
Preparation and Biological Evaluation of Two Novel Platinum(II) Complexes Based on the Ligands of Dipicolyamine Bisphosphonate Esters

Abstract
:1. Introduction
2. Results and Discussion
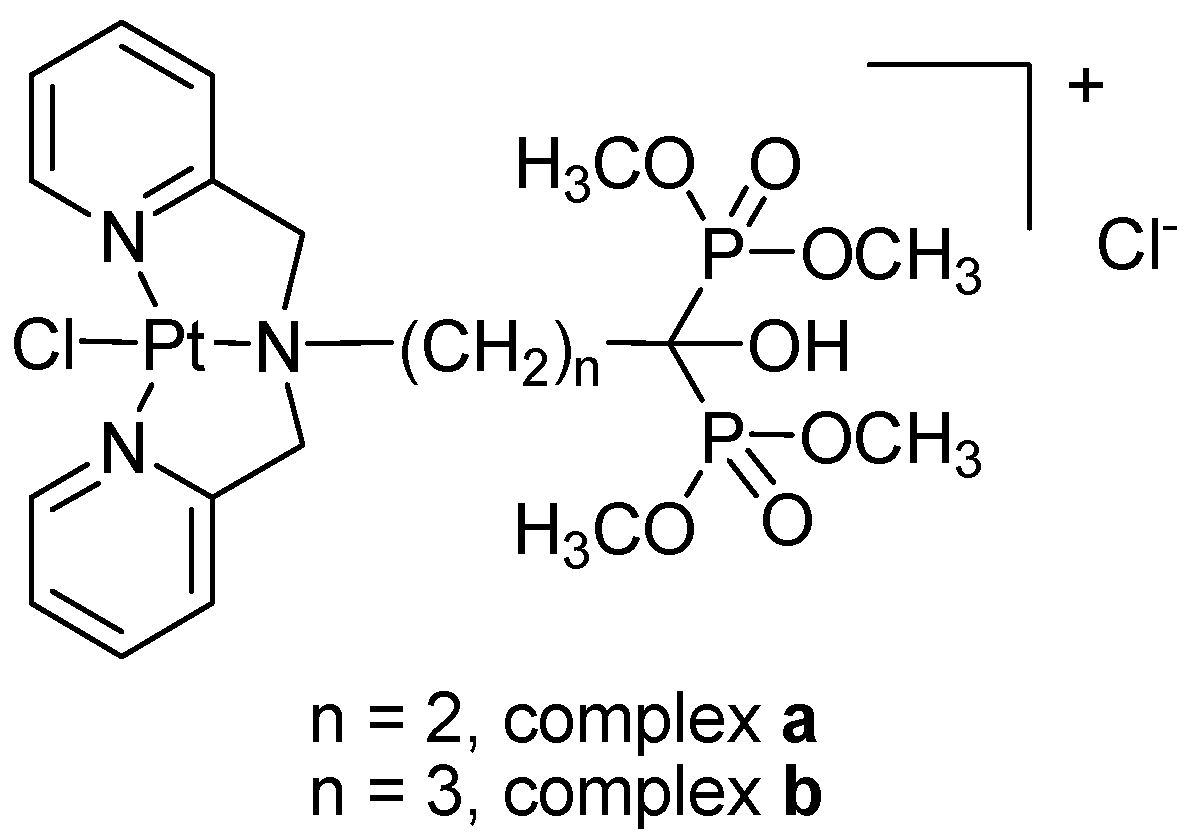
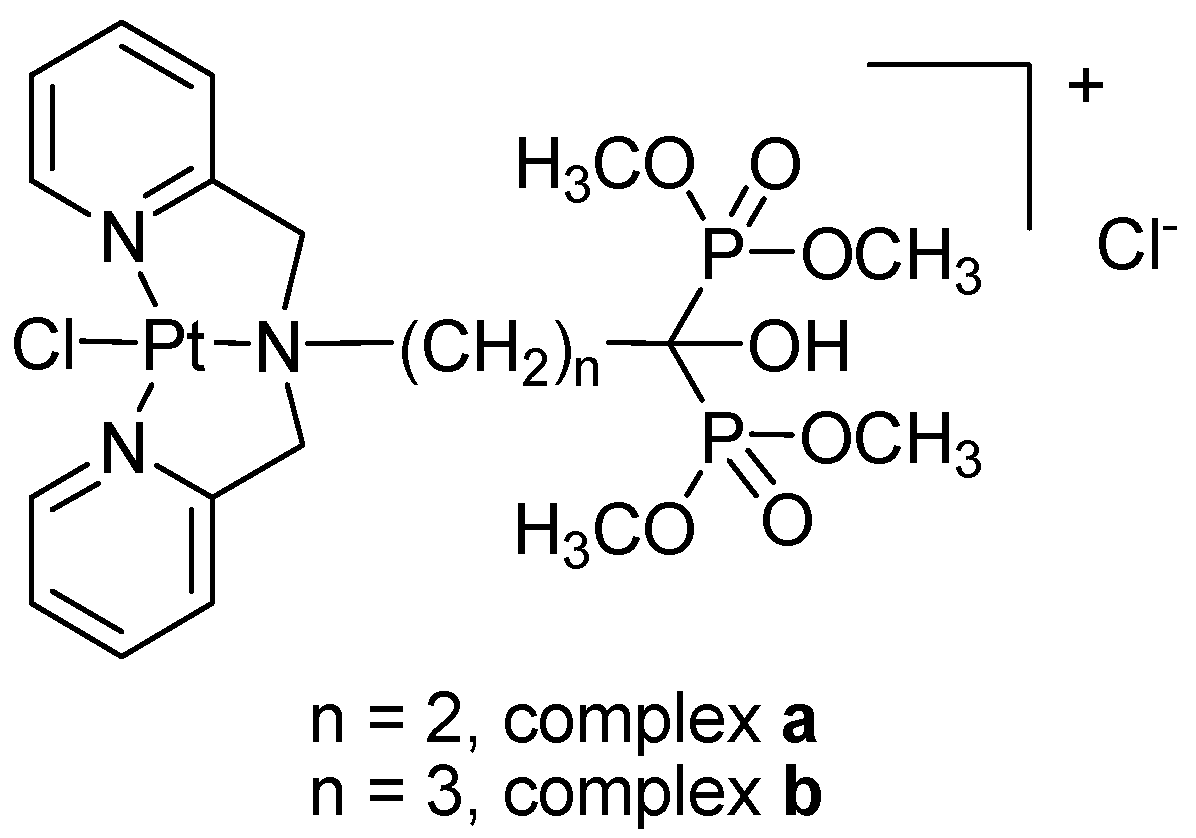
2.1. Preparation and Characterization
2.2. Lipid-Water Partition Coefficient
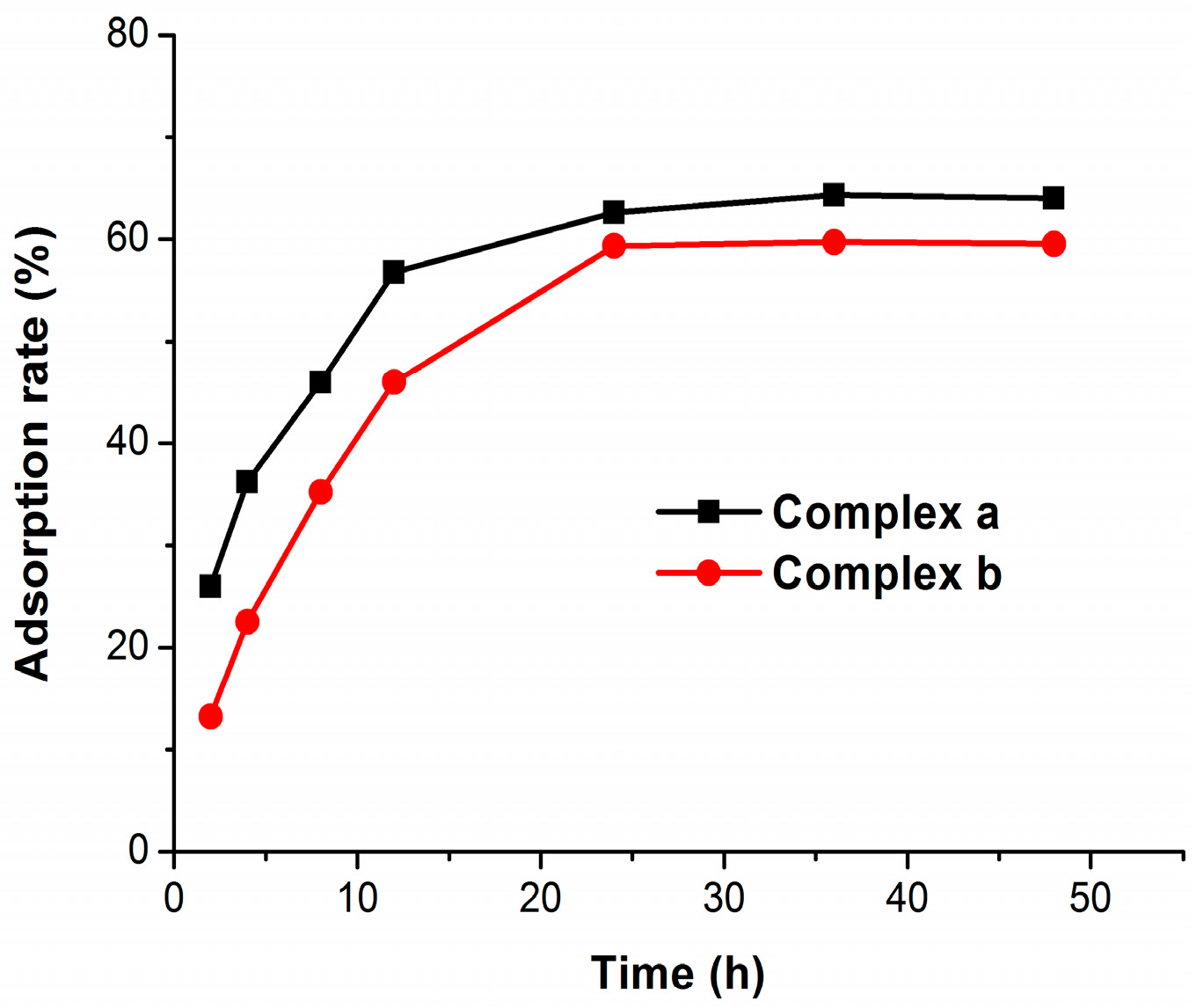
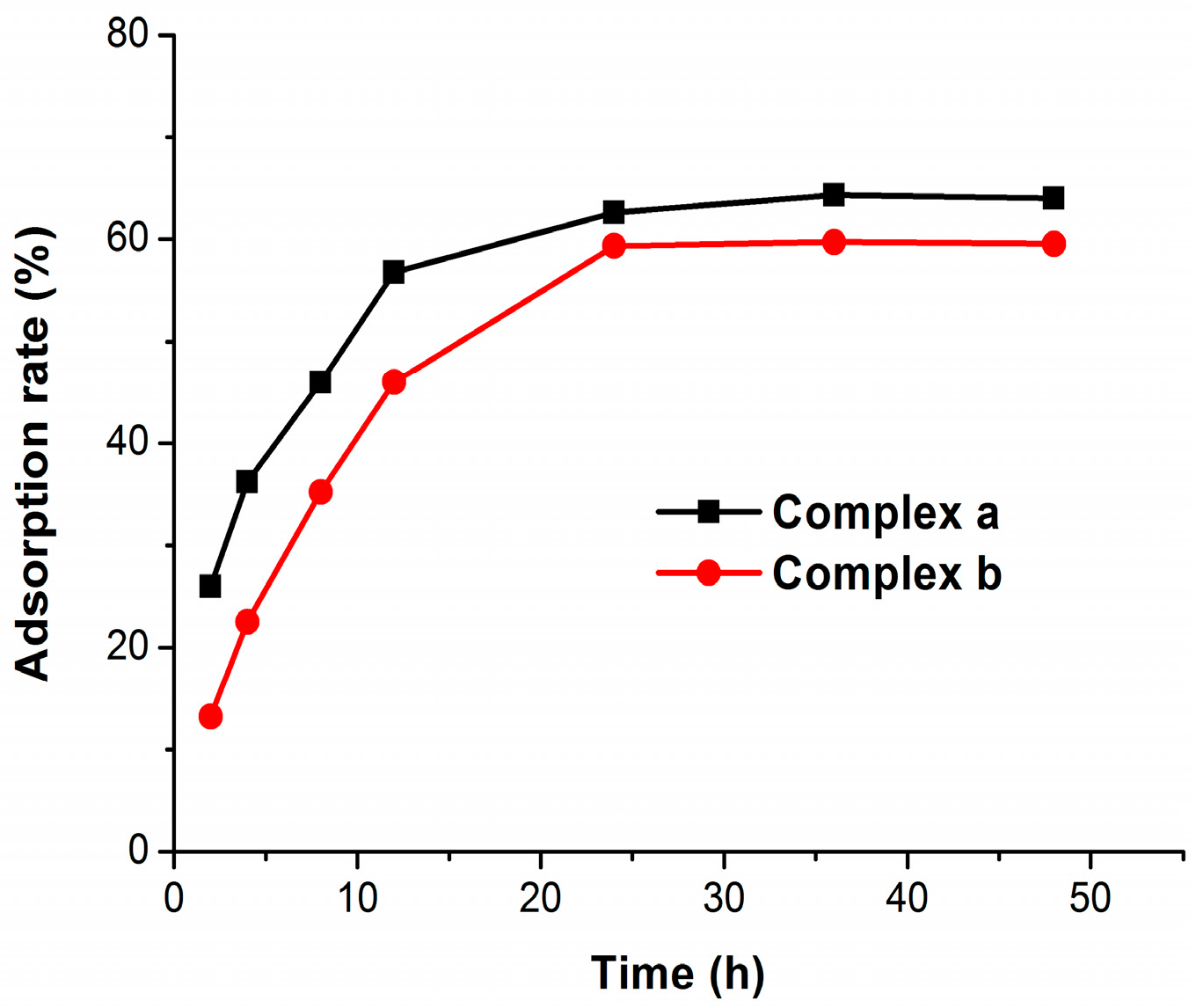
2.3. Bone Binding
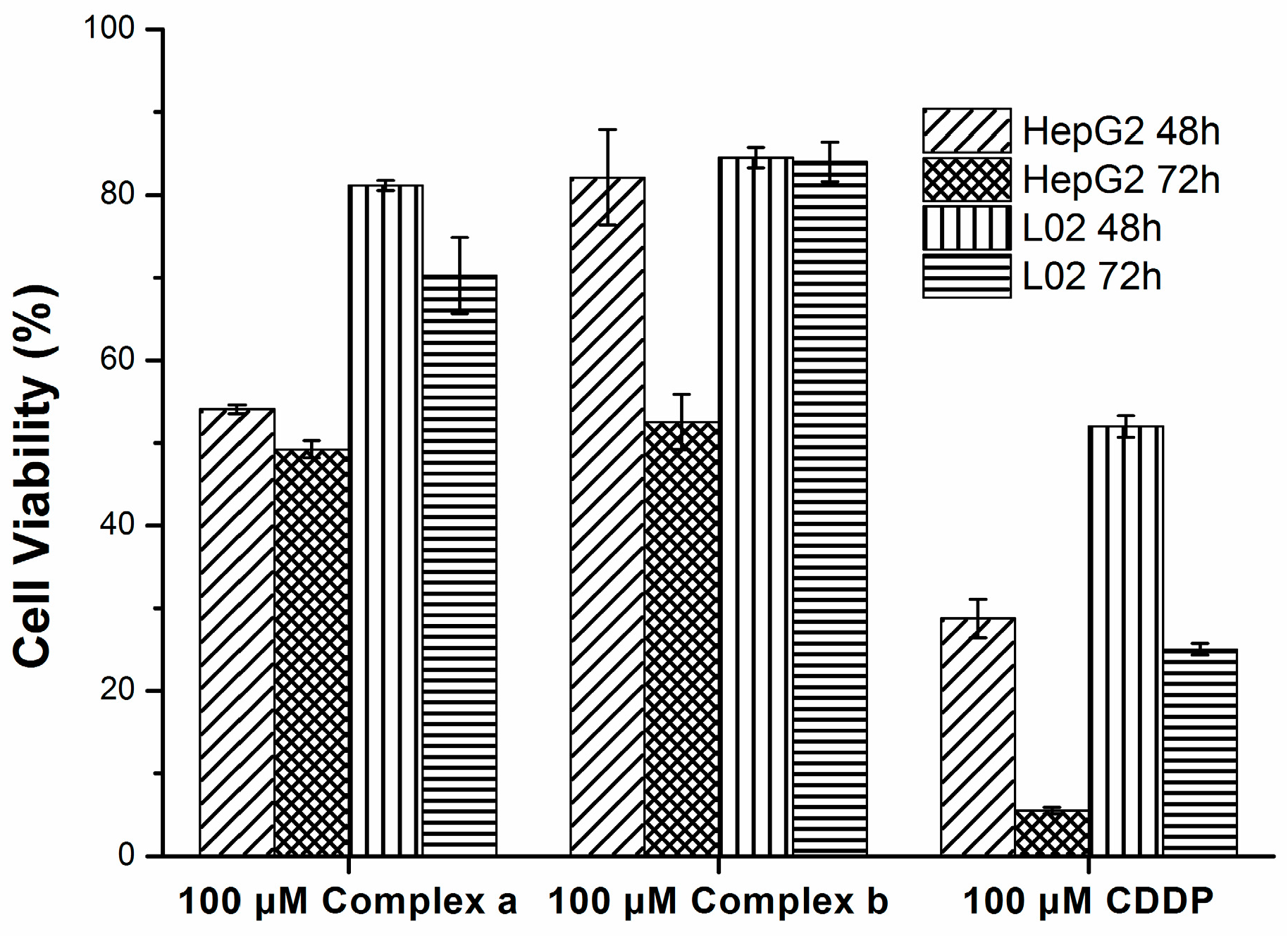
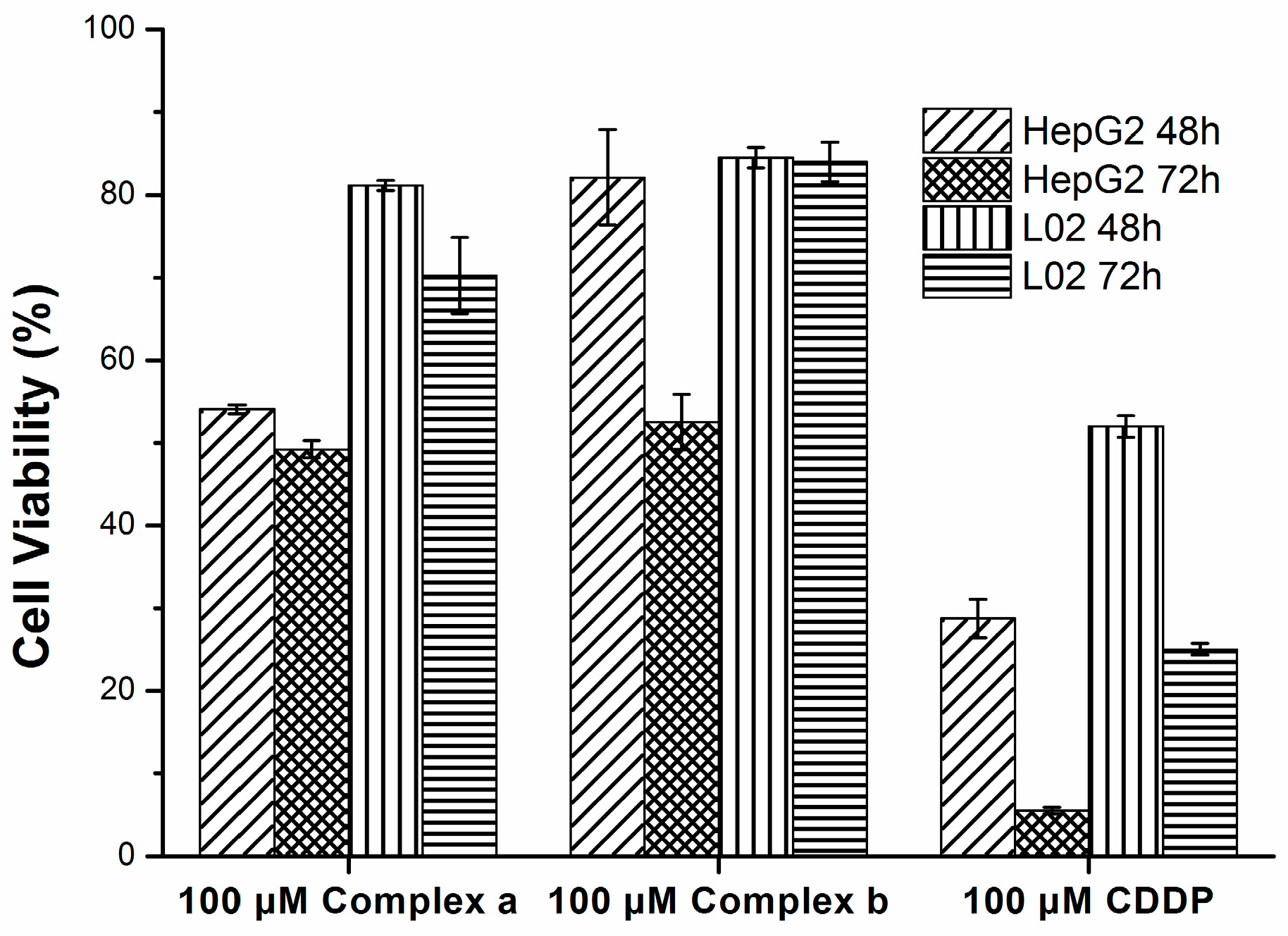
2.4. Cytotoxicity
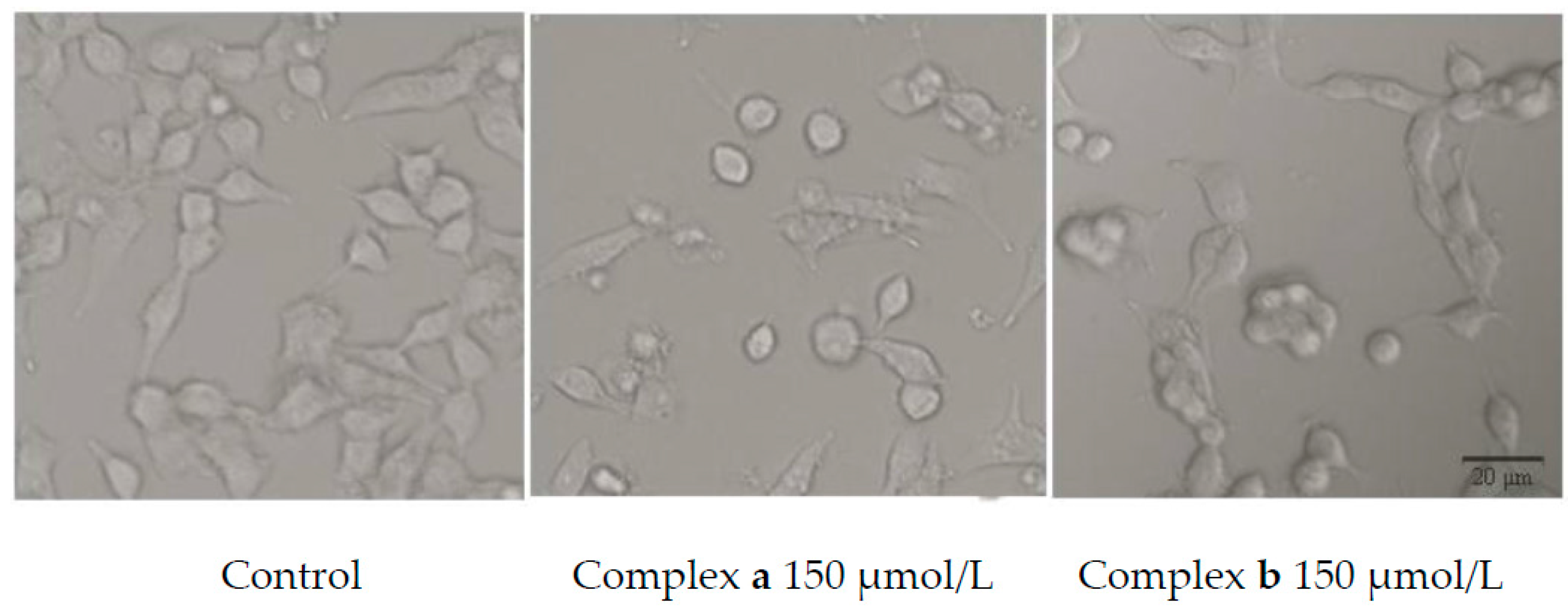
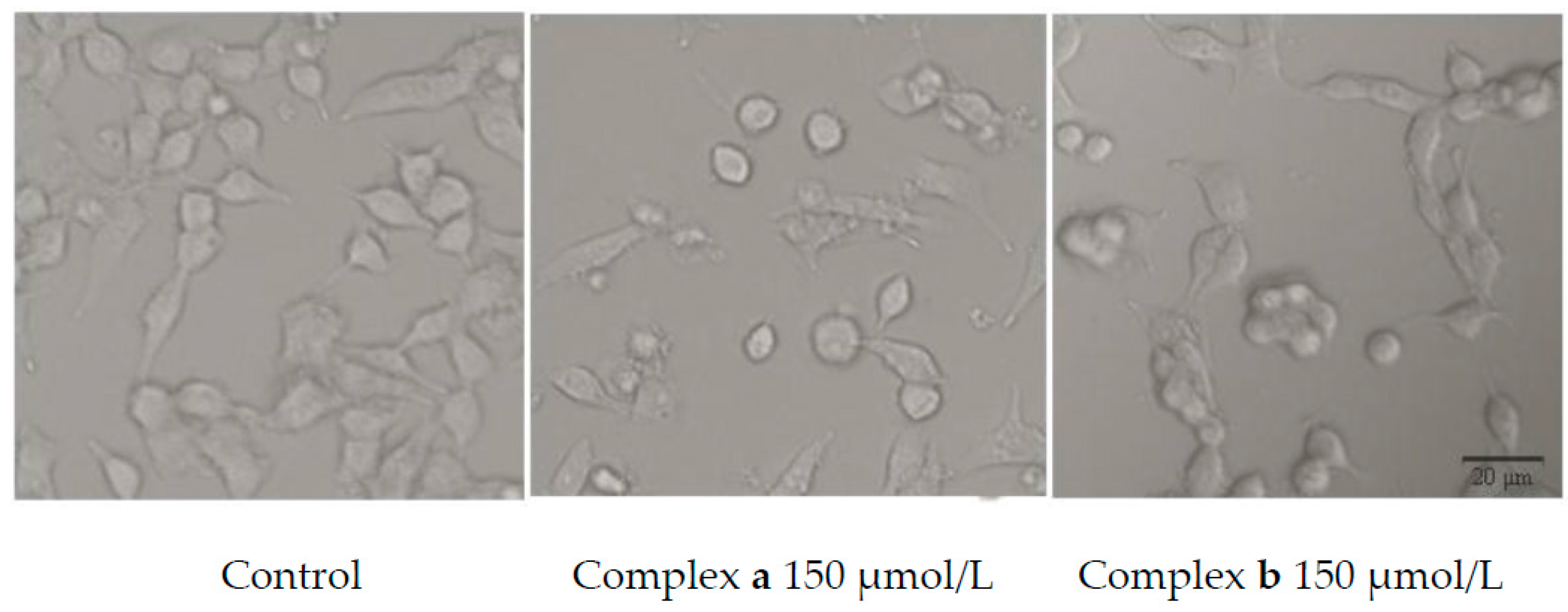
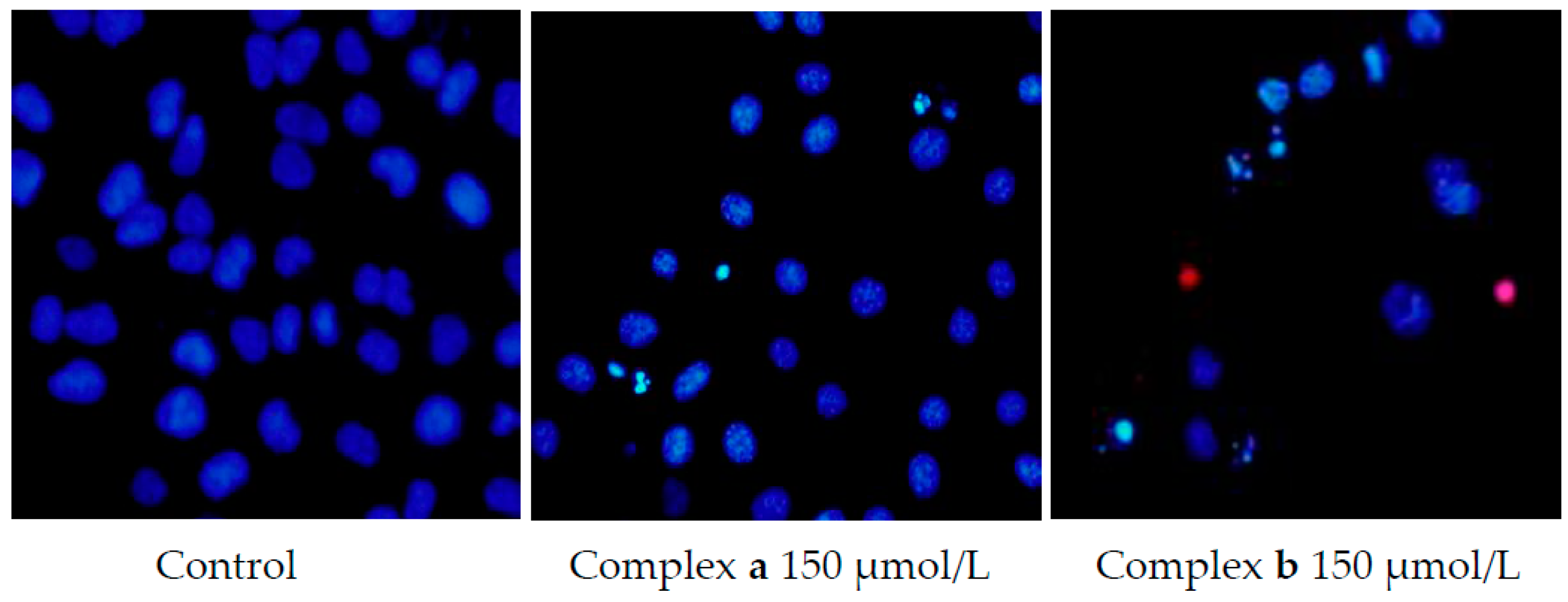
2.5. Morphology Study
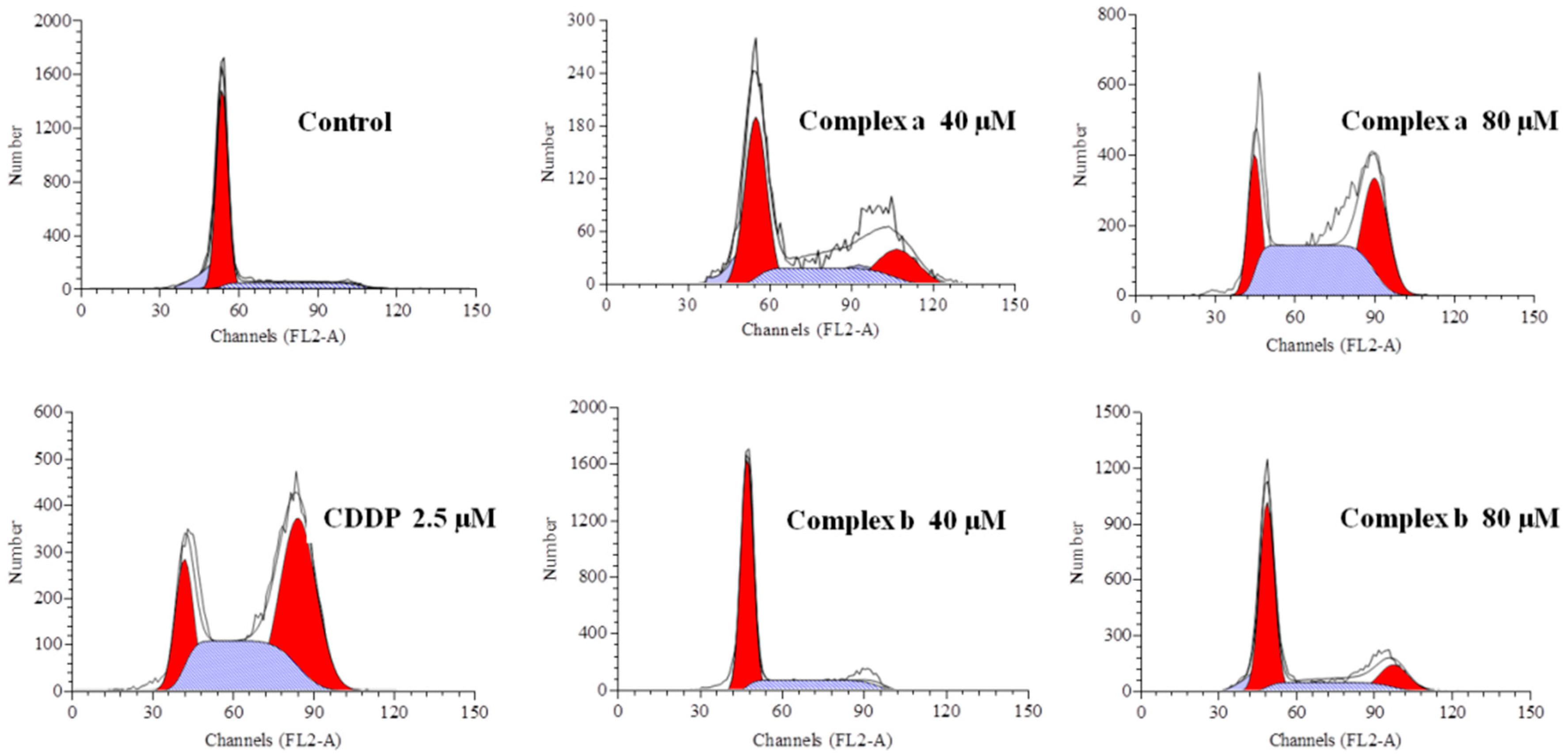
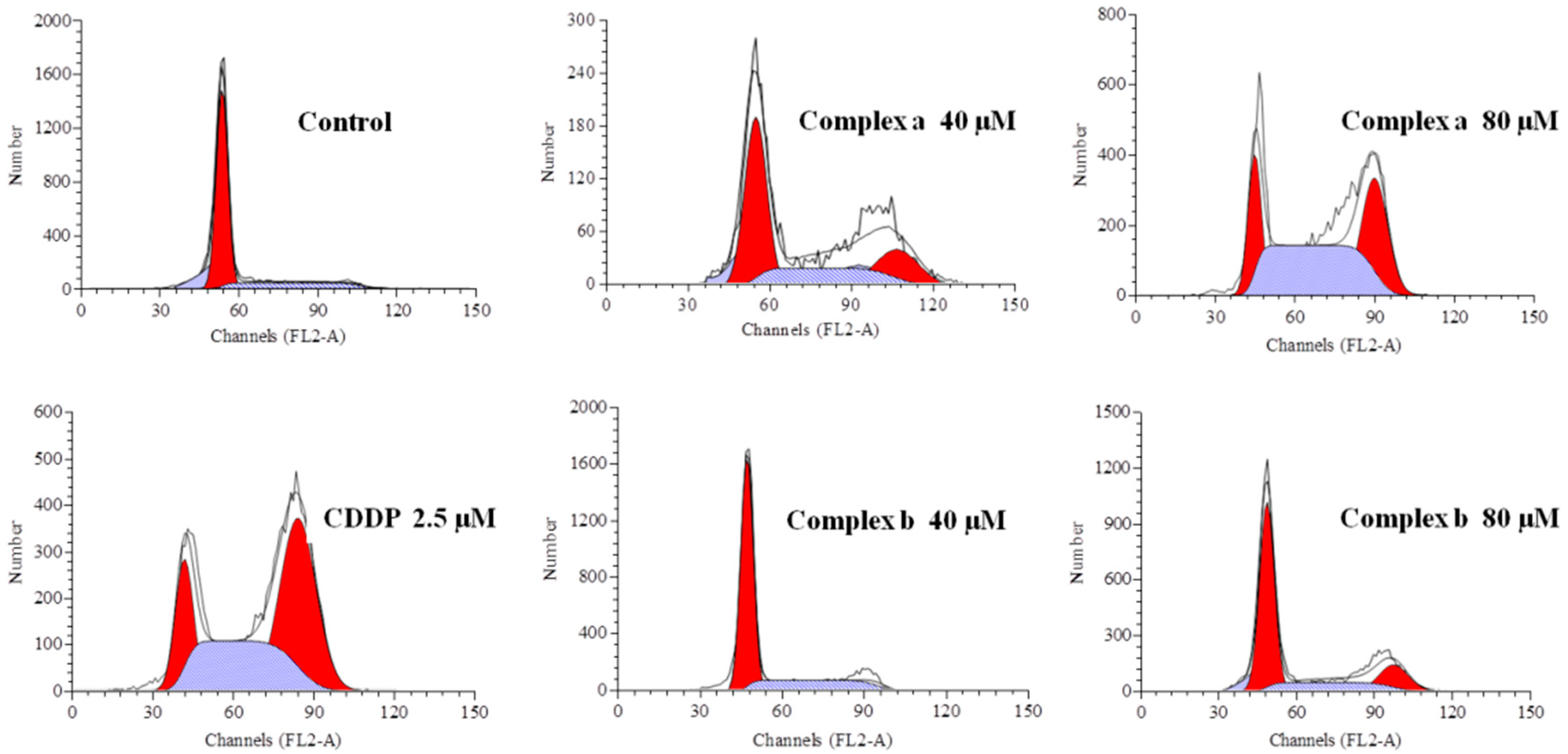
2.6. Cell Cycle Analysis
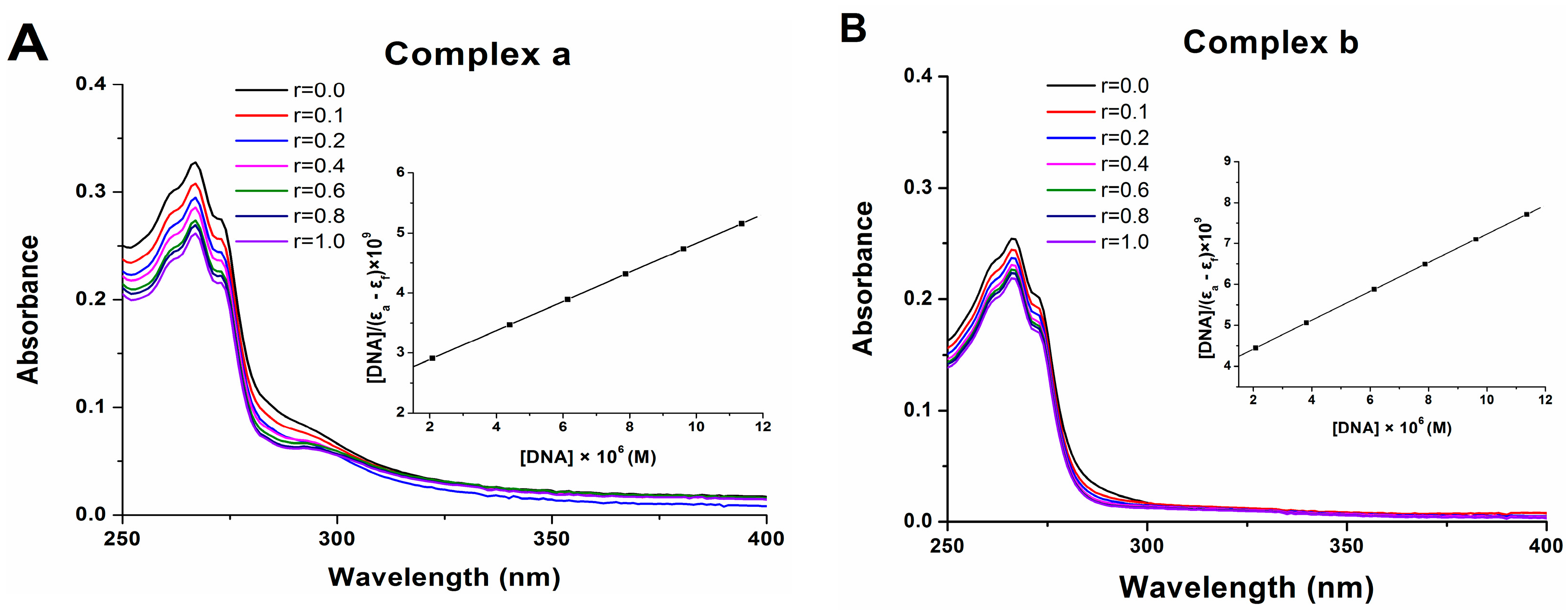
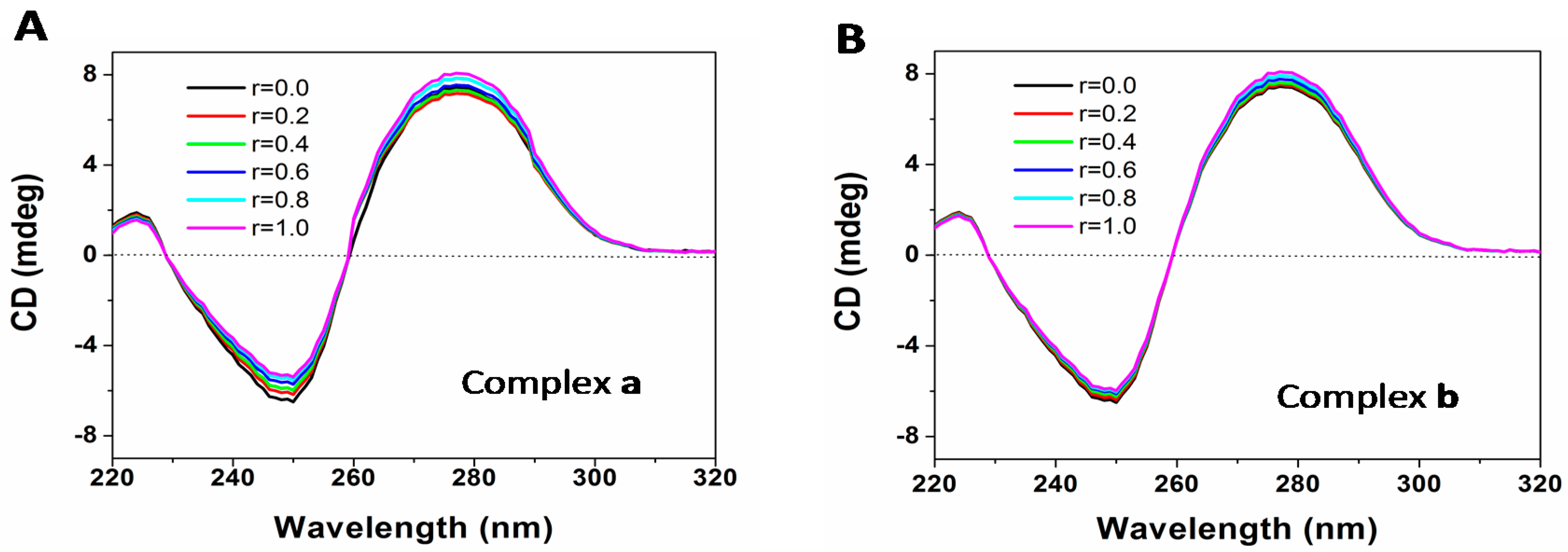
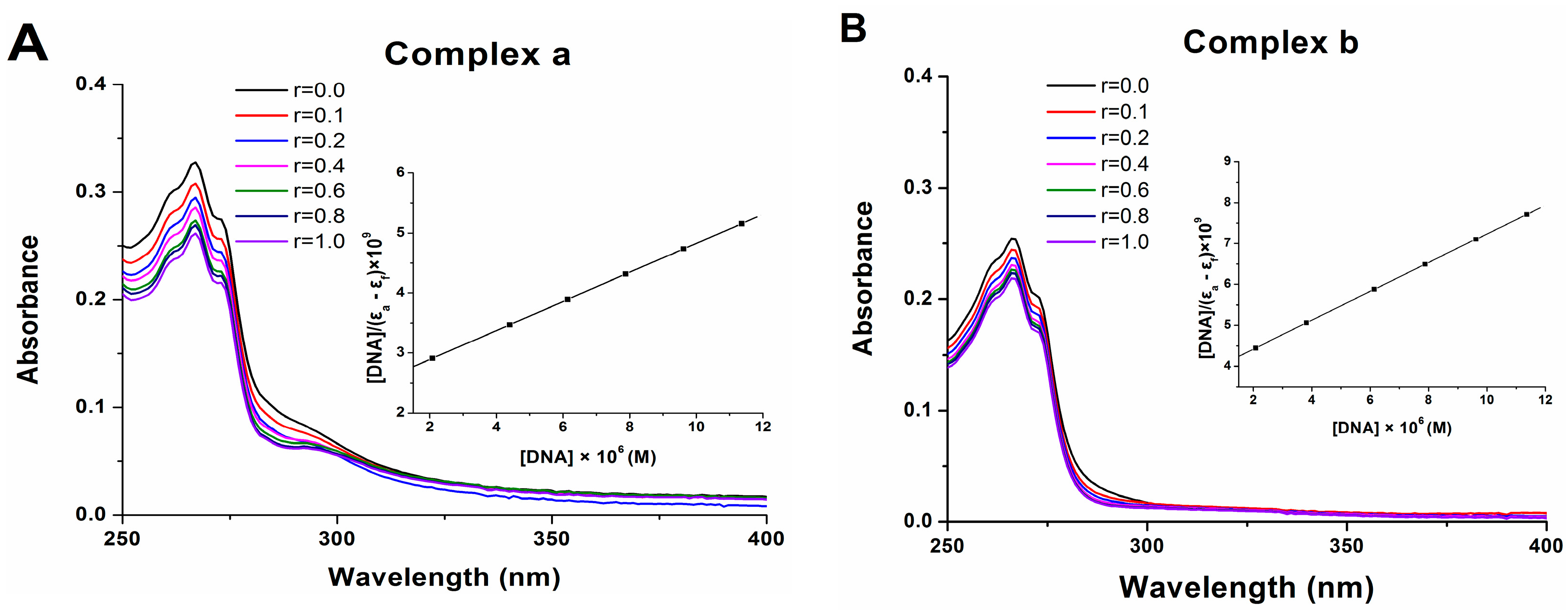
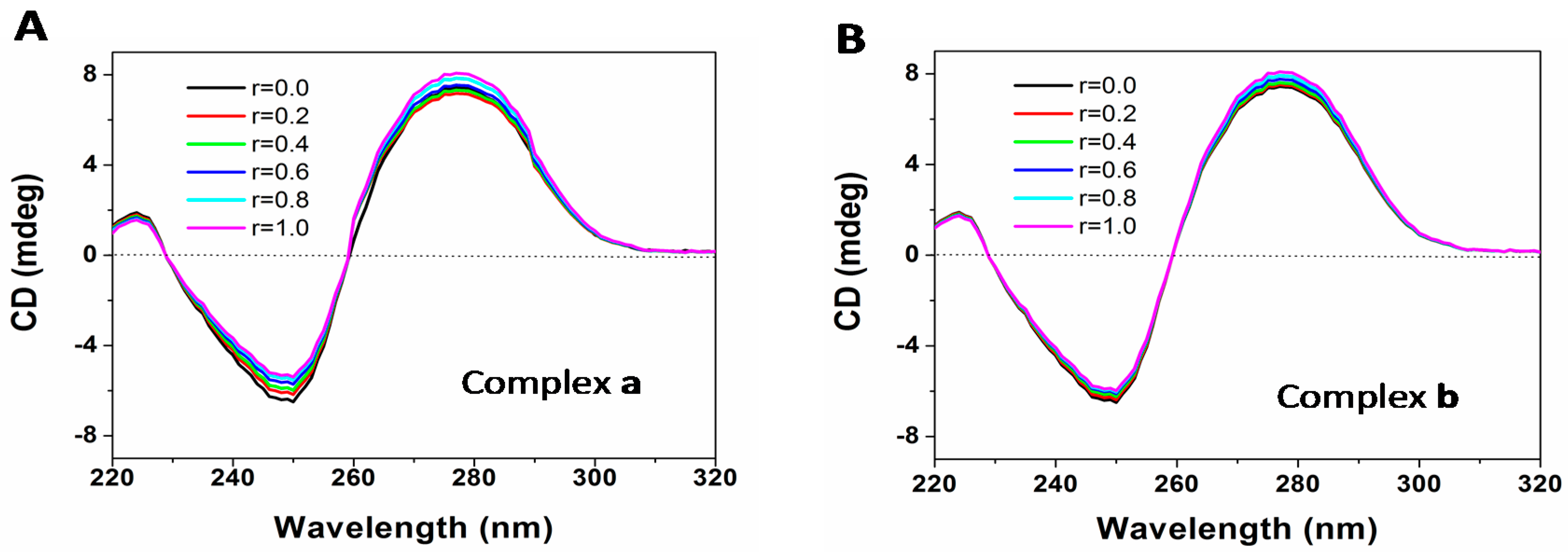
2.7. DNA Binding
3. Materials and Methods
3.1. General Information
3.2. Preparation of Ligands and Complexes
3.2.1. Preparation of Ligands
3.2.2. Preparation of Platinum Complexes
3.3. Lipid-Water Partition Coefficient
3.4. Bone Binding Assay
3.5. Cell Culture and Drug Treatment
3.6. Cytotoxicity Assay
3.7. Morphology Study
3.8. Cell Cycle Analysis
3.9. DNA Binding Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wong, E.; Giandomenico, C.M. Current status of platinum-based antitumor drugs. Chem. Rev. 1999, 99, 2451–2466. [Google Scholar] [CrossRef] [PubMed]
- Heffeter, P.; Jungwirth, U.; Jakupec, M.; Hartinger, C.; Galanski, M.; Elbling, L.; Micksche, M.; Keppler, B.; Berger, W. Resistance against novel anticancer metal compounds: Differences and similarities. Drug Resist. Updates 2008, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, Z. Targeting and delivery of platinum-based anticancer drugs. Chem. Soc. Rev. 2013, 42, 202–224. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.J.; Lippard, S.J. Synthetic methods for the preparation of platinum anticancer complexes. Chem. Rev. 2013, 114, 4470–4495. [Google Scholar] [CrossRef] [PubMed]
- Russell, R.G.G. Bisphosphonates: The first 40years. Bone 2011, 49, 2–19. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Gangal, G.; Uludağ, H. “Magic bullets” for bone diseases: Progress in rational design of bone-seeking medicinal agents. Chem. Soc. Rev. 2007, 36, 507–531. [Google Scholar] [CrossRef] [PubMed]
- Klenner, T.; Valenzuela-Paz, P.; Keppler, B.; Angres, G.; Scherf, H.; Wingen, F.; Amelung, F.; Schmähl, D. Cisplatin-linked phosphonates in the treatment of the transplantable osteosarcoma in vitro and in vivo. Cancer Treat. Rev. 1990, 17, 253–259. [Google Scholar] [CrossRef]
- Galanski, M.; Slaby, S.; Jakupec, M.A.; Keppler, B.K. Synthesis, characterization, and in vitro antitumor activity of osteotropic diam(m)ineplatinum(II) complexes bearing a N,N-bis(phosphonomethyl)glycine ligand. J. Med. Chem. 2003, 46, 4946–4951. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Ostuni, R.; Teoli, D.; Morpurgo, M.; Realdon, N.; Palazzo, B.; Natile, G. Bisphosphonate complexation and calcium doping in silica xerogels as a combined strategy for local and controlled release of active platinum antitumor compounds. Dalton Trans. 2007, 29, 3131–3139. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Ostuni, R.; Gandin, V.; Marzano, C.; Piccinonna, S.; Natile, G. Synthesis, characterization, and cytotoxicity of dinuclear platinum-bisphosphonate complexes to be used as prodrugs in the local treatment of bone tumours. Dalton Trans. 2009, 48, 10904–10913. [Google Scholar] [CrossRef] [PubMed]
- Margiotta, N.; Capitelli, F.; Ostuni, R.; Natile, G. A new dinuclear platinum complex with a nitrogen-containing geminal bisphosphonate as potential anticancer compound specifically targeted to bone tissues. J. Inorg. Biochem. 2008, 102, 2078–2086. [Google Scholar] [CrossRef] [PubMed]
- Piccinonna, S.; Margiotta, N.; Pacifico, C.; Lopalco, A.; Denora, N.; Fedi, S.; Corsini, M.; Natile, G. Dinuclear Pt (II)-bisphosphonate complexes: A scaffold for multinuclear or different oxidation state platinum drugs. Dalton Trans. 2012, 41, 9689–9699. [Google Scholar] [CrossRef] [PubMed]
- Qiu, L.; Lv, G.; Cao, Y.; Chen, L.; Yang, H.; Luo, S.; Zou, M.; Lin, J. Synthesis and biological evaluation of novel platinum complexes of imidazolyl-containing bisphosphonates as potential anticancer agents. J. Biol. Inorg. Chem. 2015, 20, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Xue, Z.; Lin, M.; Zhu, J.; Zhang, J.; Li, Y.; Guo, Z. Platinum(II) compounds bearing bone-targeting group: Synthesis, crystal structure and antitumor activity. Chem. Commun. 2010, 46, 1212–1214. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.-B.; Chen, Z.-F.; Liu, Y.-C.; Li, Z.-Q.; Wei, J.-H.; Wang, M.; Xie, X.-L.; Liang, H. Platinum(II) complexes containing aminophosphonate esters: Synthesis, characterization, cytotoxicity and action mechanism. Eur. J. Med. Chem. 2013, 64, 554–561. [Google Scholar] [CrossRef] [PubMed]
- De Rosales, R.T.M.; Finucane, C.; Mather, S.; Blower, P. Bifunctional bisphosphonate complexes for the diagnosis and therapy of bone metastases. Chem. Commun. 2009, 32, 4847–4849. [Google Scholar] [CrossRef] [PubMed]
- Tarcomnicu, I.; Gheorghea, M.C.; Silvestro, L.; Savu, S.R.; Boarua, I.; Tudoroniua, A. High-throughput HPLC-MS/MS method to determine ibandronate in human plasma for pharmacokinetic applications. J. Chromatogr. B 2009, 877, 3159–3168. [Google Scholar] [CrossRef] [PubMed]
- Price, J.H.; Williamson, A.N.; Schramm, R.F.; Wayland, B.B. Palladium (II) and platinum (II) alkyl sulfoxide complexes. Examples of sulfur-bonded, mixed sulfur-and oxygen-bonded, and totally oxygen-bonded complexes. Inorg. Chem. 1972, 11, 1280–1284. [Google Scholar] [CrossRef]
- Wu, S.; Zhu, C.; Zhang, C.; Yu, Z.; He, W.; He, Y.; Li, Y.; Wang, J.; Guo, Z. In vitro and in vivo fluorescent imaging of a monofunctional chelated platinum complex excitable using visible light. Inorg. Chem. 2011, 50, 11847–11849. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; He, W.; Shi, P.; Zhu, J.; Qiu, L.; Lin, L.; Guo, Z. A positively charged trinuclear 3N-chelated monofunctional platinum complex with high DNA affinity and potent cytotoxicity. Dalton Trans. 2006, 22, 2617–2619. [Google Scholar] [CrossRef] [PubMed]
- Foteeva, L.S.; Trofimov, D.A.; Kuznetsova, O.V.; Kowol, C.R.; Arion, V.B.; Keppler, B.K.; Timerbaev, A.R. A quantitative structure-activity approach for lipophilicity estimation of antitumor complexes of different metals using microemulsion electrokinetic chromatography. J. Pharm. Biomed. Anal. 2011, 55, 409–413. [Google Scholar] [CrossRef] [PubMed]
- Palazzo, B.; Iafisco, M.; Laforgia, M.; Margiotta, N.; Natile, G.; Bianchi, C.L.; Walsh, D.; Mann, S.; Roveri, N. Biomimetic hydroxyapatite-drug nanocrystals as potential bone substitutes with antitumor drug delivery properties. Adv. Funct. Mater. 2007, 17, 2180–2188. [Google Scholar] [CrossRef]
- Takahara, P.M.; Rosenzweig, A.C.; Frederick, C.A.; Lippard, S.J. Crystal structure of double-stranded DNA containing the major adduct of the anticancer drug cisplatin. Nature 1995, 377, 649–652. [Google Scholar] [CrossRef] [PubMed]
- Macquet, J.P.; Butour, J.L. A circular dichroism study of DNA platinum complexes. Eur. J. Biochem. 1978, 83, 375–387. [Google Scholar] [CrossRef] [PubMed]
- Carotti, S.; Marcon, G.; Marussich, M.; Mazzei, T.; Messori, L.; Mini, E.; Orioli, P. Cytotoxicity and DNA binding properties of a chloro glycylhistidinate gold(III) complex (GHAu). Chem. Biol. Interact. 2000, 125, 29–38. [Google Scholar] [CrossRef]
- Wolfe, A.; Shimer, G.H., Jr.; Meehan, T. Polycyclic aromatic hydrocarbons physically intercalate into duplex regions of denatured DNA. Biochemistry 1987, 26, 6392–6396. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Complex | IC50 (μmol/L) ± SD | ||||
|---|---|---|---|---|---|
| U2OS | A549 | HCT116 | MDA-MB-231 | HepG2 | |
| CDDP (48 h) | 11.04 ± 0.34 | 19.85 ± 1.49 | 16.20 ± 0.44 | 29.94 ± 1.65 | 6.78 ± 0.54 |
| CDDP (72 h) | 10.82 ± 0.37 | 15.38 ± 0.58 | 11.80 ± 0.26 | 14.05 ± 1.01 | 5.63 ± 0.34 |
| Complex a (48 h) | 251.74 ± 1.97 | 180.23 ± 0.93 | 273.02 ± 1.41 | 190.59 ± 0.34 | 170.72 ± 0.66 |
| Complex a (72 h) | 166.41 ± 1.04 | 108.72 ± 5.34 | 209.50 ± 0.80 | 111.13 ± 1.37 | 139.79 ± 0.42 |
| Complex b (48 h) | 262.92 ± 6.46 | 258.17 ± 3.88 | 638.47 ± 9.65 | 324.54 ± 5.83 | 271.12 ± 1.80 |
| Complex b (72 h) | 202.82 ± 0.94 | 237.31 ± 0.39 | 376.59 ± 0.43 | 245.15 ± 6.68 | 160.06 ± 3.20 |
| Phase | Control | Complex a | Complex a | Complex b | Complex b | CDDP |
|---|---|---|---|---|---|---|
| 40 μM | 80 μM | 40 μM | 80 μM | 2.5 μM | ||
| G0/G1 (%) | 75.28 | 58.79 | 17.89 | 73.30 | 45.78 | 17.49 |
| S (%) | 22.35 | 16.17 | 37.16 | 18.59 | 24.58 | 43.06 |
| G2/M (%) | 2.37 | 25.04 | 44.95 | 7.71 | 29.66 | 39.45 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qiu, L.; Liu, H.; Li, K.; Lv, G.; Yang, H.; Qin, X.; Lin, J. Preparation and Biological Evaluation of Two Novel Platinum(II) Complexes Based on the Ligands of Dipicolyamine Bisphosphonate Esters. Molecules 2016, 21, 255. https://doi.org/10.3390/molecules21030255
Qiu L, Liu H, Li K, Lv G, Yang H, Qin X, Lin J. Preparation and Biological Evaluation of Two Novel Platinum(II) Complexes Based on the Ligands of Dipicolyamine Bisphosphonate Esters. Molecules. 2016; 21(3):255. https://doi.org/10.3390/molecules21030255
Chicago/Turabian StyleQiu, Ling, Hong Liu, Ke Li, Gaochao Lv, Hui Yang, Xiaofeng Qin, and Jianguo Lin. 2016. "Preparation and Biological Evaluation of Two Novel Platinum(II) Complexes Based on the Ligands of Dipicolyamine Bisphosphonate Esters" Molecules 21, no. 3: 255. https://doi.org/10.3390/molecules21030255