
Influence of Thermal Treatment Conditions on the Properties of Dental Silicate Cements

Abstract
:1. Introduction
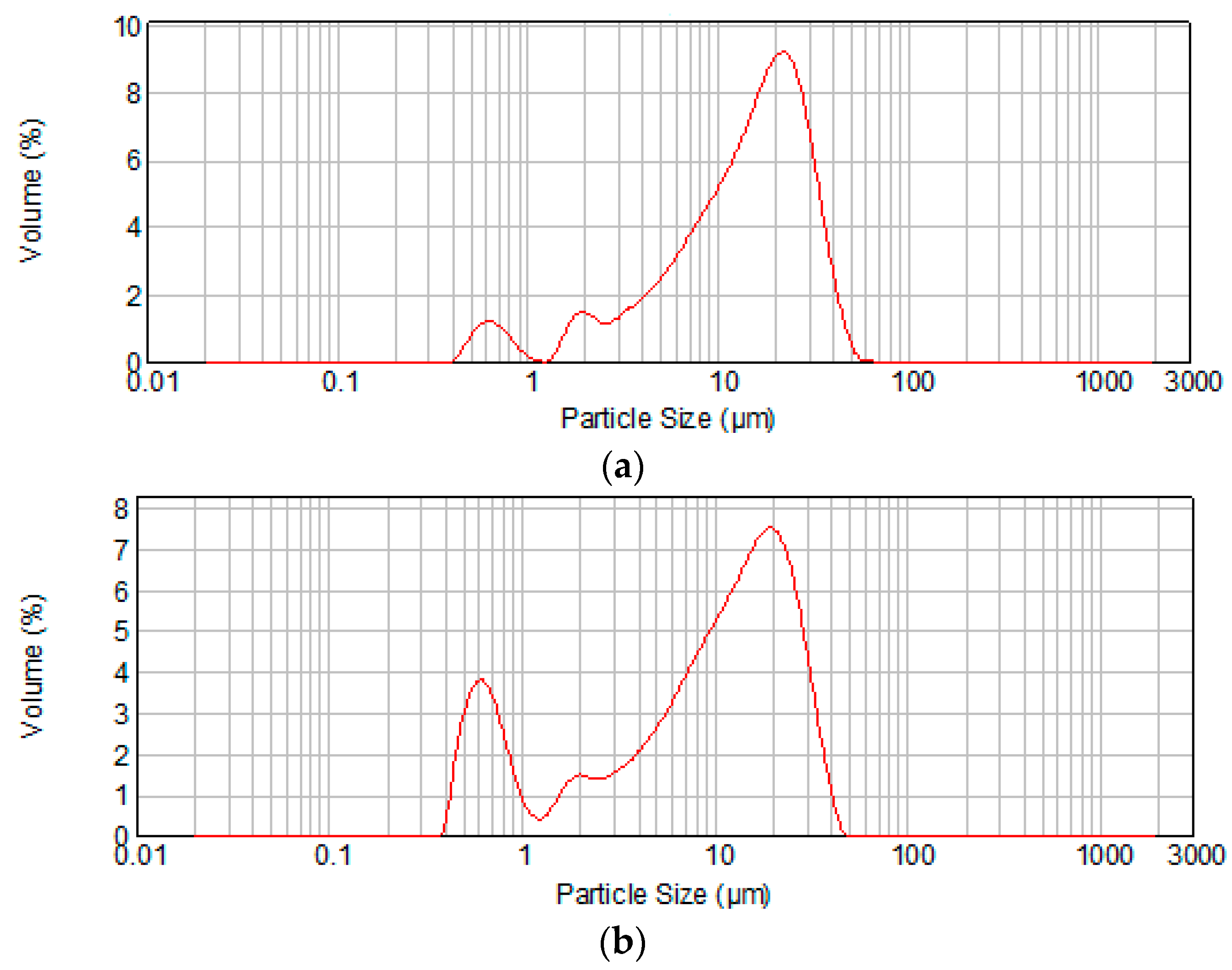
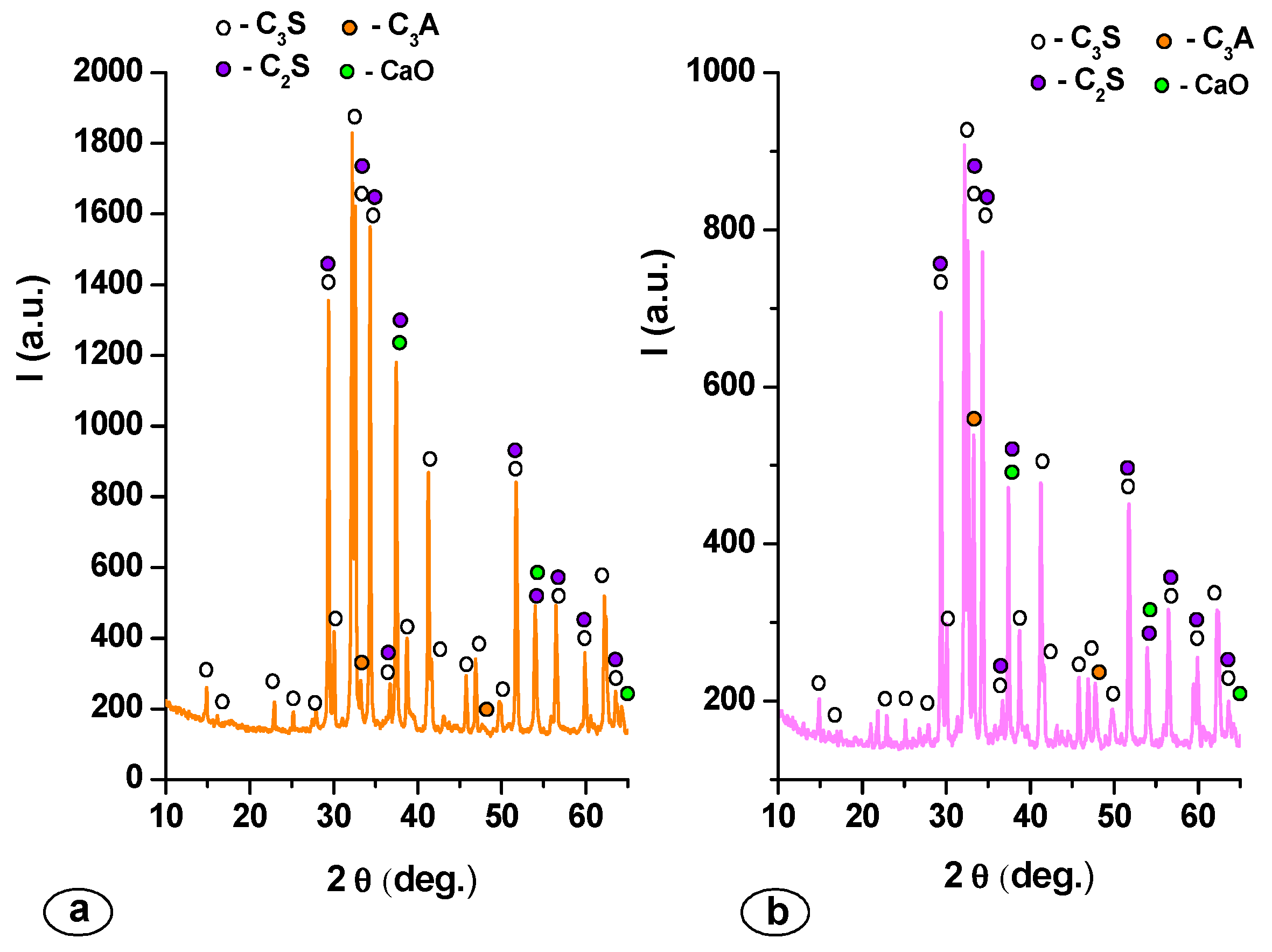
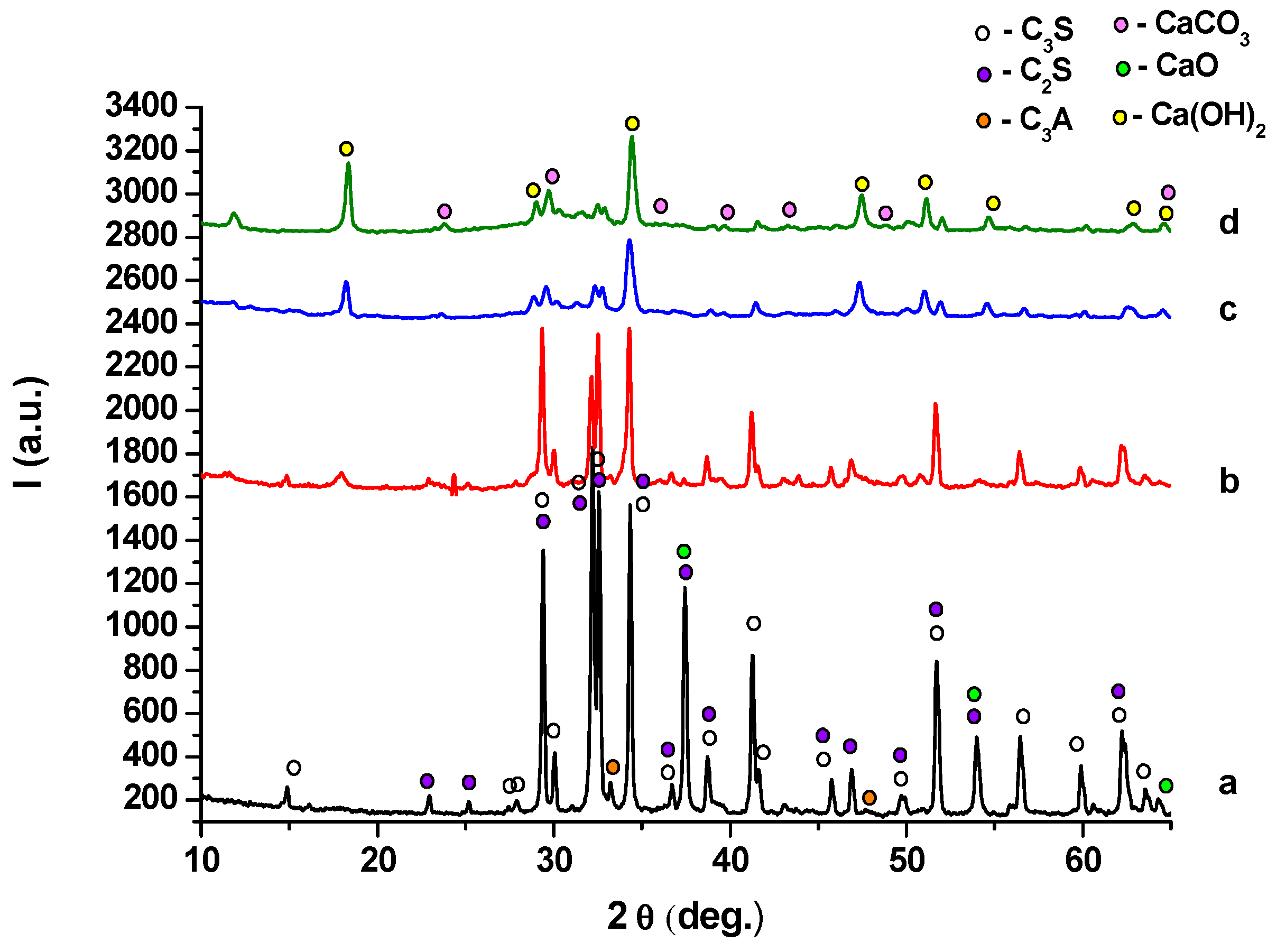
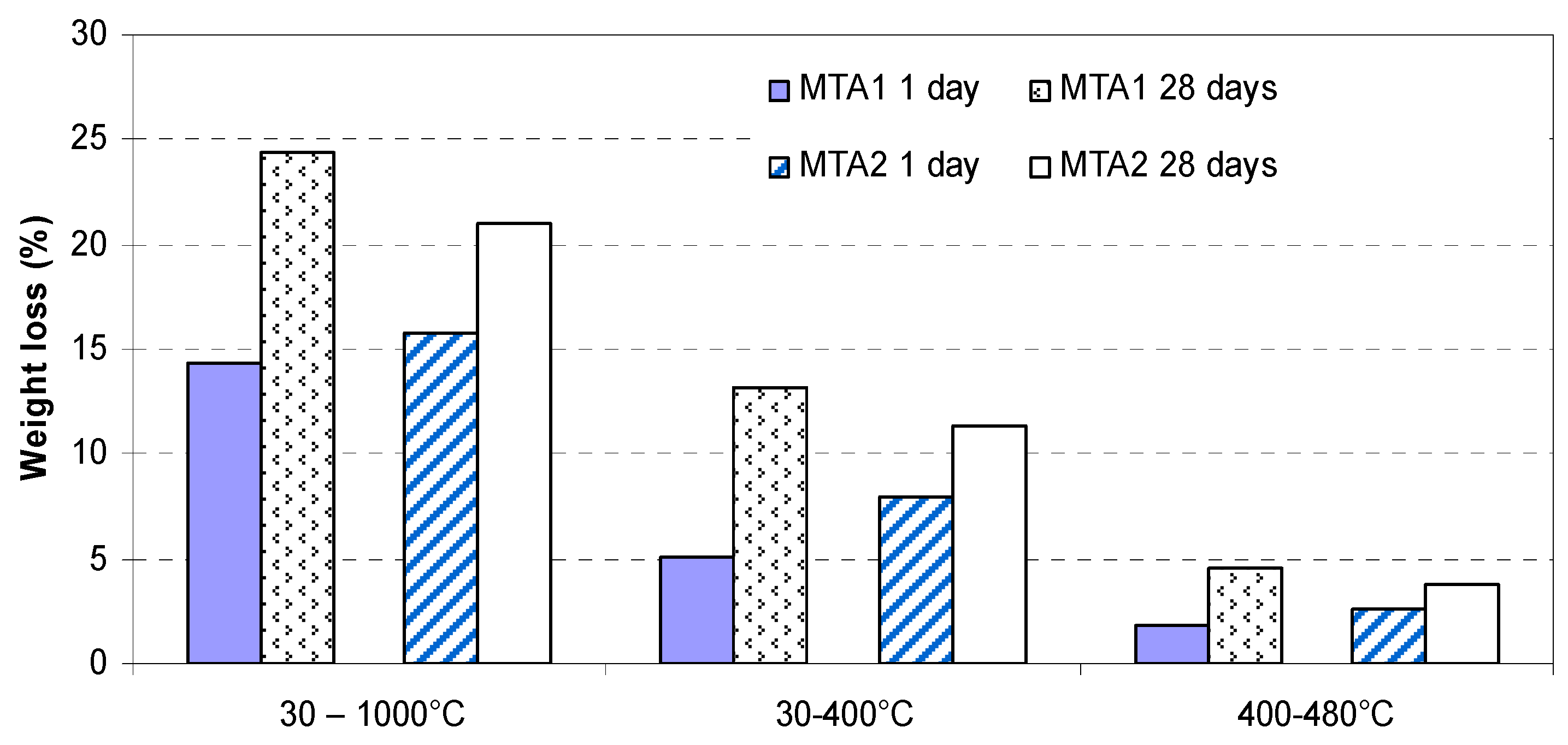
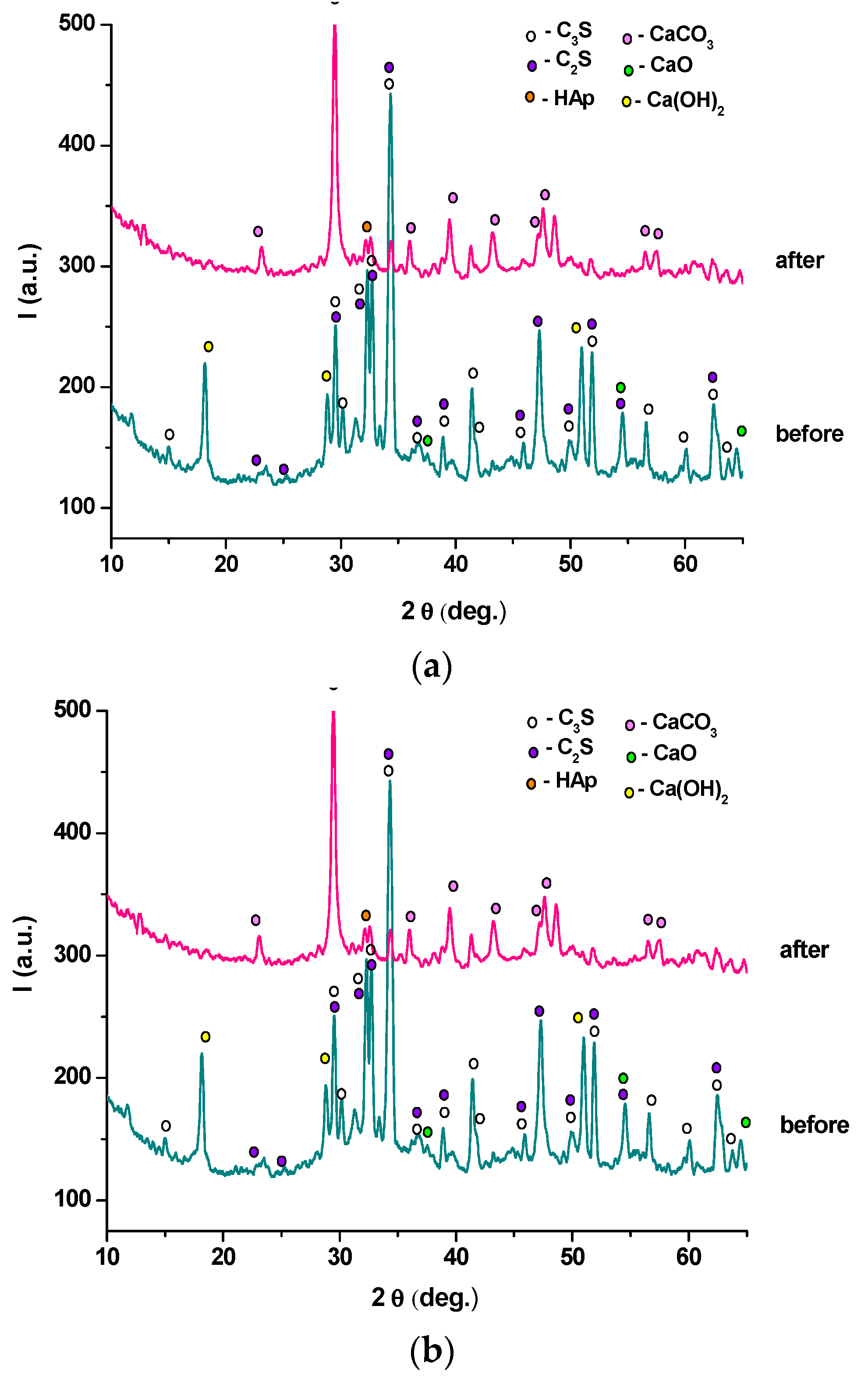
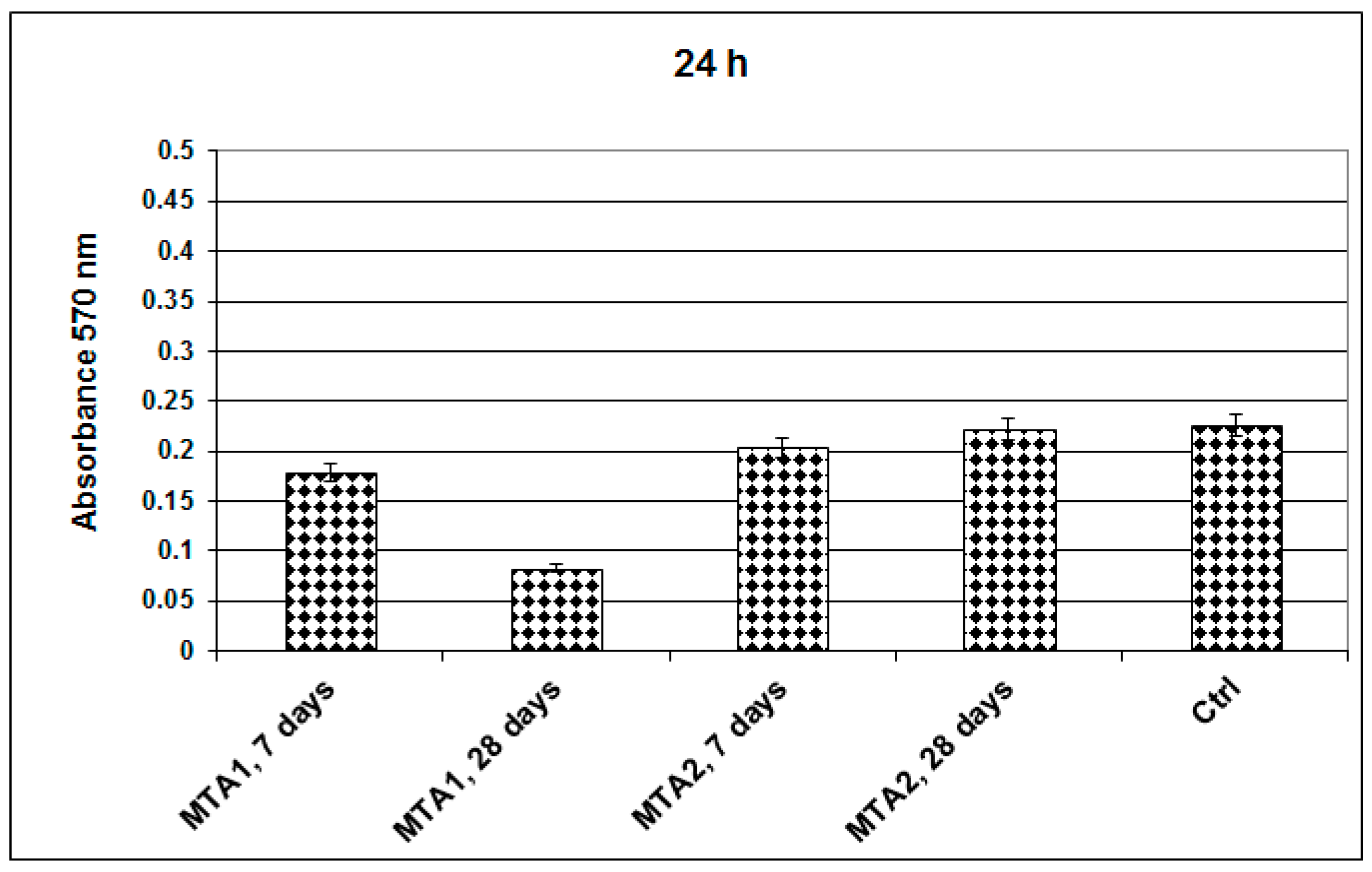
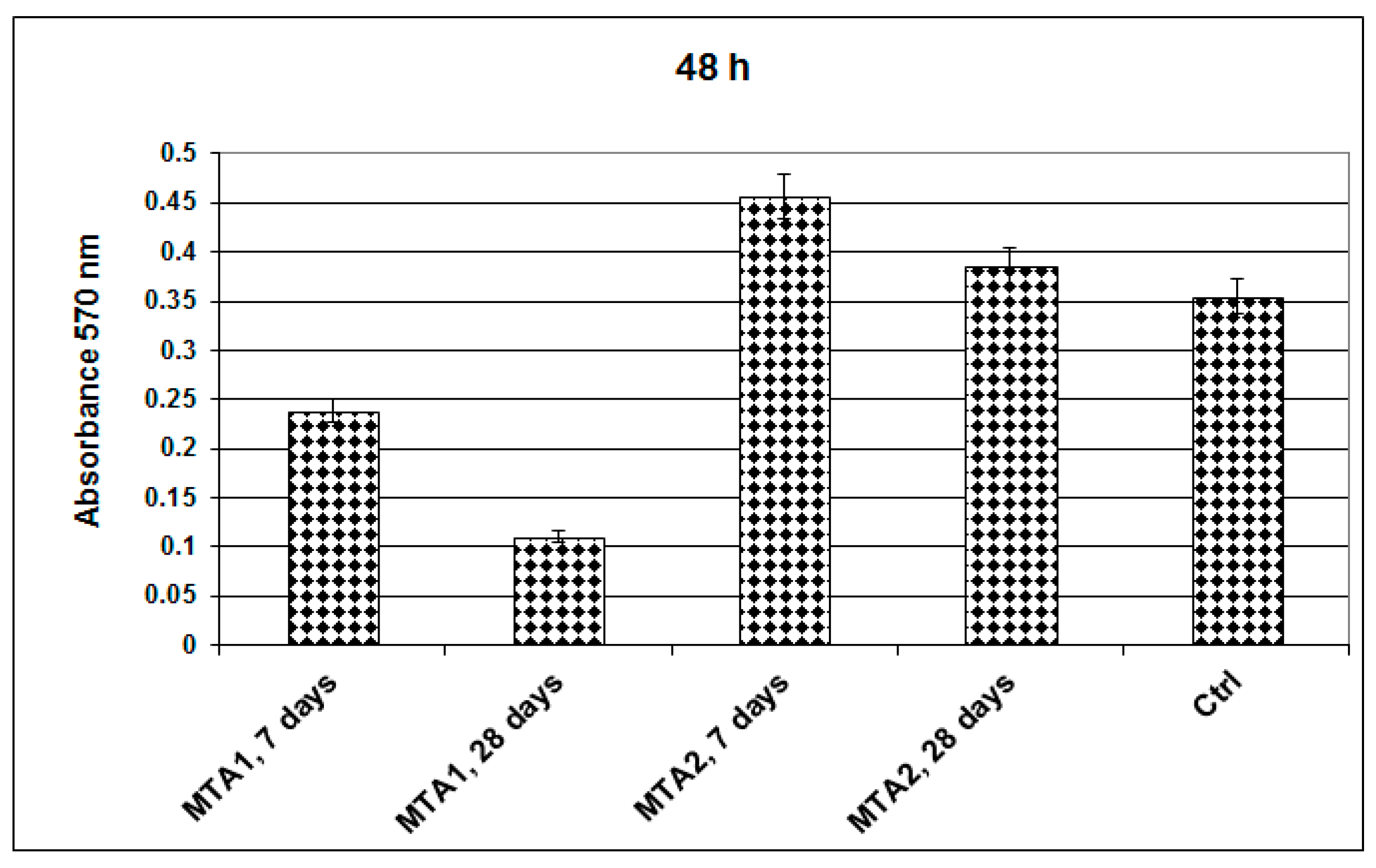
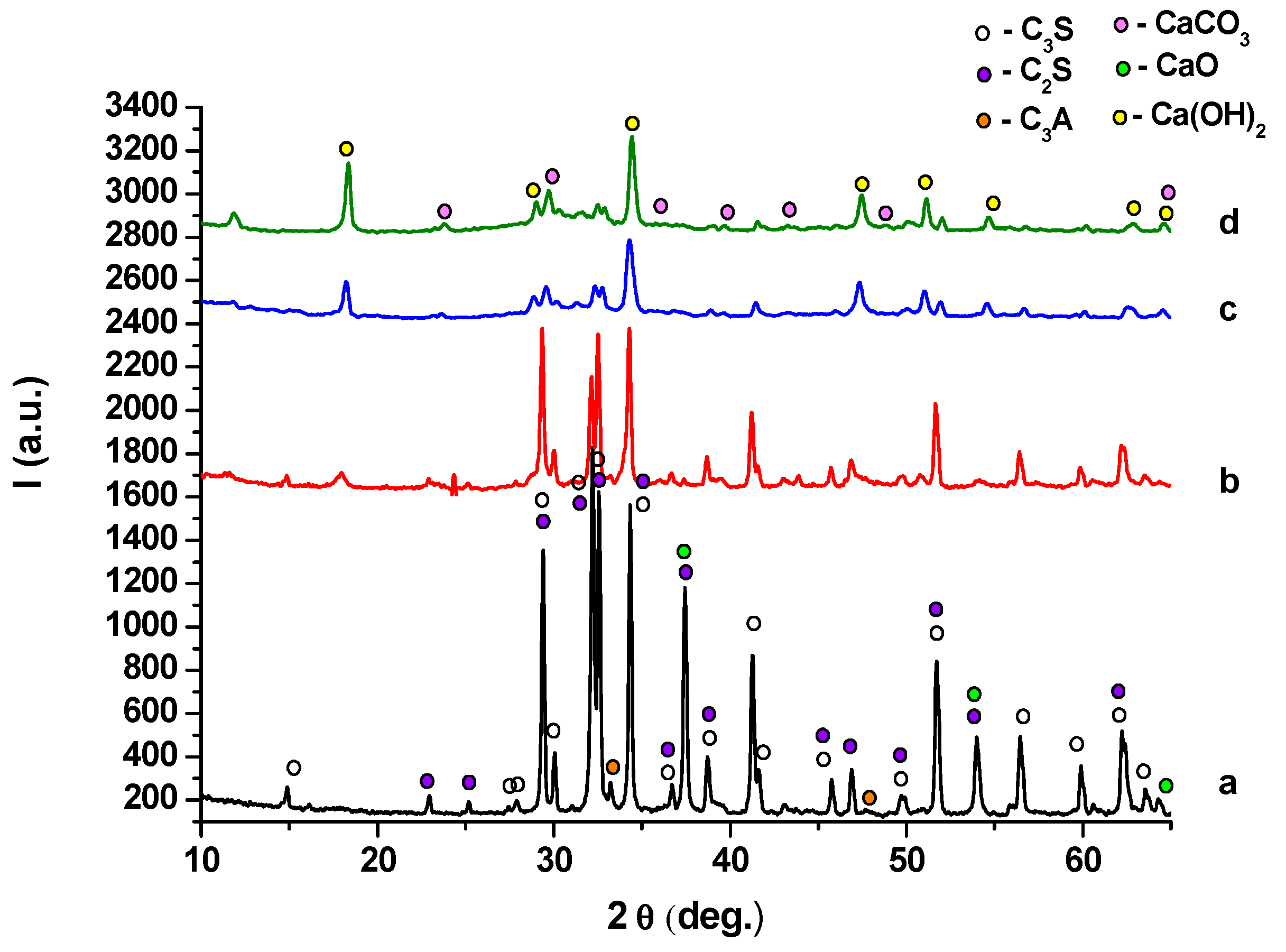
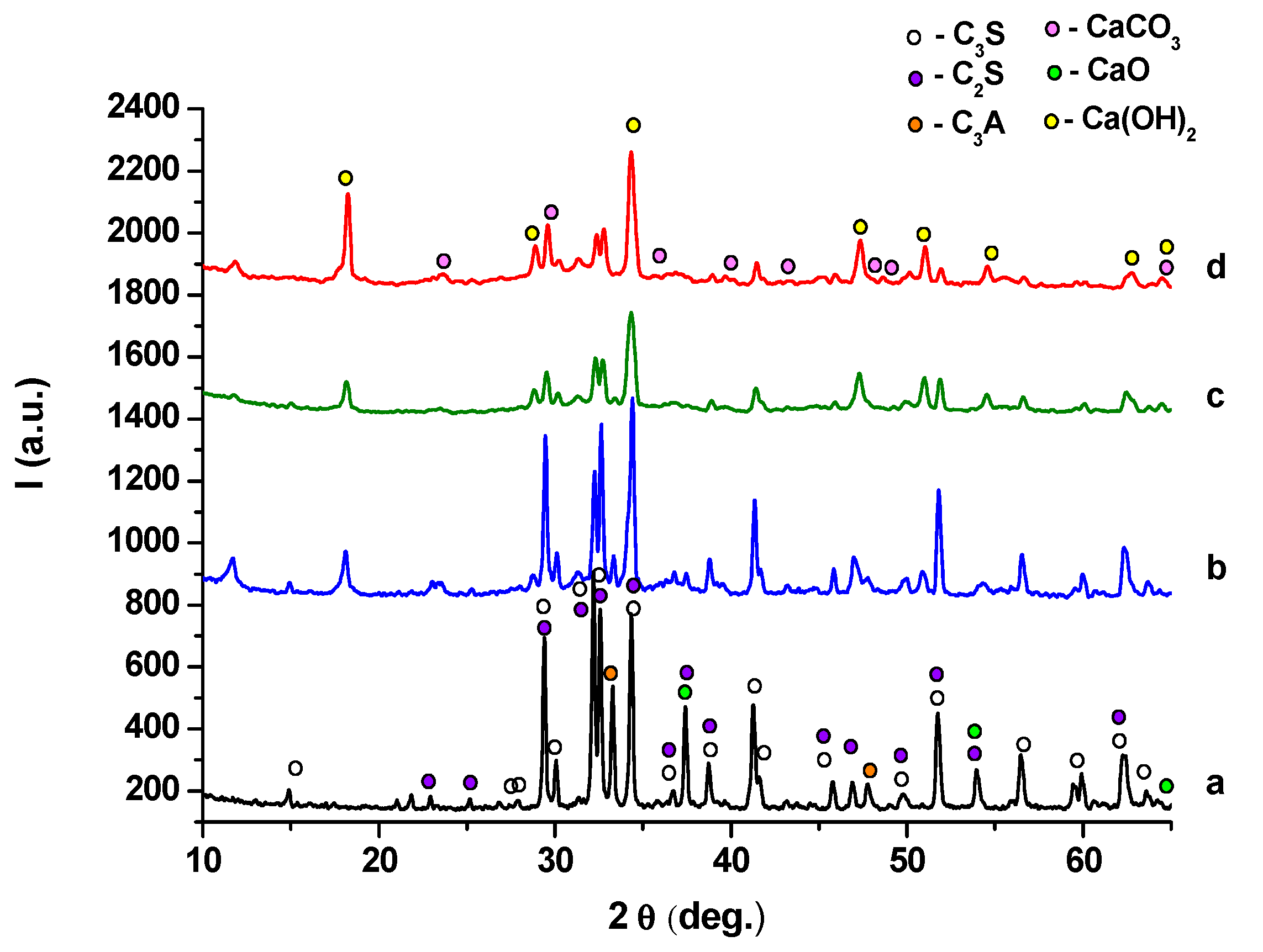
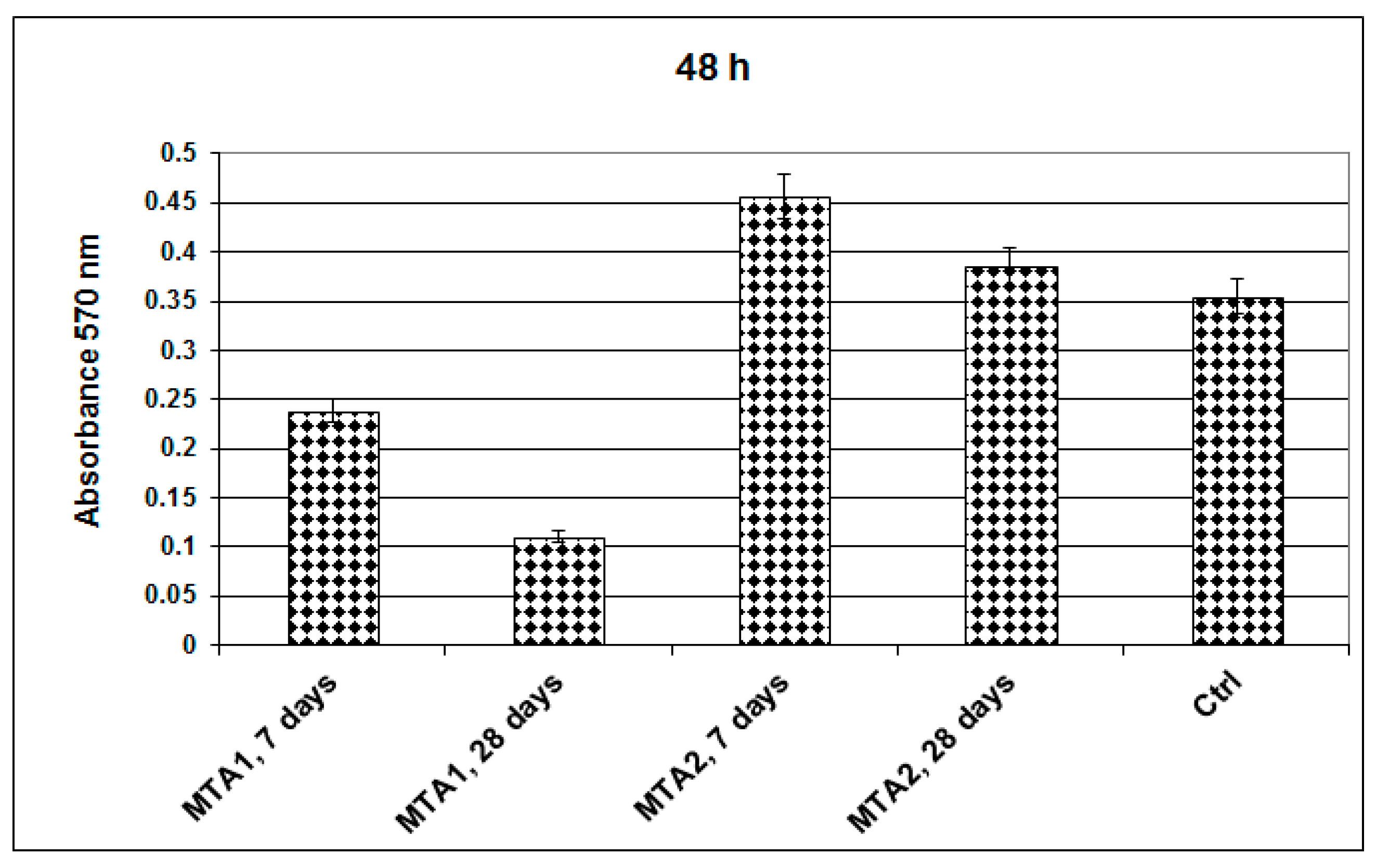
2. Results and Discussion
- (i)
- (ii)
- -
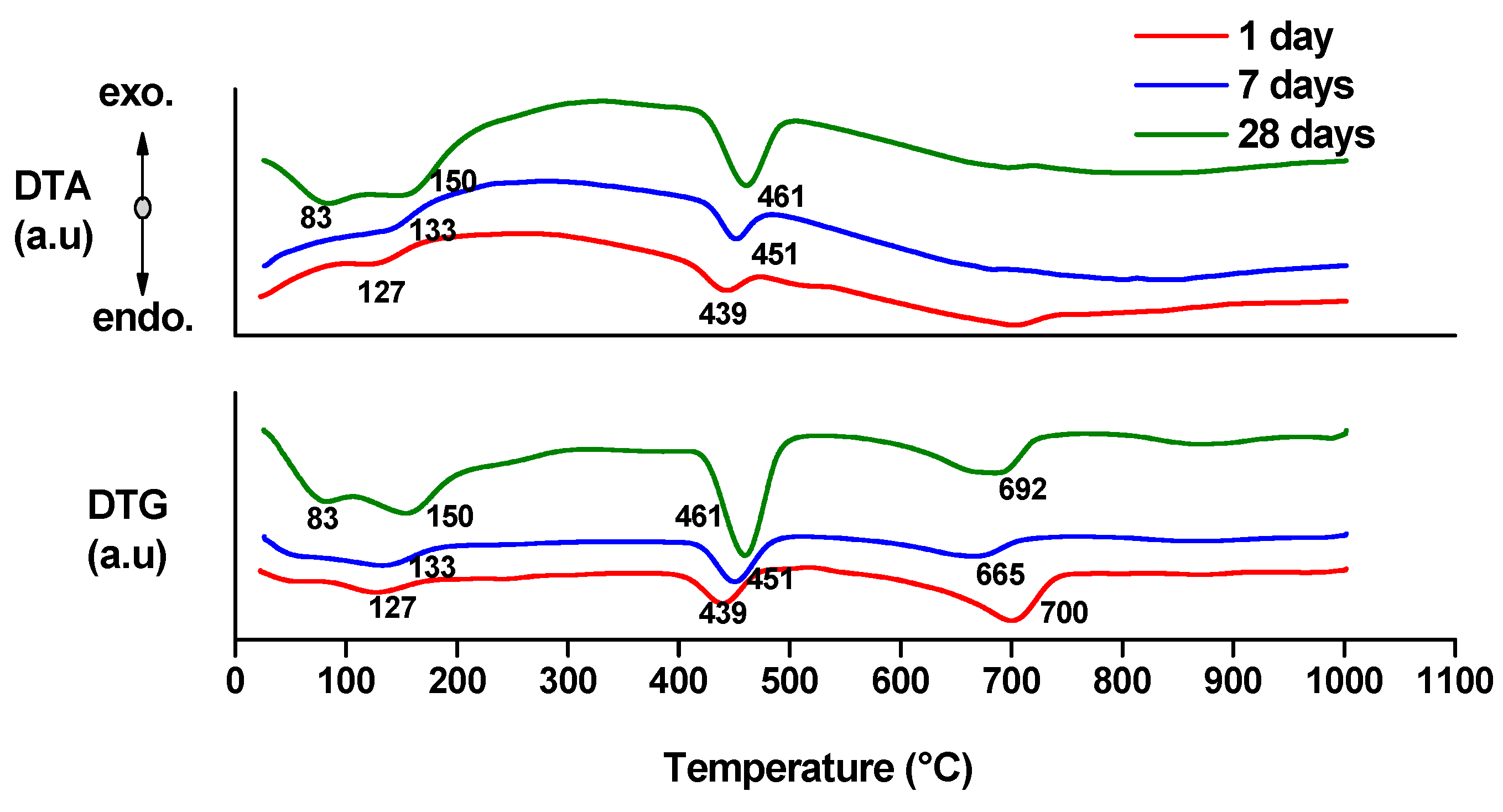
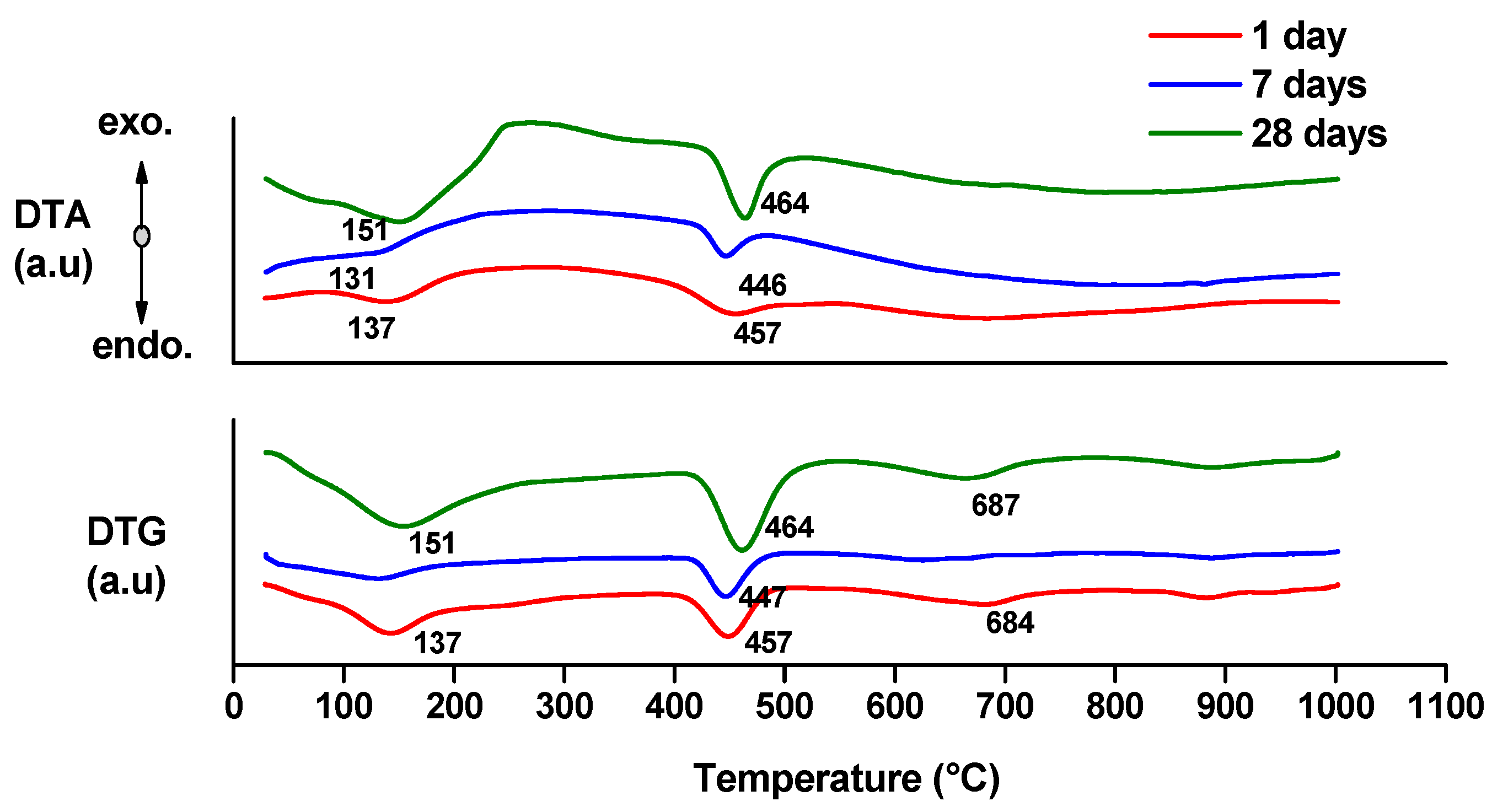
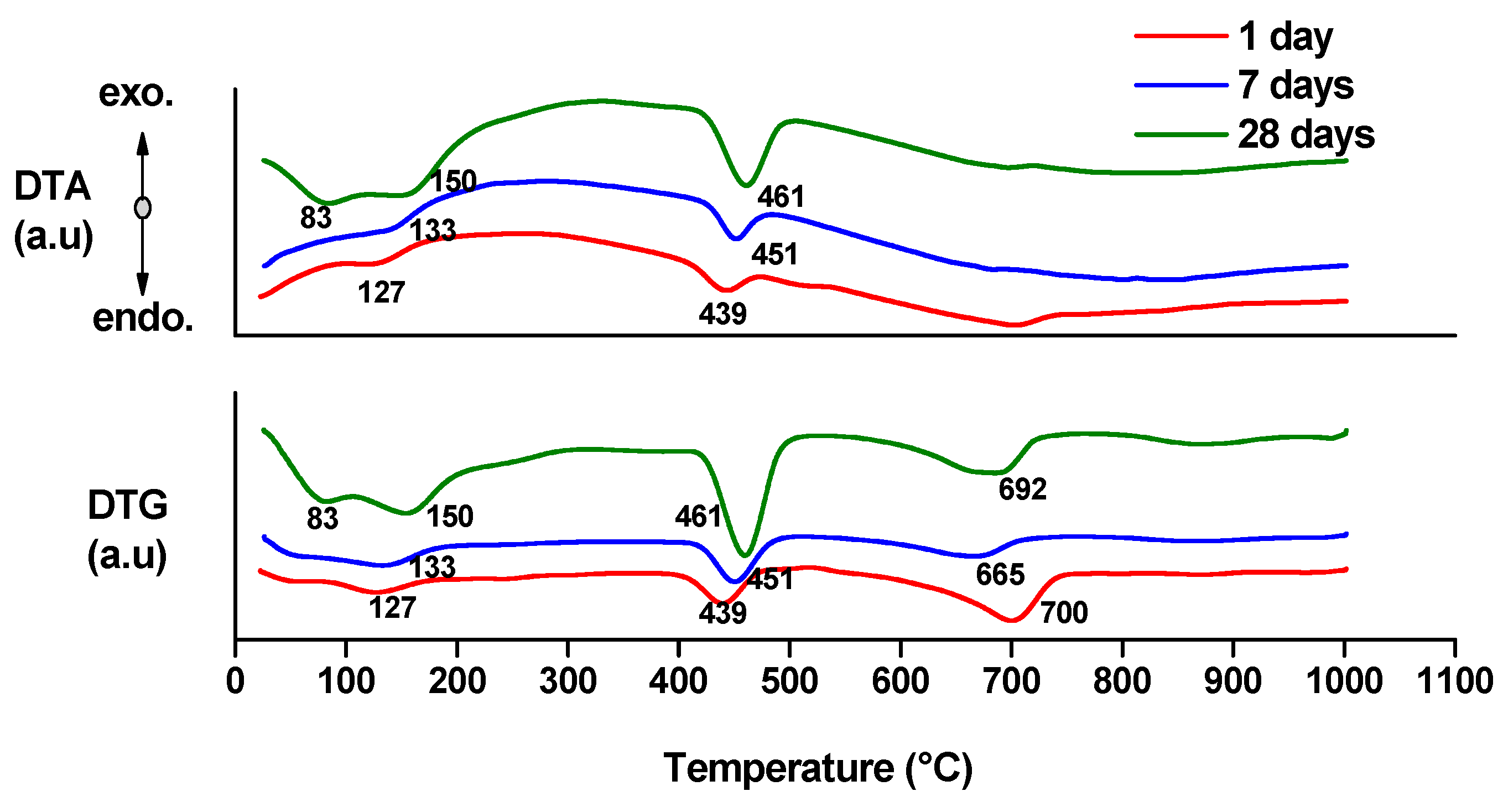
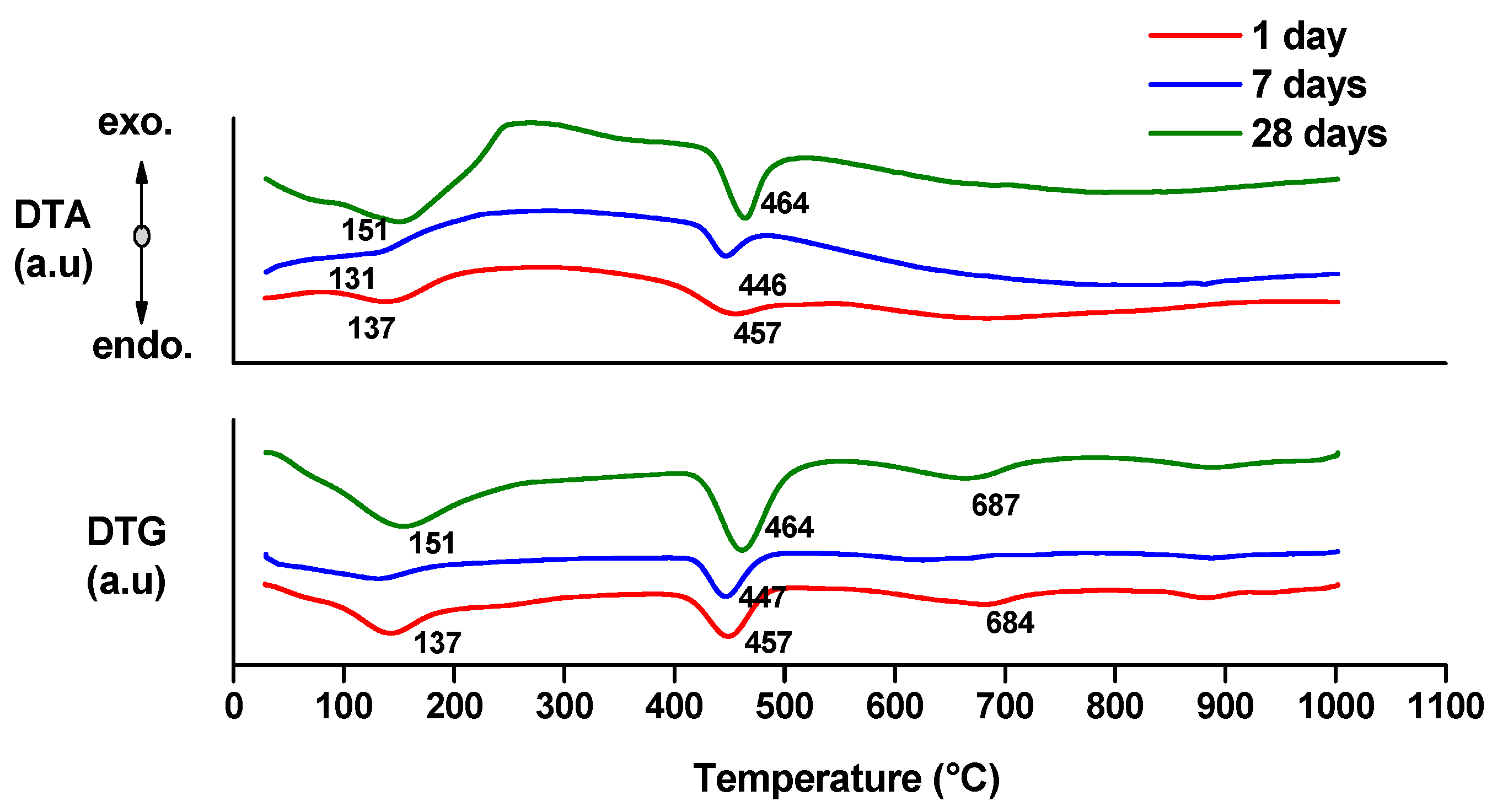
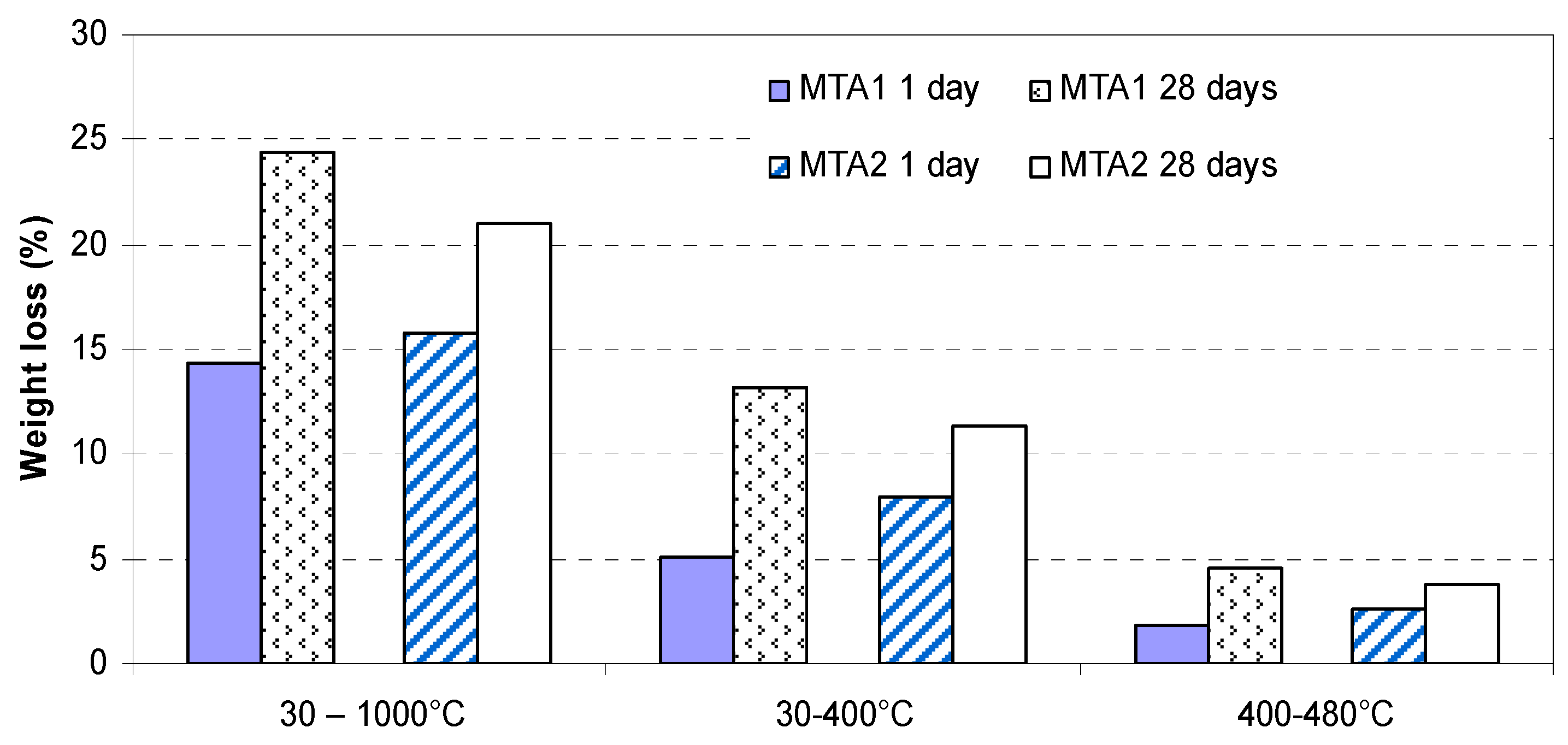
- endo-effects recorded up to 100 °C are due to the loss of moisture;
- -
- endo-effects recorded between 100 and 220 °C are due to the dehydration of gel like calcium silicates hydrates and calcium aluminate hydrates formed by cement hydration;
- -
- endo-effects recorded between 400 and 500 °C are determined by the dehydration of portlandite;
- -
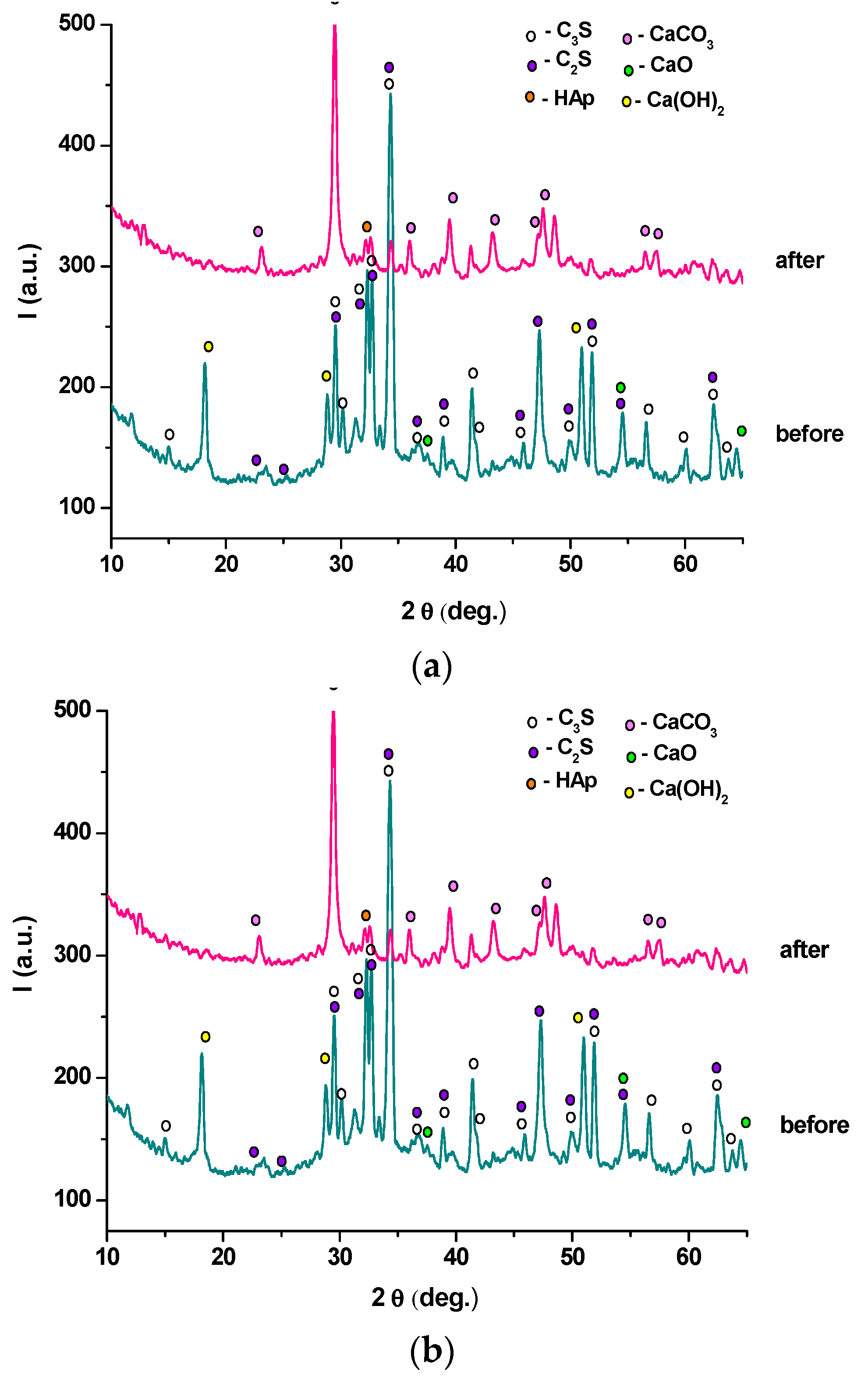
- the endo-effects recorded between 600 and 850 °C are attributed to the decarbonation of CaCO3 with different crystallization degrees; as previously presented, this compound is most probably formed due to the carbonation of portlandite with atmospheric CO2.
- -
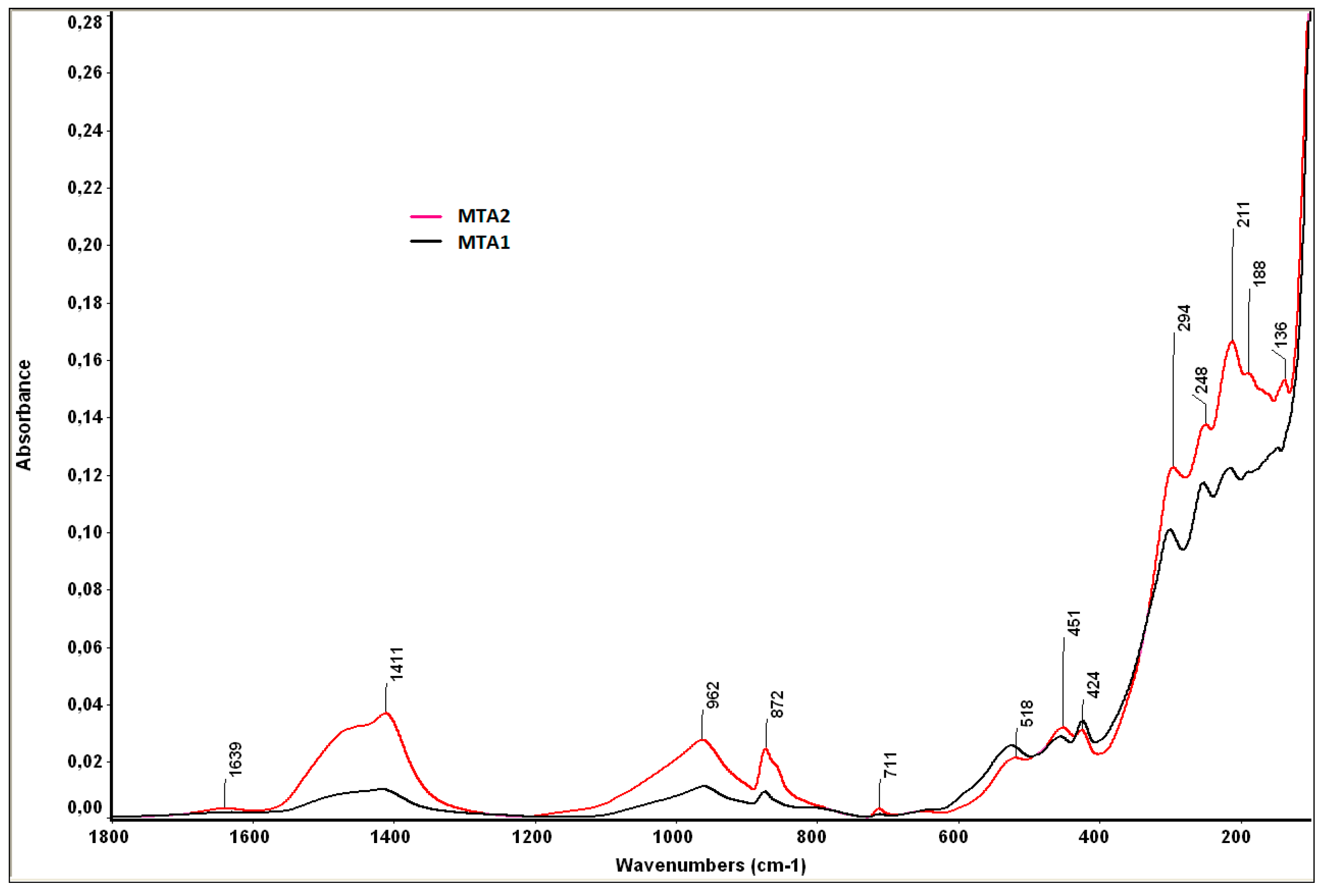
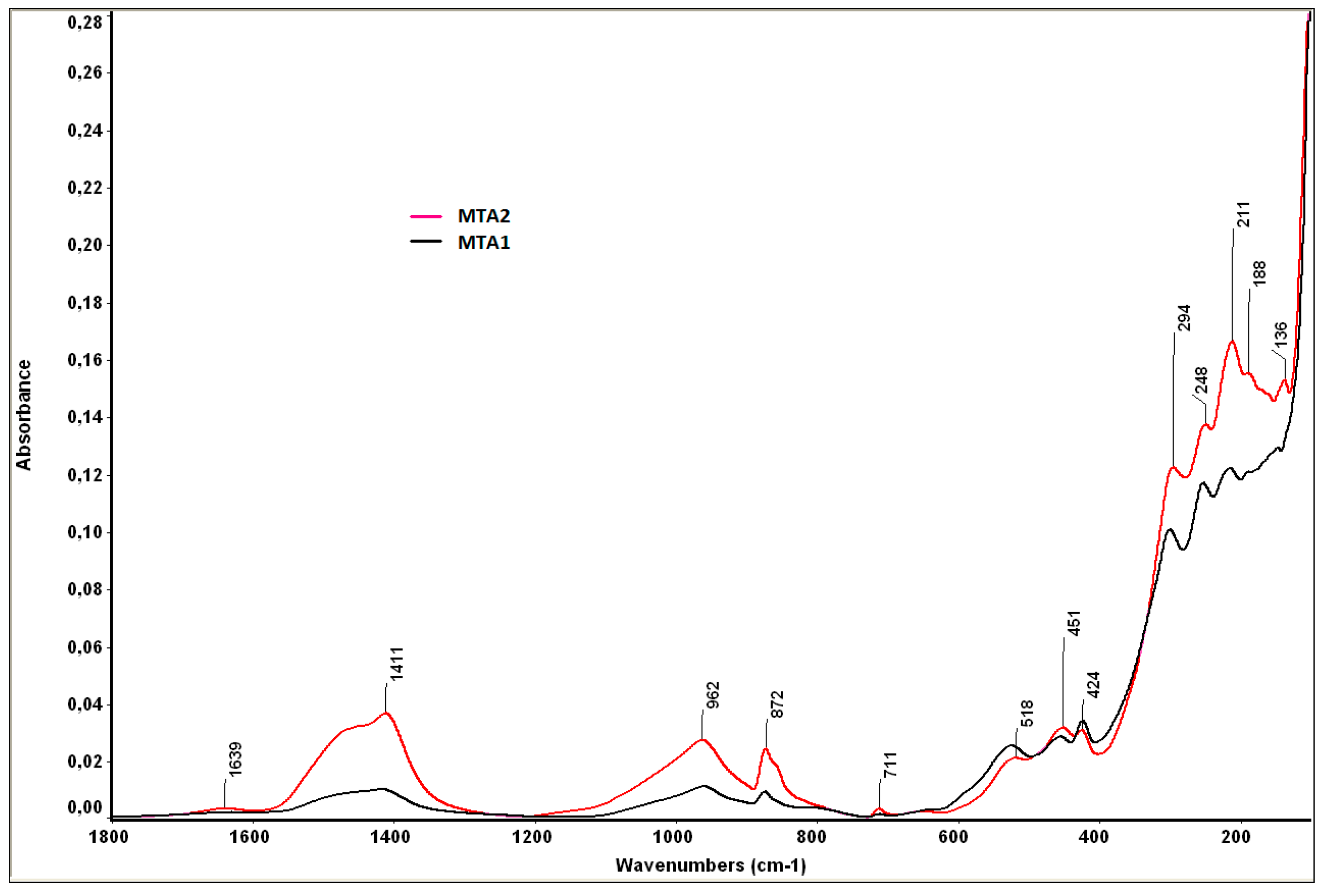
- bands at 1424 cm−1 and 872 cm−1, specific for calcium carbonate, also assessed by XRD;
- -
- bands at 962 cm−1 and 518 cm−1, specific for the phosphate group (PO43−), from HAp;
- -
- the band of 1411 cm−1 is specific for carbonated HAp formed by the substitution of PO43− with CO32−;
- -
- the band of 1639 cm−1 is specific for hydroxyl groups (moisture).
3. Experimental Section
- (a)
- aluminum butoxide and acetyl acetone (1:1 molar ratio) were magnetically stirred for 2 h;
- (b)
- calcium nitrate was dissolved in distilled water and magnetically stirred until a clear solution was obtained; then zinc acetate was added and the solution was magnetically stirred for 2 h at 80 °C; next, TEOS was added in this clear solution and the mixture was homogenized until a clear solution was obtained (molar ratio CaO:ZnO:SiO2:H2O was 1.24:0.04:0.36:10);
- (c)
- the two solutions were mixed for 3 h at room temperature and then kept at 80 °C until a gel was formed.
- a)
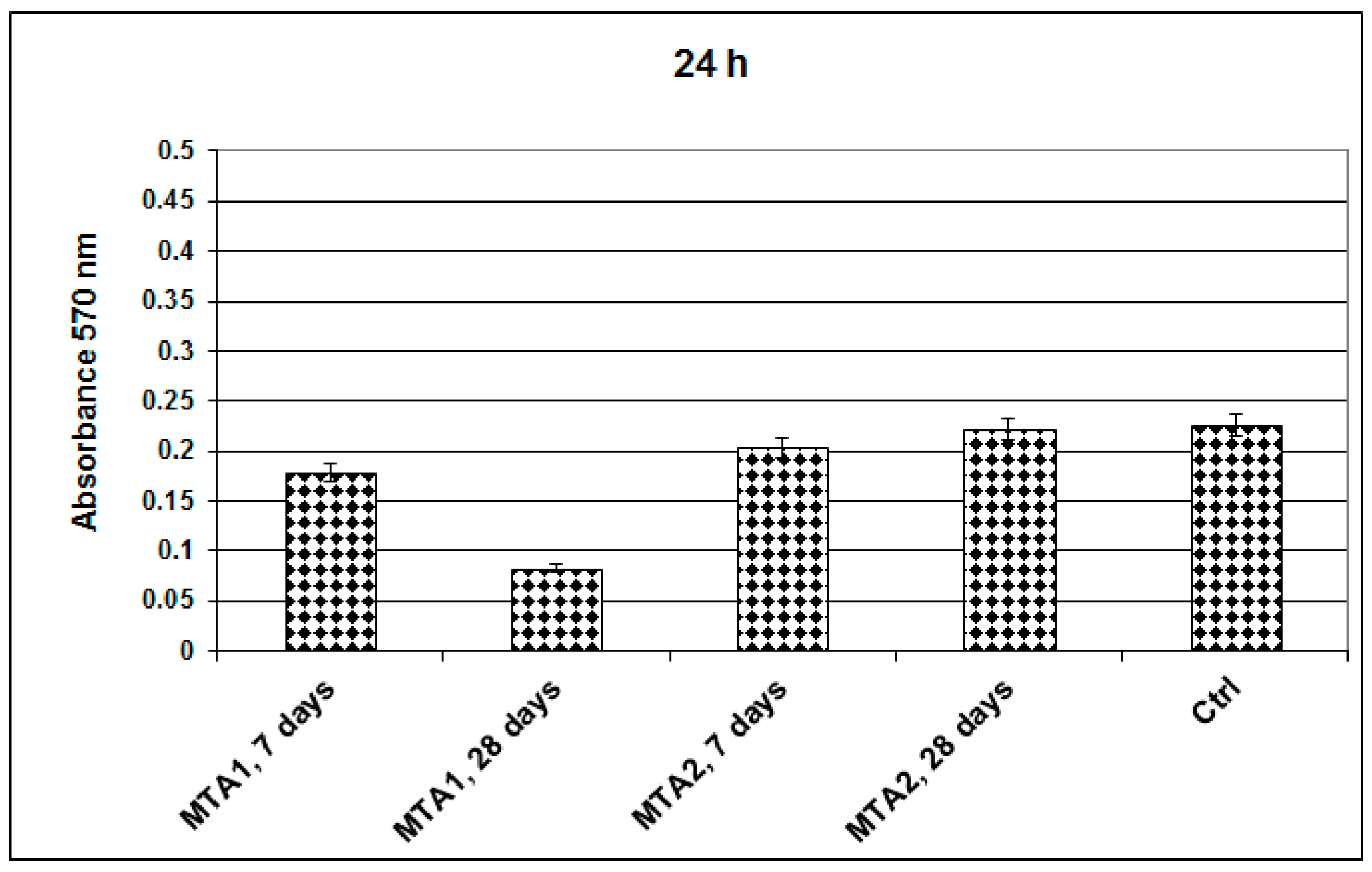
- MTT assay
- b)
- Fluorescent microscopy for tracing of living cells
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ribeiro da Silva, S.; Dias da Silva Neto, J.; Veiga, D.F.; Schnaider, T.B.; Ferreira, L.M. Portland cement vs. MTA as a root-end filling material. A pilot study 1. Acta Cir. Bras. 2015, 30, 160–164. [Google Scholar] [CrossRef] [PubMed]
- De-Deus, G.; Petruccelli, V.; Gurgel-Filho, E.; Coutinho-Filho, T. MTA vs. Portland cement as repair material for furcal perforations: A laboratory study using a polymicrobial leakage model. Int. Endod. J. 2006, 39, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Roberts, H.W.; Toth, J.M.; Beryins, D.W.; Charlton, D.G. Mineral trioxide aggregate material use in endodontic treatment: A review of the literature. Dent. Mater. 2008, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Vasudev, S.K.; Goel, B.R.; Tyagi, S. Root end filling materials—A review. Endodontology 2003, 15, 12–18. [Google Scholar]
- Camilleri, J.; Ford, P.T.R. Mineral trioxide aggregate: A review of the constituents and biological properties of the material. Int. Endod. J. 2006, 39, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Saidon, J.; He, J.; Zhu, Q.; Safavi, K.; Spångberg, L.S.W. Cell and tissue reactions to mineral trioxide aggregate and Portland cement. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2003, 95, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Casella, G.; Ferlito, S. The use of mineral trioxide aggregate in endodontics. Minerva Stomatol. 2006, 55, 123–143. [Google Scholar] [PubMed]
- Czarnecka, B.; Coleman, N.J.; Shaw, H.; Nicholson, J.W. The Use of Mineral Trioxide Aggregate in Endodontics—Status Report. Dent. Med. Probl. 2008, 45, 5–11. [Google Scholar]
- Viola, N.V.; Filho, M.T.; Cerri, P.S. MTA vs. Portland cement: Review of literature. South Braz. Dent. J. 2011, 8, 446–452. [Google Scholar]
- Gonçalves, J.L.; Viapiana, R.; Miranda, C.E.S.; Borges, A.H.; Cruz Filho, A.M. Evaluation of physico-chemical properties of Portland cements and MTA. Braz. Oral Res. 2010, 24, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Steffen, R.; Van Waes, H. Understanding mineral trioxide aggregate/Portland-cement: A review of literature and background factors. Eur. Arch. Paediatr. Dent. 2009, 10, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Voicu, G.; Ghiţulică, C.D.; Dinu, E.; Andronescu, E. In-vitro behaviour of dicalcium silicate obtained through the sol-gel method. Rev. Romana Mater. 2011, 41, 229–233. [Google Scholar]
- Voicu, G.; Ghiţulică, C.D.; Andronescu, E. Modified Pechini synthesis of tricalcium aluminate powder. Mater. Charact. 2012, 73, 89–95. [Google Scholar] [CrossRef]
- Voicu, G.; Bădănoiu, A.I.; Andronescu, E.; Chifiruc, C.M. Synthesis, characterization and bioevaluation of partially stabilized cements for medical applications. Cent. Eur. J. Chem. 2013, 11, 1657–1667. [Google Scholar] [CrossRef]
- Voicu, G.; Bădănoiu, A.I.; Andronescu, E.; Bleotu, C. Binding properties and biocompatibility of accelerated Portland cement for endodontic use. Rev. Chim. 2012, 63, 1031–1034. [Google Scholar]
- Lin, F.H.; Wang, W.H.; Lin, C.P. Transition element contained partial-stabilized cement (PSC) as a dental retrograde-filling material. Biomaterials 2003, 24, 219–233. [Google Scholar] [CrossRef]
- Taylor, H.F.W. Cement Chemistry; Academic Press: London, UK, 1997. [Google Scholar]
- Bădănoiu, A.; Paceagiu, J.; Voicu, G. Hydration and hardening processes of Portland cements obtained from clinkers mineralized with fluoride and oxides. J. Therm. Anal. Calorim. 2011, 103, 879–888. [Google Scholar] [CrossRef]
- Morrier, J.J.; Benay, G.; Hartmann, C.; Barsotti, O. Antimicrobial activity of Ca(OH)2 dental cements: An in vitro study. J. Endod. 2003, 29, 51–54. [Google Scholar] [CrossRef] [PubMed]
- Han, G.Y.; Park, S.H.; Yoon, T.C. Antimicrobial activity of Ca(OH)2 containing pastes with Enterococcus faecalis in vitro. J. Endod. 2001, 27, 328–332. [Google Scholar] [CrossRef] [PubMed]
- Mohammadi, Z.; Shalavi, S.; Yazdizadeh, M. Antimicrobial activity of calcium hydroxide in endodontics: A review. Chonnam Med. J. 2012, 48, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Saghiri, M.A.; Garcia-Godoy, F.; Asatourian, A.; Lotfi, M.; Banava, S.; Khezri-Boukani, K. Effect of pH on compressive strength of some modification of mineral trioxide aggregate. Med. Oral Patol. Oral Cir. Bucal 2013, 18, e714–e720. [Google Scholar] [CrossRef] [PubMed]
- Campbell, D.H. Microscopical Examination and Interpretation of Portland Cement and Clinker, 2nd ed.; Portland Cement Association: Skokie, IL, USA, 1999. [Google Scholar]
- Ylmén, R.; Jäglid, U.; Steenari, B.M.; Panas, I. Early hydration and setting of Portland cement monitored by IR, SEM and Vicat techniques. Cem. Concr. Res. 2009, 39, 433–439. [Google Scholar] [CrossRef]
- Ylmén, R.; Wadsö, L.; Panas, I. Insights into early hydration of Portland limestone cement from infrared spectroscopy and isothermal calorimetry. Cem. Concr. Res. 2010, 40, 1541–1546. [Google Scholar] [CrossRef]
- Mollah, M.Y.A.; Kesmez, M.; Cocke, D.L. An X-ray diffraction (XRD) and Fourier transform infrared spectroscopic (FT-IR) investigation of the long-term effect on the solidification/stabilization (S/S) of arsenic(V) in Portland cement type-V. Sci. Total Environ. 2004, 325, 255–262. [Google Scholar] [CrossRef] [PubMed]
- Alves, N.M.; Leonor, I.B.; Azevedo, H.S.; Reis, R.L.; Mano, J.F. Designing biomaterials based on biomineralization of bone. J. Mater. Chem. 2010, 20, 2911–2921. [Google Scholar] [CrossRef]
- Caridade, S.G.; Merino, E.G.; Alves, N.M.; Mano, J.F. Bioactivity and viscoelastic characterization of chitosan/bioglass® composite membranes. Macromol. Biosci. 2012, 12, 1106–1113. [Google Scholar] [CrossRef] [PubMed]
- Voicu, G.; Jinga, S.I.; Truşcă, R.; Iordache, F. Synthesis, characterization and bioevaluation of bioactive composites scaffolds based on collagen and glass ceramic. Dig. J. Nanomater. Biostr. 2014, 9, 99–108. [Google Scholar]
- Cornélio, A.L.; Rodrigues, E.M.; Salles, L.P.; Mestieri, L.B.; Faria, G.; Guerreiro-Tanomaru, J.M.; Tanomaru-Filho, M. Bioactivity of MTA Plus, Biodentine and experimental calcium silicate-based cements in human osteoblast-like cells. Int. Endod. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rathinam, E.; Rajasekharan, S.; Chitturi, R.T.; Martens, L.; De Coster, P. Gene expression profiling and molecular signaling of dental pulp cells in response to tricalcium silicate cements: A systematic review. J. Endod. 2015, 41, 1805–1817. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
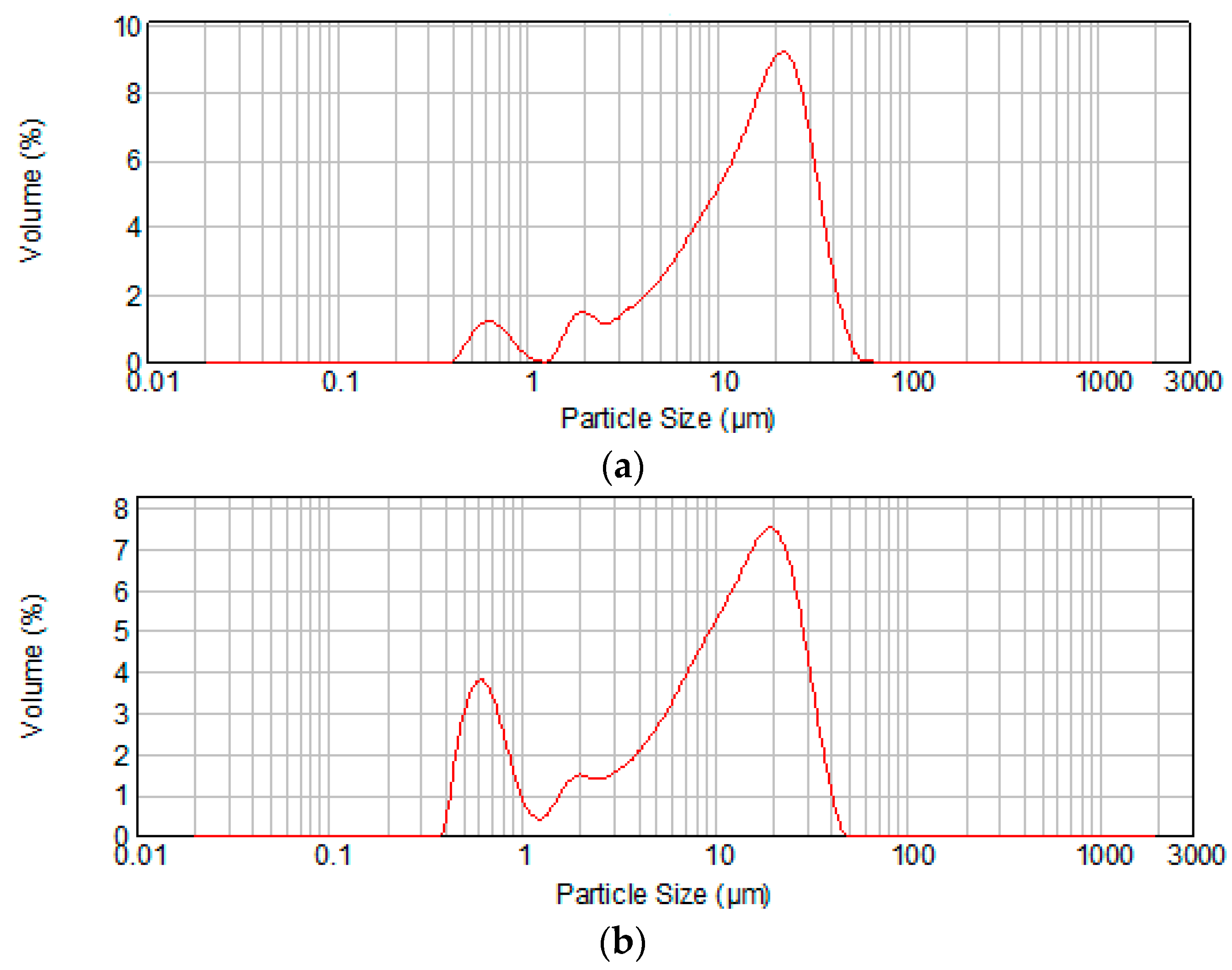
| Cement | Average Particle Size (μm) | d0.1 (μm) * | d0.9 (μm) ** |
|---|---|---|---|
| MTA1 | 15.03 | 2.867 | 31.143 |
| MTA2 | 10.77 | 0.690 | 26.652 |
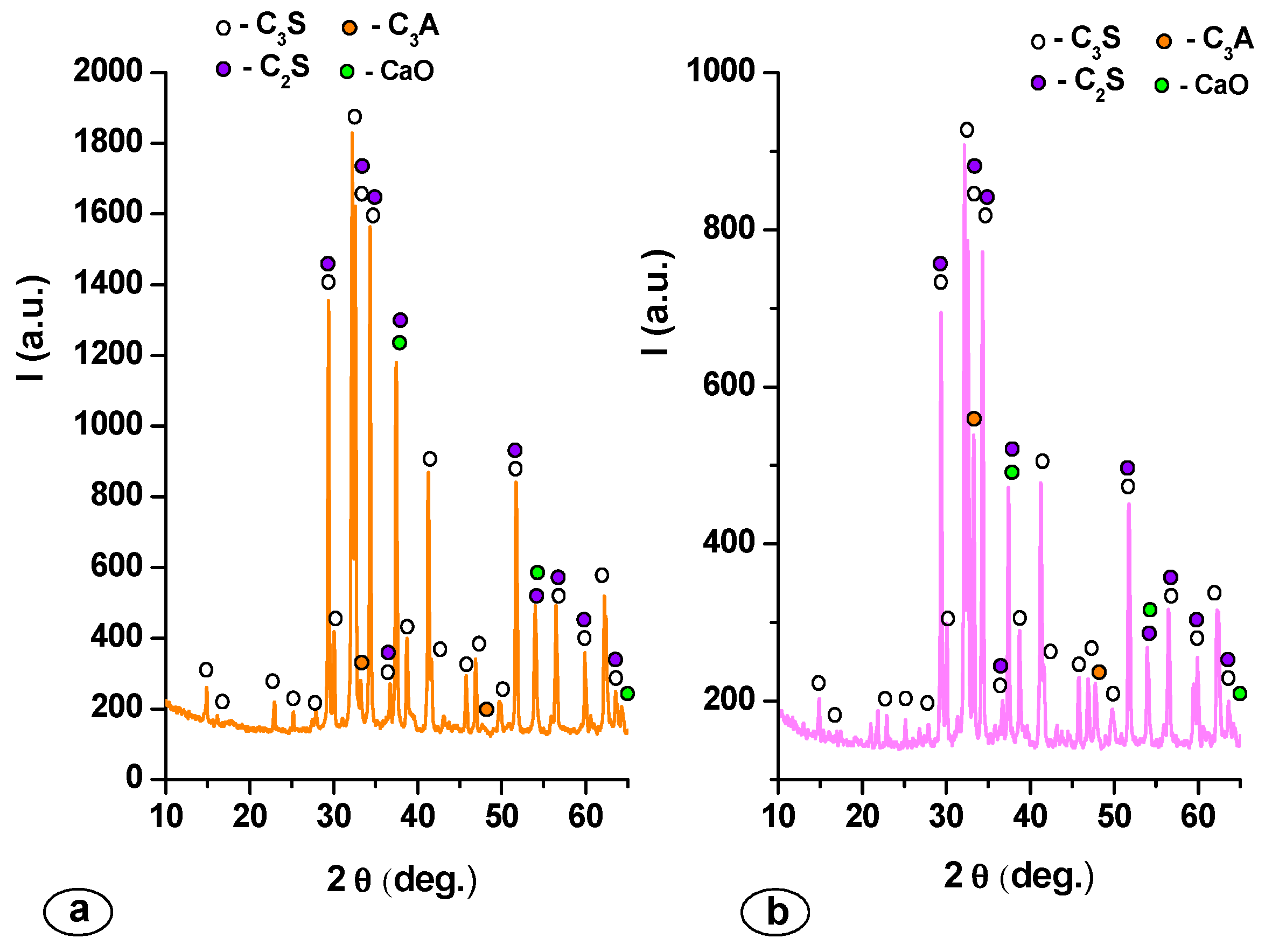
| Mineralogic Compounds | Specimen | |
|---|---|---|
| MTA1 | MTA2 | |
| C3S (%) | 68.60 | 71.40 |
| C2S (%) | 13.70 | 11.80 |
| C3A (%) | 13.40 | 15.70 |
| CaO (%) | 4.30 | 1.10 |
| Cement | Setting Time (min) | Compressive Strength after 7 Days (MPa) | Compressive Strength after 28 Days (MPa) |
|---|---|---|---|
| MTA 1 | 55 | 9.2 | 18.2 |
| MTA 2 | 15 | 12.7 | 22.9 |
- Sample Availability: Samples of the compounds presented in the manuscript are available from the authors.
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Voicu, G.; Popa, A.M.; Badanoiu, A.I.; Iordache, F. Influence of Thermal Treatment Conditions on the Properties of Dental Silicate Cements. Molecules 2016, 21, 233. https://doi.org/10.3390/molecules21020233
Voicu G, Popa AM, Badanoiu AI, Iordache F. Influence of Thermal Treatment Conditions on the Properties of Dental Silicate Cements. Molecules. 2016; 21(2):233. https://doi.org/10.3390/molecules21020233
Chicago/Turabian StyleVoicu, Georgeta, Alexandru Mihai Popa, Alina Ioana Badanoiu, and Florin Iordache. 2016. "Influence of Thermal Treatment Conditions on the Properties of Dental Silicate Cements" Molecules 21, no. 2: 233. https://doi.org/10.3390/molecules21020233
APA StyleVoicu, G., Popa, A. M., Badanoiu, A. I., & Iordache, F. (2016). Influence of Thermal Treatment Conditions on the Properties of Dental Silicate Cements. Molecules, 21(2), 233. https://doi.org/10.3390/molecules21020233