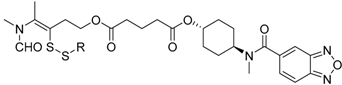
3.9. Synthesis
N,4-Dimethyl-5-(2-(hydroxy)ethyl)thiazolium iodide (9). 5-(2-Hydroxyethyl)-4-methylthiazole (100.0 g, 698.2 mmol) and methyl iodide (100.0 mL, 1500.0 mmol) were mixed and refluxed for 2 h. After evaporation of excess methyl iodide, to the residual brown syrup was added ether (100 mL) and stirred for 30 min, the precipitate was filtered to yield 9 as a pale-yellow solid (191.4 g, 96.2%). Mp: 82–84 °C. 1H-NMR (DMSO-d6, ppm): 2.43 (s, 3H), 3.03 (t, 2H, J = 5.5 Hz), 3.63 (t, 2H, J = 5.6 Hz), 4.09 (s, 3H), 9.96 (s, 1H); MS m/z [M]+ calculated for C7H12NS+: 158.1; found: 158.1.
General procedure for synthesis of S-substituted sodium thiosulfates 11a–11f. A solution of sodium thiosulfate pentahydrate (24.8 g, 100.0 mmol) in water (60 mL) was added to the solution of alkyl bromide (100 mmol) in ethanol (30 mL) under stirring. The reation mixture was heated to 110 °C and maintained for 7 h. After evaporation of the solvent, the residual white solid was dried in a vacuum desiccator to yield S-substituted sodium thiosulfates 11a–11f.
General procedure for synthesis of compounds 12a–12f. Under argon protection, to a 30 mL aqueous of quaternary ammonium salt 9 (14.3 g, 50.0 mmol) and sodium hydroxide (4.0 g, 100.0 mmol) was added S-substituted sodium thiosulfates 11a–11f (120.0 mmol). The reaction mixture was stirred for 1 h at room temperature. The resultant oily substance was extracted with ethyl acetate (450 mL) and the organic layer was dried over anhydrous Na2SO4 and concentrated under reduced pressure. Purification of the crude product by column chromatography with dichloromethane-methanol (100:1) as eluent gave a yellow oil.
N-(3-(Ethyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12a,
Figure S1). 40.9% yield as a yellow oil.
1H-NMR (CDCl
3, ppm): 1.19 (t, 3H,
J = 7.3 Hz), 1.96, 1.86 (2s, 3H), 2.64 (q, 2H,
J = 7.3 Hz), 2.70 (t, 2H,
J = 6.7 Hz), 2.98, 2.83 (2s, 3H), 3.55–3.43 (m, 2H), 7.96, 7.84 (2s, 1H). MS
m/
z [M + H]
+ calculated for C
9H
17NO
2S
2: 236.1; found: 236.2.
N-(3-(Propyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12b,
Figure S2). 64.8% yield as a yellow oil.
1H-NMR (CDCl
3, ppm): 0.97 (t, 3H,
J = 7.3 Hz), 1.69–1.63 (m, 2H), 2.01, 1.96 (2s, 3H), 2.47 (br, 1H), 2.59 (t, 2H,
J = 7.6 Hz), 2.87 (t, 2H,
J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.81 (t, 3H,
J = 6.4 Hz), 7.96, 7.92 (2s, 1H); MS
m/
z [M + H]
+ calculated for C
10H
19NO
2S
2: 250.1; found: 250.1.
N-(3-(Butyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12c,
Figure S3). 70.5% yield as a yellow oil.
1H-NMR (CDCl
3, ppm): 0.91(t, 3H,
J = 7.3 Hz), 1.43–1.33 (m, 2H), 1.65–1.57 (m, 2H), 2.01, 1.96 (2s, 3H), 2.51 (br, 1H), 2.62 (t, 2H,
J = 7.3 Hz), 2.87 (t, 2H,
J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.81 (t, 2H,
J = 6.8 Hz), 7.96, 7.92 (2s, 1H). MS
m/
z [M + H]
+ calculated for C
11H
21NO
2S
2: 264.1; found: 264.2.
N-(3-(Amyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12d,
Figure S4). 52.0% yield as a yellow oil.
1H-NMR (CDCl
3, ppm): 0.90 (t, 3H,
J = 7.3 Hz), 1.36–1.29 (m, 4H), 1.65–1.60 (m, 2H), 2.01, 1.97 (2s, 3H), 2.49 (br, 1H), 2.61 (t, 2H,
J = 7.3 Hz), 2.87 (t, 2H,
J = 6.7 Hz), 3.06, 2.95 (2s, 3H), 3.80 (t, 2H,
J = 6.8 Hz), 7.96, 7.92 (2s, 1H). MS
m/
z [M + H]
+ calculated for C
12H
23NO
2S
2: 278.1; found: 278.0.
N-(3-Isoamyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12e,
Figure S5). 76.8% yield as a yellow oil.
1H-NMR (CDCl
3, ppm): 1.19 (t, 3H,
J = 7.3 Hz), 1.53–1.47 (m, 2H), 1.68–1.61 (m, 1H), 2.01, 1.96 (2s, 3H), 2.49 (br, 1H), 2.62 (t, 2H,
J = 7.6 Hz), 2.88 (t, 2H,
J = 6.8 Hz), 3.06, 2.95 (2s, 3H), 3.80(t, 3H,
J = 6.4 Hz), 7.96, 7.92(2s, 3H). MS
m/
z [M + H]
+ calculated for C
12H
23NO
2S
2: 278.1; found: 278.0.
N-(3-(Benzyldisulfanyl)-5-hydroxypent-2-en-2-yl)-N-methylformamide (
12f,
Figure S6). 44.1% yield as a yellow oil.
1H-NMR (
d6-DMSO, ppm): 1.91, 1.85 (2s, 3H), 2.62 (t, 2H,
J = 6.7 Hz), 2.82, 2.81 (2s, 3H), 3.52–3.47 (m, 2H), 3.92 (s, 2H), 4.74–4.70 (m, 1H), 7.35–7.25 (m, 5H), 7.96, 7.81 (2s, 1H). MS
m/
z [M + H]
+ calculated for C
14H
19NO
2S
2: 298.1; found: 298.2.
General procedure for synthesis of 4-(N-methylformamido)pent-3-en-1-yl)oxy)-5-oxopentanoic acids 13a–13f. A stirred solution of intermediate 12a–12f (10.0 mmol) in dry dichloromethane (50 mL) was treated with DMAP (0.1 g, 0.8 mmol) and glutaric anhydride (3.4 g, 30.0 mmol). The mixture was refluxed for 5 h. The solvent was evaporated under vacuum and purification of the crude product by column chromatography with dichloromethane-methanol (100:1) as eluent gave 13a–13f as yellow syrup.
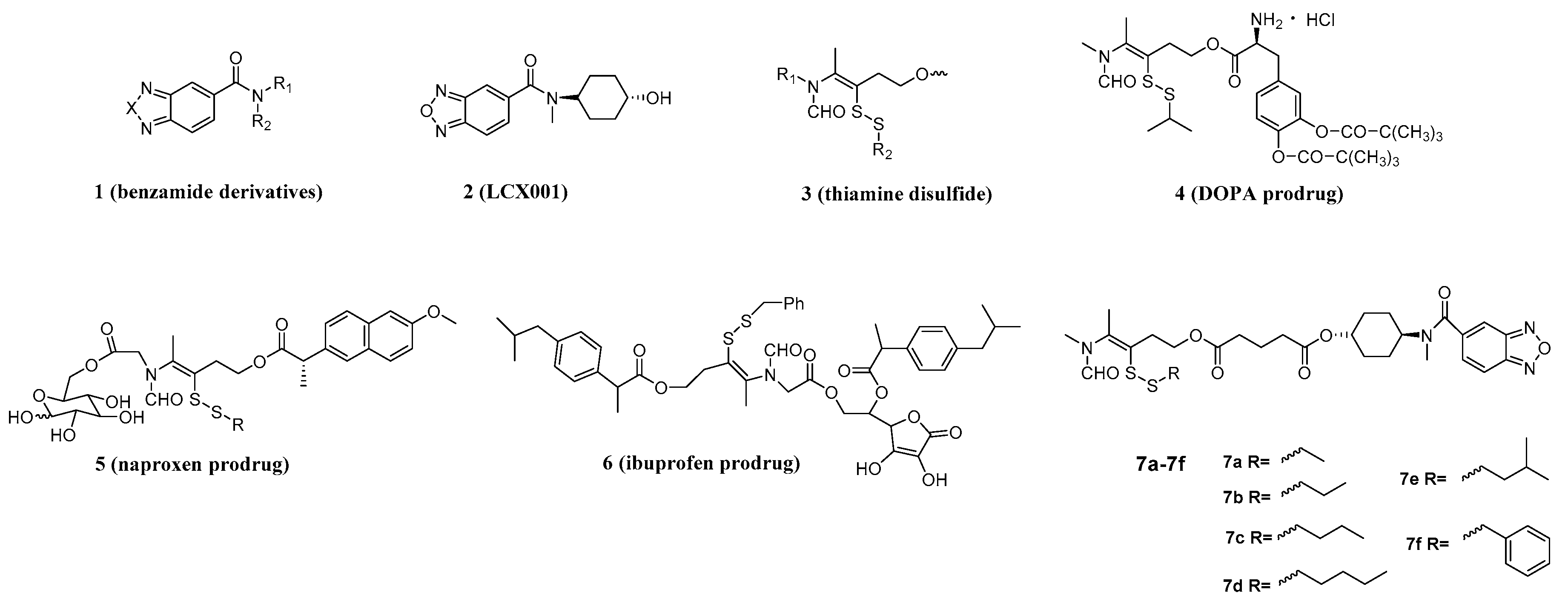
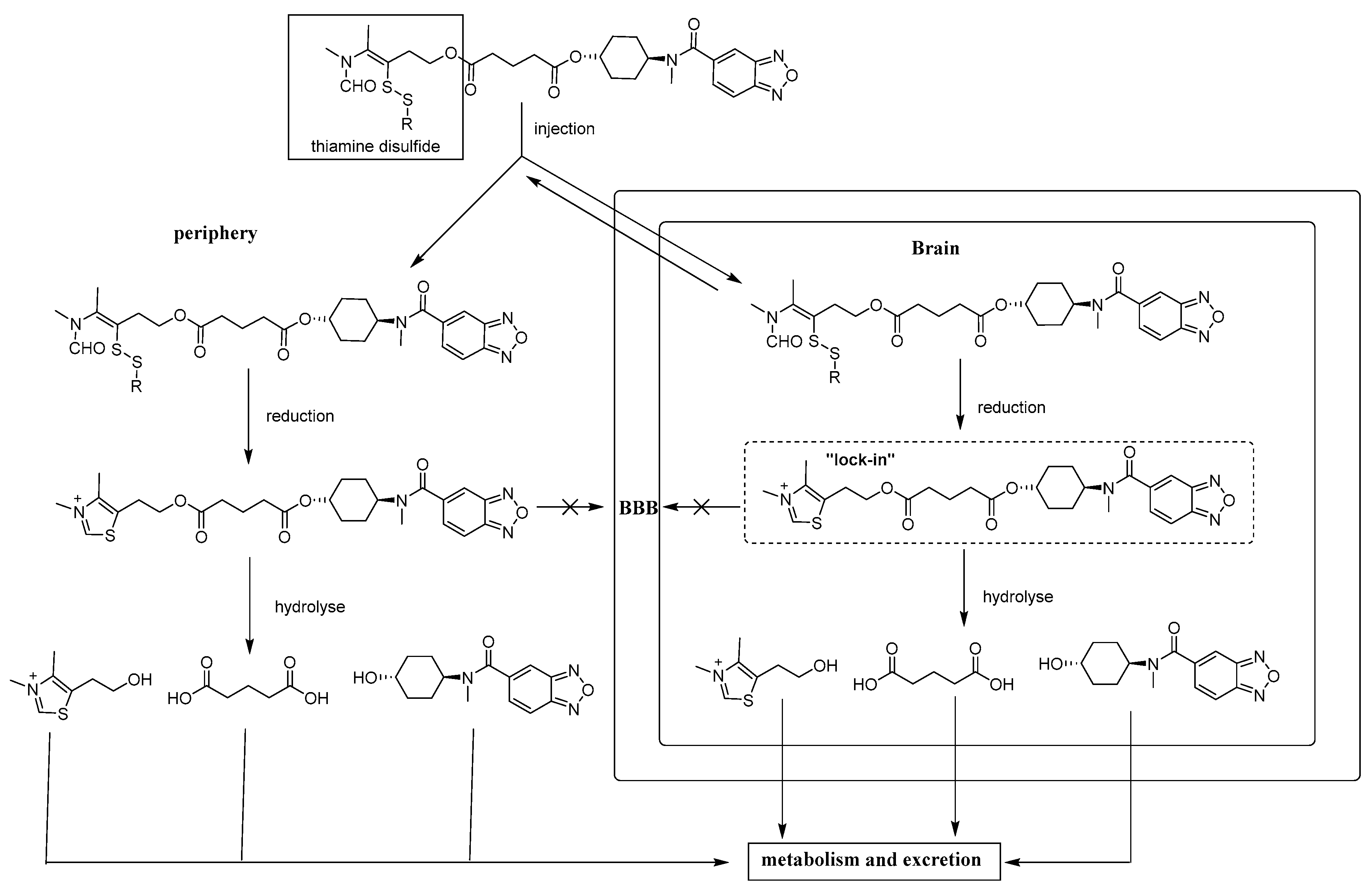
General procedure for synthesis of compounds 7a–7f. The intermediate 13a–13f (4.7 mmol), LCX001 (1.0 g, 3.6 mmol), triethylamine (0.5 mL, 3.6 mmol) and HOBT (486 mg, 3.6 mmol) were dissolved in dry dichloromethane (50 mL) in a round-bottomed flask under string for 10 min. After addition of DMAP (44 mg, 0.36 mmol) and EDCI (1.1 g, 5.4 mmol), the mixture was refluxed for 5 h. The resulting mixture was concentrated by a rotary evaporator to afford yellow oleosus residue that was purified by column chromatography on silica gel (ethyl acetate/petroleum ether, (1:1)) to provide 7a–7f as white solids.
S-3-(Ethyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7a,
Figure S7). 43.2% yield as a white solid. Mp: 78.8–79.5 °C;
1H-NMR (CDCl
3, ppm): 1.27 (t, 3H,
J = 7.3 Hz), 1.99, 1.97 (2s, 3H), 1.94–1.62 (m, 8H), 2.12–1.99 (m, 2H), 2.44–2.35 (m, 4H), 2.63 (q, 2H,
J = 7.3 Hz), 3.04–2.87 (m, 8H), 4.70–4.59, 3.51 (m, 1H), 4.24 (t, 2H,
J = 6.7 Hz), 7.41 (d, 1H,
J = 8.1 Hz);
13C-NMR (CDCl
3, ppm): 172.6, 172.2, 168.6, 162.3, 161.2, 148.4, 139.7, 136.5, 130.6, 117.6, 114.5, 113.6, 71.8, 62.0, 51.9, 33.3, 33.0, 31.8, 30.3, 29.6, 29.6, 29.0, 29.0, 28.2, 26.9, 19.8, 18.4, 14.0. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
28H
38N
4O
7S
2: 607.2182; found: 607.2255.
S-3-(Propyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7b,
Figure S8). 40.7% yield as a white solid. Mp: 78.0–79.3 °C;
1H-NMR (CDCl
3, ppm): 0.96 (t, 3H,
J = 7.3 Hz), 1.94–1.23 (m, 10H), 2.00, 1.97 (2s, 3H), 2.13–2.11 (m, 2H), 2.42–2.26 (m, 4H), 2.58 (t, 2H,
J = 6.2 Hz), 3.04–2.88 (m, 8H), 4.70–4.56, 3.50 (m, 1H), 4.25 (t, 2H,
J = 5.6 Hz), 7.42 (d, 1H,
J = 8.9 Hz), 7.98–7.83 (m, 3H);
13C-NMR (CDCl
3, ppm): 172.7, 172.3, 168.8, 162.3, 161.2, 148.4, 139.7, 136.6, 130.7, 117.5, 114.6, 113.5, 71.8, 62.1, 51.9, 41.1, 33.3, 33.1, 30.4, 29.6, 29.6, 29.0, 29.0, 28.2, 26.9, 21.9, 19.9, 18.5, 12.9. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
29H
40N
4O
7S
2: 621.2338; found: 621.2411.
S-3-(Butyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7c,
Figure S9). 38.3% yield as a white solid. Mp: 67.6–68.4 °C;
1H-NMR (CDCl
3, ppm): 0.91 (t, 3H,
J = 7.6 Hz), 2.00–1.25 (m, 12H), 2.03, 2.00 (2s, 3H), 2.15–2.09 (m, 2H), 2.40–2.26 (m, 4H), 2.61 (t, 2H,
J = 7.3 Hz), 3.03–2.87 (m, 8H), 4.72–4.53, 3.51 (m, 1H), 4.25 (t, 2H, J = 7.0 Hz), 7.42 (d, 1H,
J = 8.7 Hz), 7.94–7.83 (m, 3H);
13C-NMR (CDCl
3, ppm): 172.7, 172.3, 168.6, 162.4, 161.2, 148.3, 139.8 136.6, 130.7, 117.5, 114.6, 113.7, 71.6, 62.1, 52.0, 39.0, 33.4, 33.3, 31,7, 30.4, 29.6, 29.6, 29.0, 29.0, 28.3, 26.9, 21.5, 20.0, 18.5, 13.6. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
30H
42N
4O
7S
2: 635.2495; found: 635.2568.
S-3-(Amyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7d,
Figure S10). 45.5% yield as a white solid. Mp: 69.3–70.3 °C;
1H-NMR (CDCl
3, ppm): 0.89 (t, 3H,
J = 6.8 Hz), 1.94–1.28 (m, 14H), 2.00, 1.97 (2s, 3H), 2.14–2.09 (m, 2H), 2.41–2.27 (m, 4H), 2.60 (t, 2H,
J = 5.8 Hz), 3.04–2.88 (m, 8H), 4.71–4.59, 3.50 (m, 1H), 4.24 (t, 2H,
J = 6.8 Hz), 7.42 (d, 1H,
J = 8.4 Hz), 7.94–7.84 (m, 3H);
13C-NMR (CDCl
3, ppm): 172.7, 172.2, 168.8, 162.4, 161.2, 148.5, 139.8, 136.6, 130.4, 117.4, 114.6, 113.7, 71.9, 62.1, 51.9, 39.3, 33.4, 33.1, 31.9, 30.5, 30.2, 29.6, 29.6, 29.0, 29.0, 28.3, 26.9, 22.2, 19.2, 18.5, 13.9. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
31H
44N
4O
7S
2: 649.2651; found: 649.2724.
S-3-(Isoamyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7e,
Figure S11). 45.0% yield as a white solid. Mp: 74.0–75.4 °C;
1H-NMR (CDCl
3, ppm): 0.89 (d, 6H,
J = 6.5 Hz), 1.97–1.47 (m, 11H), 2.00,1.97 (2s, 3H), 2.15–2.09 (m, 2H), 2.41–2.26 (m, 4H), 2.62 (t, 2H,
J = 6.7 Hz), 3.04–2.88 (m, 8H), 4.71–4.57, 3.51 (m, 2H), 4.24 (d, 2H,
J = 6.4 Hz), 7.42 (d, 1H,
J = 8.1 Hz), 9.97–7.83 (m, 3H);
13C-NMR (CDCl
3, ppm): 172.7, 172.3, 168.6, 162.4, 161.3, 148.5, 139.7, 136.5, 130.8, 117.4, 114.6, 113.6, 71.8, 62.1, 51.9, 37.6, 37.4, 33.4, 33.1, 31.9, 30.2, 29.6, 29.6, 29.0, 29.0, 28.3, 28.3, 26.9, 22.2, 20.0, 18.5. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
31H
44N
4O
7S
2: 649.2651; found: 649.2723.
S-3-(Benzyldisulfanyl)-4-(N-methylformamido)pent-3-en-1-yl((1R,4R)-4-(N-methylbenzo[c][1,2,5]oxadiazole-5-carboxamido)cyclohexyl) glutarate (
7f,
Figure S12). 57.5% yield as a white solid. Mp: 72.9–73.8 °C;
1H-NMR (CDCl
3, ppm): 1.91–1.25 (m, 8H), 1.94, 1.91 (2s, 3H), 2.10–2.03 (m, 2H), 2.41–2.25 (m, 4H), 3.00–2.75 (m, 8H), 4.69–4.59, 3.50 (m, 1H), 3.86 (s, 2H), 4.15 (t, 2H,
J = 7.1 Hz), 7.42–7.28 (m, 5H), 7.97–7.83 (m, 3H).
13C-NMR (CDCl
3, ppm): 172.6, 172.3, 168.8, 162.4, 161.3, 148.4, 139.7, 136.1, 135.8, 130.4, 129.2, 129.2, 128.5, 128.5, 127.7, 117.7, 114.6, 113.6, 71.8, 62.0, 51.9, 44.2, 33.3, 33.3, 31.9, 30.4, 29.5, 29.5, 28.7, 28.7, 26.9, 19.9, 18.4. HRMS (ESI+)
m/
z [M + H]
+ calculated for C
33H
40N
4O
7S
2: 669.2338; found: 669.2411.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}