dinucleotide phosphate |
| H+/K+ ATPase | Proton/potassium ATPase |
| Ca2+ | Calcium 2+ cation |
| 4-HNE | 4-Hydroxy-2-nonenal |
| LOPAC | Library of Pharmacologically Active Compounds |
| NaH | Sodium hydride |
| CS2 | Carbon disulfide |
| BrCN | Cyanogen bromide |
| Å | Angstrom |
| PPIs | Proton pump inhibitors |
| SPR | Surface plasmon resonance |
| CM | Carboxymethylated |
| (Ph3P)2PdCl2 | Bis(triphenylphosphine)palladium chloride |
| CuI | Copper(I) iodide |
| TIPS | Triisopropylsilyl |
| TMS | Trimethylsilane |
| DPPA | Diphenylphosphoryl azide |
References
- Vallance, P.; Leiper, J. Blocking no synthesis: How, where and why? Nat. Rev. Drug Discov. 2002, 1, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Tran, C.T.; Fox, M.F.; Vallance, P.; Leiper, J.M. Chromosomal localization, gene structure, and expression pattern of DDAH1: Comparison with DDAH2 and implications for evolutionary origins. Genomics 2000, 68, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Ferrigno, A.; Demartini, C.; Zanaboni, A.; Mangione, A.S.; Blandini, F.; Nappi, G.; Vairetti, M.; Tassorelli, C. Evaluation of ADMA-DDAH-NOS axis in specific brain areas following nitroglycerin administration: Study in an animal model of migraine. J. Headache Pain 2015, 16, 74, 1–8. [Google Scholar] [CrossRef] [PubMed]
- D’Mello, R.; Sand, C.A.; Pezet, S.; Leiper, J.M.; Gaurilcikaite, E.; McMahon, S.B.; Dickenson, A.H.; Nandi, M. Dimethylarginine dimethylaminohydrolase 1 is involved in spinal nociceptive plasticity. Pain 2015, 156, 2052–2060. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, K.; Wakino, S.; Kimoto, M.; Minakuchi, H.; Fujimura, K.; Hosoya, K.; Komatsu, M.; Kaneko, Y.; Kanda, T.; Tokuyama, H.; et al. The hydrolase DDAH2 enhances pancreatic insulin secretion by transcriptional regulation of secretagogin through a sirt1-dependent mechanism in mice. FASEB J. 2013, 27, 2301–2315. [Google Scholar] [CrossRef] [PubMed]
- Amrouni, D.; Meiller, A.; Gautier-Sauvigné, S.; Piraud, M.; Bouteille, B.; Vincendeau, P.; Buguet, A.; Cespuglio, R. Cerebral changes occurring in arginase and dimethylarginine dimethylaminohydrolase (DDAH) in a rat model of sleeping sickness. PLoS ONE 2011, 6, e16891. [Google Scholar] [CrossRef] [PubMed]
- Altmann, K.S.; Havemeyer, A.; Beitz, E.; Clement, B. Dimethylarginine-dimethylaminohydrolase-2 (DDAH-2) does not metabolize methylarginines. Chembiochem 2012, 13, 2599–2604. [Google Scholar] [CrossRef] [PubMed]
- Onozato, M.L.; Tojo, A.; Leiper, J.; Fujita, T.; Palm, F.; Wilcox, C.S. Expression of ng,ng-dimethylarginine dimethylaminohydrolase and protein arginine n-methyltransferase isoforms in diabetic rat kidney: Effects of angiotensin ii receptor blockers. Diabetes 2008, 57, 172–180. [Google Scholar] [CrossRef] [PubMed]
- Tomlinson, J.A.P.; Caplin, B.; Boruc, O.; Bruce-Cobbold, C.; Cutillas, P.; Dormann, D.; Faull, P.; Grossman, R.C.; Khadayate, S.; Mas, V.R.; et al. Reduced renal methylarginine metabolism protects against progressive kidney damage. J. Am. Soc. Nephrol. 2015, 26, 3045–3059. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.; Aslam, S.; Welch, W.J.; Wilcox, C.S. Activation of NRF2 coordinates DDAH/PPAR-γ/ENOS pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension 2015, 65, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Qu, X.; Liang, Z.; Chen, W.; Xia, W.; Song, Y. Variance of DDAH/PRMT/ADMA pathway in atrial fibrillation dogs. Biochem. Biophys. Res. Commun. 2008, 377, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Park, S.; Li, Y.; Missov, E.; Hou, M.; Han, X.; Hall, J.L.; Miller, L.W.; Bache, R.J. Alterations of gene expression in failing myocardium following left ventricular assist device support. Physiol. Genom. 2003, 14, 251–260. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Wang, K.; Jin, P.; Dong, C.; Yuan, Q.; Li, Y.; Yang, Z. The association of adipose-derived dimethylarginine dimethylaminohydrolase-2 with insulin sensitivity in experimental type 2 diabetes mellitus. Acta Biochim. Biophys. Sin. 2013, 45, 641–648. [Google Scholar] [CrossRef] [PubMed]
- Ayling, L.J.; Whitley, G.S.J.; Aplin, J.D.; Cartwright, J.E. Dimethylarginine dimethylaminohydrolase (DDAH) regulates trophoblast invasion and motility through effects on nitric oxide. Hum. Reprod. 2006, 21, 2530–2537. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Chouinard, M.; Cox, A.L.; Sipes, P.; Marcelo, M.; Ficorilli, J.; Li, S.; Gao, H.; Ryan, T.P.; Michael, M.D.; et al. Farnesoid x receptor agonist reduces serum asymmetric dimethylarginine levels through hepatic dimethylarginine dimethylaminohydrolase-1 gene regulation. J. Biol. Chem. 2006, 281, 39831–39838. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sim, A.S.; Wang, X.L.; Wilcken, D.E. l-Arginine regulates asymmetric dimethylarginine metabolism by inhibiting dimethylarginine dimethylaminohydrolase activity in hepatic (HEPG2) cells. Cell. Mol. Life Sci. 2006, 63, 2838–2846. [Google Scholar] [CrossRef] [PubMed]
- Mishima, T.; Hamada, T.; Ui-Tei, K.; Takahashi, F.; Miyata, Y.; Imaki, J.; Suzuki, H.; Yamashita, K. Expression of DDAH1 in chick and rat embryos. Dev. Brain Res. 2004, 148, 223–232. [Google Scholar] [CrossRef] [PubMed]
- Matsuguma, K.; Ueda, S.; Yamagishi, S.-I.; Matsumoto, Y.; Kaneyuki, U.; Shibata, R.; Fujimura, T.; Matsuoka, H.; Kimoto, M.; Kato, S.; et al. Molecular mechanism for elevation of asymmetric dimethylarginine and its role for hypertension in chronic kidney disease. J. Am. Soc. Nephrol. 2006, 17, 2176–2183. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.M.; Santa Maria, J.; Chubb, A.; MacAllister, R.J.; Charles, I.G.; Whitley, G.S.; Vallance, P. Identification of two human dimethylarginine dimethylaminohydrolases with distinct tissue distributions and homology with microbial arginine deiminases. Biochem. J. 1999, 343, 209–214. [Google Scholar] [CrossRef] [PubMed]
- Tokuo, H.; Yunoue, S.; Feng, L.; Kimoto, M.; Tsuji, H.; Ono, T.; Saya, H.; Araki, N. Phosphorylation of neurofibromin by camp-dependent protein kinase is regulated via a cellular association of ng,ng-dimethylarginine dimethylaminohydrolase. FEBS Lett. 2001, 494, 48–53. [Google Scholar] [CrossRef]
- Hasegawa, K.; Wakino, S.; Tanaka, T.; Kimoto, M.; Tatematsu, S.; Kanda, T.; Yoshioka, K.; Homma, K.; Sugano, N.; Kurabayashi, M.; et al. Dimethylarginine dimethylaminohydrolase 2 increases vascular endothelial growth factor expression through sp1 transcription factor in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 1488–1494. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Hu, X.; Xu, X.; Chen, Y.; Bache, R.J. Dimethylarginine dimethylaminohydrolase 1 modulates endothelial cell growth through nitric oxide and akt. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 890–897. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Kakimoto, Y. Synthesis and degradation of methylated proteins of mouse organs: Correlation with protein synthesis and degradation. Metab. Clin. Exp. 1976, 25, 885–896. [Google Scholar] [CrossRef]
- Teerlink, T. Adma metabolism and clearance. Vasc. Med. 2005, 10 (Suppl. S1), S73–S81. [Google Scholar] [CrossRef] [PubMed]
- Olken, N.M.; Rusche, K.M.; Richards, M.K.; Marletta, M.A. Inactivation of macrophage nitric oxide synthase activity by NG-Methyl-l-arginine. Biochem. Biophys. Res. Commun. 1991, 177, 828–833. [Google Scholar] [CrossRef]
- Palmer, R.M.J.; Rees, D.D.; Ashton, D.S.; Moncada, S. l-Arginine is the physiological precursor for the formation of nitric oxide in endothelium-dependent relaxation. Biochem. Biophys. Res. Commun. 1988, 153, 1251–1256. [Google Scholar] [CrossRef]
- Hibbs, J.B., Jr.; Taintor, R.R.; Vavrin, Z. Macrophage cytotoxicity: Role for l-arginine deiminase and imino nitrogen oxidation to nitrite. Science 1987, 235, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Q.; Fast, W.; Marletta, M.A.; Martasek, P.; Silverman, R.B. Potent and selective inhibition of neuronal nitric oxide synthase by N omega-propyl-l-arginine. J. Med. Chem. 1997, 40, 3869–3870. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Leone, A.; Calver, A.; Collier, J.; Moncada, S. Accumulation of an endogenous inhibitor of nitric oxide synthesis in chronic renal failure. Lancet 1992, 339, 572–575. [Google Scholar] [PubMed]
- Cardounel, A.J.; Zweier, J.L. Endogenous methylarginines regulate neuronal nitric-oxide synthase and prevent excitotoxic injury. J. Biol. Chem. 2002, 277, 33995–34002. [Google Scholar] [CrossRef] [PubMed]
- Closs, E.I.; Basha, F.Z.; Habermeier, A.; Förstermann, U. Interference of l-Arginine analogues with l-Arginine transport mediated by the y+ carrier hCAT-2B. Nitric Oxide 1997, 1, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kimoto, M.; Watanabe, H.; Sasaoka, K. Metabolism of ng,ng- and ng,ng-dimethylarginine in rats. Arch. Biochem. Biophys. 1987, 252, 526–537. [Google Scholar] [CrossRef]
- Rodionov, R.N.; Murry, D.J.; Vaulman, S.F.; Stevens, J.W.; Lentz, S.R. Human alanine-glyoxylate aminotransferase 2 lowers asymmetric dimethylarginine and protects from inhibition of nitric oxide production. J. Biol. Chem. 2010, 285, 5385–5391. [Google Scholar] [CrossRef] [PubMed]
- Caplin, B.; Wang, Z.; Slaviero, A.; Tomlinson, J.; Dowsett, L.; Delahaye, M.; Salama, A.; Wheeler, D.C.; Leiper, J. Alanine-glyoxylate aminotransferase-2 metabolizes endogenous methylarginines, regulates no, and controls blood pressure. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 2892–2900. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, T.; Kimoto, M.; Sasaoka, K. Occurrence of a new enzyme catalyzing the direct conversion of ng,ng-dimethyl-l-arginine to l-citrulline in rats. Biochem. Biophys. Res. Commun. 1987, 148, 671–677. [Google Scholar] [CrossRef]
- Leiper, J.; Nandi, M. The therapeutic potential of targeting endogenous inhibitors of nitric oxide synthesis. Nat. Rev. Drug Discov. 2011, 10, 277–291. [Google Scholar] [CrossRef] [PubMed]
- Zoccali, C.; Bode-Böger, S.M.; Mallamaci, F.; Benedetto, F.A.; Tripepi, G.; Malatino, L.S.; Cataliotti, A.; Bellanuova, I.; Fermo, I.; Frölich, J.C.; et al. Plasma concentration of asymmetrical dimethylarginine and mortality in patients with end-stage renal disease: A prospective study. Lancet 2001, 358, 2113–2117. [Google Scholar] [CrossRef]
- Arrigoni, F.I. Metabolism of asymmetric dimethylarginines is regulated in the lung developmentally and with pulmonary hypertension induced by hypobaric hypoxia. Circulation 2003, 107, 1195–1201. [Google Scholar] [CrossRef] [PubMed]
- Alpoim, P.N.; Godoi, L.C.; Freitas, L.G.; Gomes, K.B.; Dusse, L.M. Assessment of l-arginine asymmetric 1 dimethyl (ADMA) in early-onset and late-onset (severe) preeclampsia. Nitric Oxide 2013, 33, 81–82. [Google Scholar] [CrossRef] [PubMed]
- Anderssohn, M.; Maass, L.M.; Diemert, A.; Luneburg, N.; Atzler, D.; Hecher, K.; Boger, R.H. Severely decreased activity of placental dimethylarginine dimethylaminohydrolase in pre-eclampsia. Eur. J. Obstet. Gynecol. Reprod. Biol. 2012, 161, 152–156. [Google Scholar] [CrossRef] [PubMed]
- Sydow, K.; Mondon, C.E.; Schrader, J.; Konishi, H.; Cooke, J.P. Dimethylarginine dimethylaminohydrolase overexpression enhances insulin sensitivity. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Jacobi, J.; Sydow, K.; von Degenfeld, G.; Zhang, Y.; Dayoub, H.; Wang, B.; Patterson, A.J.; Kimoto, M.; Blau, H.M.; Cooke, J.P. Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation 2005, 111, 1431–1438. [Google Scholar] [CrossRef] [PubMed]
- Ljubisavljevic, S.; Stojanovic, I.; Pavlovic, R.; Sokolovic, D.; Pavlovic, D.; Cvetkovic, T.; Stevanovic, I. Modulation of nitric oxide synthase by arginase and methylated arginines during the acute phase of experimental multiple sclerosis. J. Neurol. Sci. 2012, 318, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Lambden, S.; Kelly, P.; Ahmetaj-Shala, B.; Wang, Z.; Lee, B.; Nandi, M.; Torondel, B.; Delahaye, M.; Dowsett, L.; Piper, S.; et al. Dimethylarginine dimethylaminohydrolase 2 regulates nitric oxide synthesis and hemodynamics and determines outcome in polymicrobial sepsis. Arterioscler. Thromb. Vasc. Biol. 2015, 35, 1382–1392. [Google Scholar] [CrossRef] [PubMed]
- Schulze, F.; Lenzen, H.; Hanefeld, C.; Bartling, A.; Osterziel, K.J.; Goudeva, L.; Schmidt-Lucke, C.; Kusus, M.; Maas, R.; Schwedhelm, E.; et al. Asymmetric dimethylarginine is an independent risk factor for coronary heart disease: Results from the multicenter coronary artery risk determination investigating the influence of adma concentration (cardiac) study. Am. Heart J. 2006, 152, 493.e491–493.e498. [Google Scholar] [CrossRef] [PubMed]
- Maas, R.; Schulze, F.; Baumert, J.; Lowel, H.; Hamraz, K.; Schwedhelm, E.; Koenig, W.; Boger, R.H. Asymmetric dimethylarginine, smoking, and risk of coronary heart disease in apparently healthy men: Prospective analysis from the population-based monitoring of trends and determinants in cardiovascular disease/kooperative gesundheitsforschung in der region augsburg study and experimental data. Clin. Chem. 2007, 53, 693–701. [Google Scholar] [PubMed]
- Duckelmann, C.; Mittermayer, F.; Haider, D.G.; Altenberger, J.; Eichinger, J.; Wolzt, M. Asymmetric dimethylarginine enhances cardiovascular risk prediction in patients with chronic heart failure. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- Miyazaki, H.; Matsuoka, H.; Cooke, J.P.; Usui, M.; Ueda, S.; Okuda, S.; Imaizumi, T. Endogenous nitric oxide synthase inhibitor : A novel marker of atherosclerosis. Circulation 1999, 99, 1141–1146. [Google Scholar] [CrossRef] [PubMed]
- Wanby, P.; Teerlink, T.; Brudin, L.; Brattström, L.; Nilsson, I.; Palmqvist, P.; Carlsson, M. Asymmetric dimethylarginine (ADMA) as a risk marker for stroke and tia in a swedish population. Atherosclerosis 2006, 185, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Li, N.; Deb-Chatterji, M.; Dong, Q.; Kielstein, J.T.; Weissenborn, K.; Worthmann, H. Asymmetric dimethyarginine as marker and mediator in ischemic stroke. Int. J. Mol. Sci. 2012, 13, 15983–16004. [Google Scholar] [CrossRef] [PubMed]
- Isobe, C.; Abe, T.; Terayama, Y. Decrease in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in cerebrospinal fluid during elderly aging and in patients with sporadic form of amyotrophic lateral sclerosis. Neuro Signals 2010, 18, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Arlt, S.; Schulze, F.; Eichenlaub, M.; Maas, R.; Lehmbeck, J.T.; Schwedhelm, E.; Jahn, H.; Boger, R.H. Asymmetrical dimethylarginine is increased in plasma and decreased in cerebrospinal fluid of patients with Alzheimer’s disease. Dement. Geriatr. Cognit. Disord. 2008, 26, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Tohgi, H.; Murata, T.; Isobe, C.; Sato, C. Reduction in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in the cerebrospinal fluid during aging and in patients with Alzheimer’s disease. Neurosci. Lett. 2001, 312, 177–179. [Google Scholar] [CrossRef]
- Tang, X.Q.; Fang, H.R.; Li, Y.J.; Zhou, C.F.; Ren, Y.K.; Chen, R.Q.; Wang, C.Y.; Hu, B. Endogenous hydrogen sulfide is involved in asymmetric dimethylarginine-induced protection against neurotoxicity of 1-methyl-4-phenyl-pyridinium ion. Neurochem. Res. 2011, 36, 2176–2185. [Google Scholar] [CrossRef] [PubMed]
- Kostourou, V.; Robinson, S.P.; Whitley, G.S.; Griffiths, J.R. Effects of overexpression of dimethylarginine dimethylaminohydrolase on tumor angiogenesis assessed by susceptibility magnetic resonance imaging. Cancer Res. 2003, 63, 4960–4966. [Google Scholar] [PubMed]
- Kostourou, V.; Robinson, S.P.; Cartwright, J.E.; Whitley, G.S.J. Dimethylarginine dimethylaminohydrolase i enhances tumour growth and angiogenesis. Br. J. Cancer 2002, 87, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.B.; Dong, X.S.; Sun, W.Z.; Zhao, D.L.; Yang, Y. Effect of a nitric oxide synthase inhibitor NG-nitro-l-arginine methyl ester on invasion of human colorectal cancer cell line SL-174T. World J. Gastroenterol. 2005, 11, 6385–6388. [Google Scholar] [CrossRef] [PubMed]
- Punathil, T.; Katiyar, S.K. Inhibition of non-small cell lung cancer cell migration by grape seed proanthocyanidins is mediated through the inhibition of nitric oxide, guanylate cyclase, and ERK1/2. Mol. Carcinog. 2009, 48, 232–242. [Google Scholar] [CrossRef] [PubMed]
- Vanella, L.; di Giacomo, C.; Acquaviva, R.; Santangelo, R.; Cardile, V.; Barbagallo, I.; Abraham, N.G.; Sorrenti, V. The DDAH/NOS pathway in human prostatic cancer cell lines: Antiangiogenic effect of L-name. Int. J. Oncol. 2011, 39, 1303–1310. [Google Scholar] [PubMed]
- Fiedler, L.R.; Bachetti, T.; Leiper, J.; Zachary, I.; Chen, L.; Renne, T.; Wojciak-Stothard, B. The ADMA/DDAH pathway regulates vegf-mediated angiogenesis. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 2117–2124. [Google Scholar] [CrossRef] [PubMed]
- Ghebremariam, Y.T.; Erlanson, D.A.; Cooke, J.P. A novel and potent inhibitor of dimethylarginine dimethylaminohydrolase: A modulator of cardiovascular nitric oxide. J. Pharmacol. Exp. Ther. 2014, 348, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Schumer, W. Pathophysiology and treatment of septic shock. Am. J. Emerg. Med. 1984, 2, 74–77. [Google Scholar] [CrossRef]
- Thiemermann, C. Nitric oxide and septic shock. Gen. Pharmacol. Vasc. Syst. 1997, 29, 159–166. [Google Scholar] [CrossRef]
- Kilbourn, R.G.; Traber, D.L.; Szabó, C. Nitric oxide and shock. Dis. Mon. 1997, 43, 279–348. [Google Scholar] [CrossRef]
- Titheradge, M.A. Nitric oxide in septic shock. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 437–455. [Google Scholar] [CrossRef]
- Lopez, A.; Lorente, J.A.; Steingrub, J.; Bakker, J.; McLuckie, A.; Willatts, S.; Brockway, M.; Anzueto, A.; Holzapfel, L.; Breen, D.; et al. Multiple-center, randomized, placebo-controlled, double-blind study of the nitric oxide synthase inhibitor 546C88: Effect on survival in patients with septic shock. Crit. Care Med. 2004, 32, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lambden, S.; Taylor, V.; Sujkovic, E.; Nandi, M.; Tomlinson, J.; Dyson, A.; McDonald, N.; Caddick, S.; Singer, M.; et al. Pharmacological inhibition of ddah1 improves survival, haemodynamics and organ function in experimental septic shock. Biochem. J. 2014, 460, 309–316. [Google Scholar] [CrossRef] [PubMed]
- Wadham, C.; Mangoni, A.A. Dimethylarginine dimethylaminohydrolase regulation: A novel therapeutic target in cardiovascular disease. Expert Opin. Drug Metab. Toxicol. 2009, 5, 303–319. [Google Scholar] [CrossRef] [PubMed]
- Knipp, M.; Charnock, J.M.; Garner, C.D.; Vasak, M. Structural and functional characterization of the zn(ii) site in dimethylargininase-1 (DDAH-1) from bovine brain. Zn(ii) release activates ddah-1. J. Biol. Chem. 2001, 276, 40449–40456. [Google Scholar] [CrossRef] [PubMed]
- Murray-Rust, J.; Leiper, J.; McAlister, M.; Phelan, J.; Tilley, S.; Santa Maria, J.; Vallance, P.; McDonald, N. Structural insights into the hydrolysis of cellular nitric oxide synthase inhibitors by dimethylarginine dimethylaminohydrolase. Nat. Struct. Biol. 2001, 8, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Multhaup, G.; Moir, R.D.; Williamson, T.G.; Small, D.H.; Rumble, B.; Pollwein, P.; Beyreuther, K.; Masters, C.L. A novel zinc (ii) binding site modulates the function of the beta A4 amyloid protein precursor of Alzheimer’s disease. J. Biol. Chem. 1993, 268, 16109–16112. [Google Scholar] [PubMed]
- Aizenman, E.; Stout, A.K.; Hartnett, K.A.; Dineley, K.E.; McLaughlin, B.; Reynolds, I.J. Induction of neuronal apoptosis by thiol oxidation: Putative role of intracellular zinc release. J. Neurochem. 2000, 75, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Bush, A.I.; Pettingell, W.H.; Multhaup, G.; Paradis, M.D.; Vonsattel, J.P.; Gusella, J.F.; Beyreuther, K.; Masters, C.L.; Tanzi, R.E. Rapid induction of Alzheimer a beta amyloid formation by zinc. Science 1994, 265, 1464–1467. [Google Scholar] [CrossRef] [PubMed]
- Palm, F.; Onozato, M.L.; Luo, Z.; Wilcox, C.S. Dimethylarginine dimethylaminohydrolase (DDAH): Expression, regulation, and function in the cardiovascular and renal systems. Am. J. Physiol. Heart Circ. Physiol. 2007, 293, H3227–H3245. [Google Scholar] [CrossRef] [PubMed]
- Leiper, J.; Murray-Rust, J.; McDonald, N.; Vallance, P. S-Nitrosylation of dimethylarginine dimethylaminohydrolase regulates enzyme activity: Further interactions between nitric oxide synthase and dimethylarginine dimethylaminohydrolase. Proc. Natl. Acad. Sci. USA 2002, 99, 13527–13532. [Google Scholar] [CrossRef] [PubMed]
- Osanai, T.; Saitoh, M.; Sasaki, S.; Tomita, H.; Matsunaga, T.; Okumura, K. Effect of shear stress on asymmetric dimethylarginine release from vascular endothelial cells. Hypertension 2003, 42, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Stuhlinger, M.C.; Tsao, P.S.; Her, J.H.; Kimoto, M.; Balint, R.F.; Cooke, J.P. Homocysteine impairs the nitric oxide synthase pathway: Role of asymmetric dimethylarginine. Circulation 2001, 104, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.L.; Zhang, X.H.; Li, N.S.; Rang, W.Q.; Feng, Y.; Hu, C.P.; Li, Y.J.; Deng, H.W. Probucol decreases asymmetrical dimethylarginine level by alternation of protein arginine methyltransferase I and dimethylarginine dimethylaminohydrolase activity. Cardiovasc. Drugs Ther. 2006, 20, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Holden, D.P.; Cartwright, J.E.; Nussey, S.S.; Whitley, G.S.J. Estrogen stimulates dimethylarginine dimethylaminohydrolase activity and the metabolism of asymmetric dimethylarginine. Circulation 2003, 108, 1575–1580. [Google Scholar] [CrossRef] [PubMed]
- Ueda, S.; Kato, S.; Matsuoka, H.; Kimoto, M.; Okuda, S.; Morimatsu, M.; Imaizumi, T. Regulation of cytokine-induced nitric oxide synthesis by asymmetric dimethylarginine: Role of dimethylarginine dimethylaminohydrolase. Circ. Res. 2003, 92, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Jiang, D.J.; Jia, S.J.; Yan, J.; Zhou, Z.; Yuan, Q.; Li, Y.J. Involvement of DDAH/ADMA/NOS pathway in nicotine-induced endothelial dysfunction. Biochem. Biophys. Res. Commun. 2006, 349, 683–693. [Google Scholar] [CrossRef] [PubMed]
- Staab, E.B.; Weigel, J.; Xiao, F.; Madayiputhiya, N.; Wyatt, T.A.; Wells, S.M. Asymmetric dimethyl-arginine metabolism in a murine model of cigarette smoke-mediated lung inflammation. J. Immunotoxicol. 2015, 12, 273–282. [Google Scholar] [CrossRef] [PubMed]
- Tomikawa, J.; Fukatsu, K.; Tanaka, S.; Shiota, K. DNA methylation-dependent epigenetic regulation of dimethylarginine dimethylaminohydrolase 2 gene in trophoblast cell lineage. J. Biol. Chem. 2006, 281, 12163–12169. [Google Scholar] [CrossRef] [PubMed]
- Nishiyama, Y.; Ueda, M.; Otsuka, T.; Katsura, K.-I.; Abe, A.; Nagayama, H.; Katayama, Y. Statin treatment decreased serum asymmetric dimethylarginine (ADMA) levels in ischemic stroke patients. J. Atheroscler. Thromb. 2011, 18, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Serban, C.; Sahebkar, A.; Ursoniu, S.; Mikhailidis, D.P.; Rizzo, M.; Lip, G.Y.H.; Kees Hovingh, G.; Kastelein, J.J.P.; Kalinowski, L.; Rysz, J.; et al. A systematic review and meta-analysis of the effect of statins on plasma asymmetric dimethylarginine concentrations. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.M.; Ding, Y.A.; Leu, H.B.; Yin, W.H.; Sheu, W.H.; Chu, K.M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am. J. Cardiol. 2004, 94, 157–161. [Google Scholar] [CrossRef] [PubMed]
- Ivashchenko, C.Y.; Bradley, B.T.; Ao, Z.; Leiper, J.; Vallance, P.; Johns, D.G. Regulation of the ADMA-DDAH system in endothelial cells: A novel mechanism for the sterol response element binding proteins, SREBP1c and -2. Am. J. Physiol. Heart Circ. Physiol. 2010, 298, H251–H258. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.; Xia, K.; Zhao, Z.; Deng, X.; Yang, T. Atorvastatin modulates the DDAH1/ADMA system in high-fat diet-induced insulin-resistant rats with endothelial dysfunction. Vasc. Med. 2012, 17, 416–423. [Google Scholar] [CrossRef] [PubMed]
- Yin, Q.-F.; Xiong, Y. Pravastatin restores ddah activity and endothelium-dependent relaxation of rat aorta after exposure to glycated protein. J. Cardiovasc. Pharmacol. 2005, 45, 525–532. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wilson, A.; Gao, X.; Kuruba, R.; Liu, Y.; Poloyac, S.; Pitt, B.; Xie, W.; Li, S. Coordinated regulation of dimethylarginine dimethylaminohydrolase-1 and cationic amino acid transporter-1 by farnesoid x receptor in mouse liver and kidney and its implication in the control of blood levels of asymmetric dimethylarginine. J. Pharmacol. Exp. Ther. 2009, 331, 234–243. [Google Scholar] [CrossRef] [PubMed]
- Ghebremariam, Y.T.; Yamada, K.; Lee, J.C.; Johnson, C.L.C.; Atzler, D.; Anderssohn, M.; Agrawal, R.; Higgins, J.P.; Patterson, A.J.; Böger, R.H.; et al. FXR agonist INT-747 upregulates ddah expression and enhances insulin sensitivity in high-salt fed Dahl rats. PLoS ONE 2013, 8, e60653. [Google Scholar] [CrossRef] [PubMed]
- Vairappan, B.; Sharma, V.; Davies, N.; Jalan, R.; Mookherjee, R. Activation of farnesoid x receptor (FXR) by its agonist INT-747 restores hepatic endothelial nos activity and lowers portal pressure in cirrhotic rats by modulating the DDAH-ADMA pathway. J. Clin. Exp. Hepatol. 2013, 3, S11. [Google Scholar] [CrossRef]
- Jiang, J.L.; Li, N.S.; Li, Y.J.; Deng, H.W. Probucol preserves endothelial function by reduction of the endogenous nitric oxide synthase inhibitor level. Br. J. Pharmacol. 2002, 135, 1175–1182. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.-Q.; Chen, D.; Sun, D.-Q.; Zhang, H.; Li, B.; Fu, Q. Probucol improves erectile function by restoring endothelial function and preventing cavernous fibrosis in streptozotocin-induced diabetic rats. Urology 2016, 91, 241.e9–241.e16. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.-L.; Chen, M.-F.; Luo, B.-L.; Xie, Q.-Y.; Jiang, J.-L.; Li, Y.-J. Fenofibrate decreases asymmetric dimethylarginine level in cultured endothelial cells by inhibiting NF-κB activity. Naunyn-Schmiedeberg's Arch. Pharmacol. 2005, 371, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Wakino, S.; Hayashi, K.; Tatematsu, S.; Hasegawa, K.; Takamatsu, I.; Kanda, T.; Homma, K.; Yoshioka, K.; Sugano, N.; Saruta, T. Pioglitazone lowers systemic asymmetric dimethylarginine by inducing dimethylarginine dimethylaminohydrolase in rats. Hypertens. Res. 2005, 28, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Garbin, U.; Pasini, A.F.; Stranieri, C.; Manfro, S.; Boccioletti, V.; Cominacini, L. Nebivolol reduces asymmetric dimethylarginine in endothelial cells by increasing dimethylarginine dimethylaminohydrolase 2 (DDAH2) expression and activity. Pharmacol. Res. 2007, 56, 515–521. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Zhang, M.; Liu, Y.; Liu, Y.; Chen, M. The effect of nebivolol on asymmetric dimethylarginine system in spontaneously hypertension rats. Vasc. Pharmacol. 2011, 54, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Pasini, A.F.; Garbin, U.; Stranieri, C.; Boccioletti, V.; Mozzini, C.; Manfro, S.; Pasini, A.; Cominacini, M.; Cominacini, L. Nebivolol treatment reduces serum levels of asymmetric dimethylarginine and improves endothelial dysfunction in essential hypertensive patients. Am. J. Hypertens. 2008, 21, 1251–1257. [Google Scholar] [CrossRef] [PubMed]
- Knipp, M.; Braun, O.; Gehrig, P.M.; Sack, R.; Vasak, M. Zn(II)-free dimethylargininase-1 (DDAH-1) is inhibited upon specific Cys-S-nitrosylation. J. Biol. Chem. 2003, 278, 3410–3416. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, N.; Sedoris, K.C.; Steed, M.; Ovechkin, A.V.; Moshal, K.S.; Tyagi, S.C. Mechanisms of homocysteine-induced oxidative stress. Am. J. Physiol. Heart Circ. Physiol. 2005, 289, H2649–H2656. [Google Scholar] [CrossRef] [PubMed]
- Knipp, M.; Braun, O.; Vasak, M. Searching for ddah inhibitors: S-nitroso-l-homocysteine is a chemical lead. J. Am. Chem. Soc. 2005, 127, 2372–2373. [Google Scholar] [CrossRef] [PubMed]
- Duerre, J.A.; Miller, C.H. Preparation of l-homocysteine from l-homocysteine thiolactone. Anal. Biochem. 1966, 17, 310–315. [Google Scholar] [CrossRef]
- Feelisch, M.; Stamler, J. Methods in Nitric Oxide Research. 36. Preparation and Detection of S-Nitrosothiols; John Wiley & Sons Ltd: Hoboken, NJ, USA, 1996; pp. 521–539. [Google Scholar]
- Braun, O.; Knipp, M.; Chesnov, S.; Vasak, M. Specific reactions of S-nitrosothiols with cysteine hydrolases: A comparative study between dimethylargininase-1 and ctp synthetase. Protein Sci. 2007, 16, 1522–1534. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Schaller, T.H.; Bianchi, H.; Person, M.D.; Fast, W. Inactivation of two diverse enzymes in the amidinotransferase superfamily by 2-chloroacetamidine: Dimethylargininase and peptidylarginine deiminase. Biochemistry 2005, 44, 13744–13752. [Google Scholar] [CrossRef] [PubMed]
- Wharton, C.J.; Wrigglesworth, R. Inhibitors of pyrimidine biosynthesis. Part 2. The synthesis of amidine phosphonates as potential inhibitors of carbamoyl phosphate synthase. J. Chem. Soc. Perkin Trans. 1981, 1, 433–436. [Google Scholar] [CrossRef]
- Schaefer, F.C.; Peters, G.A. Base-catalyzed reaction of nitriles with alcohols. A convenient route to imidates and amidine salts. J. Org. Chem. 1961, 26, 412–418. [Google Scholar] [CrossRef]
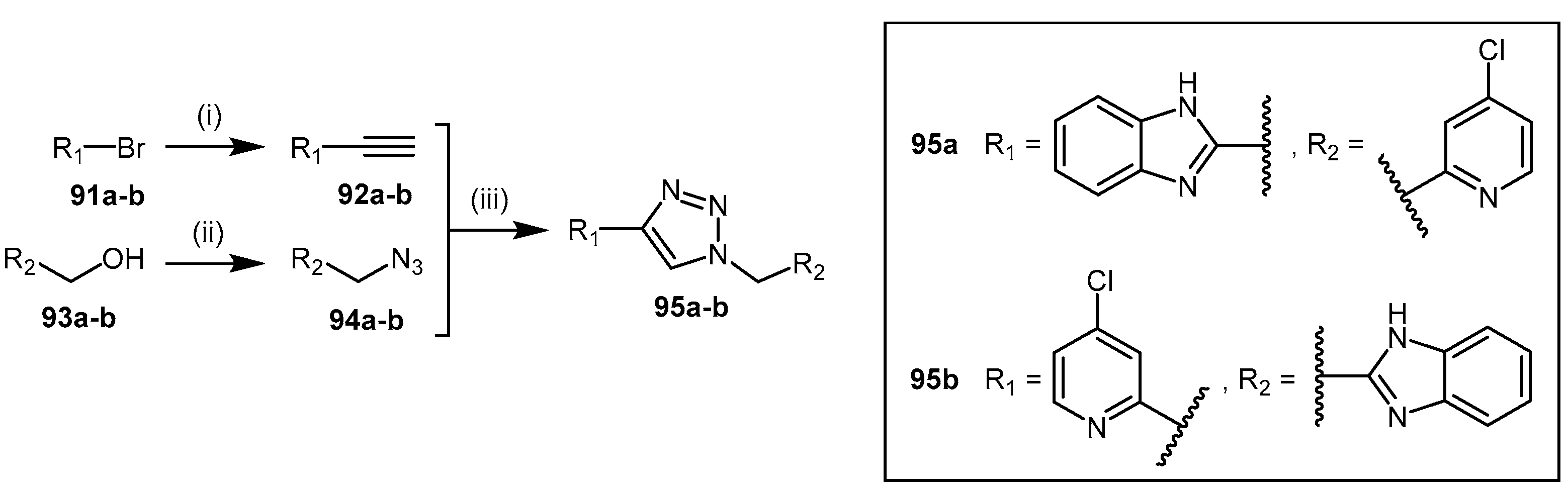
- Wang, Y.; Hu, S.; Fast, W. A click chemistry mediated in vivo activity probe for dimethylarginine dimethylaminohydrolase. J. Am. Chem. Soc. 2009, 131, 15096–15097. [Google Scholar] [CrossRef] [PubMed]
- MacAllister, R.J.; Parry, H.; Kimoto, M.; Ogawa, T.; Russell, R.J.; Hodson, H.; Whitley, G.S.; Vallance, P. Regulation of nitric oxide synthesis by dimethylarginine dimethylaminohydrolase. Br. J. Pharmacol. 1996, 119, 1533–1540. [Google Scholar] [CrossRef] [PubMed]
- Corbin, J.L.; Reporter, M. Ng-methylated arginines; a convenient preparation of ng-methylarginine. Anal. Biochem. 1974, 57, 310–312. [Google Scholar] [CrossRef]
- Rossiter, S.; Smith, C.L.; Malaki, M.; Nandi, M.; Gill, H.; Leiper, J.M.; Vallance, P.; Selwood, D.L. Selective substrate-based inhibitors of mammalian dimethylarginine dimethylaminohydrolase. J. Med. Chem. 2005, 48, 4670–4678. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.-O.; Mathew, F.; Ogbu, C. A convenient synthesis of disubstituted guanidines via the mitsunobu protocol. Synlett 1999, 1999, 193–194. [Google Scholar] [CrossRef]
- Kotthaus, J.; Schade, D.; Muschick, N.; Beitz, E.; Clement, B. Structure-activity relationship of novel and known inhibitors of human dimethylarginine dimethylaminohydrolase-1: Alkenyl-amidines as new leads. Bioorg. Med. Chem. 2008, 16, 10205–10209. [Google Scholar] [CrossRef] [PubMed]
- Kotthaus, J.; Schade, D.; Kotthaus, J.; Clement, B. Designing modulators of dimethylarginine dimethylaminohydrolase (DDAH): A focus on selectivity over arginase. J. Enzyme Inhib. Med. Chem. 2012, 27, 24–28. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Costello, A.L.; Tierney, D.L.; Fast, W. Substrate-assisted cysteine deprotonation in the mechanism of dimethylargininase (DDAH) from pseudomonas aeruginosa. Biochemistry 2006, 45, 5618–5630. [Google Scholar] [CrossRef] [PubMed]
- Tommasi, S.; Zanato, C.; Lewis, B.C.; Nair, P.C.; Dall’Angelo, S.; Zanda, M.; Mangoni, A.A. Arginine analogues incorporating carboxylate bioisosteric functions are micromolar inhibitors of human recombinant DDAH-1. Org. Biomol. Chem. 2015, 13, 11315–11330. [Google Scholar] [CrossRef] [PubMed]
- Lima, L.M.; Barreiro, E.J. Bioisosterism: A useful strategy for molecular modification and drug design. Curr. Med. Chem. 2005, 12, 23–49. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, A.O.; Bhatt, B.M.; Gitu, P.M. Solid-phase peptide synthesis of isotocin with amide of asparagine protected with 1-tetralinyl. Trifluoromethanesulphonic acid (TFMSA) deprotection, cleavage and air oxidation of mercapto groups to disulphide. Bull. Chem. Soc. Ethiop. 2001, 15, 143–149. [Google Scholar]
- Demko, Z.P.; Sharpless, K.B. Preparation of 5-substituted 1 H-tetrazoles from nitriles in water. J. Org. Chem. 2001, 66, 7945–7950. [Google Scholar] [CrossRef] [PubMed]
- Sureshbabu, V.V.; Naik, S.A.; Nagendra, G. Synthesis of boc-amino tetrazoles derived from α-amino acids. Synth. Commun. 2009, 39, 395–406. [Google Scholar] [CrossRef]
- Anderson, D.W.; Campbell, M.M.; Malik, M. Tetrazolyl peptides and the tetrazole analogue of 3-aminonocardicinic acid. Tetrahedron Lett. 1990, 31, 1755–1758. [Google Scholar] [CrossRef]
- Keil, S.; Urmann, M.; Bernardelli, P.; Glien, M.; Wendler, W.; Chandross, K.; Lee, L. Phenyl-1,2,4-Oxadiazolone Derivatives, Processes for Their Preparation and Methods for their Use as Pharmaceuticals. U.S. Patent 7,709,481 B2, 4 May 2010. [Google Scholar]
- Greenhill, J.V.; Lue, P. Amidines and guanidines in medicinal chemistry. Prog. Med. Chem. 1993, 30, 203–326. [Google Scholar] [PubMed]
- Babu, B.R.; Griffith, O.W. N5-(1-imino-3-butenyl)-l-ornithine. A neuronal isoform selective mechanism-based inactivator of nitric oxide synthase. J. Biol. Chem. 1998, 273, 8882–8889. [Google Scholar] [CrossRef] [PubMed]
- Bretscher, L.E.; Li, H.; Poulos, T.L.; Griffith, O.W. Structural characterization and kinetics of nitric-oxide synthase inhibition by novel N5-(iminoalkyl)- and N5-(iminoalkenyl)-ornithines. J. Biol. Chem. 2003, 278, 46789–46797. [Google Scholar] [CrossRef] [PubMed]
- Fast, W.; Nikolic, D.; van Breemen, R.B.; Silverman, R.B. Mechanistic studies of the inactivation of inducible nitric oxide synthase by N5-(1-iminoethyl)-l-ornithine (L-NIO). J. Am. Chem. Soc. 1999, 121, 903–916. [Google Scholar] [CrossRef]
- Wang, Y.; Monzingo, A.F.; Hu, S.; Schaller, T.H.; Robertus, J.D.; Fast, W. Developing dual and specific inhibitors of dimethylarginine dimethylaminohydrolase-1 and nitric oxide synthase: Toward a targeted polypharmacology to control nitric oxide. Biochemistry 2009, 48, 8624–8635. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Hu, S.; Gabisi, A.M., Jr.; Er, J.A.; Pope, A.; Burstein, G.; Schardon, C.L.; Cardounel, A.J.; Ekmekcioglu, S.; Fast, W. Developing an irreversible inhibitor of human DDAH-1, an enzyme upregulated in melanoma. ChemMedChem 2014, 9, 792–797. [Google Scholar] [CrossRef] [PubMed]
- Vallance, P.; Bush, H.D.; Mok, B.J.; Hurtado-Guerrero, R.; Gill, H.; Rossiter, S.; Wilden, J.D.; Caddick, S. Inhibition of dimethylarginine dimethylaminohydrolase (DDAH) and arginine deiminase (ADI) by pentafluorophenyl (PFP) sulfonates. Chem. Commun. 2005, 5563–5565. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Bush, H.D. Synthesis of functionalized sulfonamides via 1,3-dipolar cycloaddition of pentafluorophenyl vinylsulfonate. Org. Lett. 2003, 5, 2489–2492. [Google Scholar] [CrossRef] [PubMed]
- Caddick, S.; Wilden, J.D.; Bush, H.D.; Wadman, S.N.; Judd, D.B. A new route to sulfonamides via intermolecular radical addition to pentafluorophenyl vinylsulfonate and subsequent aminolysis. Org. Lett. 2002, 4, 2549–2551. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.S.; Yeung, M.L.; Chan, W.-K.; Wang, R.-J.; Mak, T.C.W. Chromium and tungsten pentacarbonyl groups as reactivity and selectivity auxiliaries in [3 + 2] cycloaddition of alkynyl fischer carbene complexes with n-alkyl nitrones. J. Org. Chem. 1995, 60, 1741–1747. [Google Scholar] [CrossRef]
- Baruah, A.K.; Prajapati, D.; Sandhu, J. Some novel aspects of regioselectivity in 1,3 dipolar cycloadditions of 4h-1-benzopyran-4-thione. Tetrahedron 1988, 44, 6137–6142. [Google Scholar] [CrossRef]
- Sims, J.; Houk, K.N. Reversal of nitrone cycloaddition regioselectivity with electron-deficient dipolarophiles. J. Am. Chem. Soc. 1973, 95, 5798–5800. [Google Scholar] [CrossRef]
- Bimanand, A.Z.; Houk, K.N. The synthesis and cycloadditions of c-cyclopropyl nitrones. Tetrahedron Lett. 1983, 24, 435–438. [Google Scholar] [CrossRef]
- Padwa, A.; Fisera, L.; Koehler, K.F.; Rodriguez, A.; Wong, G.S.K. Regioselectivity associated with the 1,3-dipolar cycloaddition of nitrones with electron-deficient dipolarophiles. J. Org. Chem. 1984, 49, 276–281. [Google Scholar] [CrossRef]
- Black, D.S.C.; Crozier, R.F.; Davis, V.C. 1,3-Dipolar cycloaddition reactions of nitrones. Synthesis 1975, 1975, 205–221. [Google Scholar] [CrossRef]
- Hartzoulakis, B.; Rossiter, S.; Gill, H.; O'Hara, B.; Steinke, E.; Gane, P.J.; Hurtado-Guerrero, R.; Leiper, J.M.; Vallance, P.; Rust, J.M.; et al. Discovery of inhibitors of the pentein superfamily protein dimethylarginine dimethylaminohydrolase (DDAH), by virtual screening and hit analysis. Bioorg. Med. Chem. Lett. 2007, 17, 3953–3956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fischer, E.; von Mering, J. Ueber eine neue klasse von schlafmitteln. Ther. Ggw. 1903, 44, 97–101. [Google Scholar]
- Nightingale, D.; Alexander, C.H. Some nitrogen substituted barbituric acids and their derivatives. J. Am. Chem. Soc. 1936, 58, 794–796. [Google Scholar] [CrossRef]
- Zhao, R.; Wang, B.; Wu, H.; Hynes, J., Jr.; Leftheri, K.; Balasubramanian, B.; Barrish, J.C.; Chen, B.-C. Improved procedure for the preparation of 7-methoxy-2-methyl-1-(2-morpholinoethyl)-1H-indole-3-carboxylic acid, key intermediate in the synthesis of novel 3-amidoindole and indolopyridone cannabinoid ligands. Arkivoc 2009, 2010, 89–95. [Google Scholar]
- Dox, A.W.; Plaisance, G.P. Condensation of thiobarbituric acid with aromatic aldehydes. J. Am. Chem. Soc. 1916, 38, 2164–2166. [Google Scholar] [CrossRef]
- Azad, G.K.; Tomar, R.S. Ebselen, a promising antioxidant drug: Mechanisms of action and targets of biological pathways. Mol. Biol. Rep. 2014, 41, 4865–4879. [Google Scholar] [CrossRef] [PubMed]
- Linsky, T.; Wang, Y.; Fast, W. Screening for dimethylarginine dimethylaminohydrolase inhibitors reveals ebselen as a bioavailable inactivator. ACS Med. Chem. Lett. 2011, 2, 592–596. [Google Scholar] [CrossRef] [PubMed]
- Bhabak, K.P.; Mugesh, G. Synthesis, characterization, and antioxidant activity of some ebselen analogues. Chem. Eur. J. 2007, 13, 4594–4601. [Google Scholar] [CrossRef] [PubMed]
- Engman, L.; Hallberg, A. Expedient synthesis of ebselen and related compounds. J. Org. Chem. 1989, 54, 2964–2966. [Google Scholar] [CrossRef]
- Chang, T.C.; Huang, M.L.; Hsu, W.L.; Hwang, J.M.; Hsu, L.Y. Synthesis and biological evaluation of ebselen and its acyclic derivatives. Chem. Pharm. Bull. 2003, 51, 1413–1416. [Google Scholar] [CrossRef] [PubMed]
- Schewe, T. Molecular actions of ebselen—An antiinflammatory antioxidant. Gen. Pharmacol. Vasc. Syst. 1995, 26, 1153–1169. [Google Scholar] [CrossRef]
- Smith, S.M.; Min, J.; Ganesh, T.; Diebold, B.; Kawahara, T.; Zhu, Y.; McCoy, J.; Sun, A.; Snyder, J.P.; Fu, H.; et al. Ebselen and congeners inhibit nadph oxidase 2-dependent superoxide generation by interrupting the binding of regulatory subunits. Chem. Biol. 2012, 19, 752–763. [Google Scholar] [CrossRef] [PubMed]
- Cotgreave, I.A.; Duddy, S.K.; Kass, G.E.N.; Thompson, D.; Moldéus, P. Studies on the anti-inflammatory activity of Ebselen: Ebselen interferes with granulocyte oxidative burst by dual inhibition of NADPH oxidase and protein kinase C? Biochem. Pharmacol. 1989, 38, 649–656. [Google Scholar] [CrossRef]
- Beil, W.; Staar, U.; Sewing, K.-F. Interaction of the anti-inflammatory seleno-organic compound ebselen with acid secretion in isolated parietal cells and gastric H+/K+-atpase. Biochem. Pharmacol. 1990, 40, 1997–2003. [Google Scholar] [CrossRef]
- Walther, M.; Holzhutter, H.G.; Kuban, R.J.; Wiesner, R.; Rathmann, J.; Kuhn, H. The inhibition of mammalian 15-lipoxygenases by the anti-inflammatory drug ebselen: Dual-type mechanism involving covalent linkage and alteration of the iron ligand sphere. Mol. Pharmacol. 1999, 56, 196–203. [Google Scholar] [PubMed]
- Zhao, R.; Masayasu, H.; Holmgren, A. Ebselen: A substrate for human thioredoxin reductase strongly stimulating its hydroperoxide reductase activity and a superfast thioredoxin oxidant. Proc. Natl. Acad. Sci. USA 2002, 99, 8579–8584. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Maret, W.; Vallee, B.L. Ebselen, a selenium-containing redox drug, releases zinc from metallothionein. Biochem. Biophys. Res. Commun. 1998, 248, 569–573. [Google Scholar] [CrossRef] [PubMed]
- Caeran Bueno, D.; Meinerz, D.F.; Allebrandt, J.; Waczuk, E.P.; dos Santos, D.B.; Mariano, D.O.; Rocha, J.B. Cytotoxicity and genotoxicity evaluation of organochalcogens in human leucocytes: A comparative study between ebselen, diphenyl diselenide, and diphenyl ditelluride. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Meotti, F.C.; Borges, V.C.; Zeni, G.; Rocha, J.B.T.; Nogueira, C.W. Potential renal and hepatic toxicity of diphenyl diselenide, diphenyl ditelluride and ebselen for rats and mice. Toxicol. Lett. 2003, 143, 9–16. [Google Scholar] [CrossRef]
- Usatyuk, P.V.; Parinandi, N.L.; Natarajan, V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J. Biol. Chem. 2006, 281, 35554–35566. [Google Scholar] [CrossRef] [PubMed]
- Benedetti, A.; Comporti, M.; Esterbauer, H. Identification of 4-hydroxynonenal as a cytotoxic product originating from the peroxidation of liver microsomal lipids. Biochim. Biophys. Acta Lipids Lipid Metab. 1980, 620, 281–296. [Google Scholar] [CrossRef]
- Uchida, K.; Stadtman, E.R. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA 1992, 89, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Uchida, K. Role of reactive aldehyde in cardiovascular diseases. Free Radic. Biol. Med. 2000, 28, 1685–1696. [Google Scholar] [CrossRef]
- Uchida, K.; Stadtman, E.R. Selective cleavage of thioether linkage in proteins modified with 4-hydroxynonenal. Proc. Natl. Acad. Sci. USA 1992, 89, 5611–5615. [Google Scholar] [CrossRef] [PubMed]
- Sayre, L.M.; Arora, P.K.; Iyer, R.S.; Salomon, R.G. Pyrrole formation from 4-hydroxynonenal and primary amines. Chem. Res. Toxicol. 1993, 6, 19–22. [Google Scholar] [CrossRef] [PubMed]
- Santaniello, E.; Repetto, A.; Chiesa, L.M.; Biondi, P.A. Synthesis and characterization of 4-hydroxy-2-nonenal derivatives for gas chromatographic analysis with electron capture detection (GC-ECD). Redox Rep. Commun. Free Radic. Res. 2007, 12, 55–58. [Google Scholar] [CrossRef] [PubMed]
- Chandra, A.; Srivastava, S.K. A synthesis of 4-hydroxy-2-trans-nonenal and 4-(3H) 4-hydroxy-2-trans-nonenal. Lipids 1997, 32, 779–782. [Google Scholar] [CrossRef] [PubMed]
- Soulère, L.; Queneau, Y.; Doutheau, A. An expeditious synthesis of 4-hydroxy-2(E)-nonenal (4-HNE), its dimethyl acetal and of related compounds. Chem. Phys. Lipids 2007, 150, 239–243. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.P.; Druhan, L.J.; Guzman, J.E.; Parinandi, N.; Zhang, L.; Green-Church, K.B.; Cardounel, A.J. Mechanism of 4-HNE mediated inhibition of hddah-1: Implications in no regulation. Biochemistry 2008, 47, 1819–1826. [Google Scholar] [CrossRef] [PubMed]
- Birck, M.R.; Holler, T.P.; Woodard, R.W. Identification of a slow tight-binding inhibitor of 3-deoxy-d-manno-octulosonic acid 8-phosphate synthase. J. Am. Chem. Soc. 2000, 122, 9334–9335. [Google Scholar] [CrossRef]
- Kosa, N.M.; Foley, T.L.; Burkart, M.D. Fluorescent techniques for discovery and characterization of phosphopantetheinyl transferase inhibitors. J. Antibiot. 2014, 67, 113–120. [Google Scholar] [CrossRef] [PubMed]
- Chamoun, A.M.; Chockalingam, K.; Bobardt, M.; Simeon, R.; Chang, J.; Gallay, P.; Chen, Z. Pd 404,182 is a virocidal small molecule that disrupts hepatitis c virus and human immunodeficiency virus. Antimicrob. Agents Chemother. 2012, 56, 672–681. [Google Scholar] [CrossRef] [PubMed]
- Chamoun-Emanuelli, A.M.; Bobardt, M.; Moncla, B.; Mankowski, M.K.; Ptak, R.G.; Gallay, P.; Chen, Z. Evaluation of PD 404,182 as an anti-HIV and anti-Herpes simplex virus microbicide. Antimicrob. Agents Chemother. 2014, 58, 687–697. [Google Scholar] [CrossRef] [PubMed]
- Hagen, H.; Fleig, H. Neue synthesen mit elementarem schwefel. Darstellung von 2-(1,3-diaza-2-cycloalken-2-yl)thiophenolen und ihre umwandlung zu 1,4-benzothiazepinen und 1,3,4-benzo-thiadiazepinen. Justus Liebigs Ann. Chem. 1975, 1975, 1994–2005. [Google Scholar] [CrossRef]
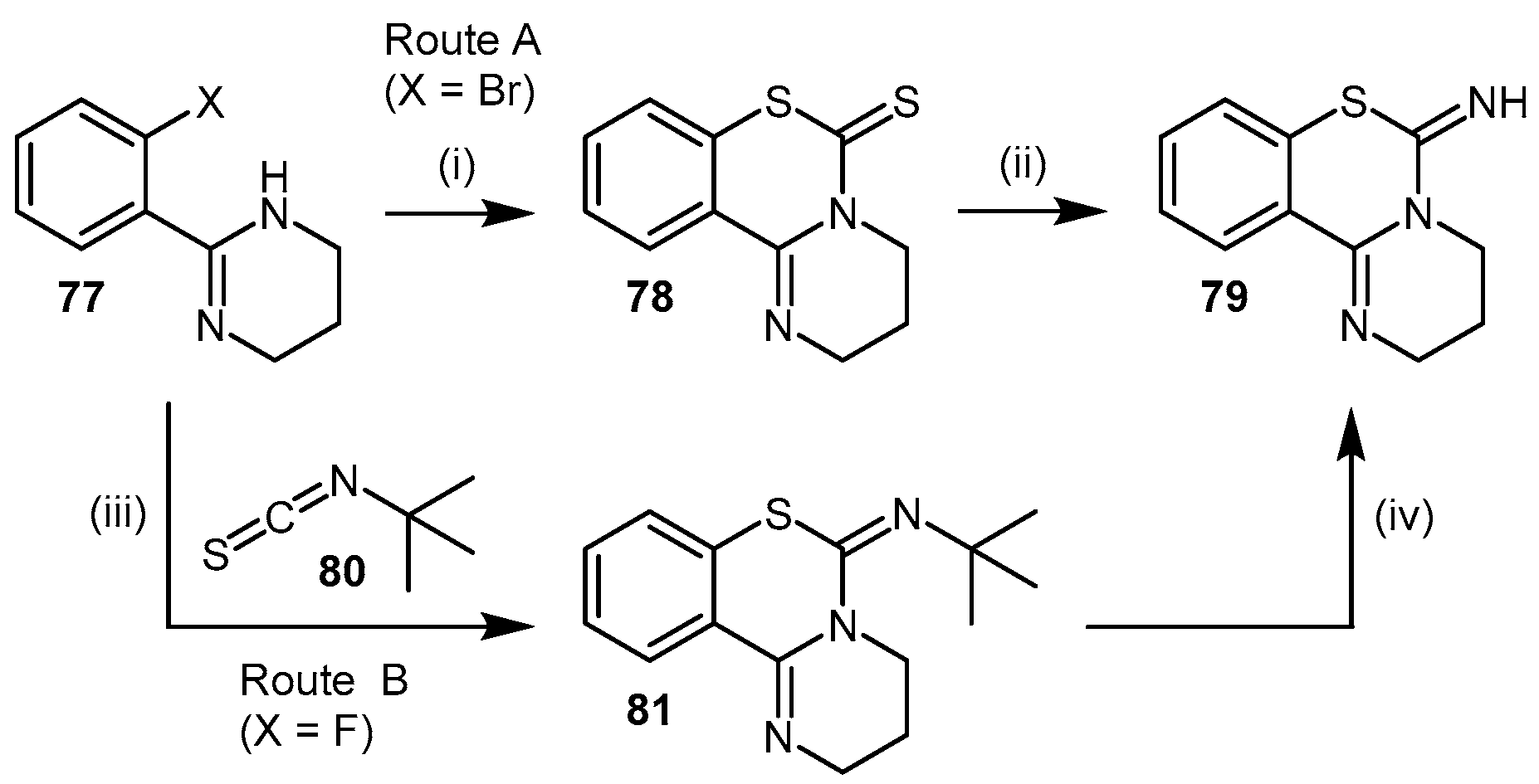
- Mizuhara, T.; Oishi, S.; Fujii, N.; Ohno, H. Efficient synthesis of pyrimido[1,2-c] [1,3]benzothiazin-6-imines and related tricyclic heterocycles by SNAr-type C-S, C-N, or C-O bond formation with heterocumulenes. J. Org. Chem. 2010, 75, 265–268. [Google Scholar] [CrossRef] [PubMed]
- Ghebremariam, Y.T.; Erlanson, D.A.; Yamada, K.; Cooke, J.P. Development of a dimethylarginine dimethylaminohydrolase (DDAH) assay for high-throughput chemical screening. J. Biomol. Screen. 2012, 17, 651–661. [Google Scholar] [CrossRef] [PubMed]
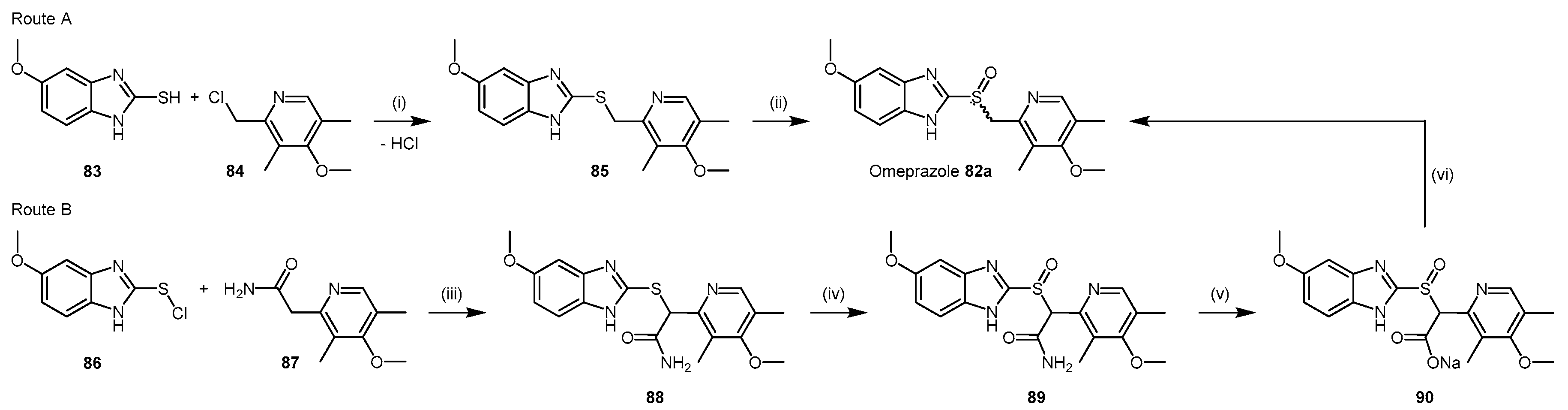
- Ghebremariam, Y.T.; LePendu, P.; Lee, J.C.; Erlanson, D.A.; Slaviero, A.; Shah, N.H.; Leiper, J.; Cooke, J.P. Unexpected effect of proton pump inhibitors: Elevation of the cardiovascular risk factor asymmetric dimethylarginine. Circulation 2013, 128, 845–853. [Google Scholar] [CrossRef] [PubMed]
- Charlot, M.; Grove, E.L.; Hansen, P.R.; Olesen, J.B.; Ahlehoff, O.; Selmer, C.; Lindhardsen, J.; Madsen, J.K.; Kober, L.; Torp-Pedersen, C.; et al. Proton pump inhibitor use and risk of adverse cardiovascular events in aspirin treated patients with first time myocardial infarction: Nationwide propensity score matched study. Br. Med. J. 2011, 342. [Google Scholar] [CrossRef] [PubMed]
- Charlot, M.; Ahlehoff, O.; Norgaard, M.L.; Jorgensen, C.H.; Sorensen, R.; Abildstrom, S.Z.; Hansen, P.R.; Madsen, J.K.; Kober, L.; Torp-Pedersen, C.; et al. Proton-pump inhibitors are associated with increased cardiovascular risk independent of clopidogrel use: A nationwide cohort study. Ann. Intern. Med. 2010, 153, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Brandstrom, A.E.; Lamm, B.R. Processes for the Preparation of Omeprazole and Intermediates Therefore. U.S. Patent 4,620,008 A, 28 October 1986. [Google Scholar]
- Crowe, A.M.; Ife, R.J.; Mitchell, M.B.; Saunders, D. The preparation of 14C, 35S and 13C labelled forms of omeprazole. J. Label. Compd. Radiopharm. 1986, 23, 21–33. [Google Scholar] [CrossRef]
- Slemon, C.; Macel, B. Preparation of Omeprazole and Lansoprazole, and Intermediates Useful Therein. U.S. Patent No. 5,470,983, 28 November 1995. [Google Scholar]
- Linsky, T.W.; Fast, W. Discovery of structurally-diverse inhibitor scaffolds by high-throughput screening of a fragment library with dimethylarginine dimethylaminohydrolase. Bioorg. Med. Chem. 2012, 20, 5550–5558. [Google Scholar] [CrossRef] [PubMed]
- Zornik, D.; Meudtner, R.M.; El Malah, T.; Thiele, C.M.; Hecht, S. Designing structural motifs for clickamers: Exploiting the 1,2,3-triazole moiety to generate conformationally restricted molecular architectures. Chem. Eur. J. 2011, 17, 1473–1484. [Google Scholar] [CrossRef] [PubMed]
- Gijsen, H.J.M.; Cleyn, M.A.J.D.; Surkyn, M.; Verbist, B.M.P. Benzimidazole Cannabinoid Agonists Bearinga Substituted Heterocyclic Group. U.S. Patent 8,524,757, 3 September 2013. [Google Scholar]
- Thompson, A.S.; Humphrey, G.R.; DeMarco, A.M.; Mathre, D.J.; Grabowski, E.J.J. Direct conversion of activated alcohols to azides using diphenyl phosphorazidate. A practical alternative to mitsunobu conditions. J. Org. Chem. 1993, 58, 5886–5888. [Google Scholar] [CrossRef]





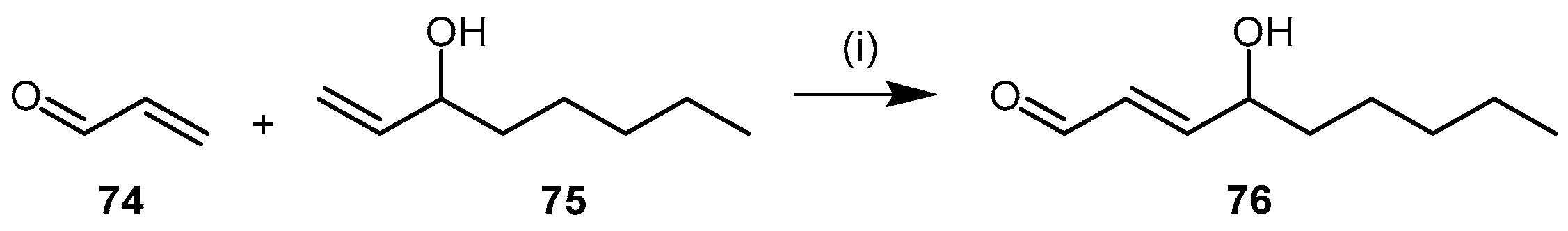
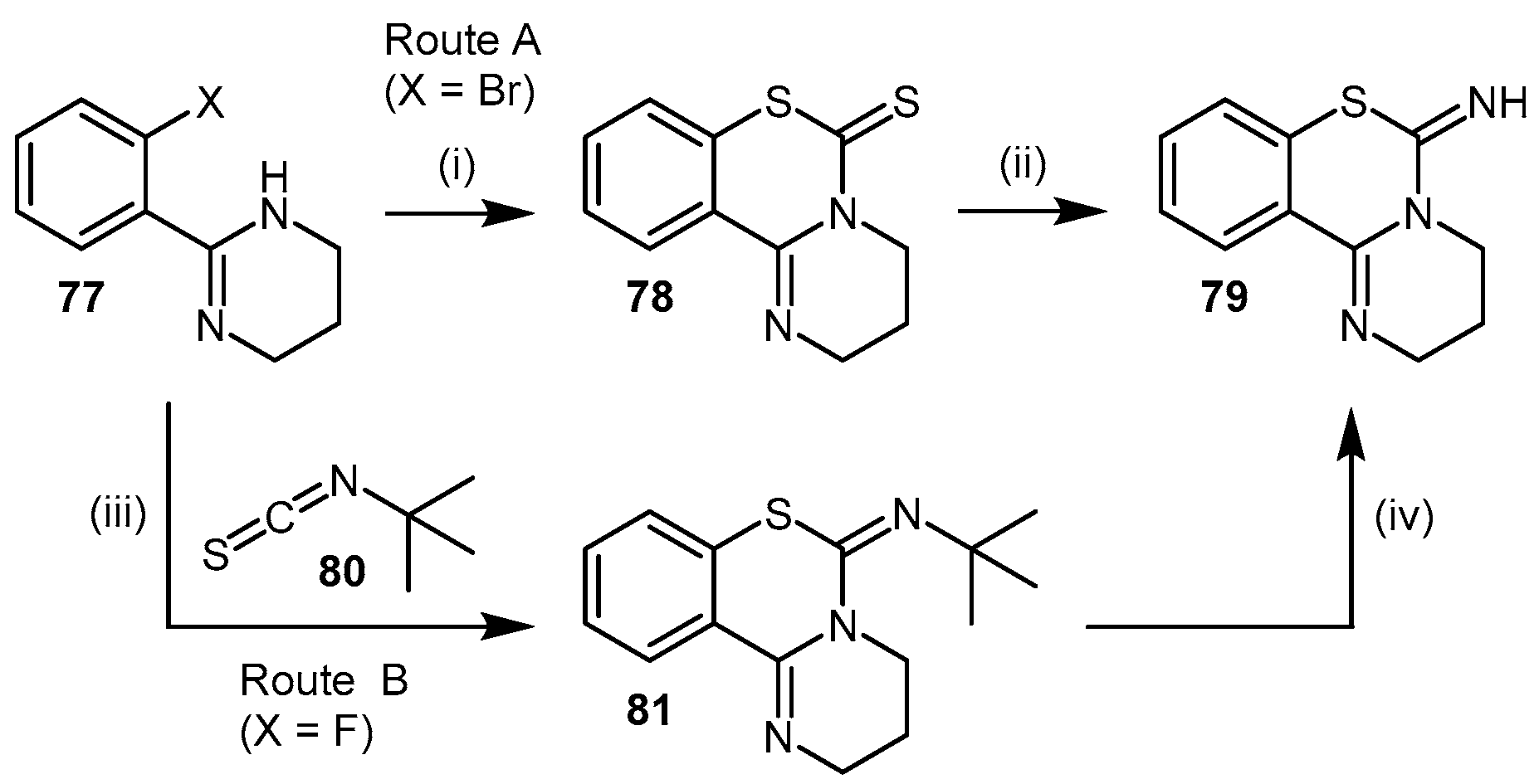
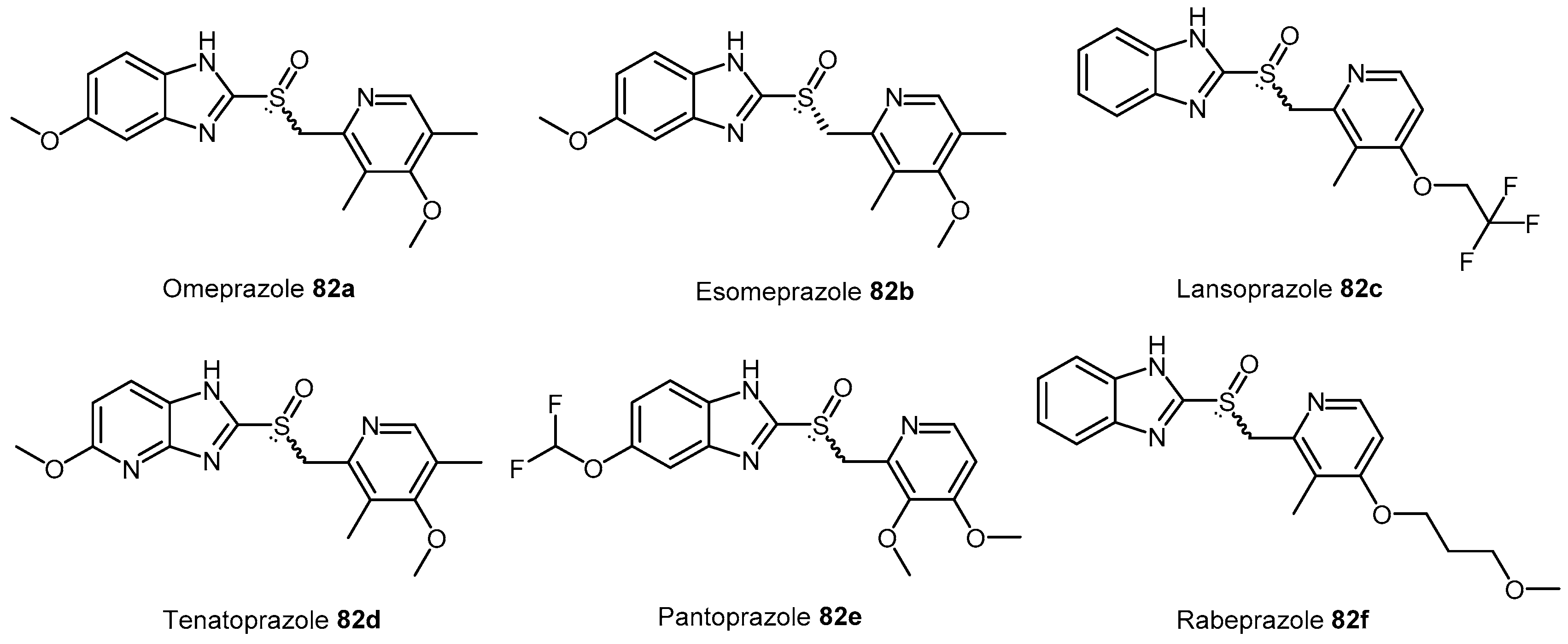
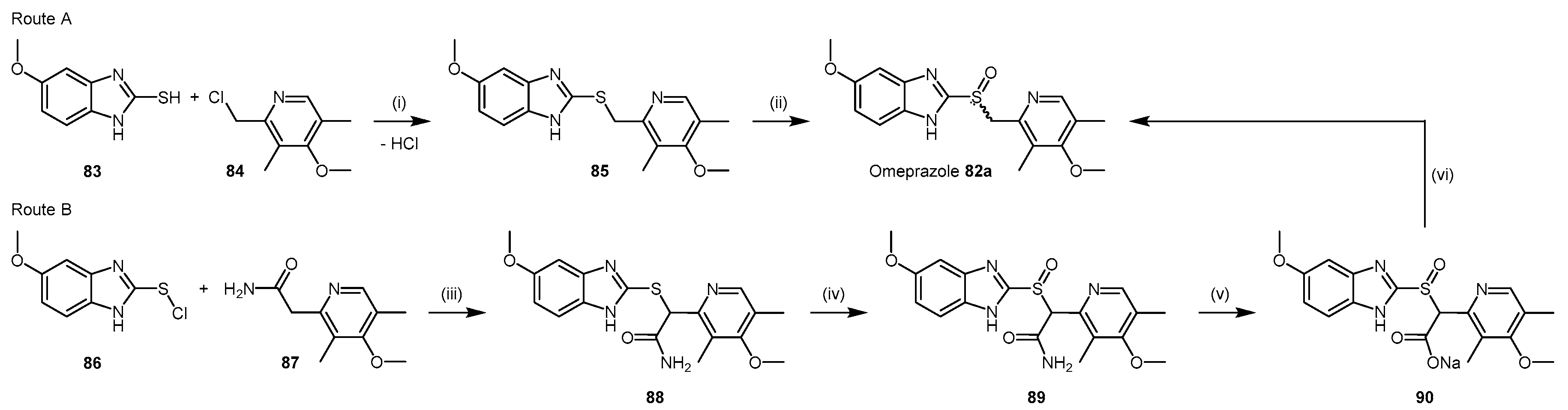

| Enzyme | Ki (mM) | Kinact (min−1) |
|---|---|---|
| PaDDAH | 3.1 ± 0.8 | 1.2 ± 0.1 |
| PAD4 | 20 ± 5 | 0.7 ± 0.1 |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | R– | Inhibition at 1 mM (%) | IC50 (μM) | Ki (μM) |
|---|---|---|---|---|
| 1 1 | * CH3OCH2CH2– | 83 | 31 | 13 |
| 2 1 | CH3NHCH2CH2– | 29 | - | - |
| 3 1 | (CH3)2NHCH2CH2– | 14 | - | - |
| 4 1 | CH3SHCH2CH2– | 80 | 408 | - |
| 5 1 | FCH2CH2– | 67 | 379 | - |
| 6 1 | BnCH2– | 55 | 866 | - |
| 7 2 | CH3CH2CH2– | 60 | 283 | 90 |
| 8 2 | CH2CHCH2– | 70 | 207 | 58 |
| 9 2 | CH2CHCH2CH2– | 71 | 189 | 57 |
| 10 2 | CHCCH2– | 83 | 55 | 17 |
| 11 2 | F3CCH2– | 41 | - | - |
| 12 2 | O2N– | 6 | - | - |
| 13 2 | NH2COCH2CH2– | 43 | - | - |
| Chemical Number | Chemical Name | Chemical Structure | Enzyme | IC50 (µM) | Ki (µM) | Kinact (min−1) | Reference |
|---|---|---|---|---|---|---|---|
| (9) | S-Nitroso-l-homocysteine (HcyNO) |  | Bovine DDAH-1 | 75 | 690 | 0.38 | [105] |
| (11) | 2-Chloroacetamidine |  | PaDDAH 1 | - | 3100 | 1.2 | [106] |
| (18) | N-But-3-ynyl-2-chloro-acetamidine |  | Human DDAH-1 | 350 | - | - | [109] |
| (21) | (S)-2-Amino-4-(3-methylguanidino)butanoic acid (4124W) |  | Rat DDAH-1 | 416 | - | - | [110] |
| (39) | NG-(2-Methoxyethyl)-l-arginine (L-257) |  | Human DDAH-1 | 31 2 | 13 2 | - | [112] |
| (40) | NG-(2-Methoxyethyl)-l-arginine methyl ester (L-291) |  | Rat DDAH-1 | 20 | - | - | [112] |
| (44a) | 2-Amino-5-(3-(2-methoxyethyl)guanidino)-N-(methylsulfonyl)-pentanamide (ZST316) |  | Human DDAH-1 | 3 | 1 | - | [117] |
| (54a) | N5-(1-Imino-3-butenyl)-l-ornithine (L-VNIO) |  | Human DDAH-1 | 13 | 2 | - | [114] |
| (54b) | N5-(1-Iminopropyl)-l-ornithine (L-IPO) |  | Human DDAH-1 | - | 52 | - | [128] |
| (54c) | N5-(1-Imino-2-chloroethyl)-l-ornithine (Cl-NIO) |  | Human DDAH-1 | - | 1.3 | 0.34 | [129] |
| 61 | Pentafluorophenyl (PFP) sulfonate esters |  | PaDDAH 1 | 16–58 | - | - | [130] |
| 68a–c | Indolylthiobarbituric acid derivatives |  | PaDDAH 1 | 2–17 | - | - | [139] |
| (73) | Ebselen |  | Human DDAH-1 | 0.33 | - | - | [145] |
| (76) | 4-Hydroxy-2-nonenal (4-HNE) |  | Human DDAH-1 | 50 | - | - | [167] |
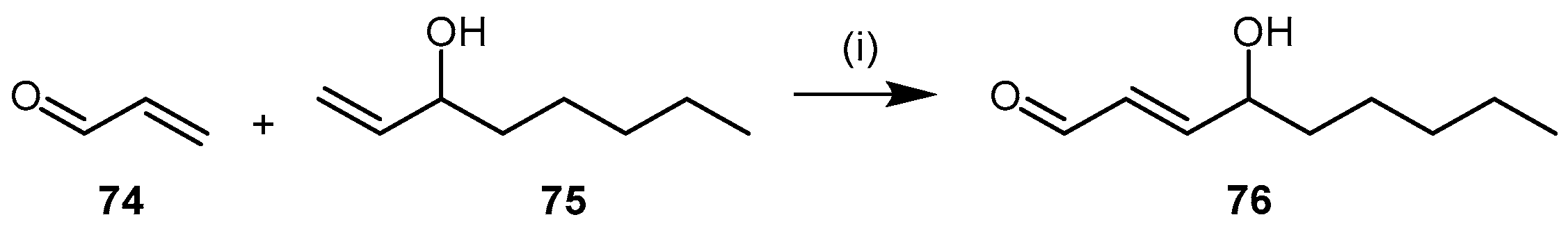
| (79) | 6H-6-Imino-(2,3,4,5-tetrahydropyrimido)[1,2-c]-[1,3]benzothiazine (PD 404182) |  | Human DDAH-1 | 9 | - | - | [61] |
| 82a–f | Proton H+/K+ ATPase pump inhibitors (PPIs) |  | Human DDAH-1 | 51–63 | - | - | [175] |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Murphy, R.B.; Tommasi, S.; Lewis, B.C.; Mangoni, A.A. Inhibitors of the Hydrolytic Enzyme Dimethylarginine Dimethylaminohydrolase (DDAH): Discovery, Synthesis and Development. Molecules 2016, 21, 615. https://doi.org/10.3390/molecules21050615
Murphy RB, Tommasi S, Lewis BC, Mangoni AA. Inhibitors of the Hydrolytic Enzyme Dimethylarginine Dimethylaminohydrolase (DDAH): Discovery, Synthesis and Development. Molecules. 2016; 21(5):615. https://doi.org/10.3390/molecules21050615
Chicago/Turabian StyleMurphy, Rhys B., Sara Tommasi, Benjamin C. Lewis, and Arduino A. Mangoni. 2016. "Inhibitors of the Hydrolytic Enzyme Dimethylarginine Dimethylaminohydrolase (DDAH): Discovery, Synthesis and Development" Molecules 21, no. 5: 615. https://doi.org/10.3390/molecules21050615