1. Introduction
One of the major current challenges of the pharmaceutical industry is related to strategies that improve the water solubility of drugs because over 40% of new drug candidates are water-insoluble compounds [
1]. Poorly water-soluble drug properties can impede the effective delivery of these drugs into humans, and affect their dissolution rate and subsequent absorption at the site of activity [
2].
Poorly soluble molecules have been successfully formulated by employing a variety of techniques to modify the physico-chemical and biopharmaceutical properties of drugs [
3,
4] such as: (i) solubilization in surfactant solutions; (ii) use of co-solvents [
5], salt formation [
6], complexation with cyclodextrins [
7], crystallization [
8], amorphization [
9], milling [
10],
etc.Milling is a technique commonly applied to produce micro- or nanosized drug crystals in order to increase the dissolution rate and absorption, and hence the bioavailability of poorly-soluble materials. According to the Noyes-Whitney equation, the reduction of the particle sizes of drug crystals increases the specific surface area, which can improve the rate of dissolution of the drug [
11]. There are many different well-known types of milling techniques with both advantages and disadvantages; dry and wet milling can be distinguished [
12,
13]. Wet milling requires less energy and time than dry milling. Thanks to the environmentally isolated system, it is a dust free process and the material is less heated up [
14]. However, wet milling has some disadvantages as well, e.g., increased wear of the grinding medium, corrosion hazards,
etc. To overcome the limitations of the conventional particle size reduction technologies for poorly-soluble drugs, new combinational methods have been developed for the production of ultra-fine suspensions. These technologies are a relatively new approach to improve the effectiveness of particle size reduction and to reduce the time of processes. In general, they can be described as a combination of a bottom-up process (the building-up of particles) [
15,
16,
17] followed by a top-down technology (disintegration) [
18,
19]. This method involves two particle size reduction steps. There is also a possibility for the combination of dry- and wet milling in one step, but literature data relating to the application of this combined method are lacking. Retsch GmbH (Haan, Germany) has recommended the combination of planetary ball milling, as dry milling, and pearl milling, as wet milling [
20] techniques.
In wet micro- and nanonization, the application of additives is required in order to retain the individuality of the particles. Different additives are used to stabilize these particles: poly(vinylpyrrolidone) (PVP), Poloxamer
® (Poloxamer 188 = poly(ethylene)–poly(propylene glycol), polysorbate (Tween 80
® = poly(oxyethylenesorbitan monooleate)), Solutol
® (Solutol HS 15 = poly(ethylene glycol 15-hydroxystearate)), PVA (poly(vinyl alcohol)),
etc. [
21].
Milling and the presence of additives during milling have the ability to decrease the crystallinity of active materials [
22,
23]. The amorphous phase transition usually results in improved dissolution rates, thereby increasing the bioavailability, with the improvement being directly related to the extent of amorphization [
24]. However, physical and chemical instability of amorphous material could be expected.
Meloxicam (MEL) is a non-steroidal anti-inflammatory drug (NSAID) with anti-inflammatory, analgesic and antipyretic effects. MEL was chosen as a model crystalline drug because of its poor aqueous solubility (4.4 µg/mL) [
25] and relatively high melting point (270 °C) [
26].
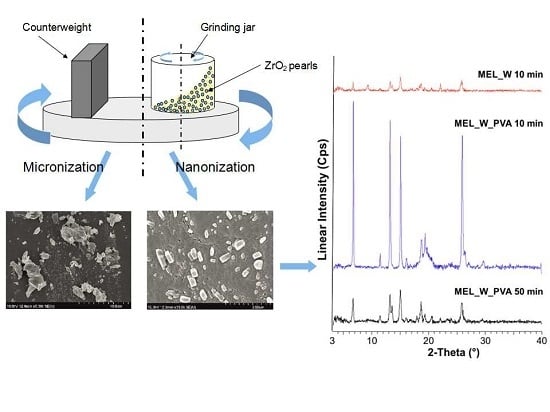
This research investigates the applicability of a wet milling method combining planetary ball and pearl milling. We set out to utilize particle size reduction to the micro- or nanometre range. The effects of milling time and the presence or absence of stabilizer (PVA) in reducing the particle size were investigated. The changes in crystallinity and morphology of MEL during milling have been studied. The effects of particle size reduction and amorphization on the dissolution rate were determined.
4. Conclusions
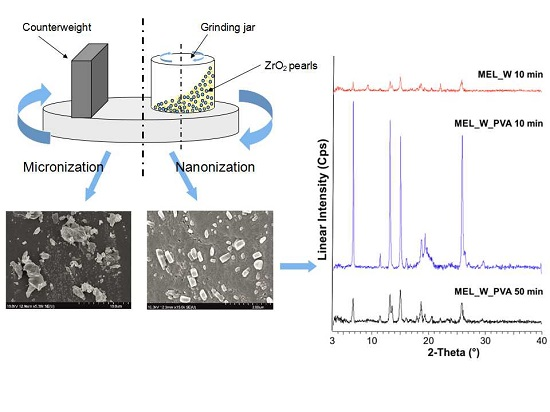
A combination of planetary ball and pearl milling (using pearls as milling media) can be applied as a wet milling procedure to decrease the particle size and change the crystal morphology of MEL. Wet milling in aqueous medium adheres to green technology principles as the product does not contain organic solvent residues. Besides several advantages of wet milling, it is necessary to calculate the wear of pearls during milling.
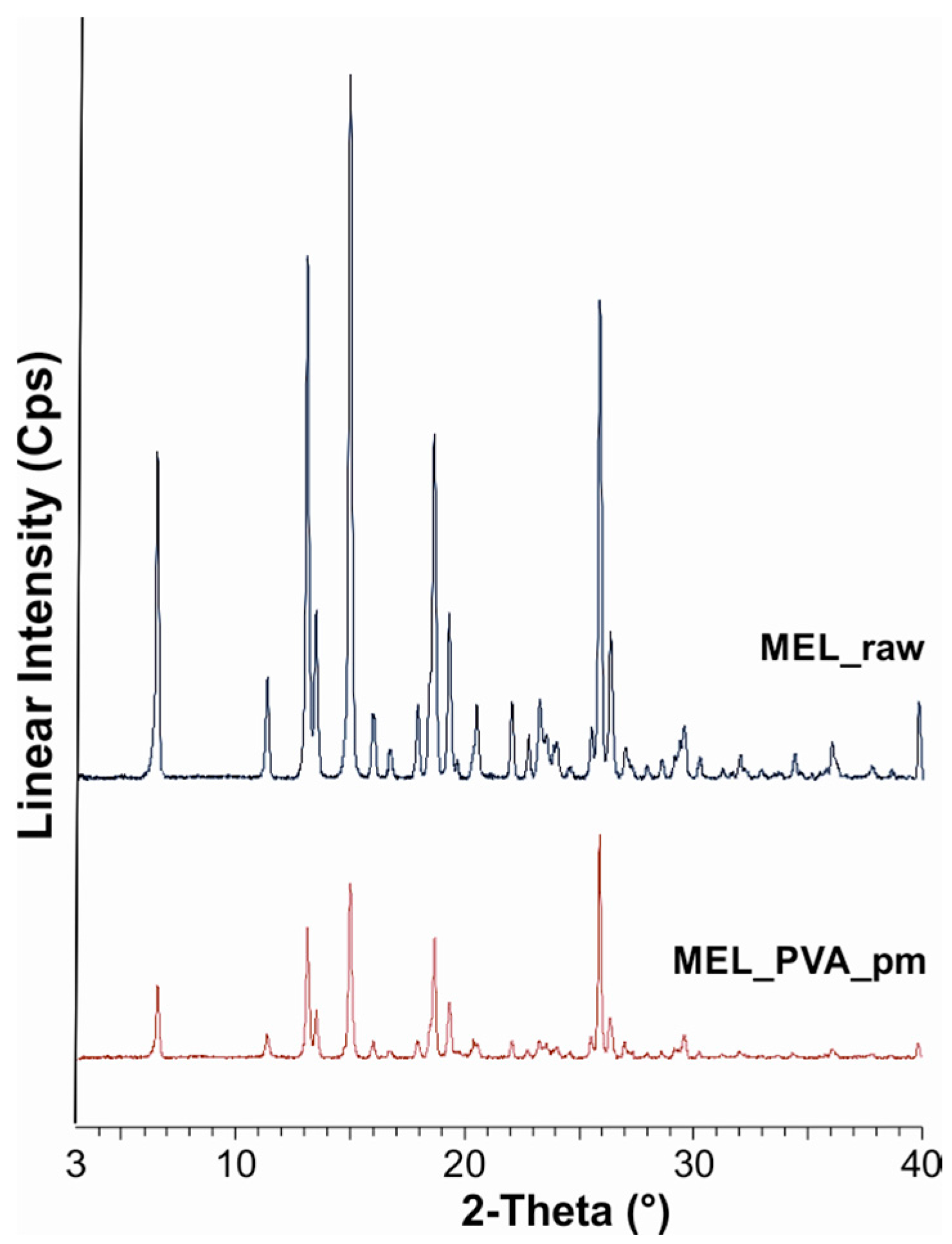
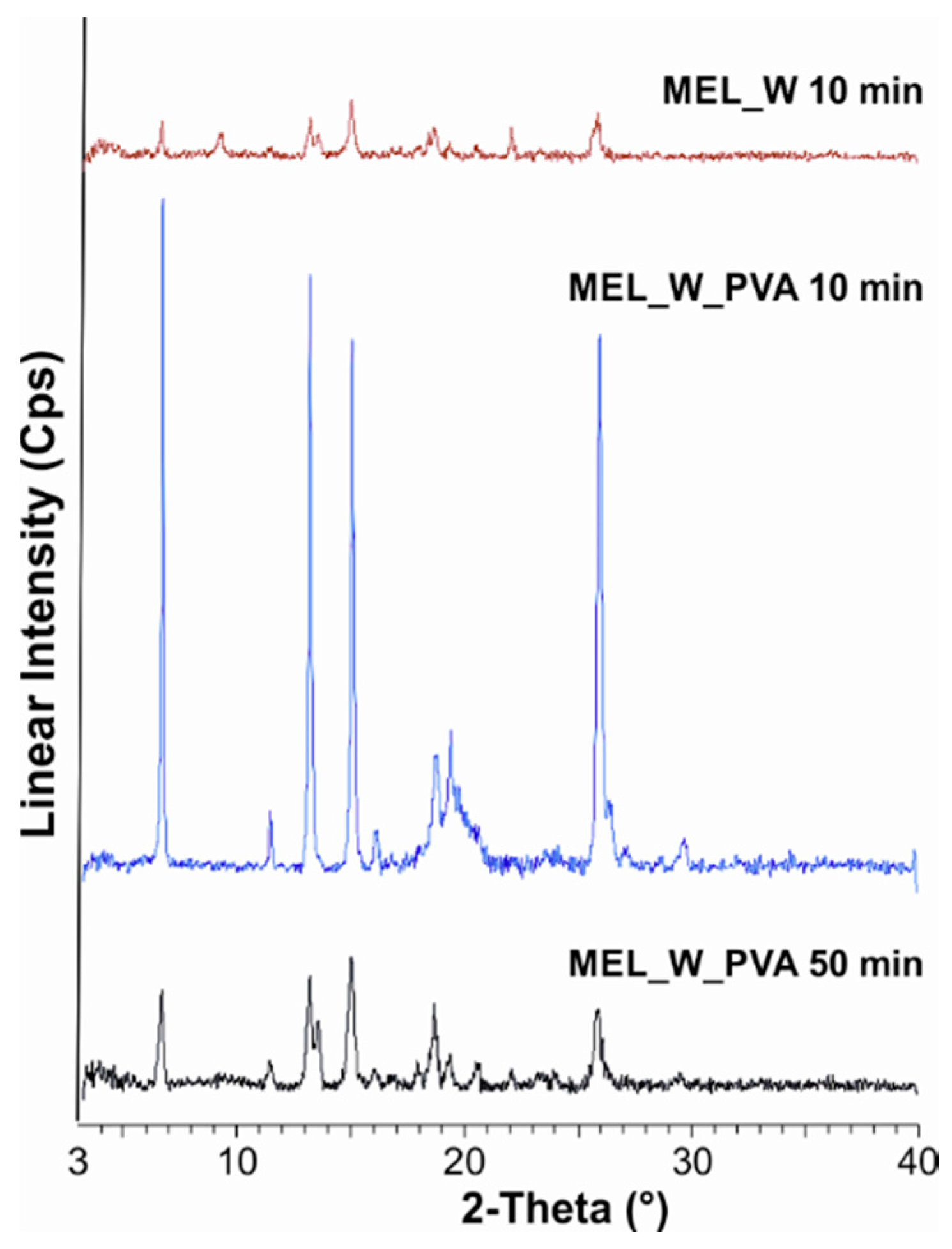
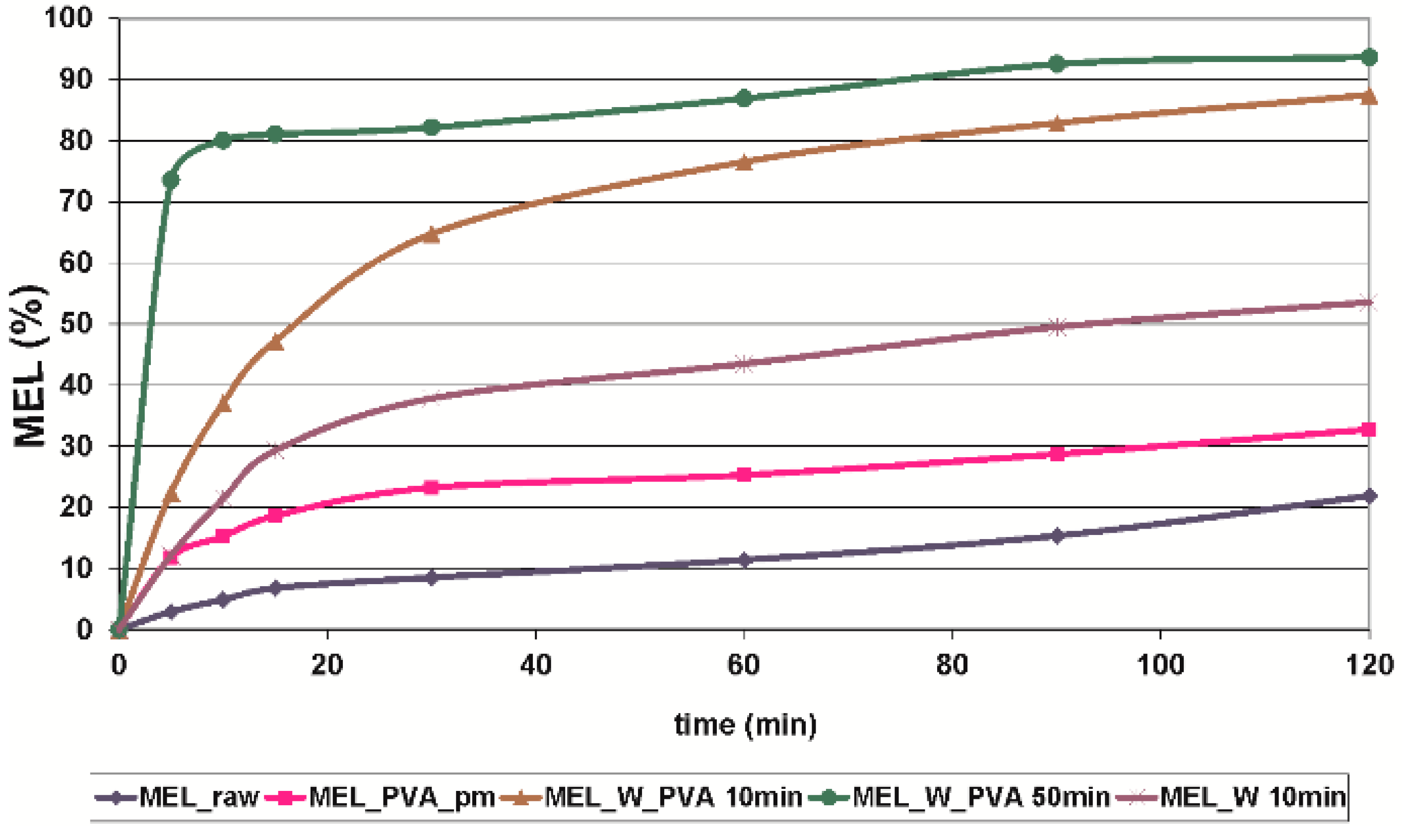
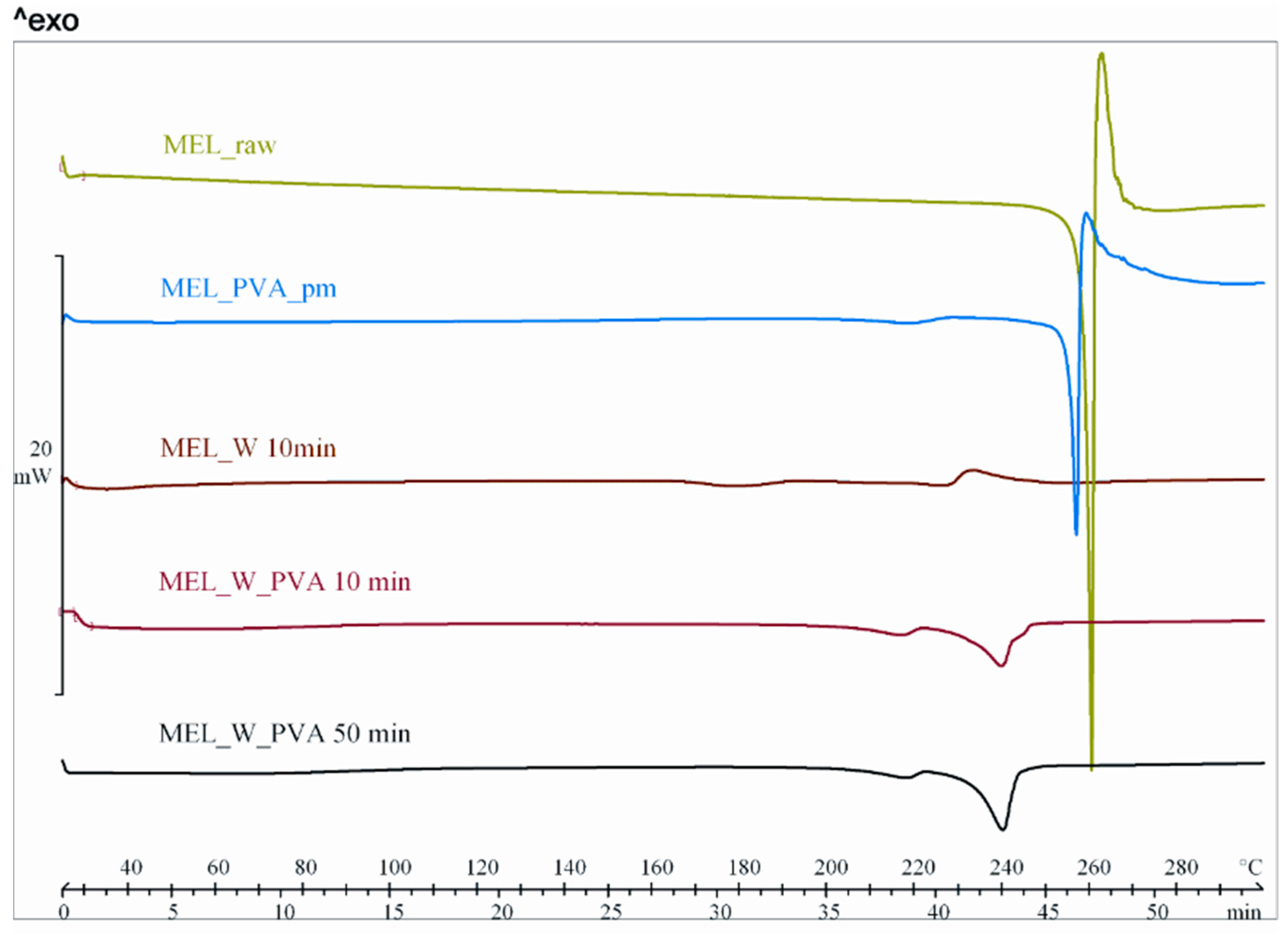
During our work at constant rotation rate in the presence and absence of a stabilizer, the effects of milling time on the particle size reduction was determined (
Table 6). Without additive, in the case of water-containing samples micronization could be achieved (D
50 = 3.55 µm). In the presence of PVA, depending on the milling time, the particle size of the drug could be reduced to the micro-(after 10 min), (D
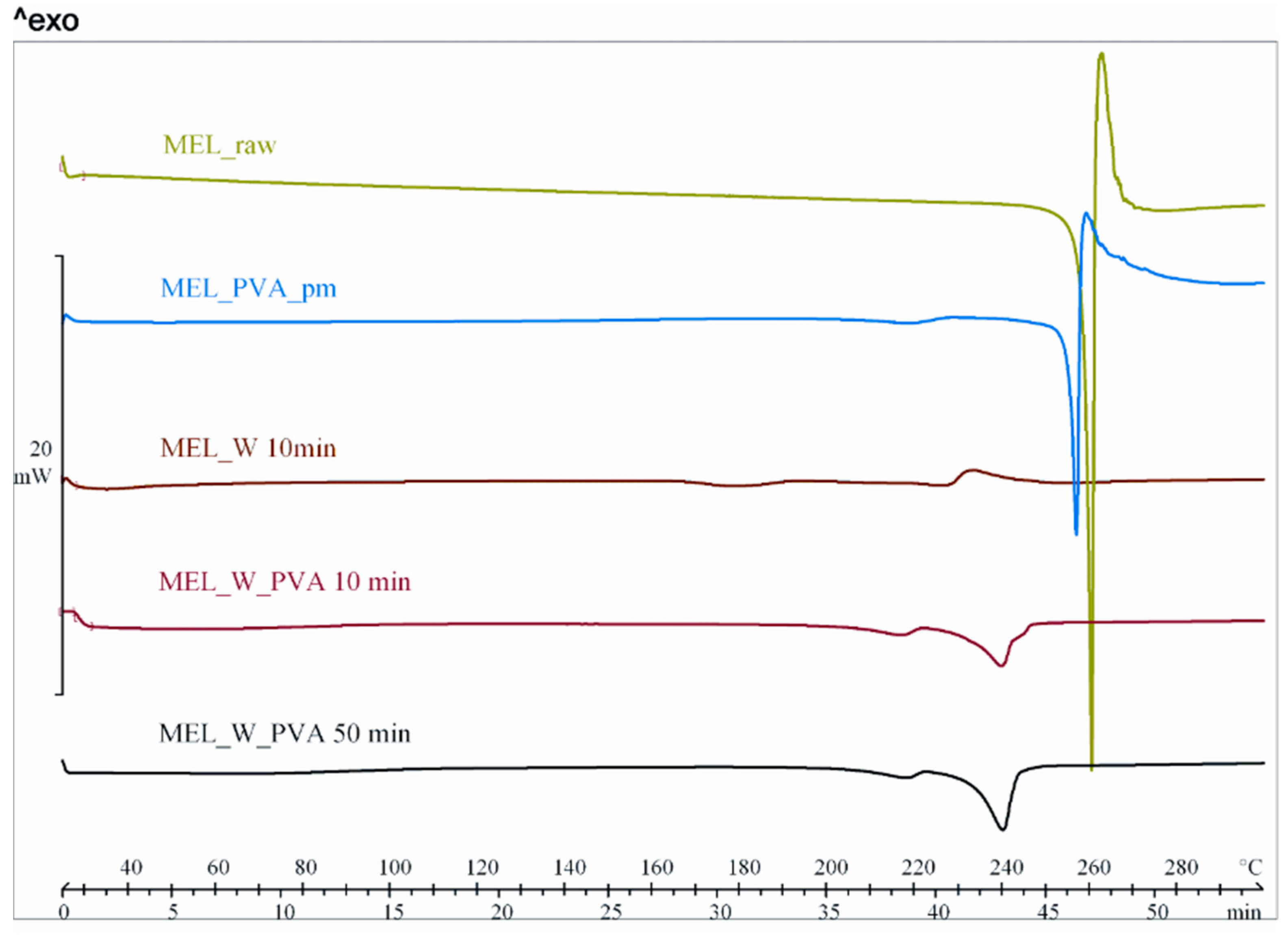
50 = 2.96 µm) or nanometre range (after 50 min, D
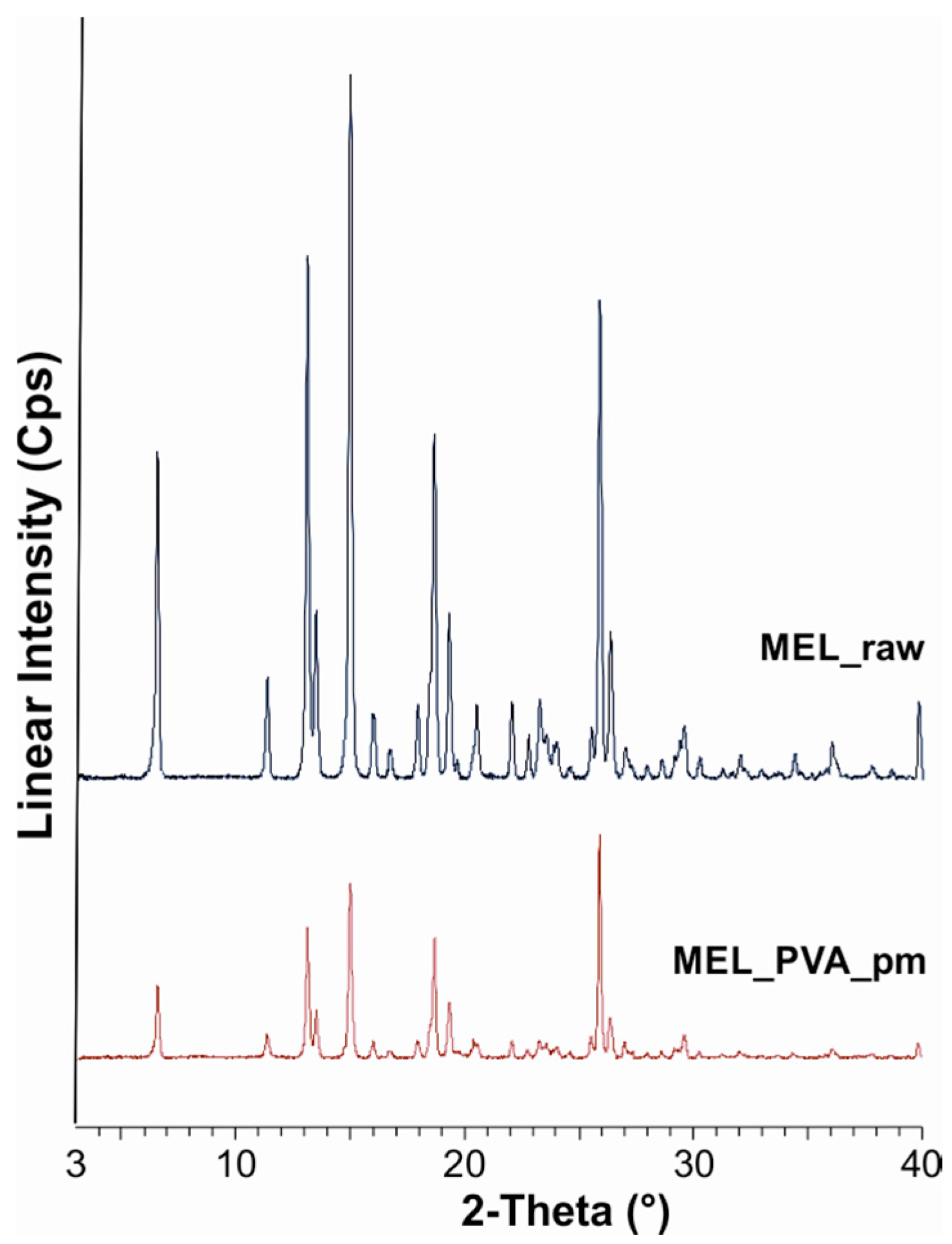
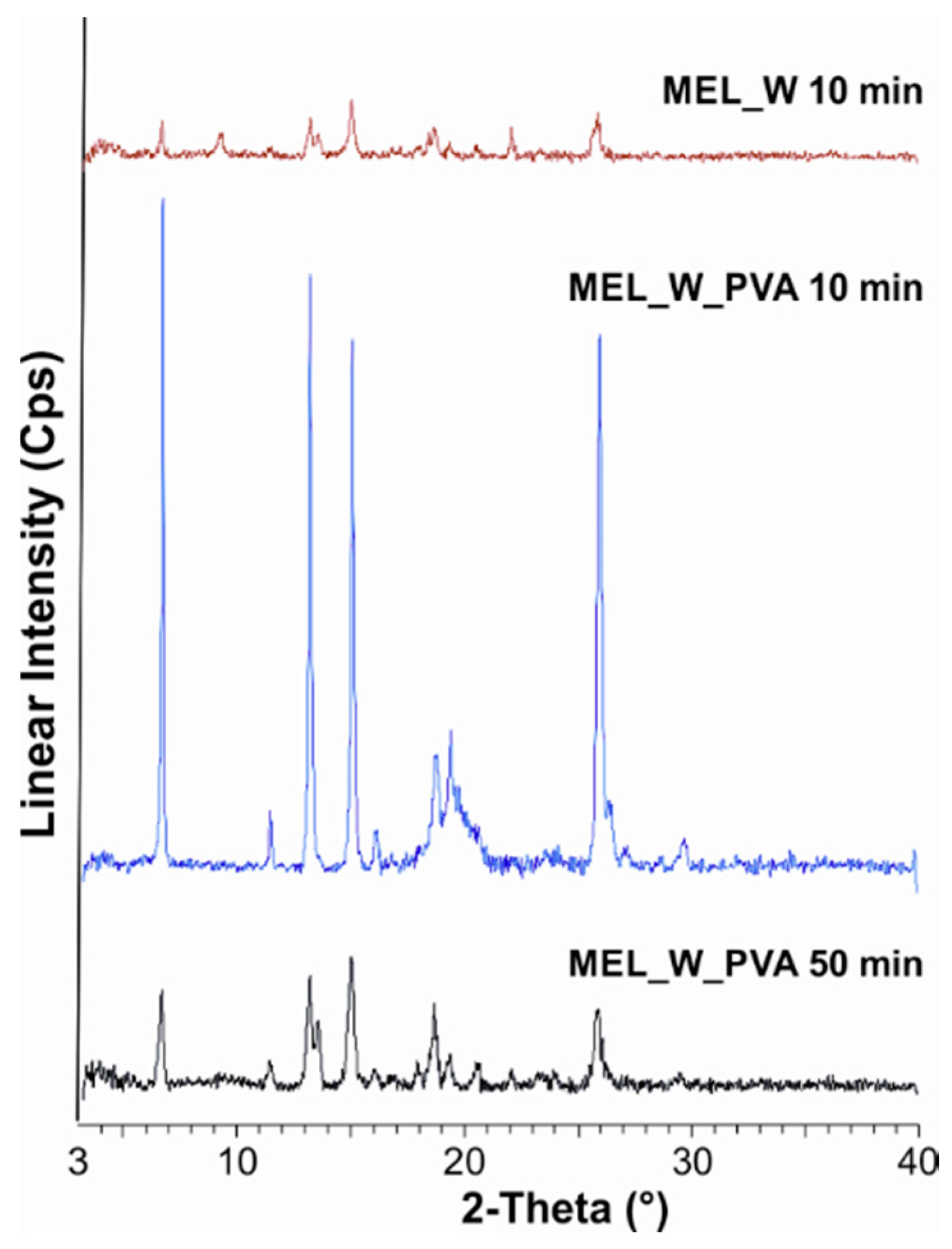
50 = 126 nm). The effect of milling on the crystallinity of MEL was investigated. XRPD and DSC examinations revealed a decrease in the crystallinity of MEL. In the case of water-containing samples (without PVA) aggregation occurred during the course of milling. In the PVA-containing samples amorphization was determined (the degree of MEL crystallinity was 2% at the end of the milling at 90 min). SEM images revealed the aggregation of nanosized particles in water-containing samples. In the presence of additive milling for 10 min resulted in irregularly shaped particles. The nanonized MEL crystals exhibited a regular shape and smooth surface. The
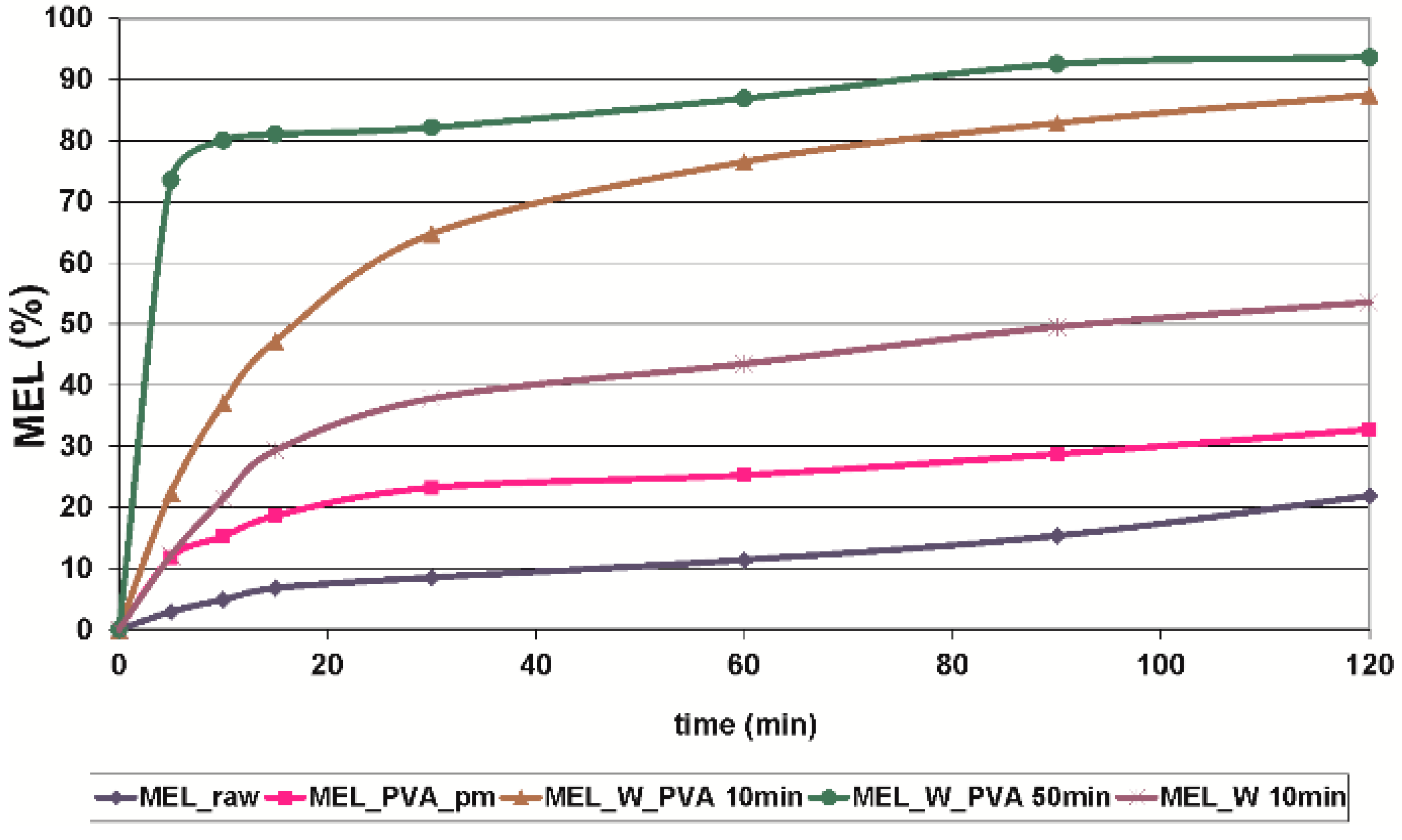
in vitro dissolution tests showed that the reduction of the particle size of MEL, the increased SSA and the structural transformation of drug resulted in a rapid dissolution in case of nanonized MEL-containing product. The amorphous form of the drug does not require lattice energy to break the bonds during the dissolution process as in the crystalline state case.
The combined wet milling technology was suitable for preparation of micronized MEL without the use of stabilizer and, depending on the milling time, of micronized and nanonized drug particles-containing pre-dispersions in the presence of PVA. Milling in the presence of additive could be the first step of pre-formulation and further formulation procedures. Decreased particle size (especially accessing the nanosize range) and the amorphization of drug could ensure higher dissolution rate and better bioavailability of poorly-water soluble drugs, though instability problems could occur in the case of amorphous forms of materials. To check the stability of the systems further investigations are needed.
Because of the low need for dispersant medium, the combined method can be used for efficient milling, and it is also suggested for the preparation of the pre-dispersions with micro- and nanosized particles, and recommended for the development of particle size-controlled therapeutic systems.


 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}