
Practical and Metal-Free Synthesis of Novel Enantiopure Amides Containing the Potentially Bioactive 5-Nitroimidazole Moiety



Abstract
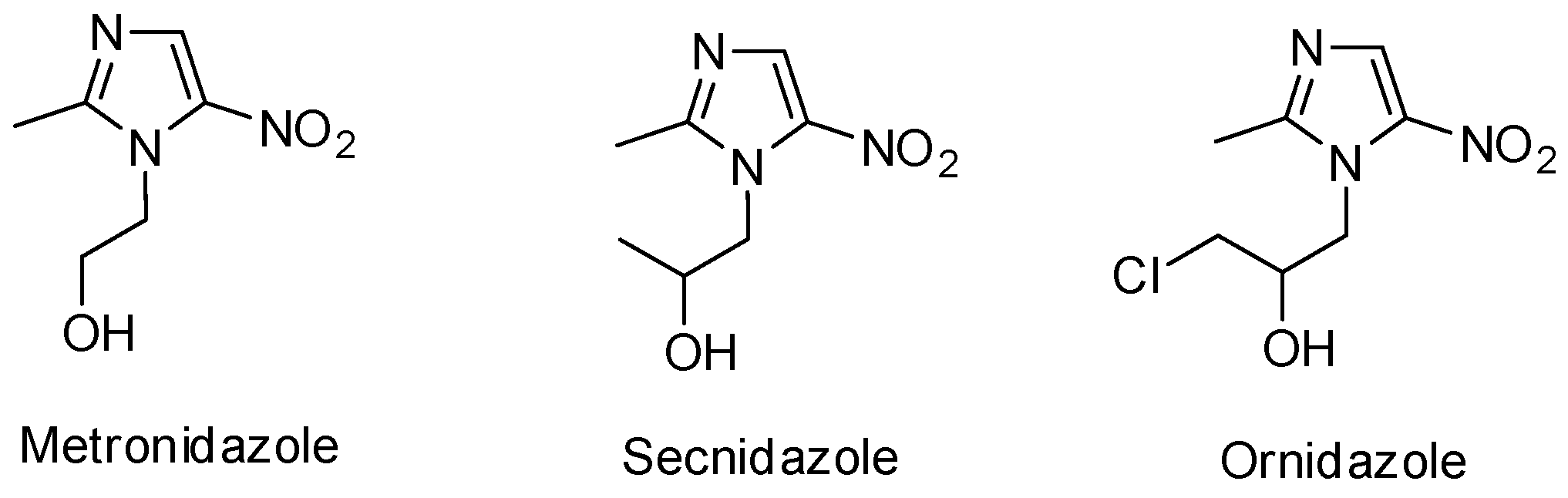
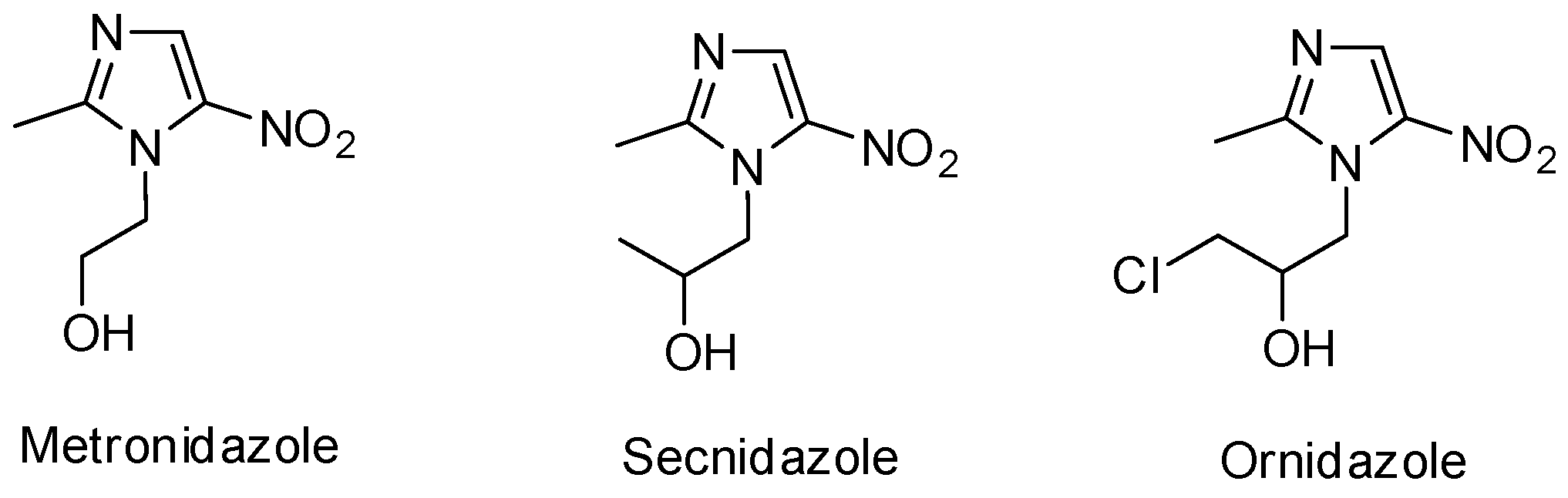
:1. Introduction
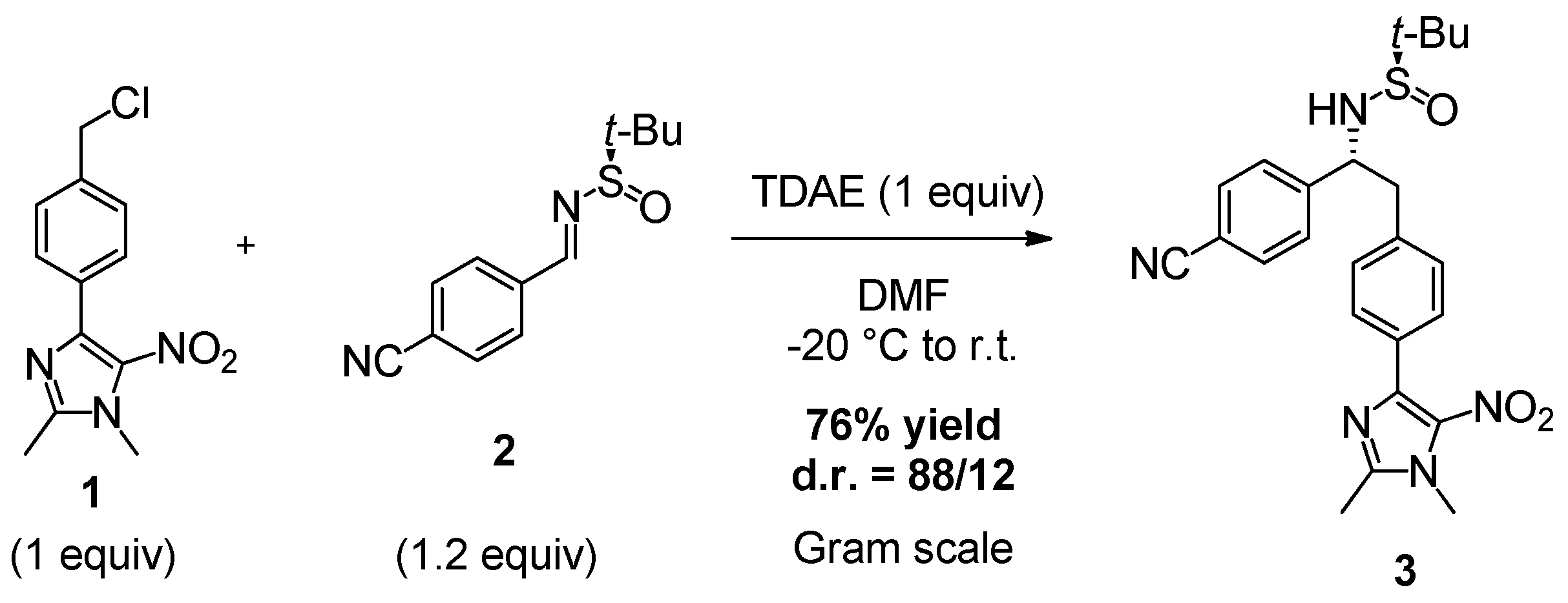
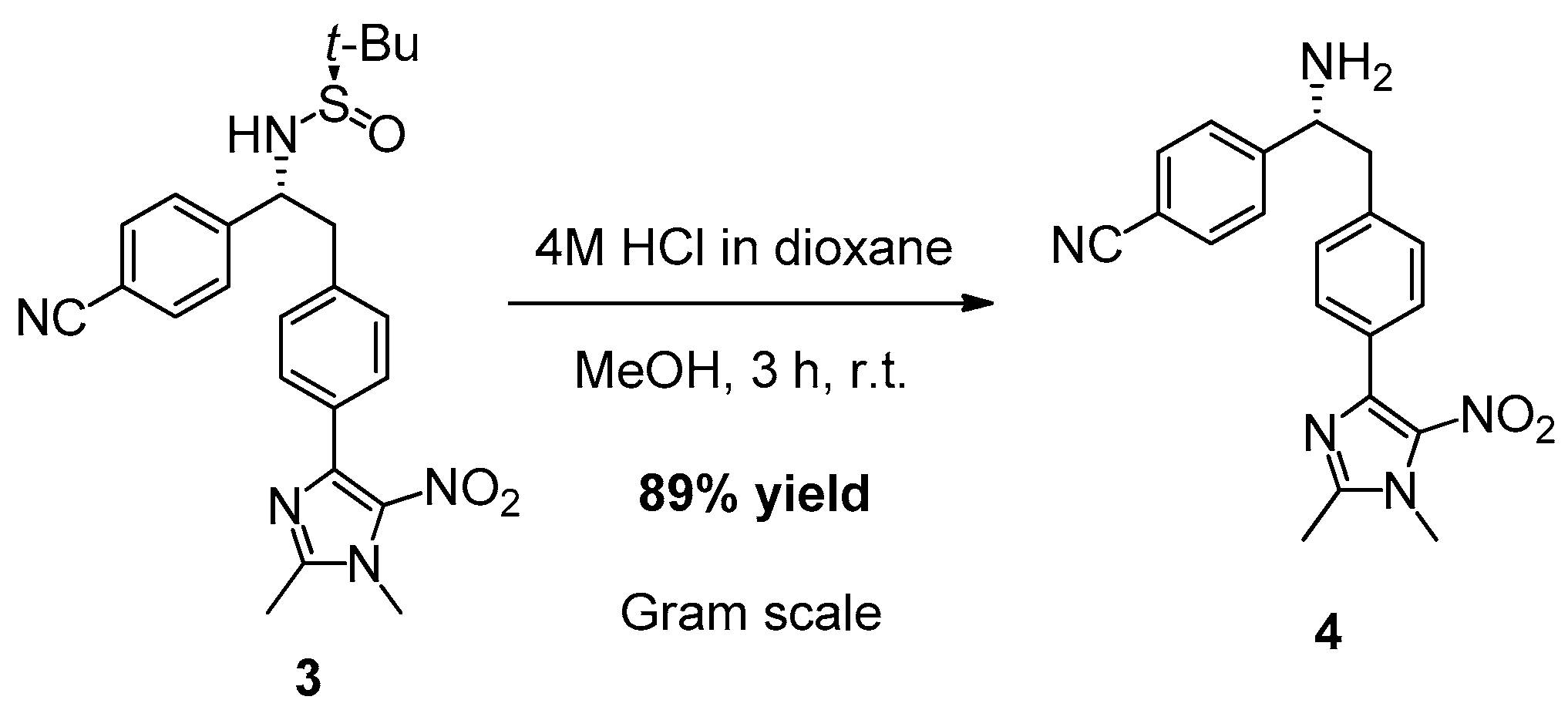
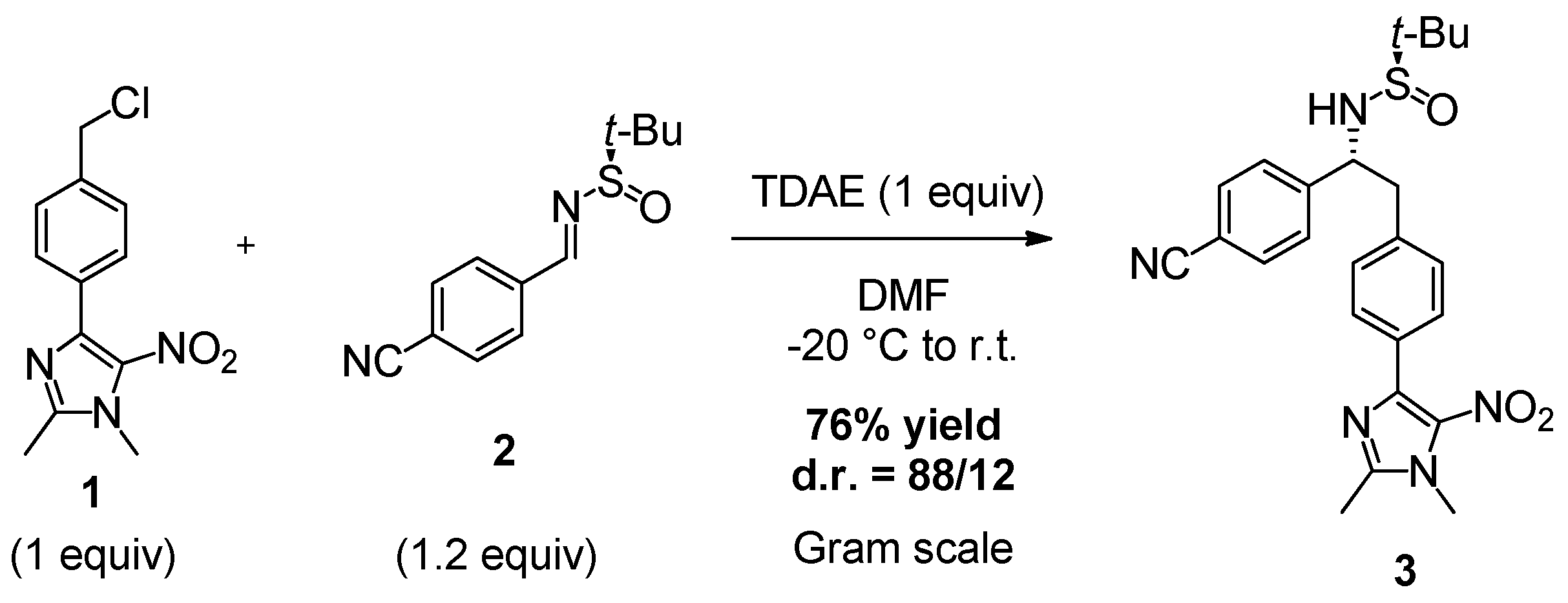
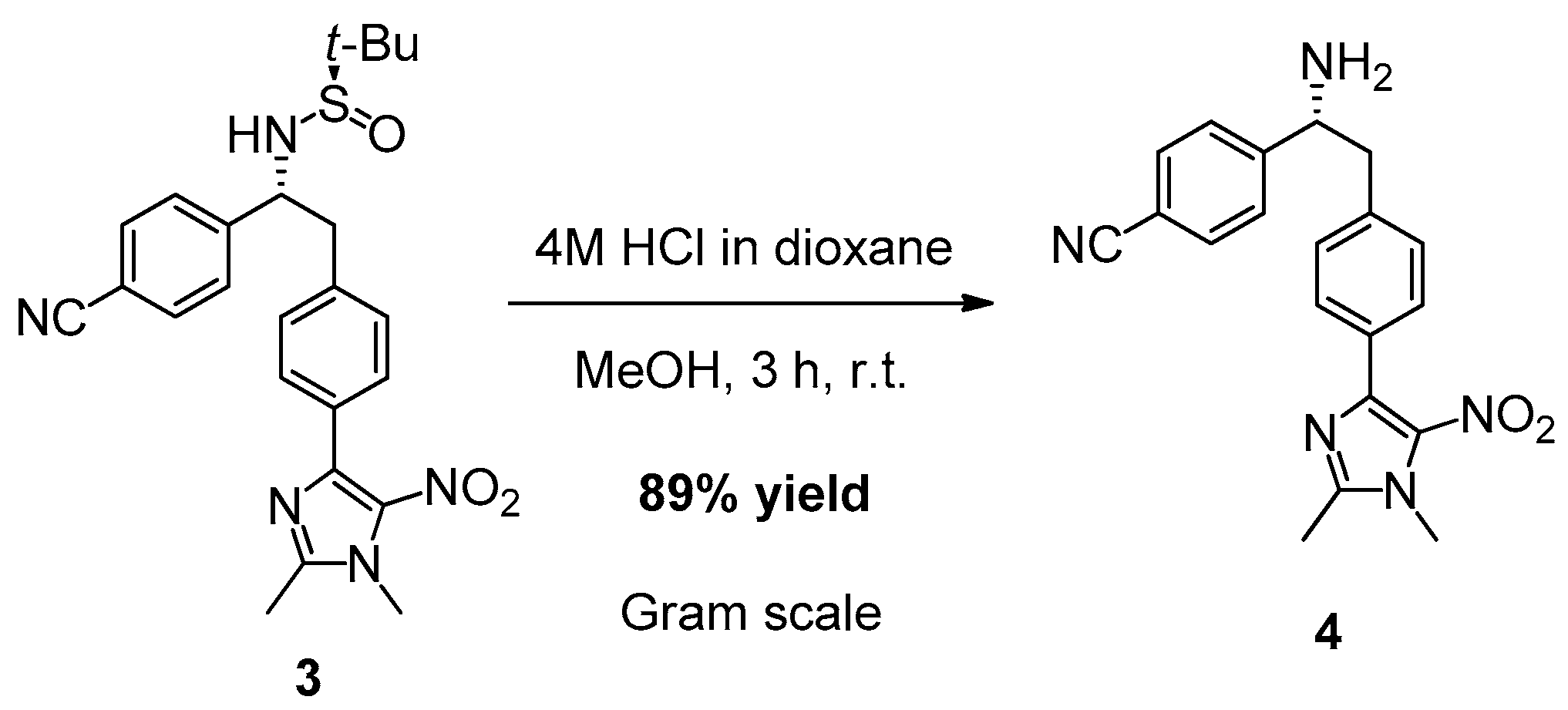
2. Results and Discussion
3. Experimental Section
3.1. General
3.2. Synthesis of (R)-N-((R)-1-(4-Cyanophenyl)-2-(4-(1,2-dimethyl-5-nitro-1H-imidazol-4-yl)phenyl)ethyl)-2-methylpropane-2-sulfinamide (3)
3.3. Synthesis of (R)-4-(1-Amino-2-(4-(1,2-dimethyl-5-nitro-1H-imidazol-4-yl)phenyl)ethyl)benzonitrile (4)
3.4. General Procedure for the Synthesis of Amides 6a–i
3.5. General Procedure for the Synthesis of Amides 6j–l
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jørgensen, M.A.; Manos, J.; Mendz, G.L.; Hazell, S.L. The mode of action of metronidazole in Helicobacter pylori: Futile cycling or reduction? J. Antimicrob. Chemother. 1998, 41, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Upcroft, J.A.; Campbell, R.W.; Benakli, K.; Upcroft, P.; Vanelle, P. Efficacy of New 5-Nitroimidazoles against Metronidazole-Susceptible and -Resistant Giardia, Trichomonas, and Entamoeba spp. Antimicrob. Agents Chemother. 1999, 43, 73–76. [Google Scholar] [PubMed]
- Citron, D.M.; Tyrrell, K.L.; Warren, Y.A.; Fernández, H.; Merriam, C.V.; Goldstein, E.J.C. In vitro activities of tinidazole and metronidazole against Clostridium difficile, Prevotella bivia and Bacteroides fragilis. Anaerobe 2005, 11, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Crozet, M.D.; Botta, C.; Gasquet, M.; Curti, C.; Rémusat, V.; Hutter, S.; Chapelle, O.; Azas, N.; De Méo, M.; Vanelle, P. Lowering of 5-nitroimidazole’s mutagenicity: Towards optimal antiparasitic pharmacophore. Eur. J. Med. Chem. 2009, 44, 653–659. [Google Scholar] [CrossRef] [PubMed]
- Kim, P.; Kang, S.; Boshoff, H.I.; Jiricek, J.; Collins, M.; Singh, R.; Manjunatha, U.H.; Niyomrattanakit, P.; Zhang, L.; Goodwin, M., III; et al. Structure-Activity Relationships of Antitubercular Nitroimidazoles. 2. Determinants of Aerobic Activity and Quantitative Structure-Activity Relationships. J. Med. Chem. 2009, 52, 1329–1344. [Google Scholar] [CrossRef] [PubMed]
- Dunn, L.A.; Burgess, A.G.; Krauer, K.G.; Eckmann, L.; Vanelle, P.; Crozet, M.D.; Gillin, F.D.; Upcroft, P.; Upcroft, J.A. A new-generation 5-nitroimidazole can induce highly metronidazole-resistant Giardia lamblia in vitro. Int. J. Antimicrob. Agents 2010, 36, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Çelik, A.; Ares Ateş, N. The frequency of sister chromatid exchanges in cultured human peripheral blood lymphocyte treated with metronidazole in vitro. Drug Chem. Toxicol. 2006, 29, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.S.; Wang, R.; Bagan, E.; Wang, C.C.; Wislocki, P.; Miwa, G.T. Structural alterations that differentially affect the mutagenic and antitrichomonal activities of 5-nitroimidazoles. J. Med. Chem. 1987, 30, 150–156. [Google Scholar] [CrossRef] [PubMed]
- Broggi, J.; Terme, T.; Vanelle, P. Organic Electron Donors as Powerful Single-Electron Reducing Agents in Organic Synthesis. Angew. Chem. Int. Ed. 2014, 53, 384–413. [Google Scholar] [CrossRef] [PubMed]
- Spitz, C.; Lin, A.; Terme, T.; Vanelle, P. Mild and Metal-Free Diastereoselective Synthesis of N-tert-Butanesulfinylamines by Using Tetrakis(dimethylamino)ethylene. Synthesis 2014, 46, 3229–3232. [Google Scholar] [CrossRef]
- Crozet, M.P.; Archaimbault, G.; Vanelle, P.; Nouguier, R. Reactions SRN1 en serie heterocyclique: IV: Reactivite des sels du dimethyl-2,2 nitro-5 dioxanne-1,3. Tetrahedron Lett. 1985, 26, 5133–5134. [Google Scholar] [CrossRef]
- Vanelle, P.; De Meo, M.P.; Maldonado, J.; Nouguier, R.; Crozet, M.P.; Laget, M.; Dumenil, G. Genotoxicity in oxazolidine derivatives: Influence of the nitro group. Eur. J. Med. Chem. 1990, 25, 241–250. [Google Scholar] [CrossRef]
- Bouhlel, A.; Curti, C.; Dumètre, A.; Laget, M.; Crozet, M.D.; Azas, N.; Vanelle, P. Synthesis and evaluation of original amidoximes as antileishmanial agents. Bioorg. Med. Chem. 2010, 18, 7310–7320. [Google Scholar] [CrossRef] [PubMed]
- Da Costa, L.; Roche, M.; Scheers, E.; Coluccia, A.; Neyts, J.; Terme, T.; Leyssen, P.; Silvestri, R.; Vanelle, P. VP1 crystal structure-guided exploration and optimization of 4,5-dimethoxybenzene-based inhibitors of rhinovirus 14 infection. Eur. J. Med. Chem. 2016, 115, 453–462. [Google Scholar] [CrossRef] [PubMed]
- Cogan, D.A.; Ellman, J.A. Asymmetric Synthesis of α,α-Dibranched Amines by the Trimethylaluminum-Mediated 1,2-Addition of Organolithiums to tert-Butanesulfinyl Ketimines. J. Am. Chem. Soc. 1999, 121, 268–269. [Google Scholar] [CrossRef]
- Juspin, T.; Zink, L.; Crozet, M.D.; Terme, T.; Vanelle, P. High Functionalization of 5-Nitro-1H-imidazole Derivatives: The TDAE Approach. Molecules 2011, 16, 6883–6893. [Google Scholar] [CrossRef] [PubMed]
- Seo, J.; Barron, A.E.; Zuckermann, R.N. Novel Peptoid Building Blocks: Synthesis of Functionalized Aromatic Helix-Inducing Submonomers. Org. Lett. 2010, 12, 492–495. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds 6a–l are available from the authors.

| Entry | R1 | Yield of 6 (%) b |
|---|---|---|
| 1 | C6H5 | 75 (6a) |
| 2 | 4-NO2C6H4 | 83 (6b) |
| 3 | 4-ClC6H4 | 80 (6c) |
| 4 | 2-BrC6H4 | 64 (6d) |
| 5 | CH3 | 79 (6e) |
| 6 | CCl3 | 74 (6f) |
| 7 | CBr3 | 73 (6g) |
| 8 | CH3(CH2)14 | 69 (6h) |
| 9 | 2-Furyl | 68 (6i) |
| 10 c | 3-ClC6H4 | 70 (6j) |
| 11 c | C6H5CH2 | 63 (6k) |
| 12 c |  | 73 (6l) |

{kind=link}
{kind=link}
{kind=link}
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Spitz, C.; Mathias, F.; Giuglio-Tonolo, A.G.; Terme, T.; Vanelle, P. Practical and Metal-Free Synthesis of Novel Enantiopure Amides Containing the Potentially Bioactive 5-Nitroimidazole Moiety. Molecules 2016, 21, 1472. https://doi.org/10.3390/molecules21111472
Spitz C, Mathias F, Giuglio-Tonolo AG, Terme T, Vanelle P. Practical and Metal-Free Synthesis of Novel Enantiopure Amides Containing the Potentially Bioactive 5-Nitroimidazole Moiety. Molecules. 2016; 21(11):1472. https://doi.org/10.3390/molecules21111472
Chicago/Turabian StyleSpitz, Cédric, Fanny Mathias, Alain Gamal Giuglio-Tonolo, Thierry Terme, and Patrice Vanelle. 2016. "Practical and Metal-Free Synthesis of Novel Enantiopure Amides Containing the Potentially Bioactive 5-Nitroimidazole Moiety" Molecules 21, no. 11: 1472. https://doi.org/10.3390/molecules21111472
APA StyleSpitz, C., Mathias, F., Giuglio-Tonolo, A. G., Terme, T., & Vanelle, P. (2016). Practical and Metal-Free Synthesis of Novel Enantiopure Amides Containing the Potentially Bioactive 5-Nitroimidazole Moiety. Molecules, 21(11), 1472. https://doi.org/10.3390/molecules21111472