
Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage

 ,
,  ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion
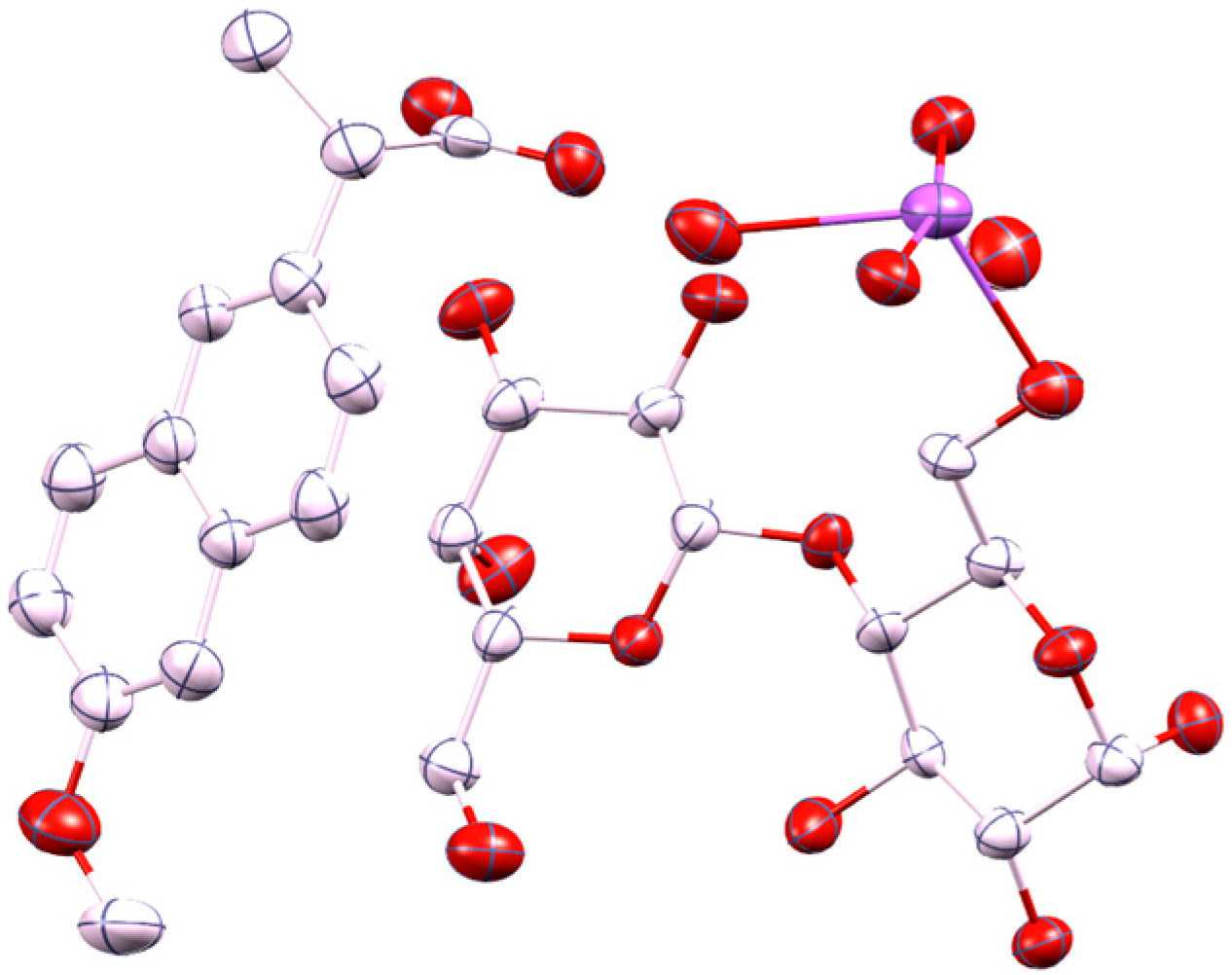
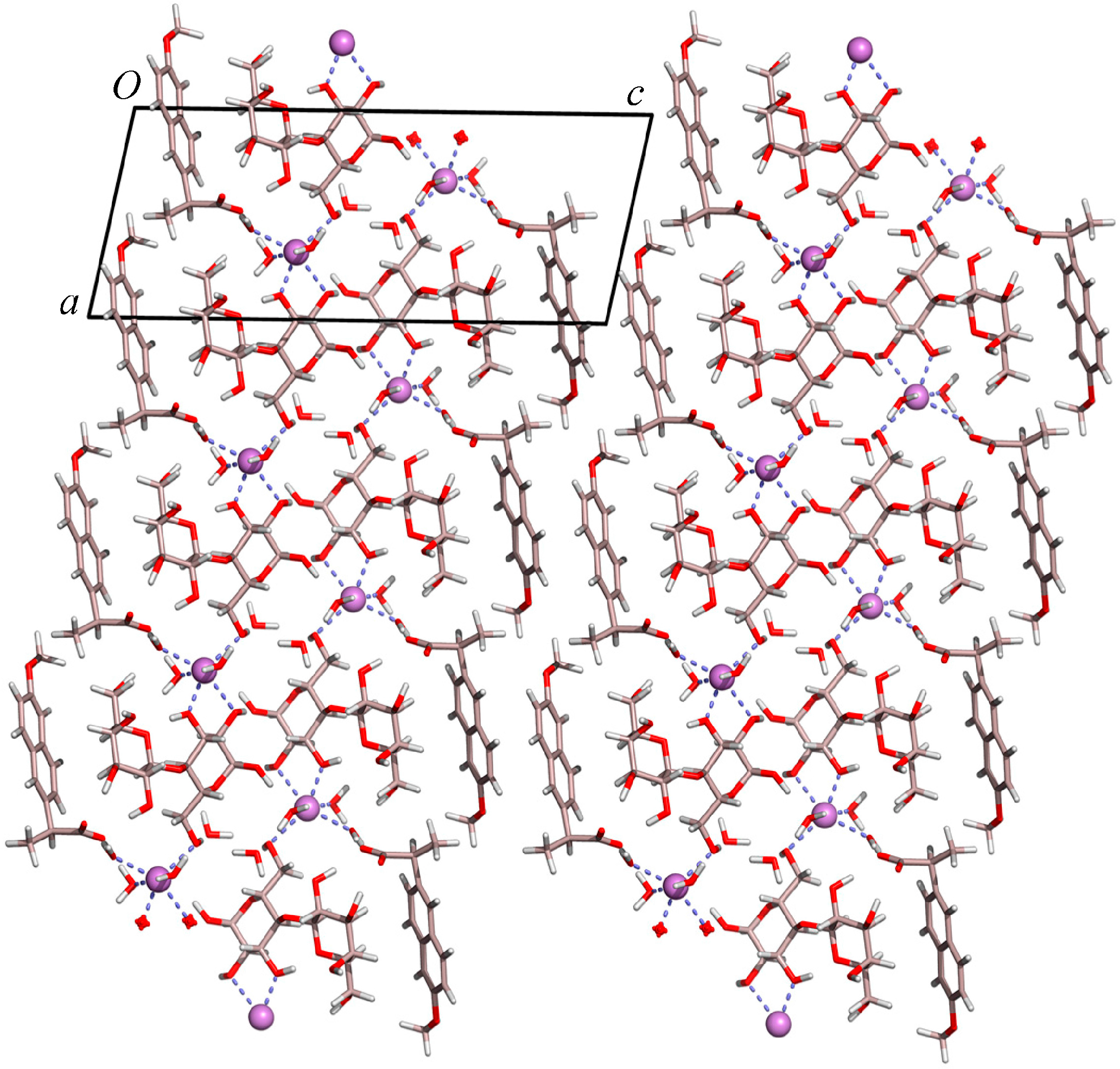
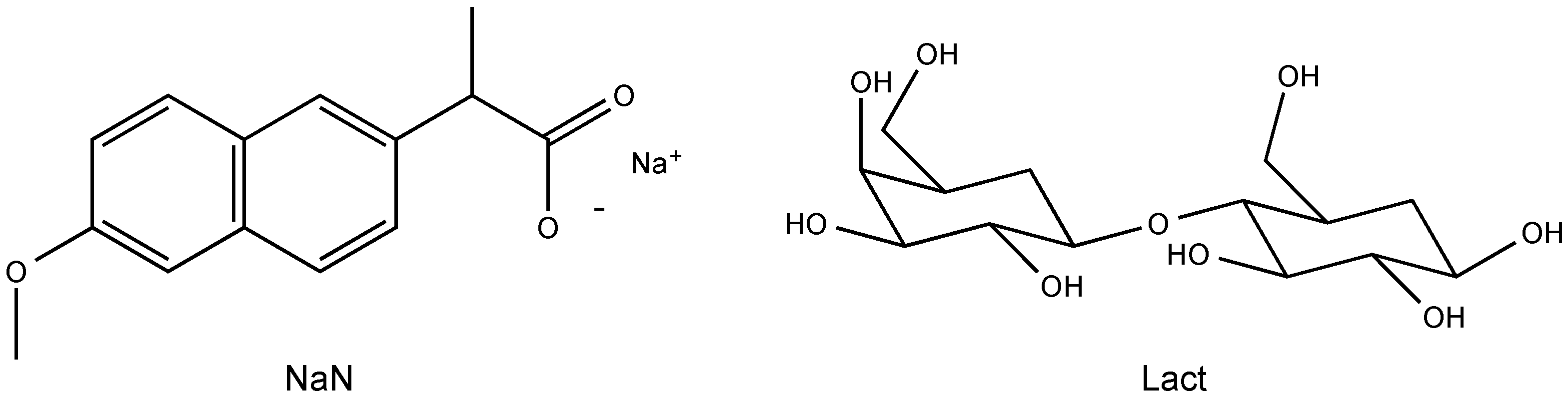
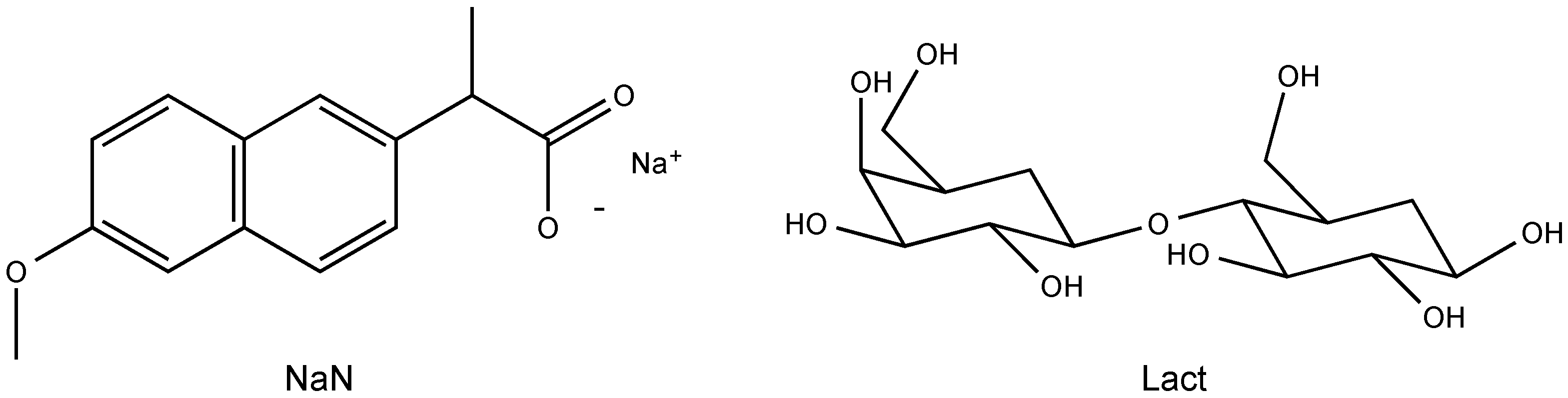
2.1. Molecular Structure of the Na-Naproxen Lactose Tetrahydrate (NaN-Lact-4H2O) Co-Crystal
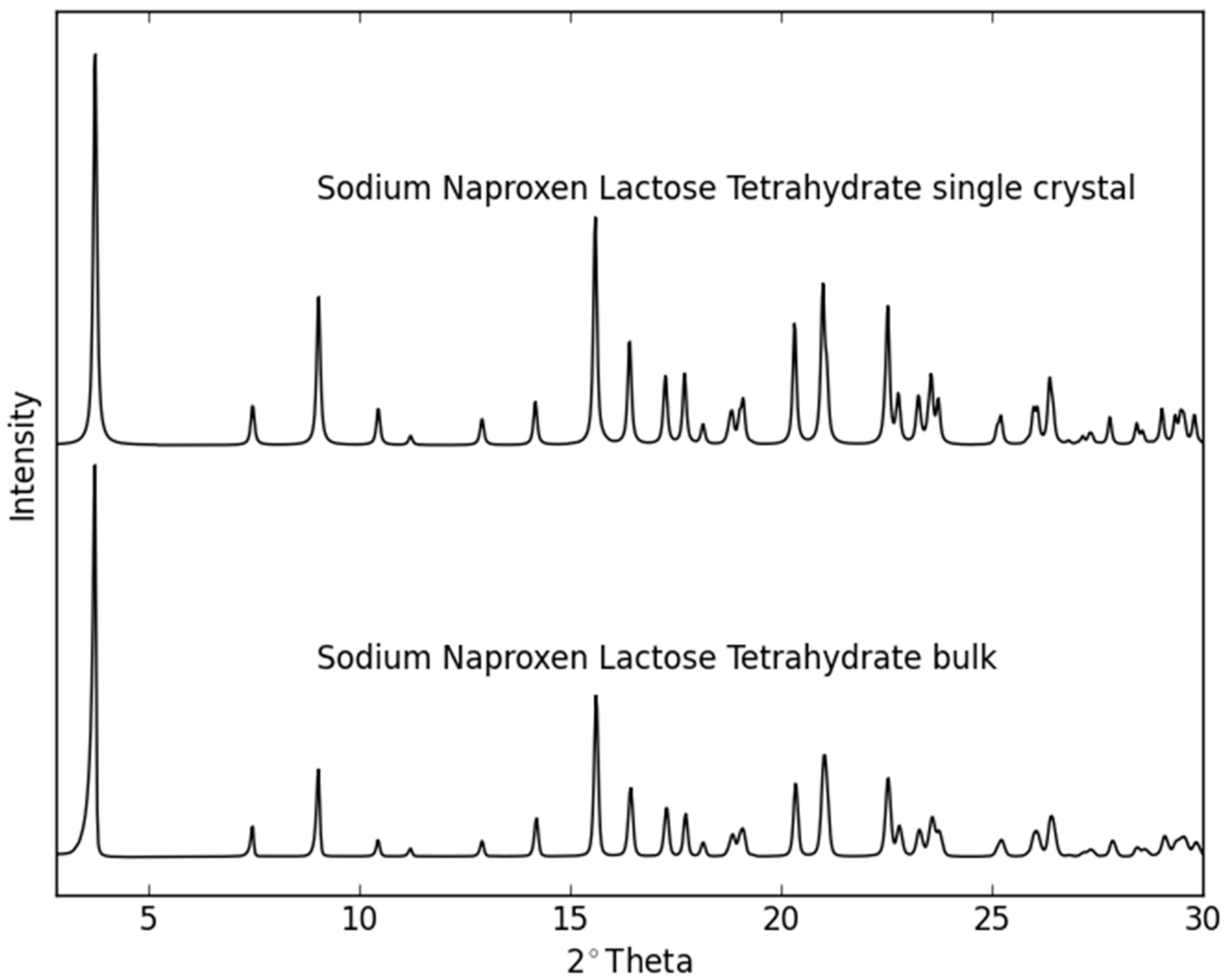
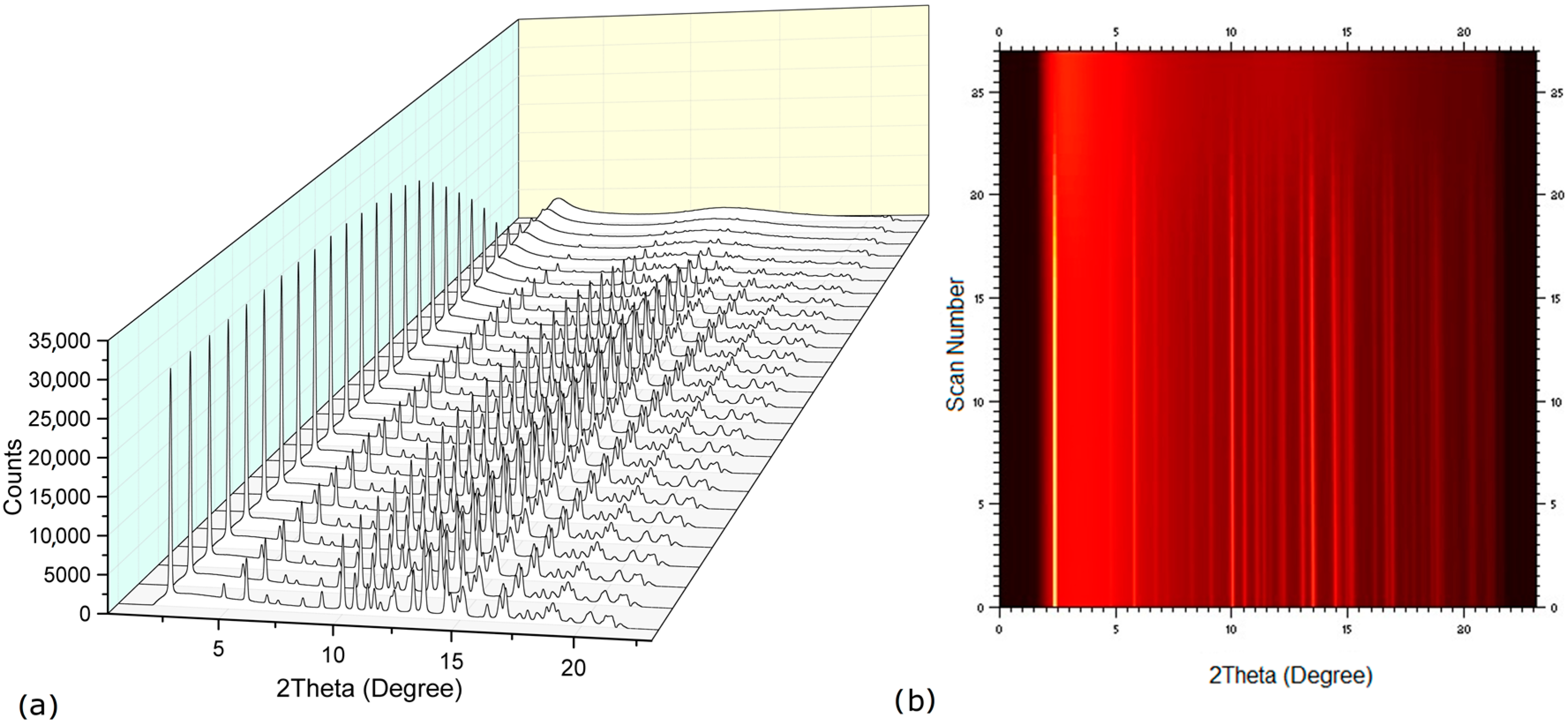
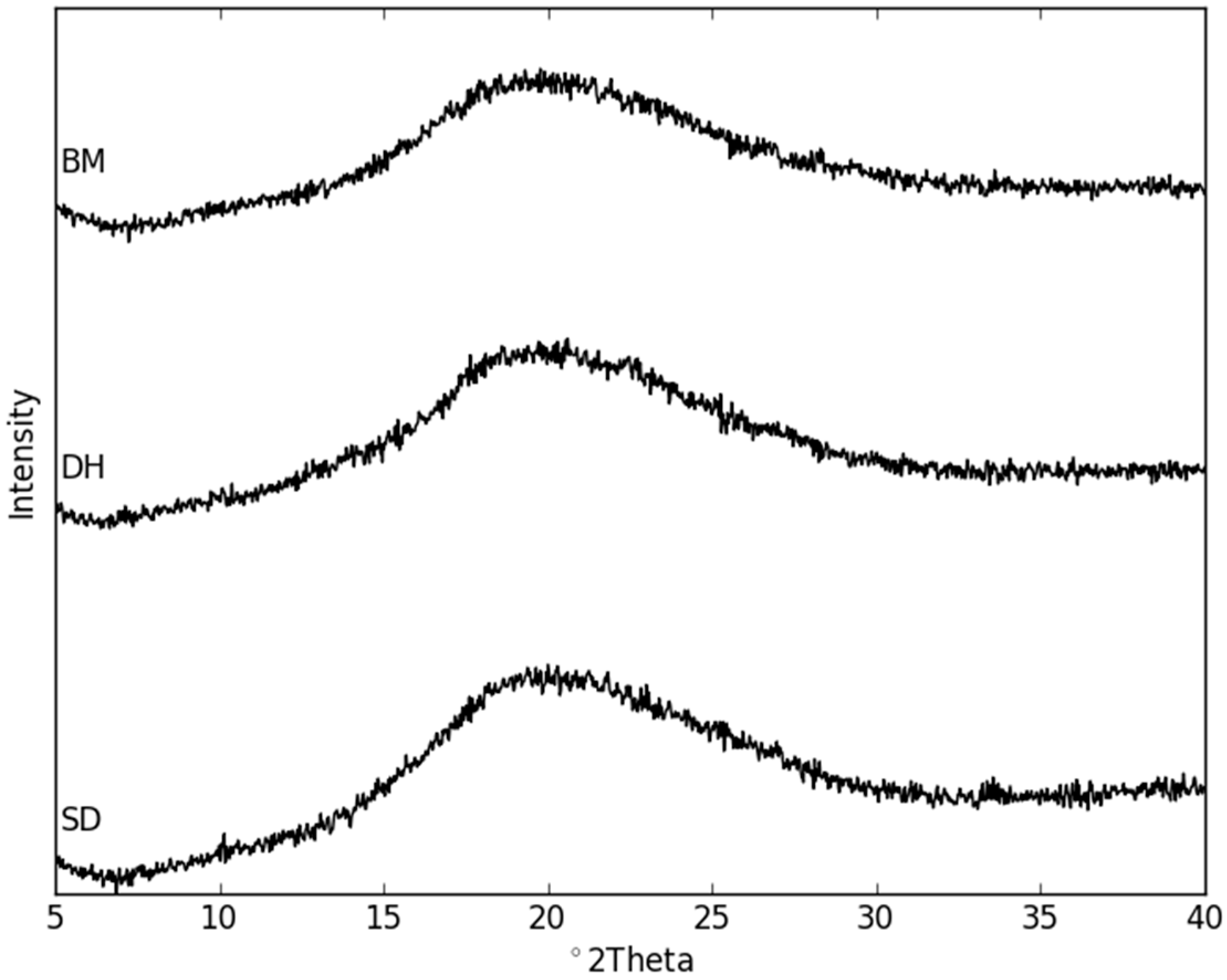
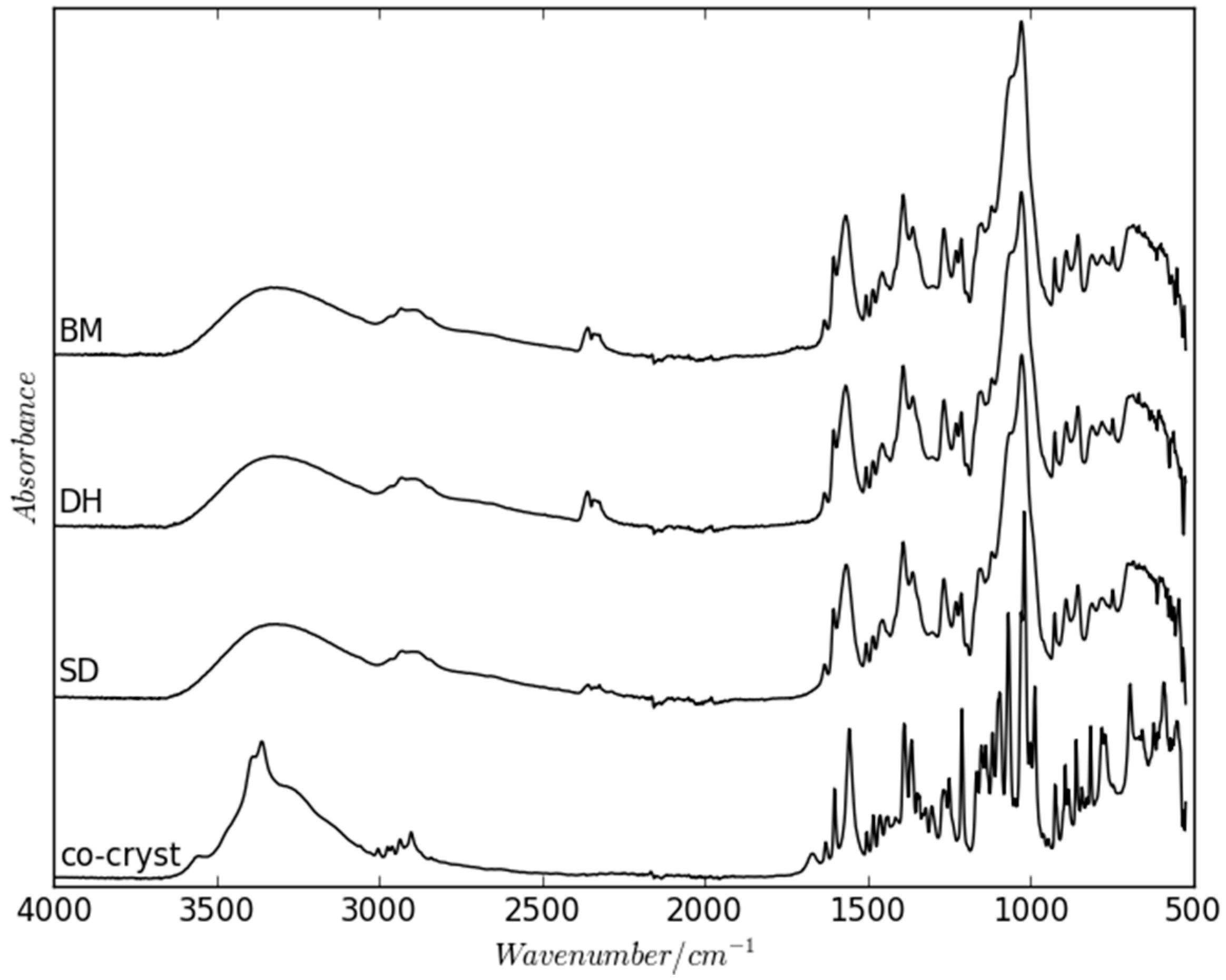
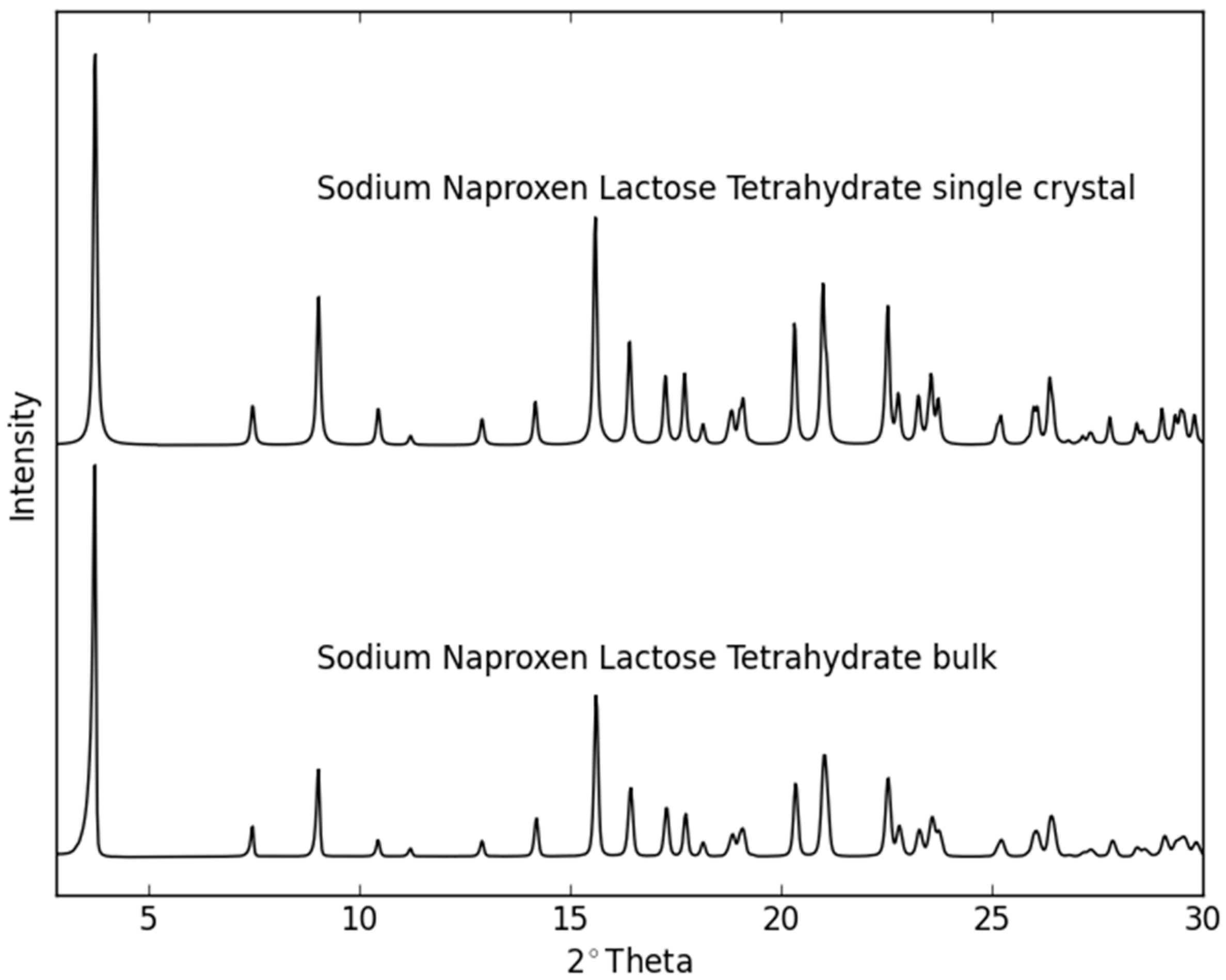
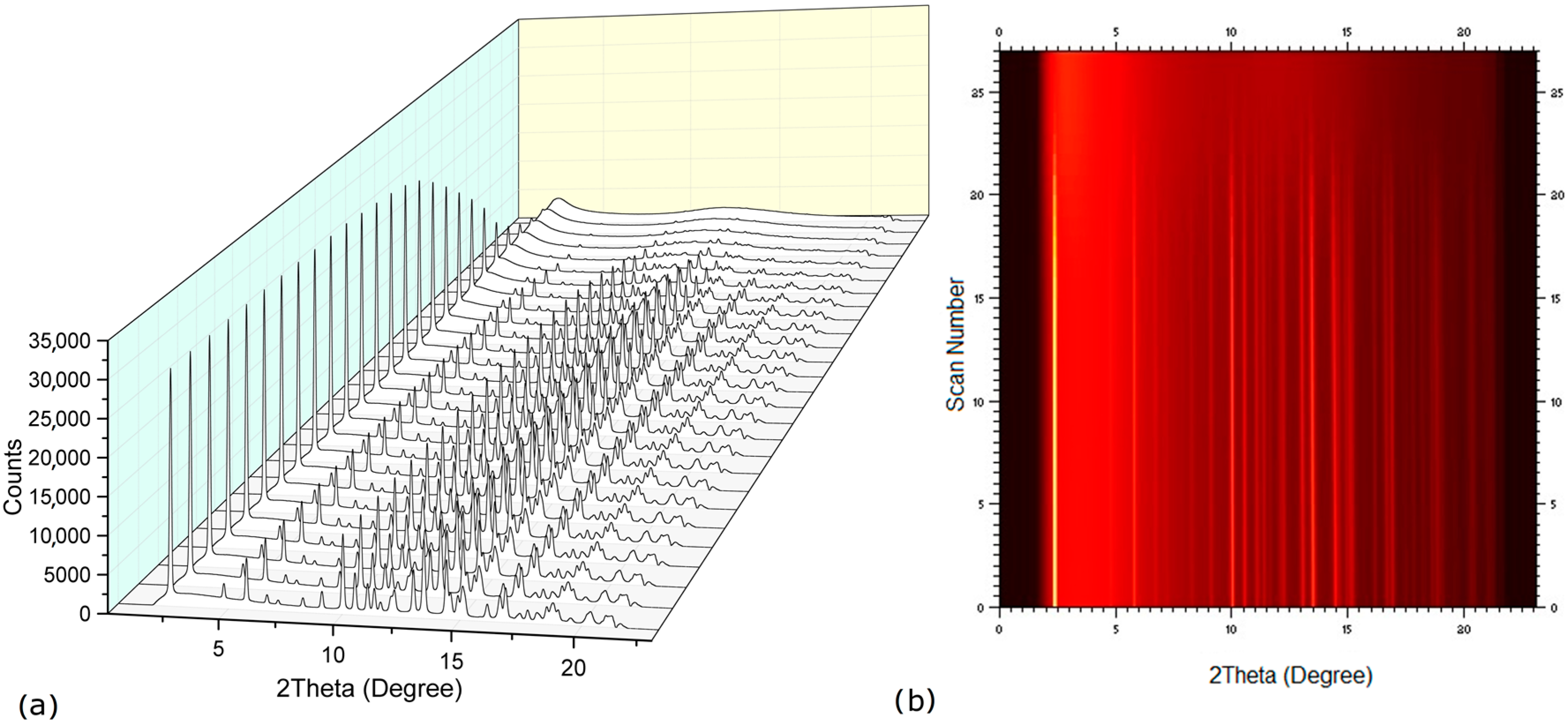
2.2. Dehydration Behavior of the NaN-Lact-4H2O Co-Crystal and Co-Amorphization
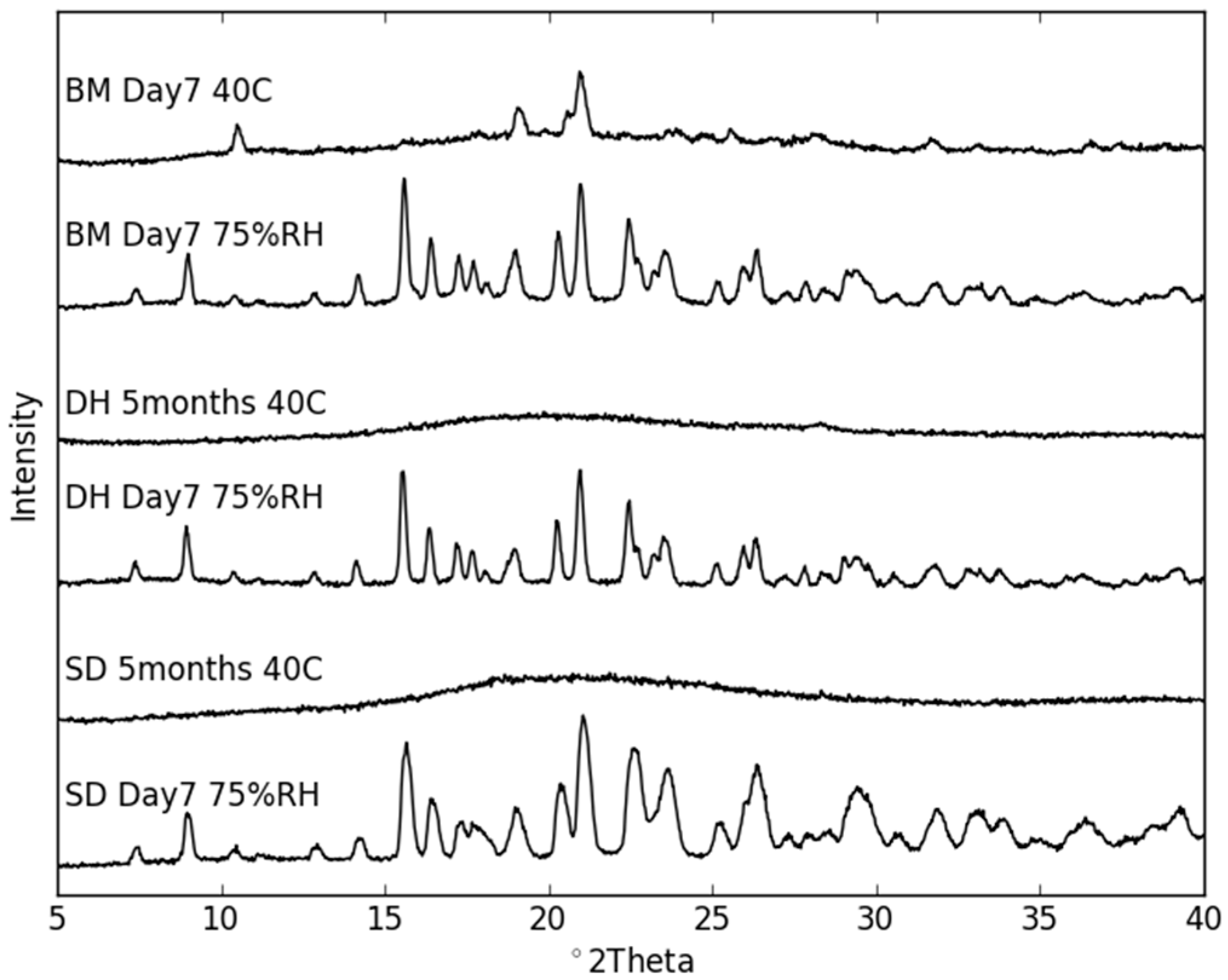
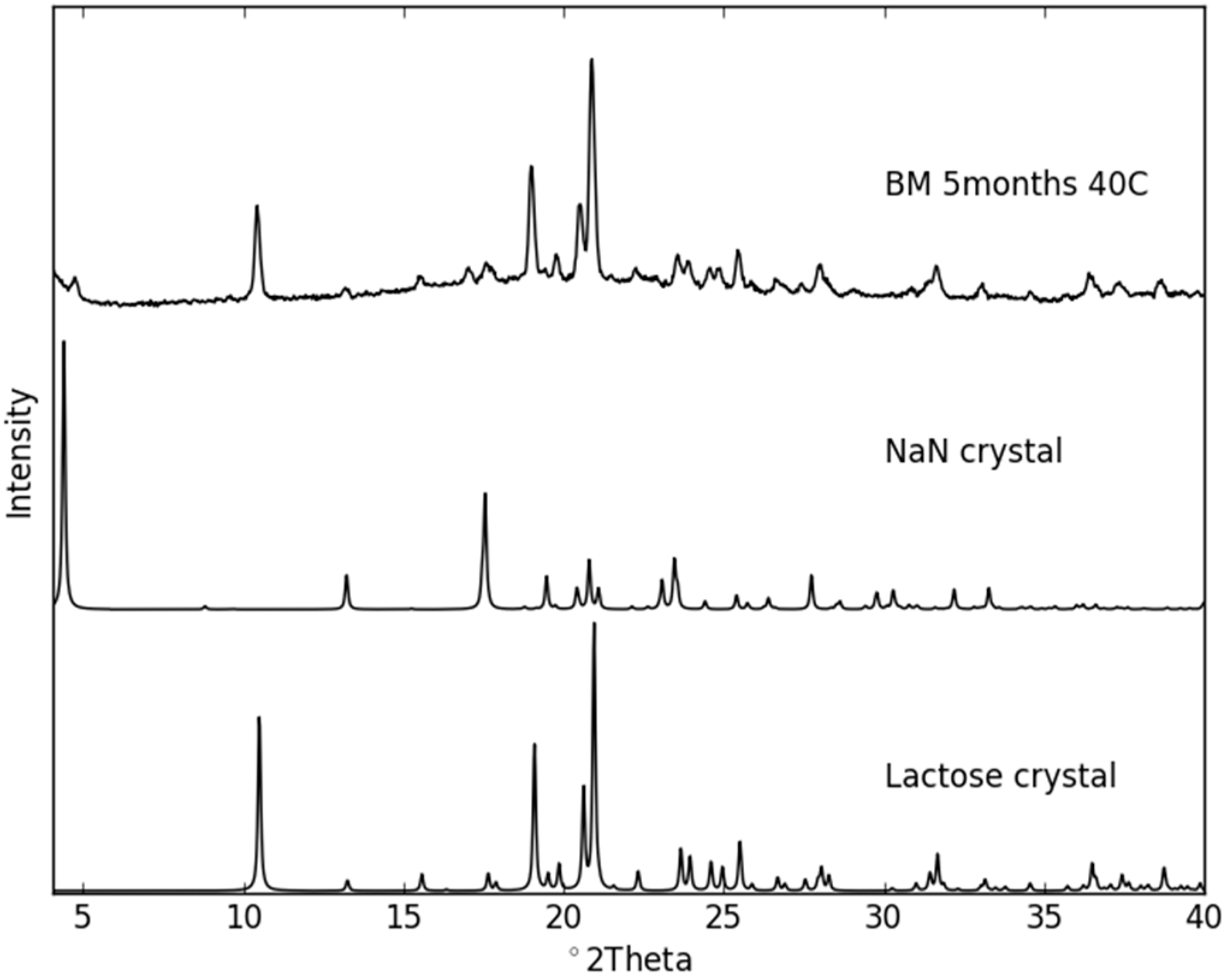
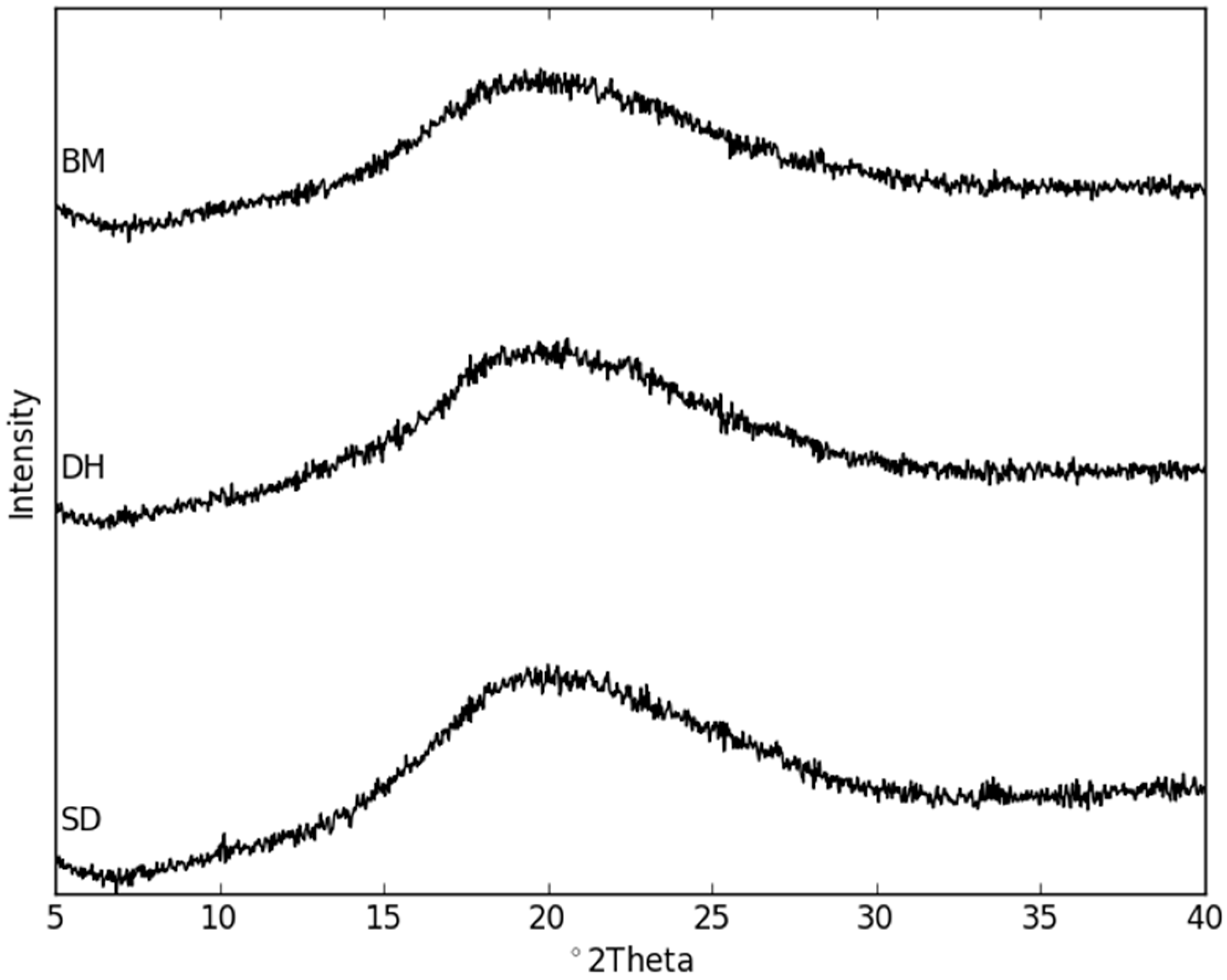
2.3. Physical Stability of Co-Amorphous NaN-Lact
3. Materials and Methods
3.1. Materials
3.2. Methods
3.2.1. Preparation of the (NaN-Lact-4H2O) Co-Crystal
3.2.2. Preparation of the NaN-Lact Co-Amorphous System
Spray Drying (SD)
Ball Milling (BM)
Dehydration of the Co-Crystal (DH)
3.2.3. Analytical Techniques
Single-Crystal X-ray Diffraction and Crystal Structure Determination
X-ray Powder Diffraction (XRPD) Measurements
Modulated Differential Scanning Calorimetry (mDSC)
Thermogravimetric Analysis (TGA)
FT-Infrared Spectroscopy (FT–IR)
Stability Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rodríguez-Hornedo, N.; Nehm, S.J.; Seefeldt, K.F.; Pagán-Torres, Y.; Falkiewicz, C.J. Reaction crystallization of pharmaceutical molecular complexes. Mol. Pharm. 2006, 3, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Trask, A.V. An overview of pharmaceutical cocrystals as intellectual property. Mol. Pharm. 2007, 4, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Thakuria, R.; Delori, A.; Jones, W.; Lipert, M.P.; Roy, L.; Rodríguez-Hornedo, N. Pharmaceutical cocrystals and poorly soluble drugs. Int. J. Pharm. 2013, 453, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Dengale, S.J.; Grohganz, H.; Rades, T.; Löbmann, K. Recent advances in co-amorphous drug formulations. Adv. Drug Deliv. Rev. 2016. [Google Scholar] [CrossRef] [PubMed]
- Laitinen, R.; Löbmann, K.; Strachan, C.J.; Grohganz, H.; Rades, T. Emerging trends in the stabilization of amorphous drugs. Int. J. Pharm. 2013, 453, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Aitipamula, S.; Banerjee, R.; Bansal, A.K.; Biradha, K.; Cheney, M.L.; Choudhury, A.R.; Desiraju, G.R.; Dikundwar, A.G.; Dubey, R.; Duggirala, N.; et al. Polymorphs, salts, and cocrystals: What’s in a name? Cryst. Growth Des. 2012, 12, 2147–2152. [Google Scholar] [CrossRef]
- Walsh, R.D.B.; Bradner, M.W.; Fleischman, S.; Morales, L.A.; Moulton, B.; Rodriguez-Hornedo, N.; Zaworotko, M.J. Crystal engineering of the composition of pharmaceutical phases. Chem. Commun. 2003, 2, 186–187. [Google Scholar] [CrossRef]
- Grohganz, H.; Priemel, P.A.; Löbmann, K.; Nielsen, L.H.; Laitinen, R.; Mullertz, A.; van den Mooter, G.; Rades, T. Refining stability and dissolution rate of amorphous drug formulations. Expert Opin. Drug Deliv. 2014, 11, 977–989. [Google Scholar] [CrossRef] [PubMed]
- Chiou, W.L.; Riegelman, S. Pharmaceutical applications of solid dispersion systems. J. Pharm. Sci. 1971, 60, 1281–1302. [Google Scholar] [CrossRef] [PubMed]
- Jie, L.; Sohrab, R. Polymorphism and crystallization of active pharmaceutical ingredients (APIs). Curr. Med. Chem. 2009, 16, 884–905. [Google Scholar]
- Chieng, N.; Aaltonen, J.; Saville, D.; Rades, T. Physical characterization and stability of amorphous indomethacin and ranitidine hydrochloride binary systems prepared by mechanical activation. Eur. J. Pharm. Biopharm. 2009, 71, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Gordon, K.C.; Strachan, C.; Rades, T. Coamorphous drug systems: Enhanced physical stability and dissolution rate of indomethacin and naproxen. Mol. Pharm. 2011, 8, 1919–1928. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Laitinen, R.; Grohganz, H.; Strachan, C.; Rades, T.; Gordon, K.C. A theoretical and spectroscopic study of co-amorphous naproxen and indomethacin. Int. J. Pharm. 2013, 453, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Löbmann, K.; Grohganz, H.; Laitinen, R.; Strachan, C.; Rades, T. Amino acids as co-amorphous stabilizers for poorly water soluble drugs—Part 1: Preparation, stability and dissolution enhancement. Eur. J. Pharm. Biopharm. 2013, 85, 873–881. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Gniado, K.; Erxleben, A.; McArdle, P. Mechanochemical reaction of sulfathiazole with carboxylic acids: Formation of a cocrystal, a salt, and coamorphous solids. Cryst. Growth Des. 2014, 14, 803–813. [Google Scholar] [CrossRef]
- Seefeldt, K.; Miller, J.; Alvarez-Núñez, F.; Rodríguez-Hornedo, N. Crystallization pathways and kinetics of carbamazepine-nicotinamide cocrystals from the amorphous state by in situ thermomicroscopy, spectroscopy, and calorimetry studies. J. Pharm. Sci. 2007, 96, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Di Martino, P.; Barthélémy, C.; Palmieri, G.F.; Martelli, S. Physical characterization of naproxen sodium hydrate and anhydrate forms. Eur. J. Pharm. Sci. 2001, 14, 293–300. [Google Scholar] [CrossRef]
- Kim, Y.-S.; Rousseau, R.W. Characterization and solid-state transformations of the pseudopolymorphic forms of sodium naproxen. Cryst. Growth Des. 2004, 4, 1211–1216. [Google Scholar] [CrossRef]
- Malaj, L.; Censi, R.; Martino, P.D. Mechanisms for dehydration of three sodium naproxen hydrates. Cryst. Growth Des. 2009, 9, 2128–2136. [Google Scholar] [CrossRef]
- Raijada, D.; Bond, A.D.; Larsen, F.H.; Cornett, C.; Qu, H.; Rantanen, J. Exploring the solid-form landscape of pharmaceutical hydrates: Transformation pathways of the sodium naproxen anhydrate-hydrate system. Pharm. Res. 2012, 30, 280–289. [Google Scholar] [CrossRef] [PubMed]
- Raijada, D.; Müllertz, A.; Cornett, C.; Munk, T.; Sonnergaard, J.; Rantanen, J. Miniaturized approach for excipient selection during the development of oral solid dosage form. J. Pharm. Sci. 2014, 103, 900–908. [Google Scholar] [CrossRef] [PubMed]
- Galwey, A.K. Structure and order in thermal dehydrations of crystalline solids. Thermochim. Acta 2000, 355, 181–238. [Google Scholar] [CrossRef]
- Li, Y.; Han, J.; Zhang, G.G.Z.; Grant, D.J.W.; Suryanarayanan, R. In Situ dehydration of carbamazepine dihydrate: A novel technique to prepare amorphous anhydrous carbamazepine. Pharm.Dev. Technol. 2000, 5, 257–266. [Google Scholar] [CrossRef] [PubMed]
- Karmwar, P.; Boetker, J.P.; Graeser, K.A.; Strachan, C.J.; Rantanen, J.; Rades, T. Investigations on the effect of different cooling rates on the stability of amorphous indomethacin. Eur. J. Pharm. Sci. 2011, 44, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Karmwar, P.; Graeser, K.; Gordon, K.C.; Strachan, C.J.; Rades, T. Investigation of properties and recrystallisation behaviour of amorphous indomethacin samples prepared by different methods. Int. J. Pharm. 2011, 417, 94–100. [Google Scholar] [CrossRef] [PubMed]
- Jensen, K.T.; Blaabjerg, L.I.; Lenz, E.; Bohr, A.; Grohganz, H.; Kleinebudde, P.; Rades, T.; Löbmann, K. Preparation and characterization of spray-dried co-amorphous drug-amino acid salts. J. Pharm. Pharmacol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Hancock, B.C.; Zografi, G. Characteristics and significance of the amorphous state in pharmaceutical systems. J. Pharm. Sci. 1997, 86, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of SHELX. Acta Cryst. 2008, A64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Clark, S.J.; Segall, M.D.; Pickard, C.J.; Hasnip, P.J.; Probert, M.I. J.; Refson, K.; Payne, M.C. First principles methods using CASTEP. Z. Kristallogr. 2005, 220, 567–570. [Google Scholar] [CrossRef]
- Allen, F. The Cambridge Structural Database: A quarter of a million crystal structures and rising. Acta Crystallogr. Sect. B 2002, 58, 380–388. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are not available from the authors.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Condition | Temperature (°C) | Humidity (% RH) |
|---|---|---|
| Condition 1 | 25 | <5 |
| Condition 2 | 25 | 75 |
| Condition 3 | 40 | <5 |
| Compound Formula | C26H43NaO18 |
| Mr | 666.59 |
| Space group | P21 |
| Crystal system | monoclinic |
| a/Å | 9.9900(6) |
| b/Å | 6.4720(4) |
| c/Å | 24.1480(17) |
| β/° | 101.632(4) |
| V/Å3 | 1529.23(17) |
| Z | 2 |
| Dcalc/g·cm−3 | 1.448 |
| λ/Å | 1.5418 |
| μ/mm−1 | 1.174 |
| Temperature/°C | 21(2) |
| Crystal size/mm | 0.20 × 0.05 × 0.05 |
| θ range/° | 7.804–54.237 |
| Max sin(θ)/λ | 0.52 |
| No. of unique data | 3728 |
| hkl range | −10 ≤ h ≤ 10 |
| −6 ≤ k ≤ 6 | |
| −25 ≤ l ≤ 25 | |
| Rint | 0.149 |
| Rσ | 0.1107 |
| No. of data in refinement | 35,347 |
| No. of refined parameters | 406 |
| R1 [I > 2σ(I)] | 0.052 |
| wR2 | 0.115 |
| Goodness of fit, S | 0.983 |
| Extreme in residual map/e·Å−3 | −0.294→0.203 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sovago, I.; Wang, W.; Qiu, D.; Raijada, D.; Rantanen, J.; Grohganz, H.; Rades, T.; Bond, A.D.; Löbmann, K. Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage. Molecules 2016, 21, 509. https://doi.org/10.3390/molecules21040509
Sovago I, Wang W, Qiu D, Raijada D, Rantanen J, Grohganz H, Rades T, Bond AD, Löbmann K. Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage. Molecules. 2016; 21(4):509. https://doi.org/10.3390/molecules21040509
Chicago/Turabian StyleSovago, Ioana, Wenbo Wang, Danwen Qiu, Dhara Raijada, Jukka Rantanen, Holger Grohganz, Thomas Rades, Andrew D. Bond, and Korbinian Löbmann. 2016. "Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage" Molecules 21, no. 4: 509. https://doi.org/10.3390/molecules21040509
APA StyleSovago, I., Wang, W., Qiu, D., Raijada, D., Rantanen, J., Grohganz, H., Rades, T., Bond, A. D., & Löbmann, K. (2016). Properties of the Sodium Naproxen-Lactose-Tetrahydrate Co-Crystal upon Processing and Storage. Molecules, 21(4), 509. https://doi.org/10.3390/molecules21040509