
The Eighth Central European Conference “Chemistry towards Biology”: Snapshot †


 ,
,  , ,
, , 
 , ,
, , 

 ,
, 

 add
Show full author list
add
Show full author list
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Disentangling Puzzles: Atomic Resolution Studies of Intrinsically Disordered Proteins
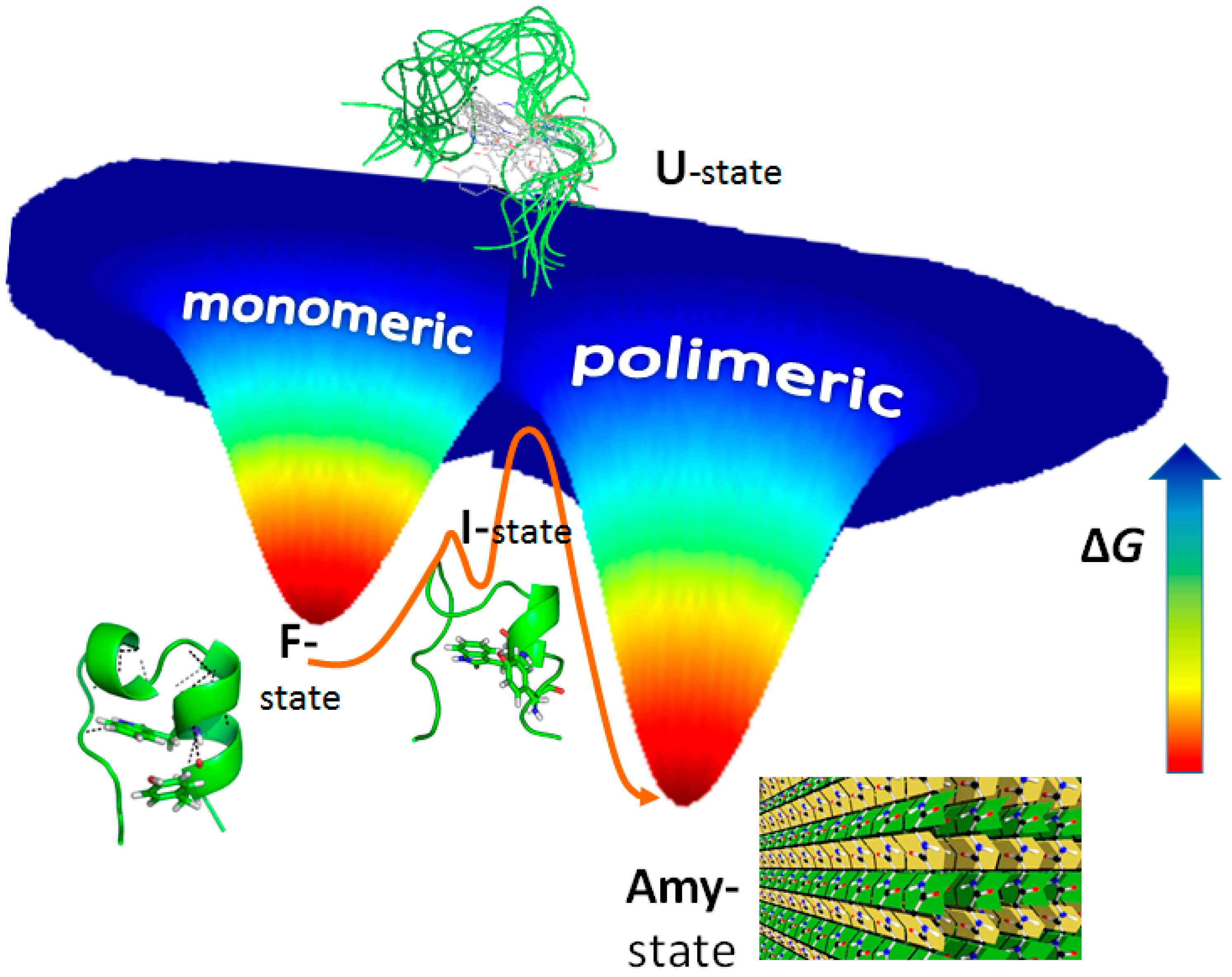
3. Amyloid Fibril Formation in Details: Dead-End Street of Protein Folding?
4. Specific Roles of Histidyl and Cysteinyl Residues in Metal Ion Binding Sites in Peptides and Proteins
5. Structural Bioinformatics: Route from 3D Biomacromolecular Structure to Biology
5.1. Introduction
5.2. Fragment Detection Tool PatternQuery
- Atoms (X) returns all atoms with the element symbol X
- Residues (R1, R2) returns all residues with the two-letter code R1 or R2
- ConnectedAtoms (F, r) returns all atoms within distance r from fragment F
- Authors (F) returns the authors of the structure containing fragment F
- Weight (F) returns the molecular weight of fragment F
5.3. Comparison Tool SiteBinder
5.4. Validation Tool MotiveValidator and Database ValidatorDB
5.5. Charge Calculation Tool AtomicChargeCalculator
5.6. Channel Detection and Characterization Tool MOLE
5.7. Integration of Software into PDBsum and PDBe
6. Simulating Ligand-Receptor Interactions: Challenge to Understand the Dynamic Process Using Static Techniques
7. From Vibrational Spectroscopy to Nanoscopy of Skin Systems with Nanoparticles
8. From Dequalinium to Mitochondria-Targeted Pharmaceutical Nanocarriers
9. ASPH Inhibitors as Second Generation NOTCH Pathway Modulators
10. Characterization and Applications of Cysteine-Histidine–Dependent Amidohydrolase Peptidase Targeting Methicillin–Resistant Staphylococcus aureus
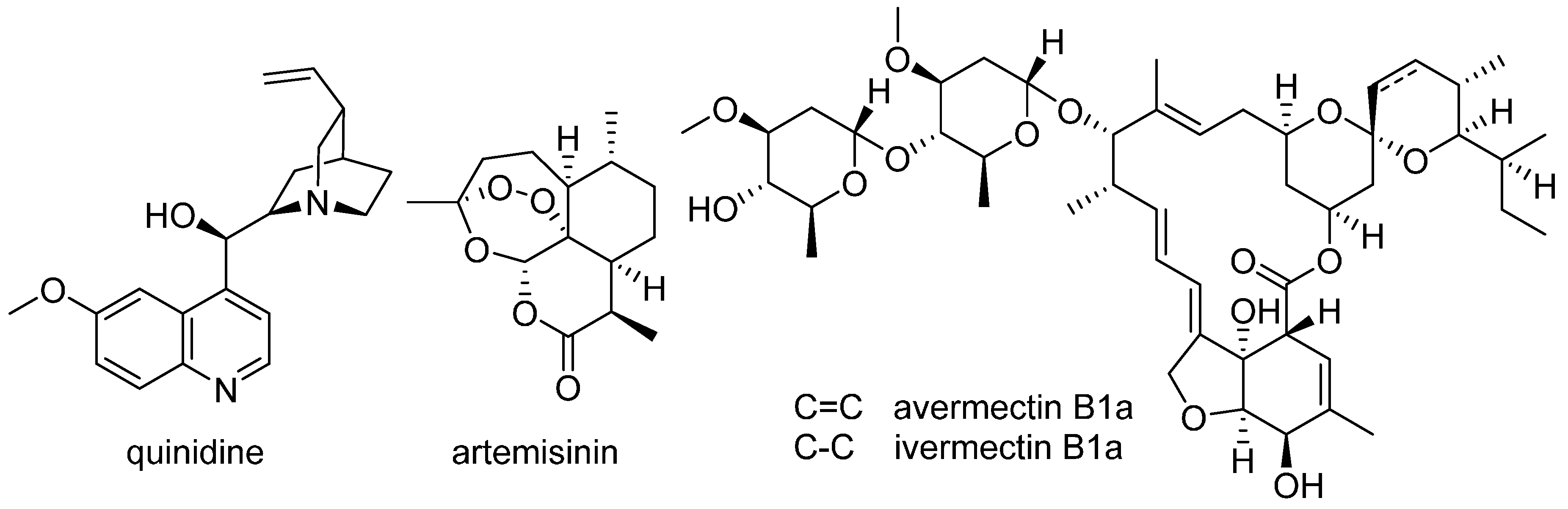
11. Natural Product–Based Drug Discovery Revival
12. Cardiac Glycosides as Novel Modulators of Cancer Cell Survival
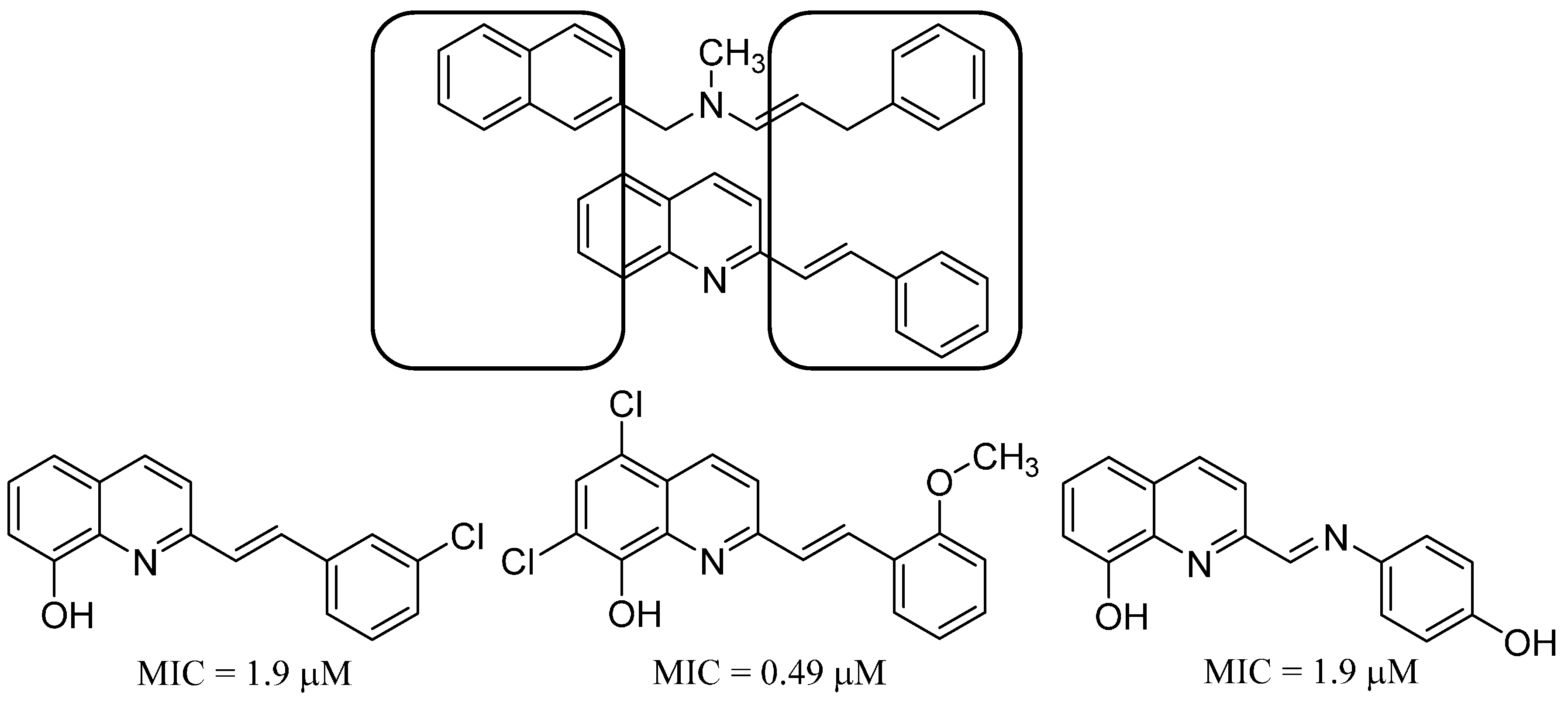
13. Antifungal Styrylquinolines as Efflux Pump Inhibitors
14. Selected Chemical and Biological Applications of Pyridine-Appended Click Triazole Derivatives
15. Big Data Problem in Drug Design and Structure-Property Studies
Acknowledgments
Author Contributions
Conflicts of Interest
References
- The 8th Central European Conference “Chemistry towards Biology”. Available online: http://sites.google.com/site/ctb2016brno (accessed on 15 October 2016).
- Dunker, A.K.; Lawson, J.D.; Brown, C.J.; Williams, R.M.; Romero, P.; Oh, J.S.; Oldfield, C.J.; Campen, A.M.; Ratliff, C.M.; Hipps, K.W.; et al. Intrinsically disordered protein. J. Mol. Graph. Model. 2001, 19, 26–59. [Google Scholar] [CrossRef]
- Dunker, A.K.; Obradovic, Z.; Romero, P.; Garner, E.C.; Brown, C.J. Intrinsic protein disorder in complete genomes. Gen. Inform. Ser. Workshop Gen. Inform. 2000, 11, 161–171. [Google Scholar]
- Motáčková, V.; Nováček, J.; Zawadzka-Kazimierczuk, A.; Kazimierczuk, K.; Žídek, L.; Šanderová, H.; Krásný, L.; Kożmiński, W.; Sklenář, V. Strategy for complete NMR assignment of disordered proteins with highly repetitive sequences based on resolution-enhanced 5D experiments. J. Biomol. NMR 2010, 48, 169–177. [Google Scholar]
- Nováček, J.; Zawadska-Kazimierczuk, A.; Motáčková, V.; Žídek, L.; Kożmiński, W.; Sklenář, V. 5D 13C-detected NMR experiments for backbone assignment of unstructured proteins with a very low signal dispersion. J. Biomol. NMR 2011, 50, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Motáčková, V.; Kadeřávek, P.; Rabatinová, A.; Šanderová, H.; Nováček, J.; Otrusinová, O.; Krásný, L.; Sklenář, V.; Žídek, L. Structural study of the partially disordered full-length δ subunit of RNA polymerasefrom Bacillus subtilis. ChemBioChem 2013, 14, 1772–1779. [Google Scholar]
- Nováček, J.; Haba, N.Y.; Chill, J.H.; Žídek, L.; Sklenář, V. 4D non-uniformly sampled HCBCACON and 1J(NCα)-selective HCBCACON experiments for the sequential assignment and chemical shift analysis of intrinsically disordered proteins. J. Biomol. NMR 2012, 53, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Nováček, J.; Janda, L.; Dopitová, R.; Žídek, L.; Sklenář, V. Efficient protocol for backbone and side-chain assignments of large, intrinsically disordered proteins: Transient secondary structure analysis of 49.2 kDa microtubule associated protein 2c. J. Biomol. NMR 2013, 56, 291–301. [Google Scholar] [CrossRef] [PubMed]
- Kadeřávek, P.; Zapletal, V.; Rabatinová, A.; Krásný, L.; Sklenář, V.; Žídek, L. Spectral density mapping protocols for analysis of molecular motions in disordered proteins. J. Biomol. NMR 2014, 58, 193–207. [Google Scholar] [CrossRef] [PubMed]
- Kadeřávek, P.; Zapletal, V.; Fiala, R.; Srb, P.; Padrta, P.; Přecechtělová, J.; Šoltésová, M.; Kowalewski, J.; Wildmalm, G.; Chmelík, J., Jr.; et al. Spectral density mapping at multiple magnetic fields suitable for 13C NMR relaxation studies. J. Magn. Reson. 2016, 266, 23–40. [Google Scholar] [CrossRef] [PubMed]
- Nováček, J.; Žídek, L.; Sklenář, V. Toward optimal-resolution NMR of intrinsically disordered proteins. J. Magn. Reson. 2014, 241, 41–52. [Google Scholar] [CrossRef] [PubMed]
- Carulla, N.; Zhou, M.; Giralt, E.; Robinson, C.V.; Dobson, C.M. Structure and intermolecular dynamics of aggregates populated during amyloid fibril formation studied by hydrogen/deuterium exchange. Acc. Chem. Res. 2010, 43, 1072–1079. [Google Scholar] [CrossRef] [PubMed]
- Perczel, A.; Hudáky, P.; Pálfi, V.K. Dead-end street of protein folding: Thermodynamic rationale of amyloid fibril formation. J. Am. Chem. Soc. 2007, 129, 14959–14965. [Google Scholar] [CrossRef] [PubMed]
- Neidigh, J.W.; Fesinmeyer, R.M.; Andersen, N.H. Designing a 20-residue protein. Nat. Struct. Biol. 2002, 9, 425–430. [Google Scholar] [CrossRef] [PubMed]
- Perczel, A.; Hollosi, M.; Tusnady, G.; Fasman, G.D. Convex constraint analysis—A natural deconvolution of circular-dichroism curves of proteins. Protein Eng. 1991, 4, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Perczel, A.; Park, K.; Fasman, G.D. Analysis of the circular-dichroism spectrum of proteins using the convex constraint algorithm - a practical guide. Anal. Biochem. 1992, 203, 83–93. [Google Scholar] [CrossRef]
- Farkas, V.; Jákli, I.; Tóth, G.K.; Perczel, A. Aromatic cluster sensor of protein folding: Near-UV electronic circular dichroism bands assigned to fold compactness. Chem. Eur. J. 2016. [Google Scholar] [CrossRef] [PubMed]
- Rovó, P.; Stráner, P.; Láng, A.; Bartha, I.; Huszár, K.; Nyitray, L.; Perczel, A. Structural insights into the Trp-Cage folding intermediate formation. Chemistry 2013, 19, 2628–2640. [Google Scholar] [CrossRef] [PubMed]
- Kardos, J.; Kiss, B.; Micsonai, A.; Rovó, P.; Menyhárd, K.D.; Kovács, J.; Tóth, K.G.; Perczel, A. Phosphorylation as conformational switch from the native to amyloid state: Trp-Cage as a protein aggregation model. J. Phys. Chem. B 2015, 119, 2946–2955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doolittle, R.F.; Fasman, G.D. Redundancies in protein sequences. In Prediction of Protein Structure and the Principles of Protein Conformation; Fasman, G.D., Ed.; Plenum Press: New York, NY, USA, 1989; pp. 599–624. [Google Scholar]
- Perutz, M.F.; Windle, A.H. Cause of neural death in neurodegenerative diseases attributable to expansion of glutamine repeats. Nature 2001, 412, 143–144. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Margolis, R.L.; Becher, M.W.; Wood, J.D.; Engelender, S.; Cooper, J.K.; Sharp, A.H. Neuronal Degeneration and Regeneration: From Basic Mechanisms to Prospects for Therapy; Van Leeuwen, F.W., Salehi, A., Giger, R.J., Holtmaat, A.J.G.D., Verhaagen, J., Eds.; Elsevier: Amsterdam, The Netherlands, 1998; pp. 397–419. [Google Scholar]
- Salichs, E.; Ledda, A.; Mularoni, L.; Alba, M.M.; de la Luna, S. Genome-wide analysis of histidine repeats reveals their role in the localization of human proteins to the nuclear speckles compartment. PLoS Genet. 2009, 5, e1000397. [Google Scholar] [CrossRef] [PubMed]
- Cheng, T.; Xia, W.; Wang, P.; Huang, F.; Wang, J.; Sun, H. Histidine-rich proteins in prokaryotes: Metal homeostasis and environmental habitat-related occurrence. Metallomics 2013, 5, 1423–1429. [Google Scholar] [CrossRef] [PubMed]
- Gaberc-Porekar, V.; Menartr, V. Perspectives of immobilized-metal affinity chromatography. J. Biochem. Biophys. Methods 2001, 49, 335–360. [Google Scholar] [CrossRef]
- Watly, J.; Simonoysky, E.; Wieczorek, R.; Barbosa, N.; Miller, Y.; Kozlowski, H. Insight into the coordination and the binding sites of Cu2+ by the histidyl-6-tag using experimental and computational tools. Inorg. Chem. 2014, 53, 6675–6683. [Google Scholar] [CrossRef] [PubMed]
- Watly, J.; Simonovsky, E.; Barbosa, N.; Spodzieja, M.; Wieczorek, R.; Rodziewicz-Motowidlo, S.; Miller, Y.; Kozlowski, H. African viper poly-his tag peptide fragment efficiently binds metal ions and is folded into an α-helical structure. Inorg. Chem. 2015, 54, 7692–7702. [Google Scholar] [CrossRef] [PubMed]
- Brasili, D.; Watly, J.; Simonovsky, E.; Guerrini, R.; Barbosa, N.A.; Wieczorek, R.; Remelli, M.; Kozlowski, H.; Miller, Y. The unusual metal ion binding ability of histidyl tags and their mutated derivatives. Dalton Trans. 2016, 45, 5629–5639. [Google Scholar] [CrossRef] [PubMed]
- Favreau, P.; Cheneval, O.; Menin, L.; Michalet, S.; Gaertner, H.; Principaud, F.; Thai, R.; Ménez, A.; Bulet, P.; Stöcklin, R. The venom of the snake genus Atheris contains a new class of peptides with clusters of histidine and glycine residues. Rapid Commun. Mass Spectrom. 2007, 21, 406–412. [Google Scholar] [CrossRef] [PubMed]
- Wagstaff, S.C.; Favreau, P.; Cheneval, O.; Laing, G.D.; Wilkinson, M.C.; Miller, R.L.; Stocklin, R.; Harrison, R.A. Molecular characterisation of endogenous snake venom metalloproteinase inhibitors. Biochem. Biophys. Res. Commun. 2008, 365, 650–656. [Google Scholar] [CrossRef] [PubMed]
- Chaturvedi, K.S.; Henderson, J.P. Pathogenic adaptations to host-derived antibacterial copper. Front. Cell. Infect. Microbiol. 2014, 4, 3. [Google Scholar] [CrossRef] [PubMed]
- Kozlowski, H.; Potocki, S.; Remelli, M.; Rowinska-Zyrek, M.; Valensin, D. Specific metal ion binding sites in unstructured regions of proteins. Coord. Chem. Rev. 2013, 257, 2625–2638. [Google Scholar] [CrossRef]
- Krzywoszynska, K.; Rowinska-Zyrek, M.; Witkowska, D.; Potocki, S.; Luczkowski, M.; Kozlowski, H. Polythiol binding to biologically relevant metal ions. Dalton Trans. 2011, 40, 10434–10439. [Google Scholar] [CrossRef] [PubMed]
- Kolkowska, P.; Krzywoszynska, K.; Potocki, S.; Chetana, P.R.; Spodzieja, M.; Rodziewicz-Motowidlo, S.; Kozlowski, H. Specificity of the Zn2+, Cd2+ and Ni2+ ion binding sites in the loop domain of the HypA protein. Dalton Trans. 2015, 44, 9887–9900. [Google Scholar] [CrossRef] [PubMed]
- Bourne, P.E.; Gu, J. Structural Bioinformatics, 2nd ed.; John Wiley & Sons: New York, NY, USA, 2009. [Google Scholar]
- Bourne, P.E.; Weissig, H. Structural Bioinformatics; Wiley: New York, NY, USA, 2003. [Google Scholar]
- Sehnal, D.; Pravda, L.; Svobodová-Vařeková, R.; Ionescu, C.M.; Koča, J. PatternQuery: Web application for fast detection of biomacromolecular fragments in entire Protein Data Bank. Nucleic Acids Res. 2015, 43, W383–W388. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Svobodová-Vařeková, R.; Huber, H.J.; Geidl, S.; Ionescu, C.M.; Wimmerová, M.; Koča, J. SiteBinder: An improved approach for comparing multiple protein structural motifs. J. Chem. Inf. Model. 2012, 52, 343–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svobodová-Vařeková, R.; Jaiswal, D.; Sehnal, D.; Ionescu, C.M.; Geidl, S.; Pravda, L.; Horský, V.; Wimmerová, M.; Koča, J. MotiveValidator: Interactive web-based validation of ligand and residue structure in biomolecular complexes. Nucleic Acids Res. 2014, 42, W227–W233. [Google Scholar]
- Sehnal, D.; Svobodová-Vařeková, R.; Pravda, L.; Ionescu, C.M.; Geidl, S.; Horský, V.; Jaiswal, D.; Wimmerová, M.; Koča, J. ValidatorDB—Database of up-to-date and comprehensive validation results for ligands and non-standard residues from the Protein Data Bank. Nucleic Acids Res. 2014, 43, D369–D375. [Google Scholar] [CrossRef] [PubMed]
- Ionescu, C.M.; Sehnal, D.; Falginella, F.L.; Pant, P.; Pravda, L.; Bouchal, T.; Svobodová-Vařeková, R.; Geidl, S.; Koča, J. AtomicChargeCalculator: Interactive web-based calculation of atomic charges in large biomolecular complexes and drug like molecules. J. Chemoinform. 2015, 7, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Berka, K.; Hanák, O.; Sehnal, D.; Banáš, P.; Navrátilová, V.; Jaiswal, D.; Ionescu, C.M.; Svobodová-Vařeková, R.; Koča, J.; Otyepka, M. MOLEonline 2.0: Interactive web-based analysis of biomacromolecular channels. Nucleic Acids Res. 2012, 40, W222–W227. [Google Scholar] [CrossRef] [PubMed]
- Sehnal, D.; Svobodová-Vařeková, R.; Berka, K.; Pravda, L.; Navrátilová, V.; Banáš, P.; Ionescu, C.M.; Otyepka, M.; Koča, J. MOLE 2.0: Advanced approach for analysis of biomacromolecular channels. J. Cheminform. 2013, 5, 39. [Google Scholar] [CrossRef] [PubMed]
- WebChemistry Services and Apps, Masaryk University. Available online: http://ncbr.muni.cz/WebChemistry (accessed on 15 October 2016).
- PatternQuery, Masaryk University. Available online: http://ncbr.muni.cz/PatternQuery (accessed on 15 October 2016).
- SiteBinder, Masaryk University. Available online: http://ncbr.muni.cz/SiteBinder (accessed on 15 October 2016).
- Wang, X.; Snoeyink, J. Defining and computing optimum RMSD for gapped and weighted multiplestructure alignment. IEEE/ACM Trans Comput. Biol. Bioinform. 2008, 5, 525–533. [Google Scholar] [CrossRef] [PubMed]
- MotiveValidator, Masaryk University. Available online: http://ncbr.muni.cz/MotiveValidator (accessed on 15 October 2016).
- Validator, D.B. Masaryk University. Available online: http://ncbr.muni.cz/ValidatorDB (accessed on 15 October 2016).
- AtomicChargeCalculator, Masaryk University. Available online: http://ncbr.muni.cz/ACC (accessed on 15 October 2016).
- Protein Data Bank in Europe: Bringing Structure to Biology. Available online: http://www.ebi.ac.uk/pdbe/coordinates (accessed on 15 October 2016).
- Andujar, S.; Suvire, F.; Angelina, E.; Peruchena, N.; Cabedo, N.; Cortes, D.; Enriz, R. Searching the “biologically relevant” conformation of dopamine. A Computational approach”. J. Chem. Inf. Model. 2012, 52, 99–112. [Google Scholar] [CrossRef] [PubMed]
- Parraga, J.; Andujar, A.; Rojas, S.; Gutierrez, L.; El Aquad, N.; Sanz, M.; Enriz, R.; Cabedo, N.; Cortes, D. Dopaminergic isoquinolines with hexahydrocyclopenta[ij]-isoquinolines as D2-like selective ligands. Eur. J. Med. Chem. 2016, 122, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Vega-Hissi, E.; Tosso, R.; Enriz, R.; Lucas Gutierrez, J. Molecular Insight into the Interaction Mechanisms of Amino-2H-Imidazole Derivatives with BACE1 Protease: A QM/MM and QTAIM Study. Int. J. Quant. Chem. 2015, 115, 389–397. [Google Scholar] [CrossRef]
- Tosso, R.; Andujar, A.; Gutierrez, L.; Angelina, E.; Rodríguez, R.; Nogueras, M.; Baldoni, H.; Suvire, F.; Cobo, J.; Enriz, R. A molecular Modeling Study of new inhibitors of Dihydrofolate Reductase. MD simulations, QM calculations and experimental corroboration. J. Chem. Inf. Model. 2013, 53, 2018–2032. [Google Scholar] [CrossRef] [PubMed]
- Ortiz, J.; Pigni, N.; Andujar, S.; Roitman, G.; Suvire, F.; Enriz, R.D.; Tapia, A.; Bastida, J.; Feresin, G. Alkaloids from Hippeastrum argentinum and their Cholinesterase Inhibitory Activities: An in vitro and in silico study. J. Nat. Prod. 2016, 79, 1241–1248. [Google Scholar] [CrossRef] [PubMed]
- Bader, R.F.W. Atoms in Molecules. A Quantum Theory; Clarendon: Oxford, UK, 1990. [Google Scholar]
- Popelier, P.L.A. Atoms in Molecules. An Introduction; Pearson Education: Harlow, UK, 1999. [Google Scholar]
- Baroli, B.M. Penetration of nanoparticles and nanomaterials in the skin: Fiction or reality? J. Pharm. Sci. 2010, 99, 21–50. [Google Scholar] [CrossRef] [PubMed]
- Barua, S.; Mitragotri, S. Challenges associated with penetration of nanoparticles across cell and tissue barriers: A review of current status and future prospects. Nano Today 2014, 9, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, R.; Rerek, M.E.; Moore, D.J. Infrared spectroscopy and microscopic imaging of stratum corneum models and skin. Phys. Chem. Chem. Phys. 2000, 2, 4651–4657. [Google Scholar] [CrossRef]
- Marcot, C.; Lo, M.; Kjoller, K.; Domanov, Y.; Balooch, G.; Luengo, G.S. Nanoscale infrared (IR) spectroscopy and imaging of structural lipids in human stratum corneum using an atomic force microscope to directly detect absorbed light from a tunable IR laser source. Exp. Dermatol. 2013, 22, 417–437. [Google Scholar] [CrossRef] [PubMed]
- Bründermann, E.; Havenith, M. SNIM: Scanning near-field infrared microscopy. Annu. Rep. Prog. Chem. Sect. C Phys. Chem. 2008, 104, 235–255. [Google Scholar] [CrossRef]
- Treffer, R.; Böhme, R.; Deckert-Gaudig, T.; Lau, K.; Tiede, S.; Lin, X.; Deckert, V. Advances in TERS (tip-enhanced Raman scattering) for biochemical applications. Biochem. Soc. Trans. 2012, 40, 609–614. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.C.; Singh, G.; Lott, M.T.; Hodge, J.A.; Schurr, T.G.; Lezza, A.M.; Elsas, L.J.; Nikoskelainen, E.K. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy. Science 1988, 242, 1427–1430. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Harding, A.E.; Morgan-Hughes, J.A. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies. Nature 1988, 331, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Brown, G.C.N.D.G.; Cooper, C.E. Mitochondria and Cell Death; Princeton University Press: Princeton, NJ, USA, 1999. [Google Scholar]
- Szewczyk, A.; Wojtczak, L. Mitochondria as a pharmacological target. Pharmacol. Rev. 2002, 54, 101–127. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Targeted drug delivery to mammalian mitochondria in living cells. Expert Opin. Drug Deliv. 2005, 2, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Weiss, M.J.; Wong, J.R.; Ha, C.S.; Bleday, R.; Salem, R.R.; Steele, G.D., Jr.; Chen, L.B. Dequalinium, a topical antimicrobial agent, displays anticarcinoma activity based on selective mitochondrial accumulation. Proc. Natl. Acad. Sci. USA 1987, 84, 5444–5448. [Google Scholar] [CrossRef] [PubMed]
- Horobin, R.W.; Trapp, S.; Weissig, V. Mitochondriotropics: A review of their mode of action, and their applications for drug and DNA delivery to mammalian mitochondria. J. Control. Release 2007, 121, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. From serendipity to mitochondria-targeted nanocarriers. Pharm. Res. 2011, 28, 2657–2668. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Vetro-Widenhouse, T.S.; Rowe, T.C. Topoisomerase II inhibitors induce cleavage of nuclear and 35-kb plastid DNAs in the malarial parasite Plasmodium falciparum. DNA Cell. Biol. 1997, 16, 1483–1492. [Google Scholar] [CrossRef] [PubMed]
- Rowe, T.C.; Weissig, V.; Lawrence, J.W. Mitochondrial DNA metabolism targeting drugs. Adv. Drug Deliv. Rev. 2001, 49, 175–187. [Google Scholar] [CrossRef]
- Weissig, V.; Lasch, J.; Erdos, G.; Meyer, H.W.; Rowe, T.C.; Hughes, J. DQAsomes: A novel potential drug and gene delivery system made from dequalinium. Pharm. Res. 1998, 15, 334–337. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V.; Torchilin, V.P. Towards mitochondrial gene therapy: DQAsomes as a strategy. J. Drug Target. 2001, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Lyrawati, D.; Trounson, A.; Cram, D. Expression of GFP in the mitochondrial compartment using DQAsome-mediated delivery of an artificial mini-mitochondrial genome. Pharm. Res. 2011, 28, 2848–2862. [Google Scholar] [CrossRef] [PubMed]
- Weissig, V. Mitochondria-specific nanocarriers for improving the proapoptotic activity of small molecules. Methods Enzymol. 2012, 508, 131–155. [Google Scholar] [PubMed]
- Weissig, V. DQAsomes as the prototype of mitochondria-targeted pharmaceutical nanocarriers: Preparation, characterization, and use. Methods Mol. Biol. 2015, 1265, 1–11. [Google Scholar] [PubMed]
- Jang, M.S.; Zlobin, A.; Kast, W.M.; Miele, L. Notch signaling as a target in multimodality cancer therapy. Curr. Opin. Mol. Ther. 2000, 2, 55–65. [Google Scholar] [PubMed]
- Kumar, R.; Juillerat-Jeanneret, L.; Golshayan, D. Notch antagonists: Potential modulators of cancer and inflammatory diseases. J. Med. Chem. 2016, 59, 7719–7737. [Google Scholar] [CrossRef] [PubMed]
- Takebe, N.; Nguyen, D.; Yang, S.X. Targeting notch signaling pathway in cancer: Clinical development advances and challenges. Pharmacol. Ther. 2014, 141, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Lobry, C.; Oh, P.; Mansour, M.R.; Look, A.T.; Aifantis, I. Notch signaling: Switching an oncogene to a tumor suppressor. Blood 2014, 123, 2451–2459. [Google Scholar] [CrossRef] [PubMed]
- Loenarz, C.; Schofield, C.J. Physiological and biochemical aspects of hydroxylations and demethylations catalyzed by human 2-oxoglutarate oxygenases. Trends Biochem. Sci. 2011, 36, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Lavaissiere, L.; Jia, S.; Nishiyama, M.; de la Monte, S.; Stern, A.M.; Wands, J.R.; Friedman, P.A. Overexpression of human aspartyl(asparaginyl)beta-hydroxylase in hepatocellular carcinoma and cholangiocarcinoma. J. Clin. Invest. 1996, 98, 1313–1323. [Google Scholar] [CrossRef] [PubMed]
- Aihara, A.; Huang, C.K.; Olsen, M.J.; Lin, Q.; Chung, W.; Tang, Q.; Dong, X.; Wands, J.R. A cell-surface beta-hydroxylase is a biomarker and therapeutic target for hepatocellular carcinoma. Hepatology 2014, 60, 1302–1313. [Google Scholar] [CrossRef] [PubMed]
- Iwagami, Y.; Huang, C.K.; Olsen, M.J.; Thomas, J.M.; Jang, G.; Kim, M.; Lin, Q.; Carlson, R.I.; Wagner, C.E.; Dong, X.; et al. Aspartate beta-hydroxylase modulates cellular senescence through glycogen synthase kinase 3beta in hepatocellular carcinoma. Hepatology 2016, 63, 1213–1226. [Google Scholar] [CrossRef] [PubMed]
- von Eiff, C.; Becker, K.; Machka, K.; Stammer, H.; Peters, G. Nasal carriage as a source of Staphylococcus aureus bacteremia. Study group. N. Engl. J. Med. 2001, 344, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Keary, R.; Sanz-Gaitero, M.; van Raaij, M.J.; O’Mahony, J.; Fenton, M.; McAuliffe, O.; Hill, C.; Ross, R.P.; Coffey, A. Characterization of a bacteriophage-derived murein peptidase for elimination of antibiotic-resistant Staphylococcus aureus. Curr. Protein Pept. Sci. 2015, 17, 183–190. [Google Scholar] [CrossRef]
- Sanz-Gaitero, M.; Keary, R.; García-Doval, C.; Coffey, A.; van Raaij, M.J. Crystal structure of the lytic CHAP(K) domain of the endolysin LysK from Staphylococcus aureus bacteriophage K. Virol. J. 2014, 11, 133. [Google Scholar] [CrossRef] [PubMed]
- Atanasov, A.G.; Waltenberger, B.; Pferschy-Wenzig, E.M.; Linder, T.; Wawrosch, C.; Uhrin, P.; Temml, V.; Wang, L.; Schwaiger, S.; Heiss, E.H.; et al. Discovery and resupply of pharmacologically active plant-derived natural products: A review. Biotechnol. Adv. 2015, 33, 1582–1614. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Wolfender, J.L.; Dias, D.A. The pharmaceutical industry and natural products: Historical status and new trends. Phyochem. Rev. 2014, 14, 299–315. [Google Scholar] [CrossRef]
- Amirkia, V.; Heinrich, M. Natural products and drug discovery: A survey of stakeholders in industry and academia. Front Pharmacol. 2015, 6, 237. [Google Scholar] [CrossRef] [PubMed]
- Waltenberger, B.; Mocan, A.; Smejkal, K.; Heiss, E.H.; Atanasov, A.G. Natural products to counteract the epidemic of cardiovascular and metabolic disorders. Molecules 2016, 21, 807. [Google Scholar] [CrossRef] [PubMed]
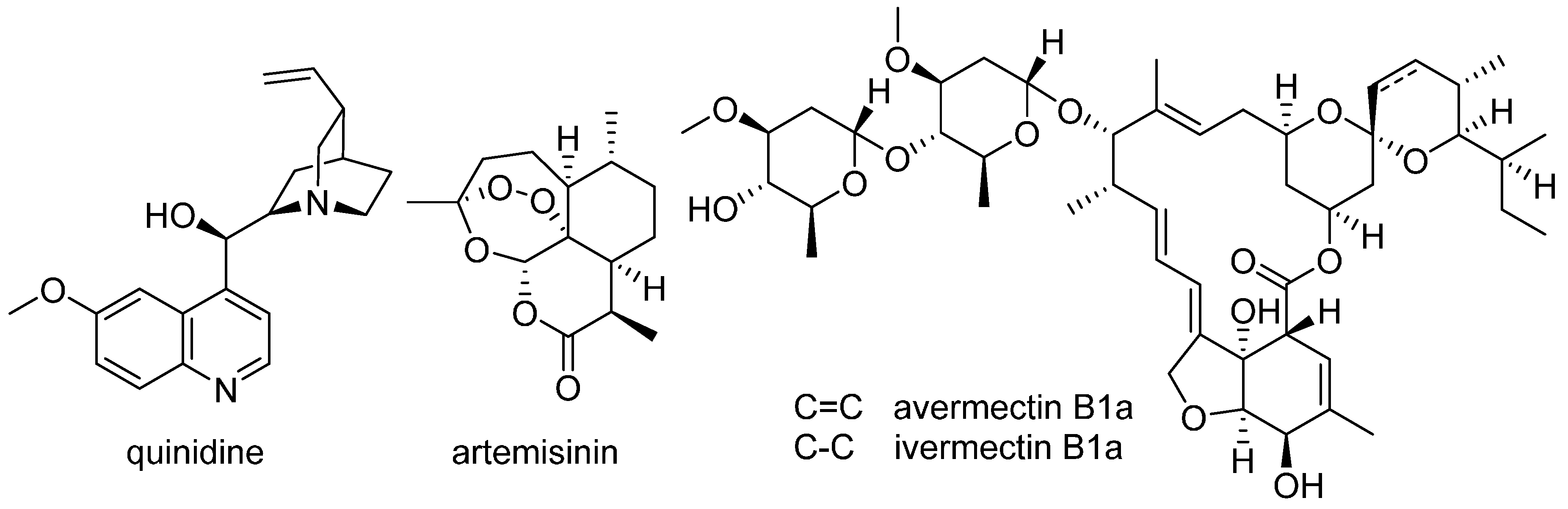
- Andersson, J.; Forssberg, H.; Zierath, J.R. Avermectin and artemisinin—Revolutionary therapies against parasitic diseases. The Nobel Assembly at Karolinska Institutet. 2015. Available online: http://www.nobelprize.org/nobel_prizes/medicine/laureates/2015/advanced-medicineprize2015.pdf (accessed on 20 September 2016).
- World Health Organization. World malaria report 2015; WHO Press: Geneva, Switzerland, 2015. [Google Scholar]
- Wiesner, J.; Ortmann, R.; Jomaa, H.; Schlitzer, M. New antimalarial drugs. Angew. Chem. Int. Ed. 2003, 42, 5274–5293. [Google Scholar] [CrossRef] [PubMed]
- Efferth, T.; Zacchino, S.; Georgiev, M.I.; Liu, L.; Wagner, H.; Panossian, A. Nobel prize for artemisinin brings phytotherapy into the spotlight. Phytomedicine 2015, 22, A1–A3. [Google Scholar] [CrossRef] [PubMed]
- Hertweck, C. Natural products as source of therapeutics against parasitic diseases. Angew. Chem. Int. Ed. 2015, 54, 14622–14624. [Google Scholar] [CrossRef] [PubMed]
- Paddon, C.J.; Westfall, P.J.; Pitera, D.J.; Benjamin, K.; Fisher, K.; McPhee, D.; Leavell, M.D.; Tai, A.; Main, A.; Eng, D.; et al. High-level semi-synthetic production of the potent antimalarial artemisinin. Nature 2013, 496, 528–532. [Google Scholar] [CrossRef] [PubMed]
- Diederich, M.; Muller, F.; Cerella, C. Cardiac glycosides: From molecular targets to immunogenic cell death. Biochem. Pharmacol. 2016. [Google Scholar] [CrossRef] [PubMed]
- Radogna, F.; Cerella, C.; Gaigneaux, A.; Christov, C.; Dicato, M.; Diederich, M. Cell type-dependent ROS and mitophagy response leads to apoptosis or necroptosis in neuroblastoma. Oncogene 2016, 35, 3839–3853. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sudbery, P. Candida albicans, a major human fungal pathogen. J. Microbiol. 2011, 49, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Sanguinetti, M.; Posteraro, B.; Fiori, B.; Ranno, S.; Torelli, R.; Fadda, G. Mechanisms of azole resistance in clinical isolates of Candida glabrata collected during a hospital survey of antifungal resistance. Antimicrob. Agents Chemother. 2005, 49, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Kowalczyk, W. Azole antimycotics—A highway to new drugs or a dead end? Curr. Med. Chem. 2012, 19, 1378–1388. [Google Scholar] [CrossRef] [PubMed]
- Holmes, A.R.; Lin, Y.H.; Niimi, K.; Lamping, E.; Keniya, M.; Niimi, M.; Tanabe, K.; Monk, B.C.; Cannon, R.D. ABC transporter Cdr1p contributes more than Cdr2p does to fluconazole efflux in fluconazole-resistant Candida albicans clinical isolates. Antimicrob. Agents Chemother. 2008, 52, 3851–3862. [Google Scholar] [CrossRef] [PubMed]
- Polanski, J.; Kurczyk, A.; Bak, A.; Musiol, R. Privileged structures—dream or reality: Preferential organization of azanaphthalene scaffold. Curr. Med. Chem. 2012, 19, 1921–1945. [Google Scholar] [CrossRef] [PubMed]
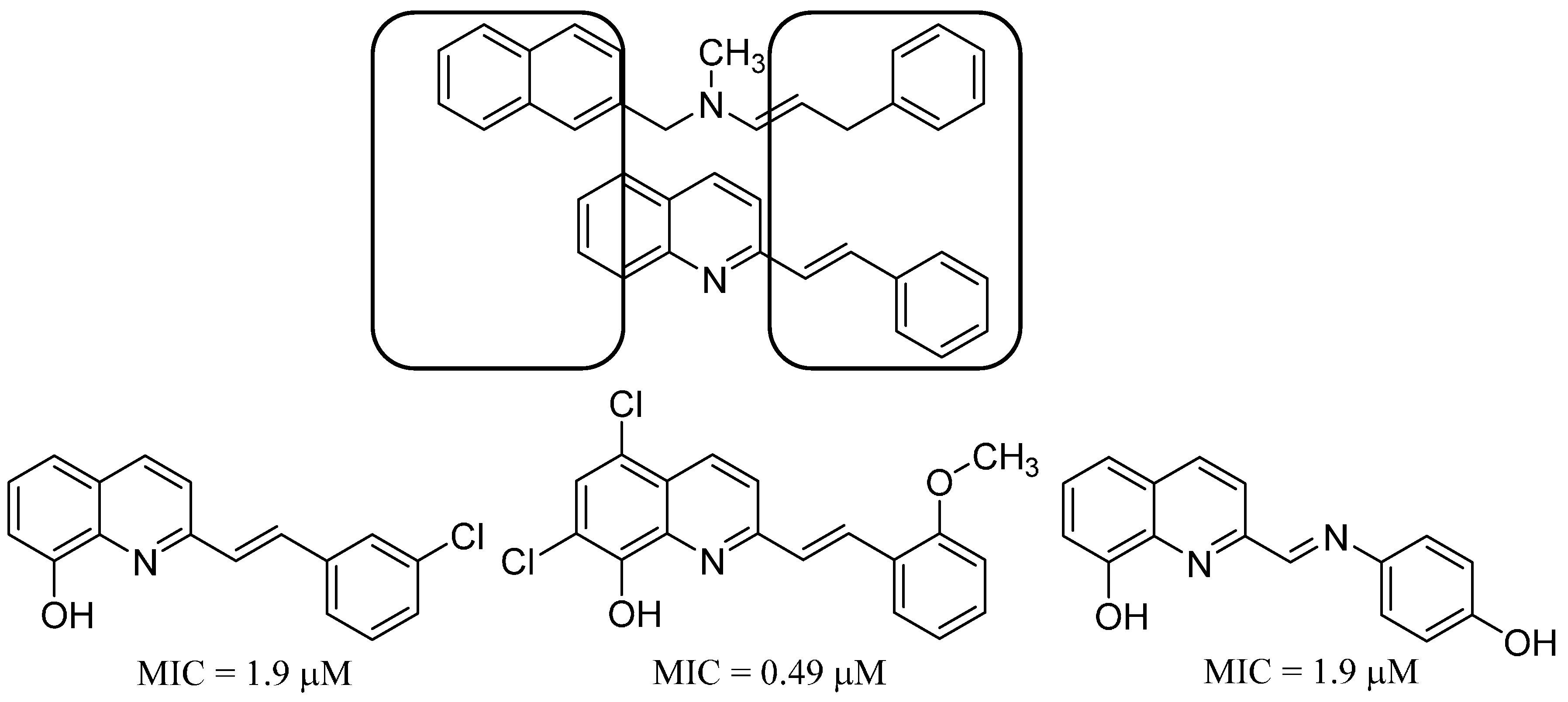
- Musiol, R.; Jampilek, J.; Buchta, V.; Silva, L.; Niedbala, H.; Podeszwa, B.; Palka, A.; Majerz-Maniecka, K.; Oleksyn, B.; Polanski, J. Antifungal properties of new series of quinoline derivatives. Bioorg. Med. Chem. 2006, 14, 3592–3598. [Google Scholar] [CrossRef] [PubMed]
- Cieslik, W.; Musiol, R.; Nycz, J.E.; Jampilek, J.; Vejsova, M.; Wolff, M.; Machura, B.; Polanski, J. Contribution to investigation of antimicrobial activity of styrylquinolines. Bioorg. Med. Chem. 2012, 20, 6960–6968. [Google Scholar] [CrossRef] [PubMed]
- Musiol, R.; Serda, M.; Hensel-Bielowka, S.; Polanski, J. Quinoline-based antifungals. Curr. Med. Chem. 2010, 17, 1960–1973. [Google Scholar] [CrossRef] [PubMed]
- Mrozek-Wilczkiewicz, A.; Spaczynska, E.; Malarz, K.; Cieslik, W.; Rams-Baron, M.; Kryštof, V.; Musiol, R. Design, synthesis and in vitro activity of anticancer styrylquinolines. The p53 independent mechanism of action. PLoS ONE 2015, 10, e0142678. [Google Scholar] [CrossRef] [PubMed]
- Huisgen, R. 1,3-Dipolar Cycloaddition Chemistry; Padwa, A., Ed.; Wiley: New York, NY, USA, 1984; Volume 1. [Google Scholar]
- Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-Triazoles by regiospecific copper(I)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 2002, 67, 3057–3064. [Google Scholar]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A Stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Buckley, B.R.; Heaney, H. Mechanistic investigations of copper(I)-catalysed alkyne–azide cycloaddition reactions. Top. Heterocycl. Chem. 2012, 28, 1–30. [Google Scholar]
- Košmrlj, J. Click Triazoles, Topics in Heterocyclic Chemistry; Springer: Berlin/Heidelberg, Germany, 2012. [Google Scholar]
- Zheng, T.; Rouhanifard, S.; Jalloh, A.; Wu, P. Click triazoles for bioconjugation. Top. Heterocycl. Chem. 2012, 28, 163–184. [Google Scholar] [PubMed]
- Watkinson, M. Click triazoles as chemosensors. Top. Heterocycl. Chem. 2012, 28, 109–136. [Google Scholar]
- Lee, S.; Flood, A.H. Binding anions in rigid and reconfigurable triazole receptors. Top. Heterocycl. Chem. 2012, 28, 85–108. [Google Scholar]
- Chow, H.F.; Lo, C.M.; Chen, Y. Triazole-based polymer gels. Top. Heterocycl. Chem. 2012, 28, 137–162. [Google Scholar]
- Mignani, S.; Zhou, Y.; Lecourt, T.; Micouin, L. Recent developments in the synthesis 1,4,5-trisubstituted triazoles. Top. Heterocycl. Chem. 2012, 28, 185–232. [Google Scholar]
- Crowley, J.D.; McMorran, D.A. “Click-Triazole” coordination chemistry: Exploiting 1,4-disubstituted-1,2,3-triazoles as ligands. Top. Heterocycl. Chem. 2012, 28, 31–84. [Google Scholar]
- Urankar, D.; Pinter, B.; Pevec, A.; de Proft, F.; Turel, I.; Košmrlj, J. Click-Triazole N2 coordination to transition-metal ions is assisted by a pendant pyridine substituent. Inorg. Chem. 2010, 49, 4820–4829. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Urankar, D.; Košmrlj, J. Synthesis and NMR analysis of 1,4-disubstituted 1,2,3-triazoles tethered to pyridine, pyrimidine, and pyrazine rings. Eur. J. Org. Chem. 2014, 8167–8181. [Google Scholar] [CrossRef]
- Kafka, S.; Hauke, S.; Salčinović, A.; Soidinsalo, O.; Urankar, D.; Košmrlj, J. Copper(I)-catalyzed [3+2]cycloaddition of 3-azidoquinoline-2,4(1H,3H)-diones with terminal alkynes. Molecules 2011, 16, 4070–4081. [Google Scholar] [CrossRef]
- Urankar, D.; Pevec, A.; Turel, I.; Košmrlj, J. Pyridyl conjugated 1,2,3-triazole is a versatile coordination ability ligand enabling supramolecular associations. Cryst. Growth Des. 2010, 10, 4920–4927. [Google Scholar] [CrossRef]
- Pinter, B.; Demšar, A.; Urankar, D.; de Proft, F.; Košmrlj, J. Conformational fluxionality in a palladium(II) complex of flexible click chelator 4-phenyl-1-(2-picolyl)-1,2,3-triazole: A dynamic NMR and DFT study. Polyhedron 2011, 30, 2368–2373. [Google Scholar] [CrossRef]
- Bratsos, I.; Urankar, D.; Zangrando, E.; Genova, P.; Košmrlj, J.; Alessio, E.; Turel, I. 1-(2-Picolyl)-substituted 1,2,3-triazole as novel chelating ligand for the preparation of ruthenium complexes with potential anticancer activity. Dalton Trans. 2011, 40, 5188–5199. [Google Scholar] [CrossRef] [PubMed]
- Urankar, D.; Košmrlj, J. Concise and diversity-oriented synthesis of ligand arm-functionalized azoamides. J. Comb. Chem. 2008, 10, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Urankar, D.; Steinbücher, M.; Kosjek, J.; Košmrlj, J. N-(Propargyl)diazenecarboxamides for ‘click’ conjugation and their 1,3-dipolar cycloadditions with azidoalkylamines in the presence of Cu(II). Tetrahedron 2010, 66, 2602–2613. [Google Scholar] [CrossRef]
- Urankar, D.; Pevec, A.; Košmrlj, J. Synthesis and characterization of platinum(II) complexes with a diazenecarboxamide-appended picolyl-triazole ligand. Eur. J. Inorg. Chem. 2011, 1921–1929. [Google Scholar] [CrossRef]
- Urankar, D.; Košmrlj, J. Preparation of diazenecarboxamide-carboplatin conjugates by click chemistry. Inorg. Chim. Acta 2010, 363, 3817–3822. [Google Scholar] [CrossRef]
- Stojanović, N.; Urankar, D.; Brozović, A.; Ambriović-Ristov, A.; Osmak, M.; Košmrlj, J. Design and evaluation of biological activity of diazenecarboxamide-extended cisplatin and carboplatin analogues. Acta Chim. Slov. 2013, 60, 368–374. [Google Scholar] [PubMed]
- Bolje, A.; Košmrlj, J. A Selective approach to pyridine appended 1,2,3-triazolium salts. Org. Lett. 2013, 15, 5084–5087. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Hohloch, S.; Urankar, D.; Pevec, A.; Gazvoda, M.; Sarkar, B.; Košmrlj, J. Exploring the scope of pyridyl- and picolyl-functionalized 1,2,3-triazol-5-ylidenes in bidentate coordination to ruthenium(II) cymene chloride complexes. Organometallics 2014, 33, 2588–2598. [Google Scholar] [CrossRef]
- Hohloch, S.; Kaiser, S.; Duecker, F.L.; Bolje, A.; Maity, R.; Košmrlj, J.; Sarkar, B. Catalytic oxygenation of sp3 “C–H” bonds with Ir(III) complexes of chelating triazoles and mesoionic carbenes. Dalton Trans. 2015, 44, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Hohloch, S.; van der Meer, M.; Košmrlj, J.; Sarkar, B. RuII, OsII, and IrIII Complexes with chelating pyridyl–mesoionic carbene ligands: Structural characterization and applications in transfer hydrogenation catalysis. Chem. Eur. J. 2015, 21, 6756–6764. [Google Scholar] [CrossRef] [PubMed]
- Bolje, A.; Hohloch, S.; Košmrlj, J.; Sarkar, B. RuII, IrIII and OsII mesoionic carbene complexes: Efficient catalysts for transfer hydrogenation of selected functionalities. Dalton Trans. 2016, 45, 15983–15993. [Google Scholar] [CrossRef] [PubMed]
- Gazvoda, M.; Virant, M.; Pevec, A.; Urankar, D.; Bolje, A.; Kočevar, M.; Košmrlj, J. A mesoionic bis(Py-tzNHC) palladium(II) complex catalyses “green” Sonogashira reaction through an unprecedented mechanism. Chem. Commun. 2016, 52, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Wagner, F.F.; Comins, D.L. Expedient five-step synthesis of SIB-1508Y from natural nicotine. J. Org. Chem. 2006, 71, 8673–8675. [Google Scholar] [CrossRef] [PubMed]
- Kilpin, K.J.; Crot, S.; Riedel, T.; Kitchen, J.A.; Dyson, P.J. Ruthenium(II) and osmium(II) 1,2,3-triazolylidene organometallics: A preliminary investigation into the biological activity of ‘click’ carbene complexes. Dalton Trans. 2014, 43, 1443–1448. [Google Scholar] [CrossRef] [PubMed]
- Steiner, I.; Stojanović, N.; Bolje, A.; Brozovic, A.; Polančec, D.; Ambriović-Ristov, A.; Radić Stojković, M.; Piantanida, I.; Eljuga, D.; Košmrlj, J.; et al. Discovery of ‘click’ 1,2,3-triazolium salts as potential anticancer drugs. Radiol. Oncol. 2016, 50, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Laney, D. 3-D Data Management: Controlling Data Volume, Velocity; META Group Inc.: Stamford, CT, USA, 2001. [Google Scholar]
- Szlezák, N.; Evers, M.; Wang, J.; Pérez, L. The role of big data and advanced analytics in drug discovery, development, and commercialization. Clin. Pharmacol. Ther. 2014, 95, 492–4955. [Google Scholar] [CrossRef] [PubMed]
- Todeschini, R.; Consonni, V. Handbook of Molecular Descriptors; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Polanski, J.; Gasteiger, J. Computer Representation of Chemical Compounds. In Handbook of Computational Chemistry; Leszczynski, J., Puzyn, T., Eds.; Springer: Dordrecht, Germany, 2016. [Google Scholar]
- Polanski, J. Chemoinformatics. In Comprehensive Chemometrics; Walczak, B., Tauler, R., Brown, S., Eds.; Elsevier: Amsterdam, The Netherlands, 2009; Volume 4, pp. 459–505. [Google Scholar]
- Polanski, J. Big data in structure-property studies—From definitions to models. In Advances in QSAR Modeling with Applications in Pharmaceutical, Chemical, Food, Agricultural, and Environmental Sciences; Leszczynski, J., Roy, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2017; In press. [Google Scholar]
- PASS, Prediction of Activity Spectra for Substances. Available online: www.pharmaexpert.ru/passonline (accessed on 20 September 2016).
- Polanski, J.; Kucia, U.; Duszkiewicz, R.; Kurczyk, A.; Magdziarz, T.; Gasteiger, J. Molecular descriptor data explains market prices of the large commercial chemical compound library. Sci. Rep. 2016, 6, 28521. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perczel, A.; Atanasov, A.G.; Sklenář, V.; Nováček, J.; Papoušková, V.; Kadeřávek, P.; Žídek, L.; Kozłowski, H.; Wątły, J.; Hecel, A.; et al. The Eighth Central European Conference “Chemistry towards Biology”: Snapshot. Molecules 2016, 21, 1381. https://doi.org/10.3390/molecules21101381
Perczel A, Atanasov AG, Sklenář V, Nováček J, Papoušková V, Kadeřávek P, Žídek L, Kozłowski H, Wątły J, Hecel A, et al. The Eighth Central European Conference “Chemistry towards Biology”: Snapshot. Molecules. 2016; 21(10):1381. https://doi.org/10.3390/molecules21101381
Chicago/Turabian StylePerczel, András, Atanas G. Atanasov, Vladimír Sklenář, Jiří Nováček, Veronika Papoušková, Pavel Kadeřávek, Lukáš Žídek, Henryk Kozłowski, Joanna Wątły, Aleksandra Hecel, and et al. 2016. "The Eighth Central European Conference “Chemistry towards Biology”: Snapshot" Molecules 21, no. 10: 1381. https://doi.org/10.3390/molecules21101381
APA StylePerczel, A., Atanasov, A. G., Sklenář, V., Nováček, J., Papoušková, V., Kadeřávek, P., Žídek, L., Kozłowski, H., Wątły, J., Hecel, A., Kołkowska, P., Koča, J., Svobodová-Vařeková, R., Pravda, L., Sehnal, D., Horský, V., Geidl, S., Enriz, R. D., Matějka, P., ... Jampílek, J. (2016). The Eighth Central European Conference “Chemistry towards Biology”: Snapshot. Molecules, 21(10), 1381. https://doi.org/10.3390/molecules21101381