
Development of Mushroom-Based Cosmeceutical Formulations with Anti-Inflammatory, Anti-Tyrosinase, Antioxidant, and Antibacterial Properties



 , and
, and 
Abstract
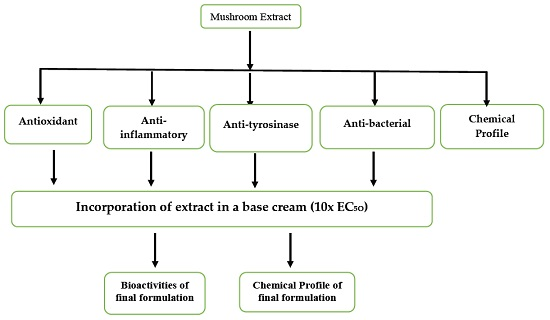
: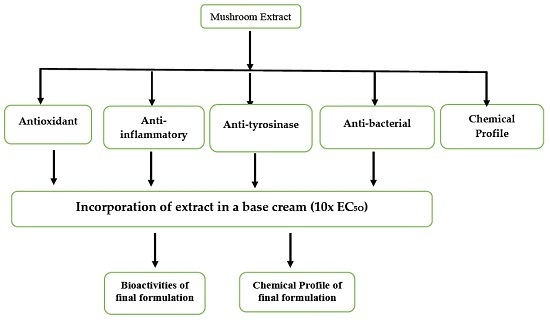
1. Introduction
2. Results and Discussion
2.1. Chemical Characterization of the Mushroom Ethanolic Extracts
2.2. Bioactive Properties of the Mushroom Ethanolic Extracts
2.2.1. Anti-Inflammatory Activity
2.2.2. Anti-Tyrosinase Activity
2.2.3. Antioxidant Activity
2.2.4. Antibacterial Activity
2.3. Chemical Profile and Bioactivity of the Final Cosmeceutical Formulations
3. Materials and Methods
3.1. Standards and Reagents
3.2. Mushroom Species and Preparation of the Ethanolic Extracts
3.3. Chemical Characterization of the Extracts
3.3.1. Analysis of Phenolic Acids
3.3.2. Analysis of Ergosterol
3.4. Evaluation of the Anti-Inflammatory Activity
3.5. Evaluation of the Anti-Tyrosinase Activity
3.6. Evaluation of the Antioxidant Activity
3.6.1. Reducing Power
3.6.2. DPPH Scavenging Activity
3.7. Evaluation of the Antibacterial Activity
3.7.1. Bacteria Strains
3.7.2. Characterization of the Antibiotic Susceptibility of the Bacteria Strains
3.7.3. Determination of the Minimal Inhibitory Concentration (MIC)
3.8. Development of the Cosmeceutical Formulations Based on Mushroom Extracts
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Papakonstantinou, E.; Roth, M.; Karakiulakis, G. Hyaluronic acid: A key molecule in skin aging. Dermato-Endocrinology 2012, 4, 253–258. [Google Scholar] [CrossRef] [PubMed]
- Leem, K.H. Effects of Olibanum extracts on the collagenase activity and procollagen synthesis in Hs68 human fibroblasts and tyrosinase activity. Adv. Sci. Technol. Lett. 2015, 88, 172–175. [Google Scholar]
- Yadav, T.; Mishra, S.; Das, S.; Aggarwal, S.; Rani, V. Anticedants and natural prevention of environmental toxicants induced accelerated aging of skin. Environ. Toxicol. Pharmacol. 2015, 39, 384–391. [Google Scholar] [CrossRef] [PubMed]
- Gündoğdu, A.; Kılıç, H.; Ulu-Kılıç, A.; Kutateladze, M. Susceptibilities of multidrug-resistant pathogens responsible for complicated skin and soft tissue infections to standard bacteriophage cocktails. Mikrobiyol. Bulteni 2016, 2, 215–223. [Google Scholar] [CrossRef]
- Tamsyn, S.A.T.; Pauline, H.; Declan, P.N. Anti-collagenase, anti-elastase and anti-oxidant activities of extracts from 21 plants. BMC Complement. Altern. Med. 2009, 9, 1–11. [Google Scholar]
- Soto, M.L.; Falqué, E.; Domínguez, H. Relevance of natural phenolics from grape and derivative products in the formulation of cosmetics—Review. Cosmetics 2015, 2, 259–276. [Google Scholar] [CrossRef]
- Taofiq, O.; González-Paramás, A.M.; Martins, A.; Barreiro, M.F.; Ferreira, I.C.F.R. Mushrooms extracts and compounds in cosmetics, cosmeceuticals and nutricosmetics—A review. Ind. Crops Prod. 2016, 90, 38–48. [Google Scholar] [CrossRef]
- Kamarudzaman, A.N.; Chay, T.C.; Amir, A.; Talib, S.A. Biosorption of Mn(II) ions from Aqueous Solution by Pleurotus Spent Mushroom Compost in a Fixed-Bed Column. Procedia Soc. Behav. Sci. 2015, 195, 2709–2716. [Google Scholar] [CrossRef]
- Stojković, D.; Reis, F.S.; Glamočlija, J.; Ćirić, A.; Barros, L.; van Griensven, L.J.L.D.; Ferreira, I.C.F.R.; Soković, M. Cultivated strains of Agaricus bisporus and A. brasiliensis: Chemical characterization and evaluation of antioxidant and antimicrobial properties for final healthy product—Natural preservatives in yoghurt. Food Funct. 2014, 5, 1602–1612. [Google Scholar] [CrossRef] [PubMed]
- Baba, E.; Uluköy, G.; Öntaş, C. Effects of feed supplemented with Lentinula edodes mushroom extract on the immune response of rainbow trout, Oncorhynchus mykiss, and disease resistance against Lactococcus garvieae. Aquaculture 2015, 448, 476–482. [Google Scholar] [CrossRef]
- Cheah, I.K.; Halliwell, B. Ergothioneine: Antioxidant potential, physiological function and role in disease. Biochim. Biophys. Acta 2012, 1822, 784–793. [Google Scholar] [CrossRef] [PubMed]
- Zembron-Lacny, A.; Gajewski, M.; Naczk, M.; Siatkowski, I. Effect of shiitake (Lentinus edodes) extract on antioxidant and inflammatory response to prolonged eccentric exercise. J. Physiol. Pharmacol. 2013, 64, 249–254. [Google Scholar] [PubMed]
- Mizuno, M.; Nishitani, Y.; Hashimoto, T.; Kanazawa, K. Different suppressive effects of fucoidan and lentinan on IL-8 mRNA expression in in vitro gut inflammation. Biosci. Biotechnol. Biochem. 2009, 3, 2324–2325. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Barros, L.; Martins, A.; Herbert, P.; Ferreira, I.C.F.R. Nutritional characterisation of Pleurotus ostreatus (Jacq. ex Fr.) P. Kumm. Produced using paper scraps as substrate. Food Chem. 2015, 169, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Taofiq, O.; Calhelha, R.C.; Heleno, S.A.; Barros, L.; Martins, A.; Santos-Buelga, C.; Queiroz, M.J.R.P.; Ferreira, I.C.F.R. The contribution of phenolic acids to the anti-inflammatory activity of mushrooms: Screening in phenolic extracts, individual parent molecules and synthesized glucuronated and methylated derivatives. Food Res. Int. 2015, 76, 821–827. [Google Scholar] [CrossRef] [Green Version]
- Stefan, R.I.; Vamanu, E.; Angelescu, G.C. Antioxidant activity of crude methanolic extracts from Pleurotus ostreatus. Res. J. Phytochem. 2015, 9, 25–32. [Google Scholar]
- Facchini, J.M.; Alves, E.P.; Aguilera, C.; Gern, R.M.M.; Silveira, M.L.L.; Wisbeck, E.; Furlan, S.A. Antitumor activity of Pleurotus ostreatus polysaccharide fractions on Ehrlich tumor and Sarcoma 180. Int. J. Biol. Macromol. 2014, 68, 72–77. [Google Scholar] [CrossRef] [PubMed]
- Carneiro, A.A.; Ferreira, I.C.F.R.; Dueñas, M.; Barros, L.; da Silva, R.; Gomes, E.; Santos-Buelga, C. Chemical composition and antioxidant activity of dried powder formulations of Agaricus blazei and Lentinus edodes. Food Chem. 2013, 138, 2168–2173. [Google Scholar] [CrossRef] [PubMed]
- Barreira, J.C.M.; Oliveira, M.B.P.P.; Ferreira, I.C.F.R. Development of a novel methodology for the analysis of ergosterol in mushrooms. Food Anal. Methods 2014, 7, 217–223. [Google Scholar] [CrossRef]
- Taofiq, O.; Martins, A.; Barreiro, M.F.; Ferreira, I.C.F.R. Anti-inflammatory potential of mushroom extracts and isolated metabolites. Trends Food Sci. Technol. 2016, 50, 193–210. [Google Scholar] [CrossRef]
- Alam, N.; Yoon, K.N.; Lee, K.R.; Shin, P.G.; Cheong, J.C.; Yoo, Y.B.; Shim, M.J.; Lee, M.W.; Lee, U.Y.; Lee, T.S. Antioxidant activities and tyrosinase inhibitory effects of different extracts from Pleurotus ostreatus fruiting bodies. Mycobiology 2010, 38, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Miyake, M.; Yamamoto, S.; Sano, O.; Fujii, M.; Kohno, K.; Ushio, S.; Iwaki, K.; Fukuda, S. Inhibitory effects of 2-amino-3H-phenoxazin-3one on the melanogenesis of murine B16 melanoma cell line. Biosci. Biotechnol. Biochem. 2010, 74, 753–758. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.N.; Alam, N.; Lee, J.S.; Lee, K.R.; Lee, T.S. Detection of phenolic compounds concentration and evaluation of antioxidant and antityrosinase activity of various extract from Lentinus edodes. World Appl. Sci. J. 2011, 12, 1851–1859. [Google Scholar]
- Yan, Z.F.; Yang, Y.; Tian, F.H.; Mao, X.X.; Li, Y.; Li, C.T. Inhibitory and acceleratory effects of Inonotus obliquus on tyrosinase activity and melanin formation in B16 melanoma cells. Evid. Based Complement. Altern. Med. 2014. [Google Scholar] [CrossRef] [PubMed]
- Meng, T.X.; Zhang, C.F.; Miyamoto, T.; Ishikawa, H.; Shimizu, K.; Ohga, S.; Kondo, R. The melanin biosynthesis stimulating compounds isolated from the fruiting bodies of Pleurotus citrinopileatus. J. Cosmet. Dermatol. Sci. Appl. 2012, 2, 151–157. [Google Scholar]
- Ali, S.A.; Choudhary, R.K.; Naaz, I.; Ali, A.S. Understanding the Challenges of Melanogenesis: Key role of bioactive compounds in the treatment of hyperpigmentory disorders. J. Pigment. Dis. 2015, 2, 1–9. [Google Scholar]
- Reis, F.S.; Martins, A.; Barros, L.; Ferreira, I.C.F.R. Antioxidant properties and phenolic profile of the most widely appreciated cultivated mushrooms: A comparative study between in vivo and in vitro samples. Food Chem. Toxicol. 2012, 50, 1201–1207. [Google Scholar] [CrossRef] [PubMed]
- Heleno, S.A.; Ferreira, R.C.; Antonio, A.L.; Queiroz, M.J.R.P.; Barros, L.; Ferreira, I.C.F.R. Nutritional value, bioactive compounds and antioxidant properties of three edible mushrooms from Poland. Food Biosci. 2015, 11, 48–55. [Google Scholar] [CrossRef] [Green Version]
- Masaki, H. Role of antioxidants in the skin: Anti-aging effects. J. Dermatol. Sci. 2010, 58, 85–90. [Google Scholar] [CrossRef] [PubMed]
- Barros, L.; Cruz, T.; Baptista, P.; Estevinho, L.M.; Ferreira, I.C.F.R. Wild and commercial mushrooms as source of nutrients and nutraceuticals. Food Chem. Toxicol. 2008, 46, 2742–2747. [Google Scholar] [CrossRef] [PubMed]
- Alves, M.J.; Ferreira, I.C.F.R.; Martins, A.; Pintado, M. Antimicrobial activity of wild mushroom extracts against clinical isolates resistant to different antibiotics. J. Appl. Microbiol. 2012, 115, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, F.; Palmeira-de-Oliveira, A.; das Neves, J.; Sarmento, B.; Amaral, M.H.; Oliveira, M.B. Medicago spp. extracts as promising ingredients for skin care products. Ind. Crops Prod. 2013, 49, 634–644. [Google Scholar] [CrossRef]
- Russo, A.; Concia, E.; Cristini, F.; de Rosa, F.G.; Esposito, S.; Menichetti, F.; Petrosillo, N.; Tumbarello, M.; Venditti, M.; Viale, P.; et al. Current and future trends in antibiotic therapy of acute bacterial skin and skin-structure infections. Clin. Microbiol. Infect. 2016, 22, 27–36. [Google Scholar] [CrossRef]
- Martins, N.; Barros, L.; Henriques, M.; Silva, S.; Ferreira, I.C.F.R. In vivo anti-candida activity of phenolic extracts and compounds: Future perspectives focusing on effective clinical interventions. BioMed Res. Int. 2015, 247382, 1–14. [Google Scholar]
- Heleno, S.A.; Diz, P.; Prieto, M.A.; Barros, L.; Rodrigues, A.; Barreiro, M.A.; Ferreira, I.C.F.R. Optimization of ultrasound-assisted extraction to obtain mycosterols from Agaricus bisporus L. by response surface methodology and comparison with conventional Soxhlet extraction. Food Chem. 2016, 197, 1054–1063. [Google Scholar] [CrossRef] [PubMed]
- Yoon, K.N.; Alam, N.; Lee, K.R.; Shin, P.G.; Cheong, J.C.; Yoo, Y.B.; Lee, T.S. Antioxidant and antityrosinase activities of various extracts from the fruiting bodies of Lentinus lepideus. Molecules 2011, 16, 2334–2347. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, A.; Barreira, J.C.M.; Antonio, A.L.; Oliveira, M.B.P.P.; Martins, A.; Ferreira, I.C.F.R. Effects of gamma irradiation on chemical composition and antioxidant potential of processed samples of the wild mushroom Macrolepiota procera. Food Chem. 2014, 149, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Performance Standards for Antimicrobial Susceptibility Testing; CLSI Document M100-S18; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2008.
- Comité de L’Antibiogramme de la Société Française de Microbiologie; Communiqué; Société Française de Microbiologie: Paris, France, 2012.
- European Committee on Antimicrobial Susceptibility Testing (EUCAST); European Society of Clinical Microbiology and Infectious Diseases (ESCMID): Stockholm, Sweden, 2013.
- Kuete, V.; Ango, P.Y.; Fotso, G.W.; Kapche, G.D.; Dzoyem, J.P.; Wouking, A.G.; Ngadjui, B.T.; Abegaz, B.M. Antimicrobial activities of the methanol extract and compounds from Artocarpus communis (Moraceae). BMC Complement. Altern. Med. 2011, 25, 11–42. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Mushroom samples are available from the authors.

{kind=link}
| Phenolic Acids (μg/g) | Mushroom Extracts | Cosmeceutical Formulations | ||||
|---|---|---|---|---|---|---|
| A. bisporus | P. ostreatus | L. edodes | A. bisporus cream | P. ostreatus Cream | L. edodes Cream | |
| Cinnamic acid | 90.06 ± 0.74 b | 362.7 ± 1.28 a | 7.31 ± 0.14 c | 87.73 ± 1.63 b | 317.43 ± 1.32 a | 6.10 ± 0.56 c |
| p-Hydroxybenzoic acid | nd | 157.78 ± 4.13 | 83.05 ± 2.15 | nd | 138.88 ± 2.30 | 73.67 ± 1.56 |
| p-Coumaric acid | nd | 63.74 ± 0.15 | nd | nd | 53.16 ± 2.94 | nd |
| Protocatechuic acid | nd | nd | 52.45 ± 0.38 | nd | nd | 39.85 ± 1.53 |
| Total (μg/g) | 90.06 ± 0.74 c | 584.24 ± 3.01 a | 142.81 ± 2.39 b | 87.73 ± 1.63 c | 509.47 ± 3.93 a | 119.61 ± 2.54 b |
| Ergosterol (mg/g) | 44.79 ± 0.37 b | 78.20 ± 0.54 a | 8.94 ± 0.04 c | 45.43 ± 1.38 b | 71.62 ± 0.29 a | 8.60 ± 0.25 c |
| Bioactivities | Mushroom Extracts | Cosmeceutical Formulations | ||||
|---|---|---|---|---|---|---|
| A. bisporus | P. ostreatus | L. edodes | A. bisporus Cream | P. ostreatus Cream | L. edodes Cream | |
| Anti-inflammatory (EC50 value, mg/mL) | 0.18 ± 0.01 b | 0.29 ± 0.03 a | 0.16 ± 0.01 b | 2.52 ± 0.23 b | 3.81 ± 0.23 a | 2.59 ± 0.23 b |
| Anti-tyrosinase (EC50 value, mg/mL) | 0.16 ± 0.01 b | 0.86 ± 0.07 a | 0.82 ± 0.08 a | 3.22 ± 0.37 b | 11.01 ± 0.35 a | 11.89 ± 0.85 a |
| Antioxidant (EC50 value, mg/mL) | ||||||
| DPPH radical-scavenging activity | 7.04 ± 0.32 b | 7.69 ± 0.20 b | 23.36 ± 1.11 a | 234.3 ± 10.9 b | 239.5 ± 10.4 b | 321.8 ± 16.4 a |
| Reducing power | 2.34 ± 0.05 b | 2.36 ± 0.08 b | 3.03 ± 0.04 a | 35.91 ± 0.31 b | 32.18 ± 2.73 c | 48.90 ± 0.64 a |
| Bacteria Strains | Mushroom Extracts | Cosmeceutical Formulations | ||||
|---|---|---|---|---|---|---|
| A. bisporus | P. ostreatus | L. edodes | A. bisporus Cream | P. ostreatus Cream | L. edodes Cream | |
| Gram-positive | ||||||
| Enterococcus faecalis | >20 | 10 | 5 | >200 | 200 | 100 |
| Methicillin sensitive Staphylococcus aureus | 10 | 2.5 | 2.5 | 200 | 50 | 50 |
| Methicillin resistant Staphylococcus aureus | 10 | 2.5 | 2.5 | 200 | 50 | 50 |
| Gram-negative | ||||||
| Escherichia coli | >20 | >20 | >20 | >200 | >200 | >200 |
| Pseudomonas aeruginosa | >20 | >20 | >20 | >200 | >200 | >200 |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Taofiq, O.; Heleno, S.A.; Calhelha, R.C.; Alves, M.J.; Barros, L.; Barreiro, M.F.; González-Paramás, A.M.; Ferreira, I.C.F.R. Development of Mushroom-Based Cosmeceutical Formulations with Anti-Inflammatory, Anti-Tyrosinase, Antioxidant, and Antibacterial Properties. Molecules 2016, 21, 1372. https://doi.org/10.3390/molecules21101372
Taofiq O, Heleno SA, Calhelha RC, Alves MJ, Barros L, Barreiro MF, González-Paramás AM, Ferreira ICFR. Development of Mushroom-Based Cosmeceutical Formulations with Anti-Inflammatory, Anti-Tyrosinase, Antioxidant, and Antibacterial Properties. Molecules. 2016; 21(10):1372. https://doi.org/10.3390/molecules21101372
Chicago/Turabian StyleTaofiq, Oludemi, Sandrina A. Heleno, Ricardo C. Calhelha, Maria José Alves, Lillian Barros, Maria Filomena Barreiro, Ana M. González-Paramás, and Isabel C. F. R. Ferreira. 2016. "Development of Mushroom-Based Cosmeceutical Formulations with Anti-Inflammatory, Anti-Tyrosinase, Antioxidant, and Antibacterial Properties" Molecules 21, no. 10: 1372. https://doi.org/10.3390/molecules21101372