
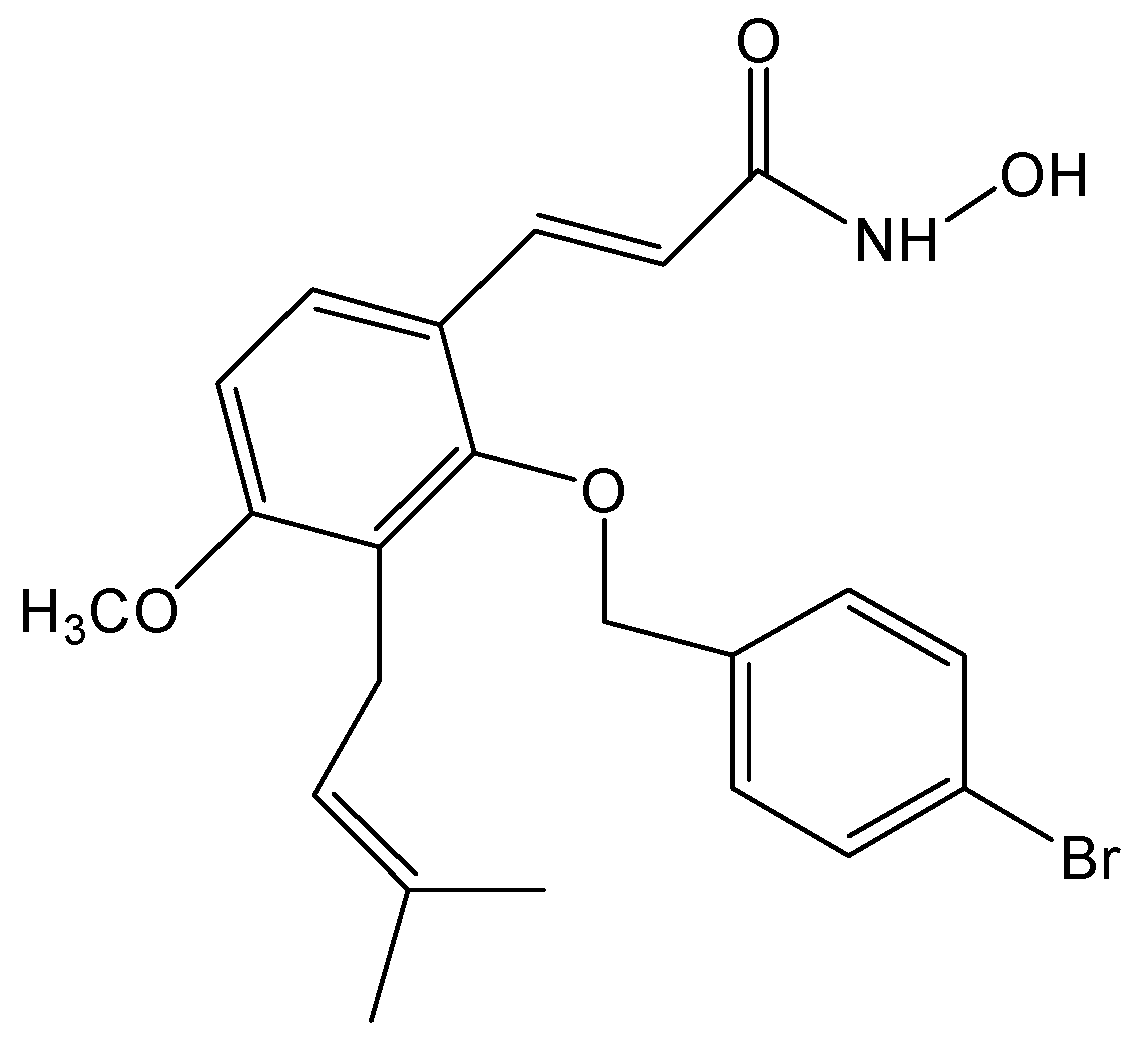
NBM-T-BBX-OS01, Semisynthesized from Osthole, Induced G1 Growth Arrest through HDAC6 Inhibition in Lung Cancer Cells

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
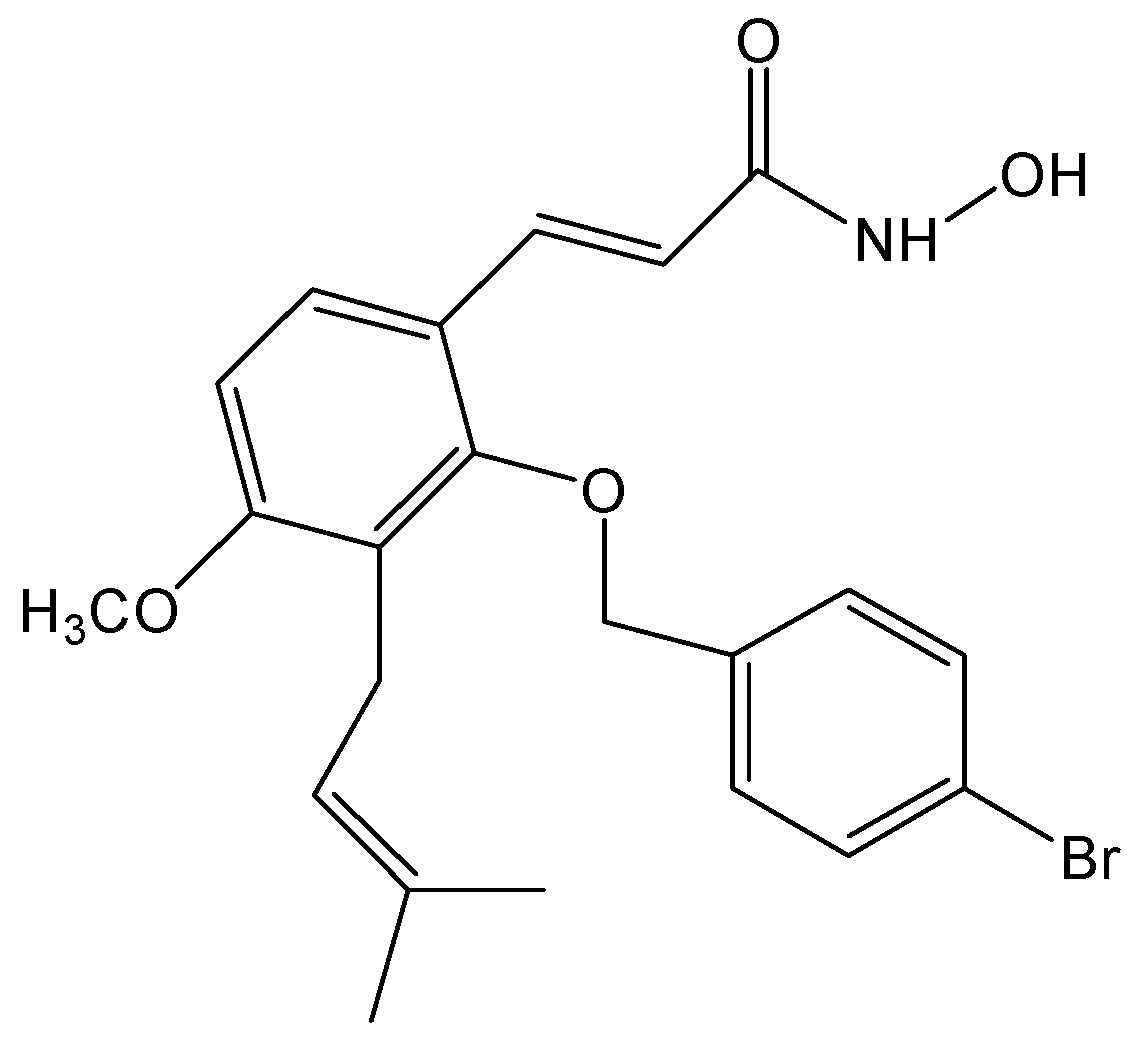
2. Results and Discussion
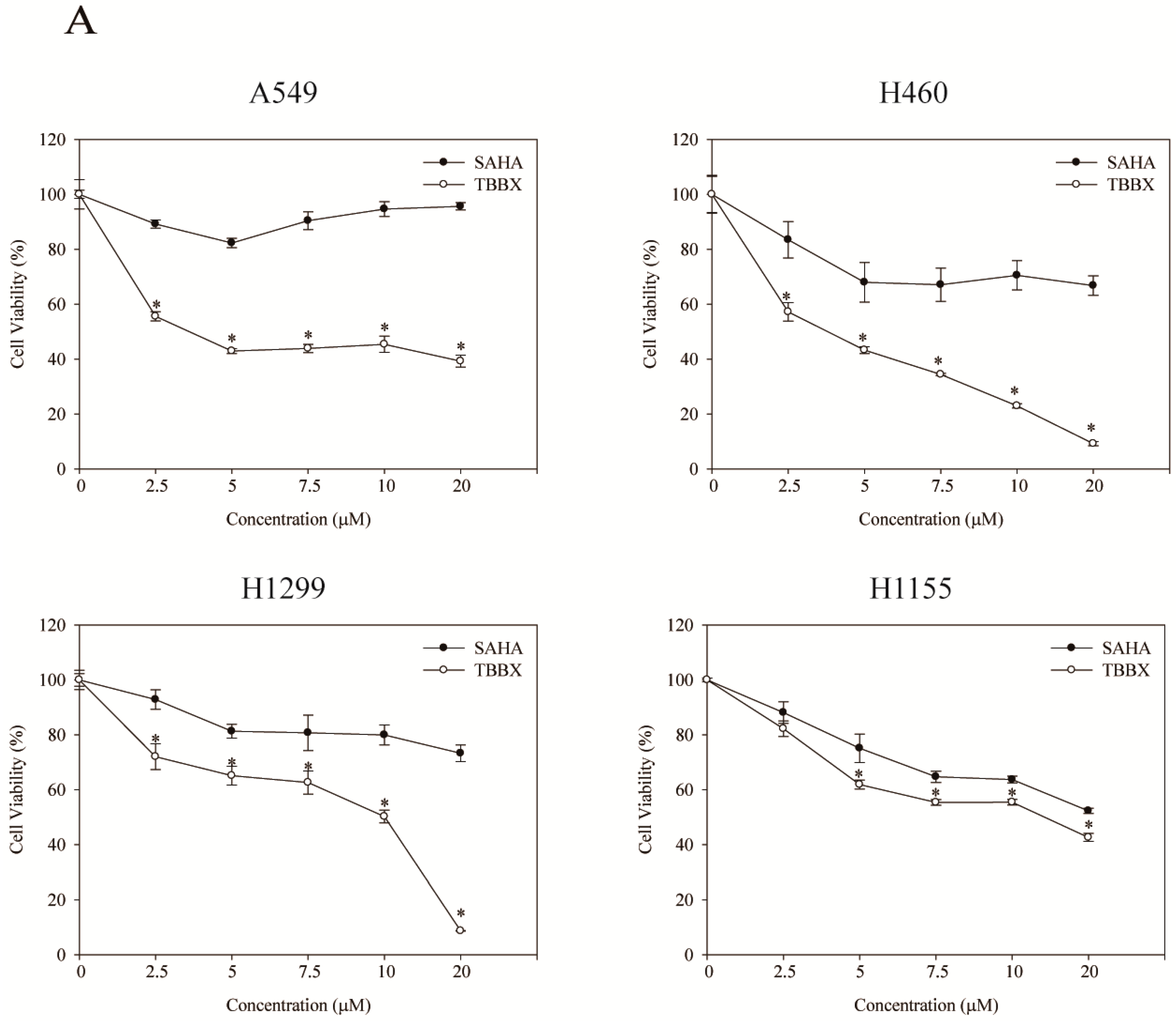
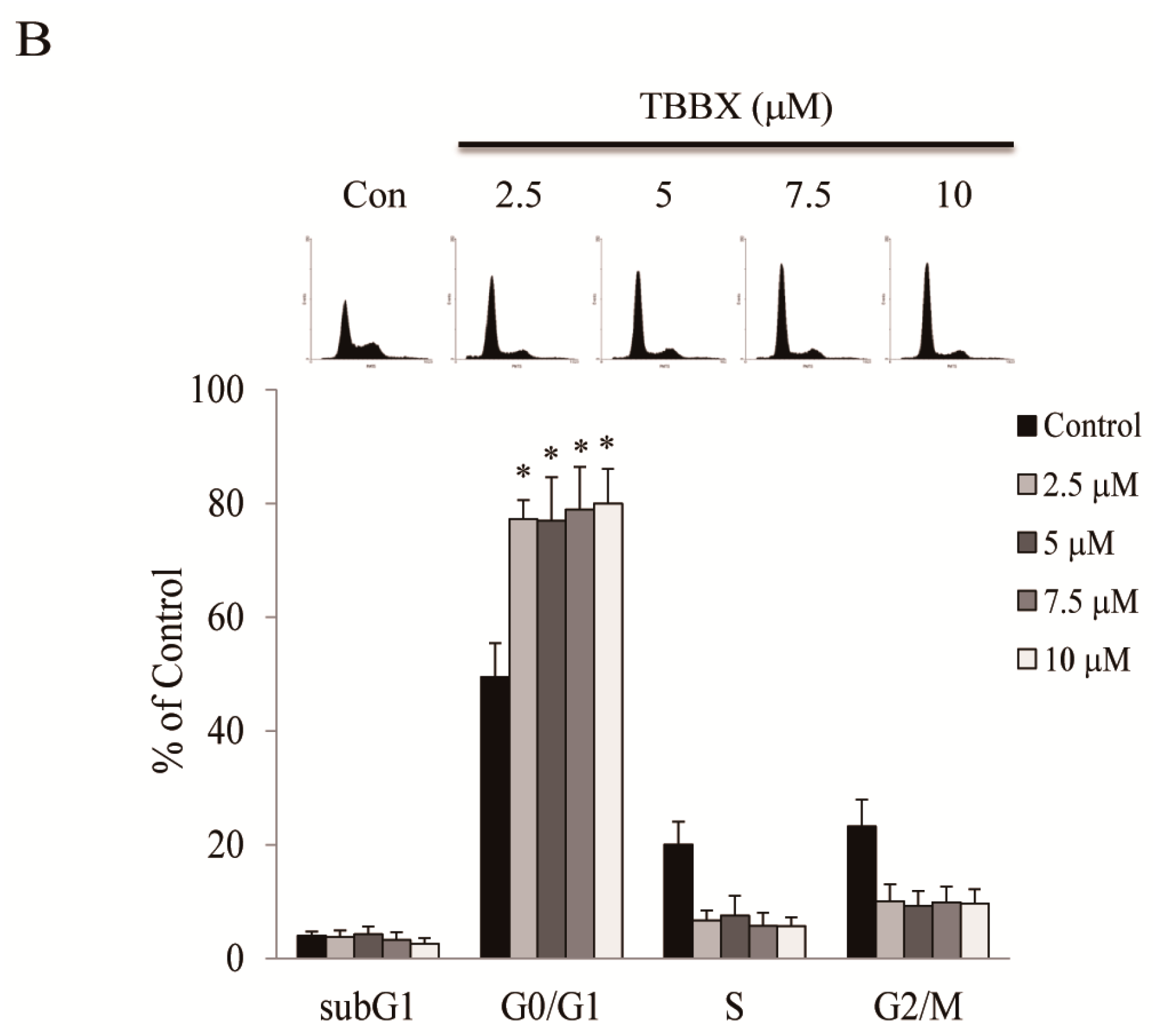
2.1. TBBX Induced G1 Growth Arrest in Human Lung Cancer Cells


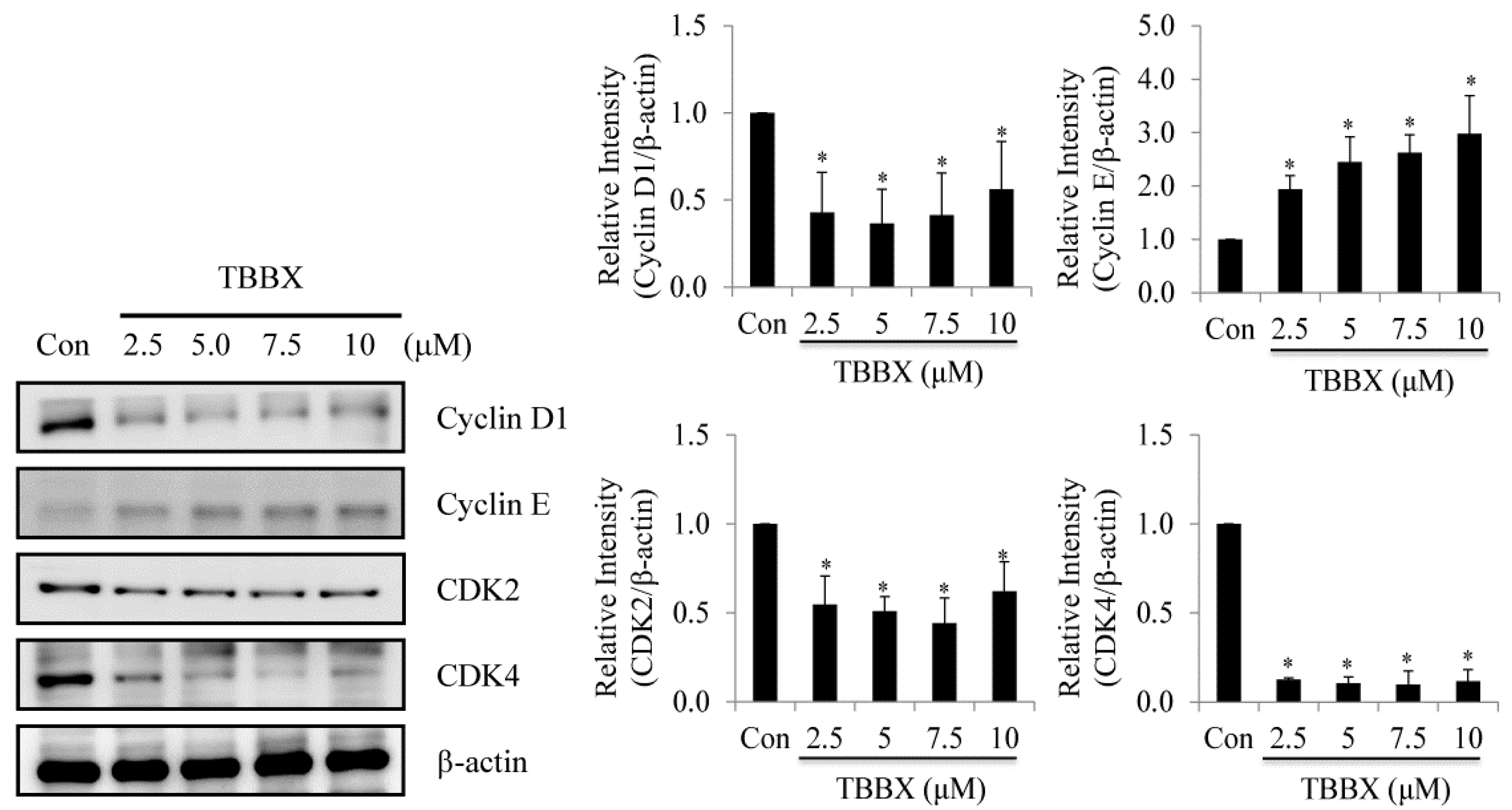
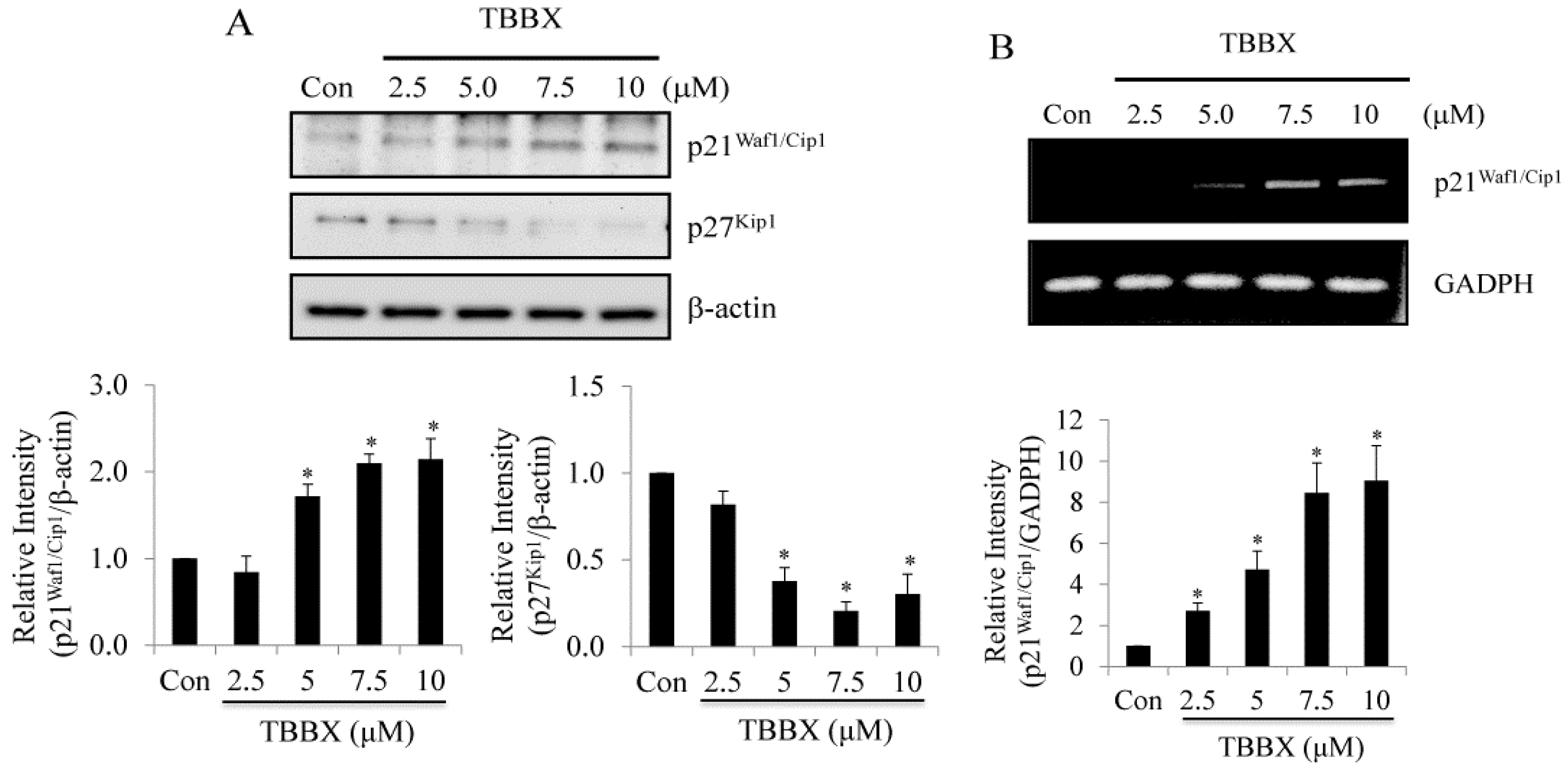
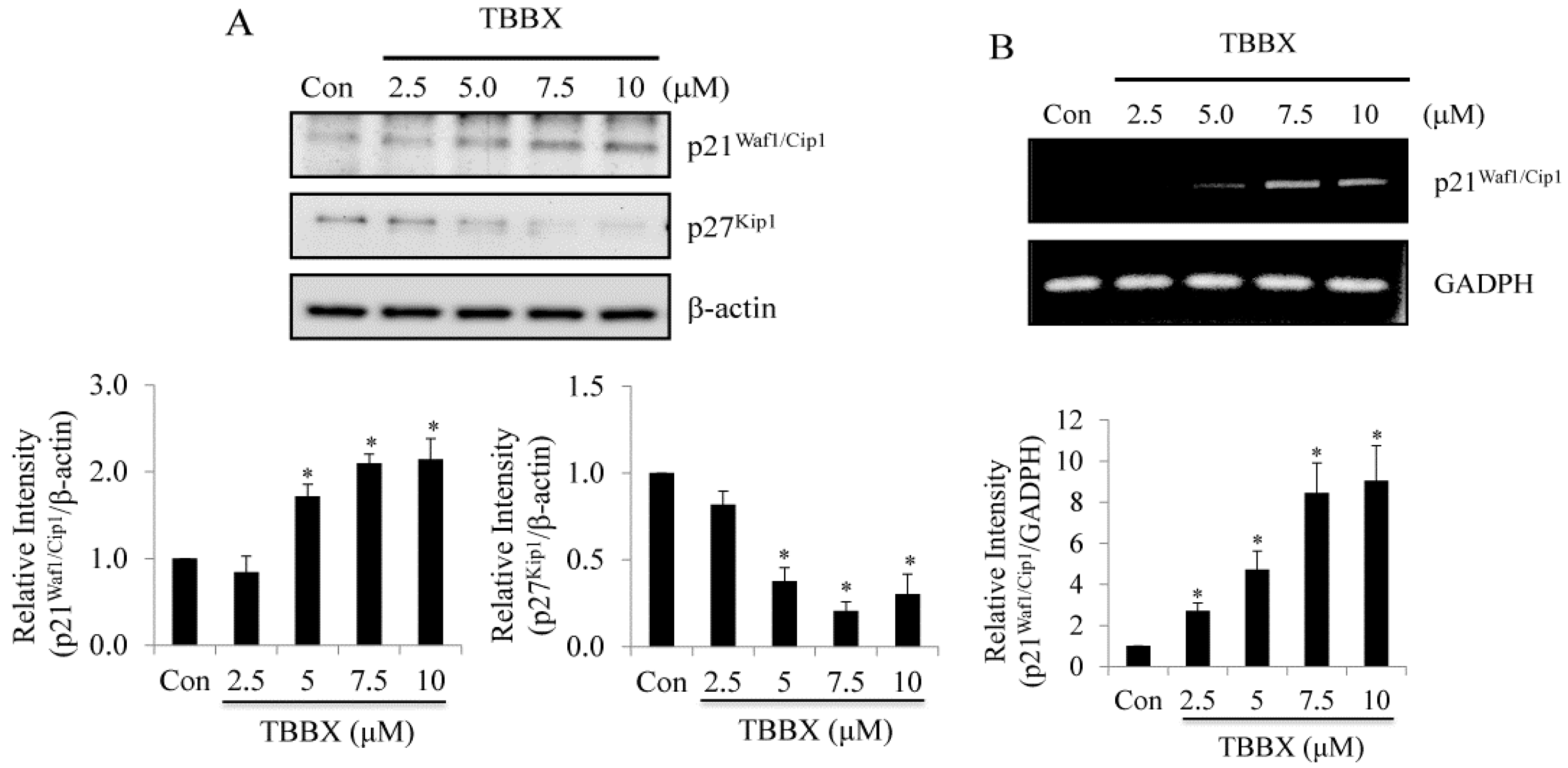
2.2. Up-Regulation of CDK Inhibitors Was Observed in TBBX-Treated H1299 Lung Cancer Cells
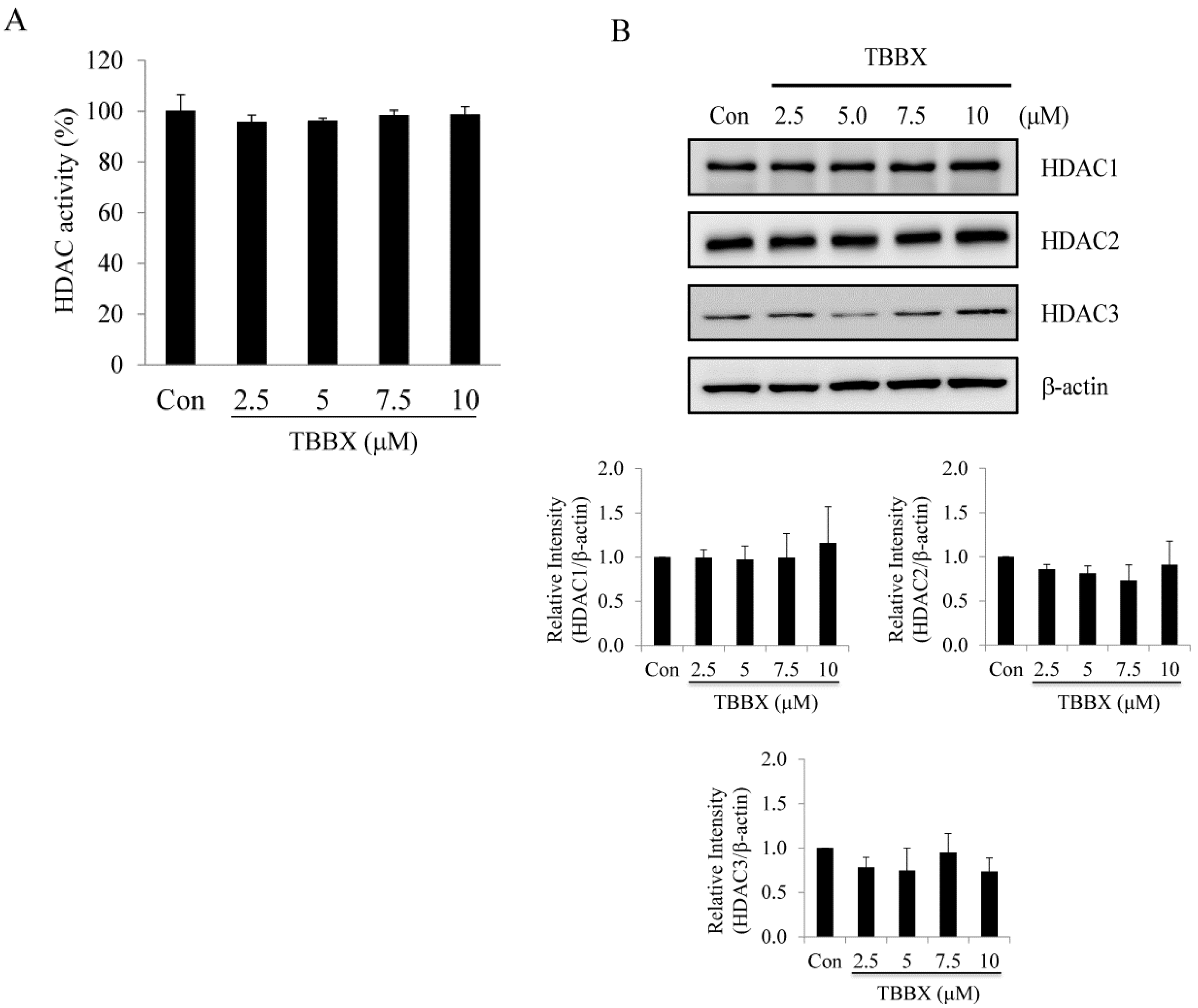
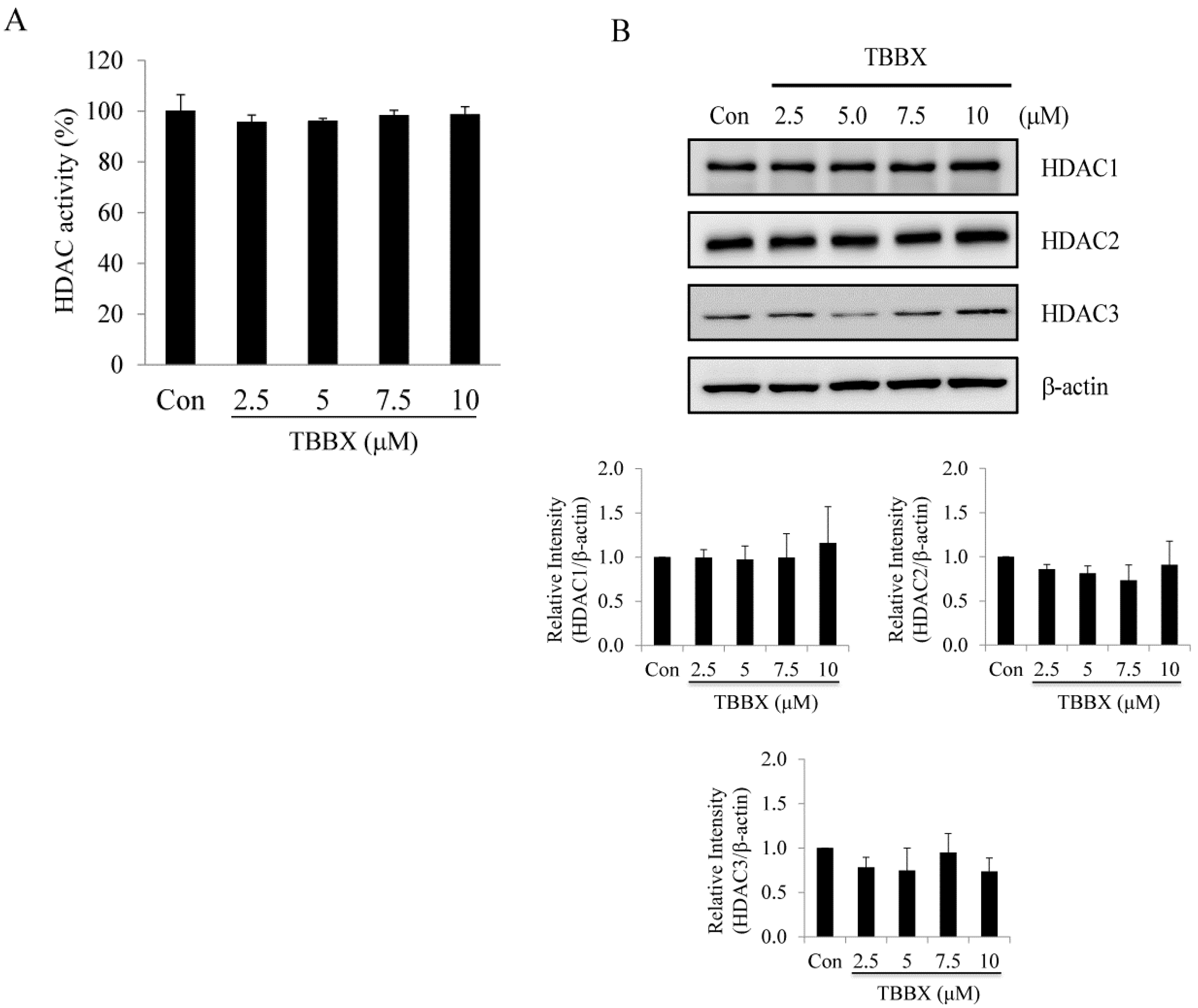
2.3. Class I HDACs Were Not Involved in TBBX-Induced Growth Arrest in H1299 Lung Cancer Cells
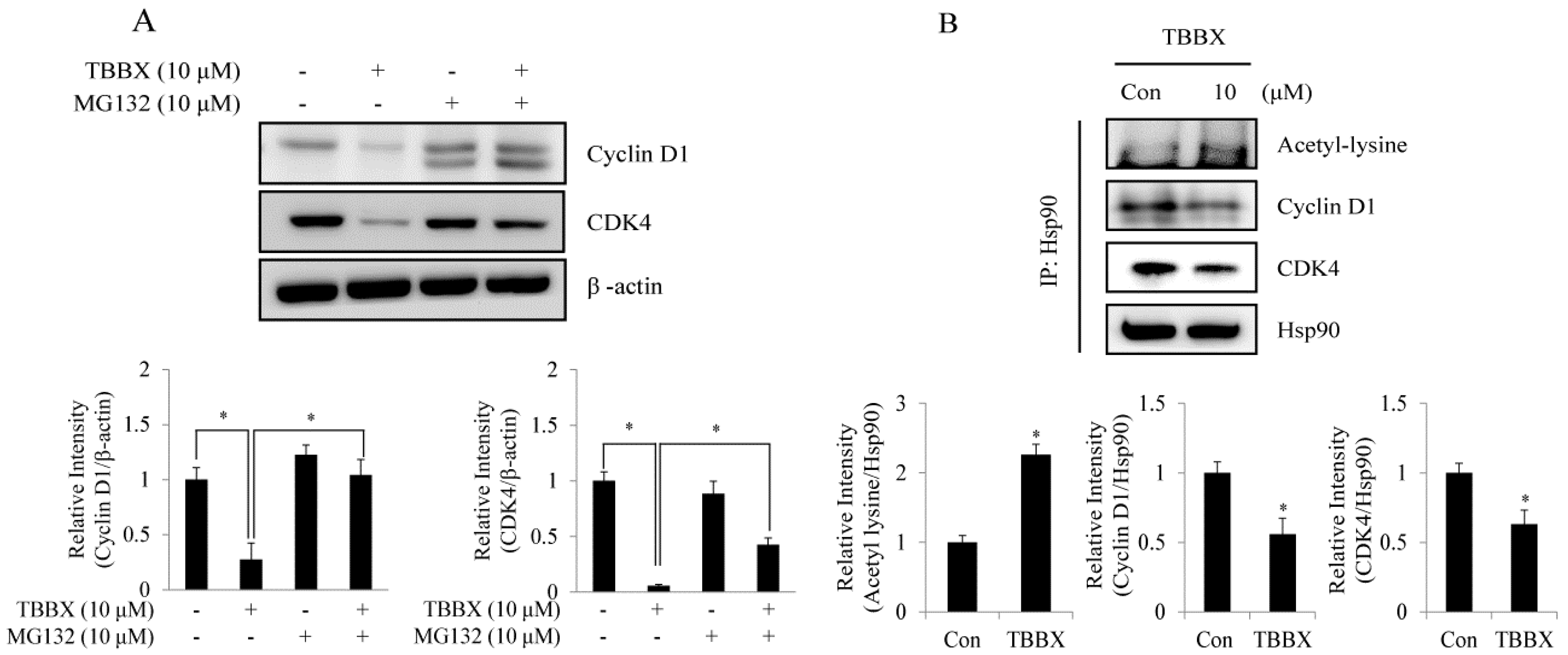
2.4. TBBX-Prompted Cyclin D1 and CDK4 Degradation Was through Interruption of Hsp90 with Cyclin D1 and CDK4 Association

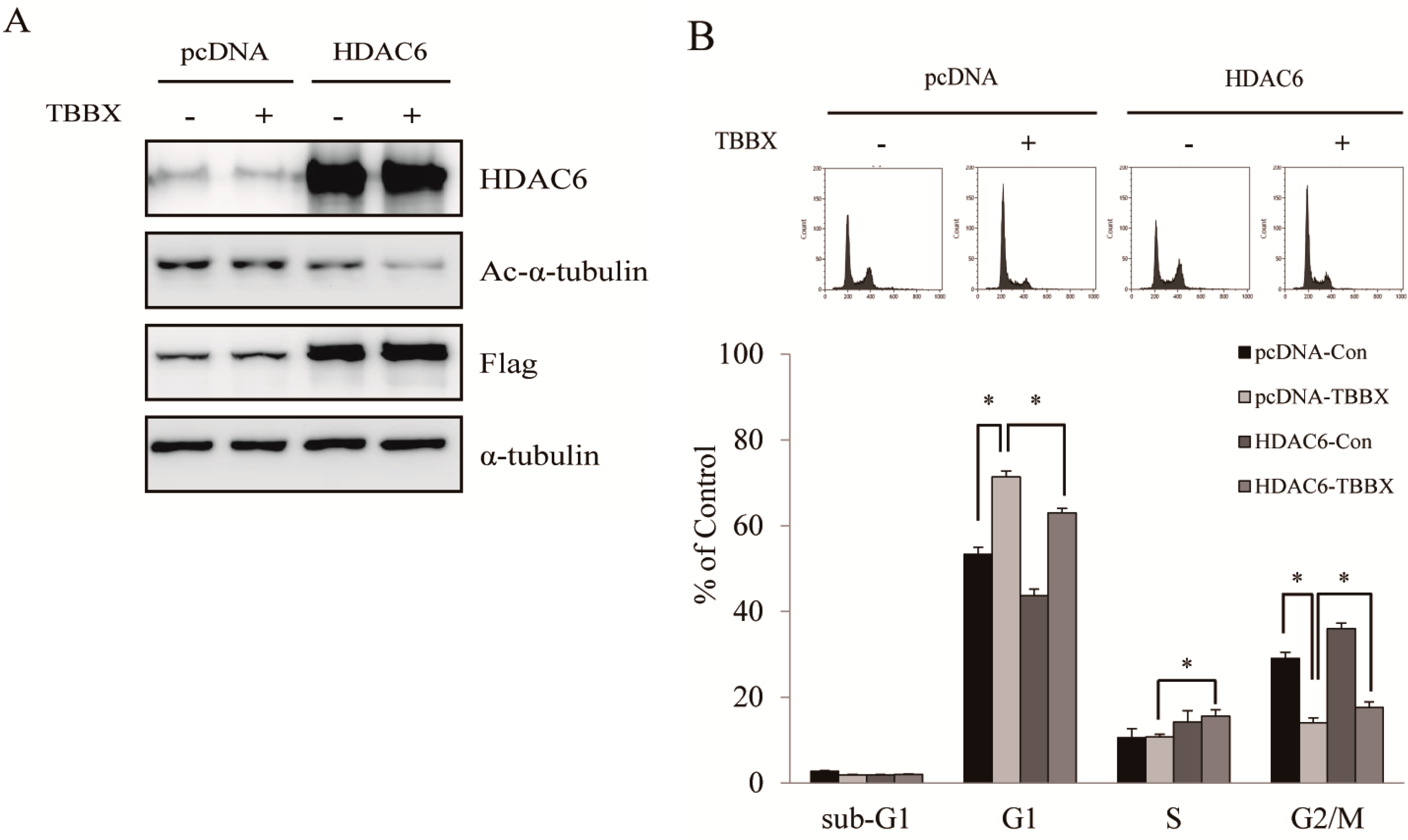
2.5. TBBX-Induced G1 Growth Arrest Was Mediated by HDAC6-Regulated Hsp90
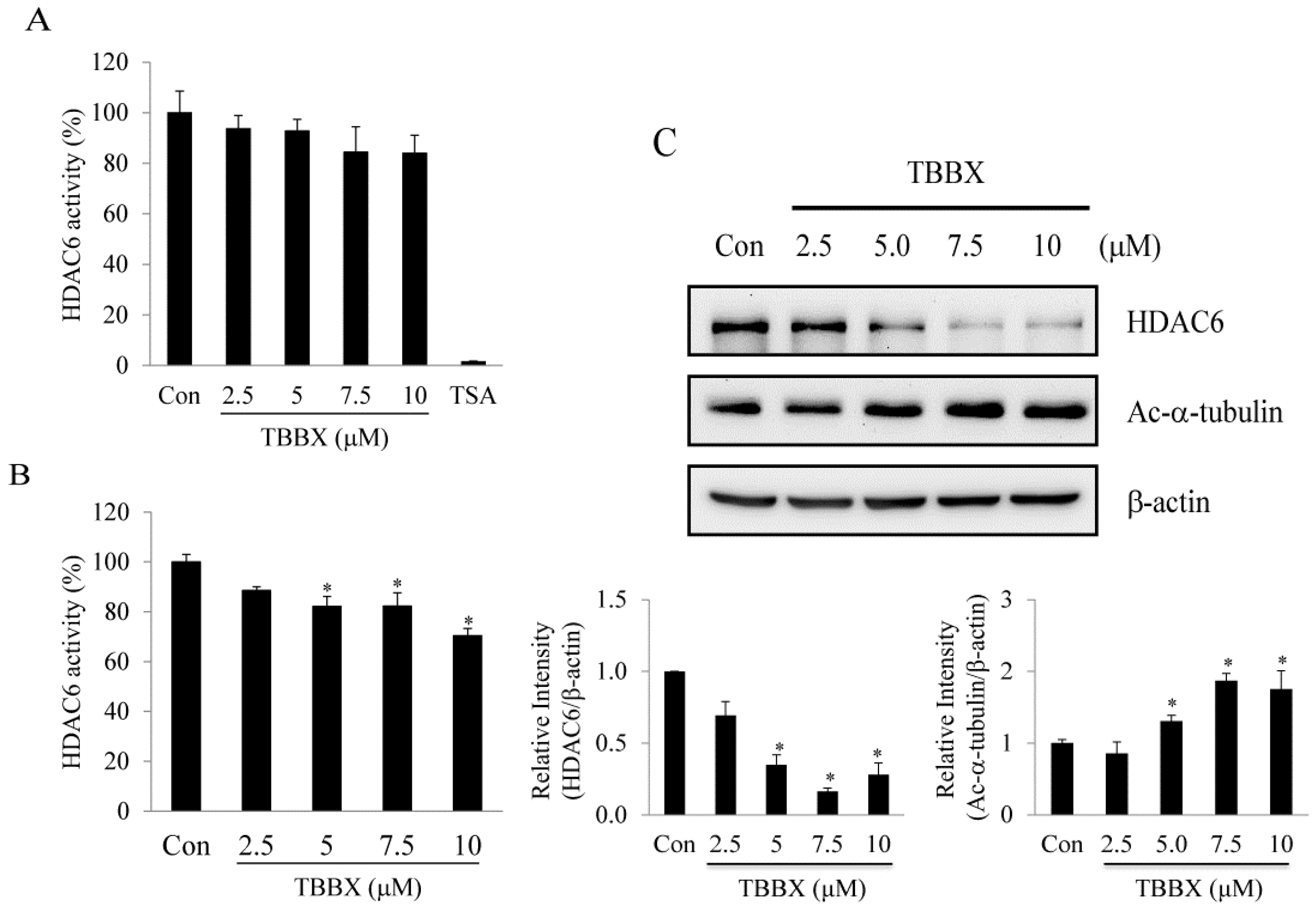

2.6. Discussion
3. Experimental Section
3.1. Chemicals and Reagents
3.2. Cell Culture and Cytotoxicity Assay
3.3. Cell Cycle Analysis
3.4. Western Blot Analysis
3.5. Immuno-Precipitation
3.6. Reverse Transcription Polymerase Chain Reaction (RT-PCR)
3.7. HDAC Activity Detection
3.8. Ectopic Overexpression of HDAC6
3.9. Statistical Analysis
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [PubMed]
- Pao, W.; Hutchinson, K.E. Chipping away at the lung cancer genome. Nat. Med. 2012, 18, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Viktorsson, K.; de Petris, L.; Lewensohn, R. The role of p53 in treatment responses of lung cancer. Biochem. Biophys. Res. Commun. 2005, 331, 868–880. [Google Scholar] [CrossRef] [PubMed]
- Steels, E.; Paesmans, M.; Berghmans, T.; Branle, F.; Lemaitre, F.; Mascaux, C.; Meert, A.P.; Vallot, F.; Lafitte, J.J.; Sculier, J.P. Role of p53 as a prognostic factor for survival in lung cancer: A systematic review of the literature with a meta-analysis. Eur. Respir. J. 2001, 18, 705–719. [Google Scholar] [CrossRef] [PubMed]
- Fraga, M.F.; Ballestar, E.; Paz, M.F.; Ropero, S.; Setien, F.; Ballestar, M.L.; Heine-Suner, D.; Cigudosa, J.C.; Urioste, M.; Benitez, J.; et al. Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. USA 2005, 102, 10604–10609. [Google Scholar] [CrossRef] [PubMed]
- Crea, F.; Nobili, S.; Paolicchi, E.; Perrone, G.; Napoli, C.; Landini, I.; Danesi, R.; Mini, E. Epigenetics and chemoresistance in colorectal cancer: An opportunity for treatment tailoring and novel therapeutic strategies. Drug Resist. Update. 2011, 14, 280–296. [Google Scholar] [CrossRef]
- Hrabeta, J.; Stiborova, M.; Adam, V.; Kizek, R.; Eckschlager, T. Histone deacetylase inhibitors in cancer therapy. A review. Biomed. Pap. 2014, 158, 161–169. [Google Scholar]
- West, A.C.; Johnstone, R.W. New and emerging HDAC inhibitors for cancer treatment. J. Clin. Investig. 2014, 124, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Aldana-Masangkay, G.I.; Sakamoto, K.M. The role of HDAC6 in cancer. J. Biomed. Biotechnol. 2010, 2011, 875824–875833. [Google Scholar] [PubMed]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef] [PubMed]
- Cress, W.D.; Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 2000, 184, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.; Chin, Y.E.; Weisiger, E.; Malter, C.; Tawara, I.; Toubai, T.; Gatza, E.; Mascagni, P.; Dinarello, C.A.; Reddy, P. Cutting edge: Negative regulation of dendritic cells through acetylation of the nonhistone protein STAT-3. J. Immunol. 2009, 182, 5899–5903. [Google Scholar] [CrossRef] [PubMed]
- Bali, P.; Pranpat, M.; Bradner, J.; Balasis, M.; Fiskus, W.; Guo, F.; Rocha, K.; Kumaraswamy, S.; Boyapalle, S.; Atadja, P.; et al. Inhibition of histone deacetylase 6 acetylates and disrupts the chaperone function of heat shock protein 90: A novel basis for antileukemia activity of histone deacetylase inhibitors. J. Biol. Chem. 2005, 280, 26729–26734. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, J.J.; Murphy, P.J.; Gaillard, S.; Zhao, X.; Wu, J.T.; Nicchitta, C.V.; Yoshida, M.; Toft, D.O.; Pratt, W.B.; Yao, T.P. HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 2005, 18, 601–607. [Google Scholar] [CrossRef] [PubMed]
- Scroggins, B.T.; Robzyk, K.; Wang, D.; Marcu, M.G.; Tsutsumi, S.; Beebe, K.; Cotter, R.J.; Felts, S.; Toft, D.; Karnitz, L.; Rosen, N.; Neckers, L. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 2007, 25, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Spange, S.; Wagner, T.; Heinzel, T.; Kramer, O.H. Acetylation of non-histone proteins modulates cellular signalling at multiple levels. Int. J. Biol. Chem. 2009, 41, 185–198. [Google Scholar]
- Marks, P.A. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs 2010, 19, 1049–1066. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Xiao, W.; Chen, W.; Luo, L.; Ye, S.; Liu, Y. The epigenetic modifier trichostatin A, a histone deacetylase inhibitor, suppresses proliferation and epithelial-mesenchymal transition of lens epithelial cells. Cell Death Dis. 2013, 4, e884. [Google Scholar] [CrossRef] [PubMed]
- Singh, T.; Prasad, R.; Katiyar, S.K. Inhibition of class I histone deacetylases in non-small cell lung cancer by honokiol leads to suppression of cancer cell growth and induction of cell death in vitro and in vivo. Epigenetics 2013, 8, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, H.; Moriyama, S.; Nakashima, Y.; Kobayashi, Y.; Kiriyama, M.; Fukai, I.; Yamakawa, Y.; Fujii, Y. Histone deacetylase 1 mRNA expression in lung cancer. Lung Cancer 2004, 46, 171–178. [Google Scholar] [CrossRef] [PubMed]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Mitsui, M.; Motoyama, S.; Ogawa, J. Expression of histone deacetylase 1 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Lung Cancer 2011, 74, 300–304. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, T.; Cubizolles, F.; Zhang, Y.; Reichert, N.; Kohler, H.; Seiser, C.; Matthias, P. Histone deacetylases 1 and 2 act in concert to promote the G1-to-S progression. Genes Dev. 2010, 24, 455–469. [Google Scholar] [CrossRef] [PubMed]
- Wilson, A.J.; Byun, D.S.; Popova, N.; Murray, L.B.; L’Italien, K.; Sowa, Y.; Arango, D.; Velcich, A.; Augenlicht, L.H.; Mariadason, J.M. Histone deacetylase 3 (HDAC3) and other class I HDACs regulate colon cell maturation and p21 expression and are deregulated in human colon cancer. J. Biol. Chem. 2006, 281, 13548–13558. [Google Scholar] [CrossRef] [PubMed]
- Yin, D.; Ong, J.M.; Hu, J.; Desmond, J.C.; Kawamata, N.; Konda, B.M.; Black, K.L.; Koeffler, H.P. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor: Effects on gene expression and growth of glioma cells in vitro and in vivo. Clin. Cancer Res. 2007, 13, 1045–1052. [Google Scholar] [CrossRef] [PubMed]
- Vallo, S.; Xi, W.; Hudak, L.; Juengel, E.; Tsaur, I.; Wiesner, C.; Haferkamp, A.; Blaheta, R.A. HDAC inhibition delays cell cycle progression of human bladder cancer cells in vitro. Anti-Cancer Drugs 2011, 22, 1002–1009. [Google Scholar] [PubMed]
- Lee, Y.S.; Lim, K.H.; Guo, X.; Kawaguchi, Y.; Gao, Y.; Barrientos, T.; Ordentlich, P.; Wang, X.F.; Counter, C.M.; Yao, T.P. The cytoplasmic deacetylase HDAC6 is required for efficient oncogenic tumorigenesis. Cancer Res. 2008, 68, 7561–7569. [Google Scholar] [CrossRef] [PubMed]
- Kamemura, K.; Ito, A.; Shimazu, T.; Matsuyama, A.; Maeda, S.; Yao, T.P.; Horinouchi, S.; Khochbin, S.; Yoshida, M. Effects of downregulated HDAC6 expression on the proliferation of lung cancer cells. Biochem. Biophys. Res. Commun. 2008, 374, 84–89. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Kaluza, D.; Kroll, J.; Gesierich, S.; Yao, T.P.; Boon, R.A.; Hergenreider, E.; Tjwa, M.; Rossig, L.; Seto, E.; Augustin, H.G.; et al. Class IIb HDAC6 regulates endothelial cell migration and angiogenesis by deacetylation of cortactin. EMBO J. 2011, 30, 4142–4156. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Kim, S.H.; Choi, M.C.; Lee, J.; Oh, D.Y.; Im, S.A.; Bang, Y.J.; Kim, T.Y. Class II histone deacetylases play pivotal roles in heat shock protein 90-mediated proteasomal degradation of vascular endothelial growth factor receptors. Biochem. Biophys. Res. Commun. 2008, 368, 318–322. [Google Scholar] [CrossRef] [PubMed]
- Sawai, A.; Chandarlapaty, S.; Greulich, H.; Gonen, M.; Ye, Q.; Arteaga, C.L.; Sellers, W.; Rosen, N.; Solit, D.B. Inhibition of Hsp90 down-regulates mutant epidermal growth factor receptor (EGFR) expression and sensitizes EGFR mutant tumors to paclitaxel. Cancer Res. 2008, 68, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Stepanova, L.; Leng, X.; Parker, S.B.; Harper, J.W. Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 1996, 10, 1491–1502. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Marchenko, N.D.; Moll, U.M. SAHA shows preferential cytotoxicity in mutant p53 cancer cells by destabilizing mutant p53 through inhibition of the HDAC6-Hsp90 chaperone axis. Cell Death Differ. 2011, 18, 1904–1913. [Google Scholar] [CrossRef] [PubMed]
- Malumbres, M.; Barbacid, M. Cell cycle, CDKs and cancer: A changing paradigm. Nat. Rev. Cancer 2009, 9, 153–166. [Google Scholar] [CrossRef] [PubMed]
- Eymin, B.; Gazzeri, S. Role of cell cycle regulators in lung carcinogenesis. Cell Adhes. Migr. 2010, 4, 114–123. [Google Scholar] [CrossRef]
- Gautschi, O.; Ratschiller, D.; Gugger, M.; Betticher, D.C.; Heighway, J. Cyclin D1 in non-small cell lung cancer: A key driver of malignant transformation. Lung Cancer 2007, 55, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Inoue, S.; Umemura, T.; Moriya, J.; Arakawa, M.; Nagashima, K.; Kato, H. Cyclin D1, p16 and retinoblastoma gene product expression as a predictor for prognosis in non-small cell lung cancer at stages I and II. Lung Cancer 2001, 34, 207–218. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev. 1995, 9, 1149–1163. [Google Scholar] [CrossRef] [PubMed]
- Donjerkovic, D.; Scott, D.W. Regulation of the G1 phase of the mammalian cell cycle. Cell Res. 2000, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.M.; Kim, C.S.; Lee, J.H.; Jang, S.J.; Hwang, J.J.; Ro, S.; Hyun, Y.L.; Choi, J. CG0006, a novel histone deacetylase inhibitor, induces breast cancer cell death via histone-acetylation and chaperone-disrupting pathways independent of ER status. Breast Cancer Res. Treat. 2011, 130, 365–375. [Google Scholar] [CrossRef] [PubMed]
- Gryder, B.E.; Sodji, Q.H.; Oyelere, A.K. Targeted cancer therapy: Giving histone deacetylase inhibitors all they need to succeed. Future Med. Chem. 2012, 4, 505–524. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.C.; Chen, C.N.; Wu, C.I.; Huang, W.J.; Kuo, T.Y.; Kuan, M.C.; Tsai, T.H.; Huang, J.S.; Huang, C.Y. NBM-T-L-BMX-OS01, semisynthesized from osthole, is a novel inhibitor of histone deacetylase and enhances learning and memory in rats. Evid.-Based Complement. Altern. 2013, 2013. [Google Scholar] [CrossRef]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Rev. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Sherr, C.J.; Roberts, J.M. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev. 1999, 13, 1501–1512. [Google Scholar] [CrossRef] [PubMed]
- Mollapour, M.; Neckers, L. Post-translational modifications of Hsp90 and their contributions to chaperone regulation. Biochim. Biophys. Acta 2012, 1823, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Rao, R.; Shen, J.; Tang, Y.; Fiskus, W.; Nechtman, J.; Atadja, P.; Bhalla, K. Role of acetylation and extracellular location of heat shock protein 90alpha in tumor cell invasion. Cancer Res. 2008, 68, 4833–4842. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, A.; Ramanand, S.G.; Whitehead, C.; Hiniker, S.M.; Rehemtulla, A.; Pratt, W.B.; Jolly, S.; Gouveia, C.; Truong, K.; Van Waes, C.; et al. Wild-type EGFR is stabilized by direct interaction with HSP90 in cancer cells and tumors. Neoplasia 2012, 14, 670–677. [Google Scholar] [CrossRef] [PubMed]
- Shimamura, T.; Lowell, A.M.; Engelman, J.A.; Shapiro, G.I. Epidermal growth factor receptors harboring kinase domain mutations associate with the heat shock protein 90 chaperone and are destabilized following exposure to geldanamycins. Cancer Res. 2005, 65, 6401–6408. [Google Scholar] [PubMed]
- Kobayashi, N.; Toyooka, S.; Soh, J.; Yamamoto, H.; Dote, H.; Kawasaki, K.; Otani, H.; Kubo, T.; Jida, M.; Ueno, T.; et al. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor. Lung Cancer 2012, 75, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Yamaki, H.; Nakajima, M.; Shimotohno, K.W.; Tanaka, N. Molecular basis for the actions of Hsp90 inhibitors and cancer therapy. J. Antibiot. 2011, 64, 635–644. [Google Scholar] [CrossRef] [PubMed]
- Erlandsson, F.; Wahlby, C.; Ekholm-Reed, S.; Hellstrom, A.C.; Bengtsson, E.; Zetterberg, A. Abnormal expression pattern of cyclin E in tumour cells. Int. J. Cancer 2003, 104, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Carcagno, A.L.; Ogara, M.F.; Sonzogni, S.V.; Marazita, M.C.; Sirkin, P.F.; Ceruti, J.M.; Canepa, E.T. E2F1 transcription is induced by genotoxic stress through ATM/ATR activation. IUBMB Life 2009, 61, 537–543. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.S.; Ho, Y.S.; Lin, J.K. Chrysin induces G1 phase cell cycle arrest in C6 glioma cells through inducing p21Waf1/Cip1 expression: Involvement of p38 mitogen-activated protein kinase. Biochem. Pharmacol. 2005, 69, 1815–1827. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.H.; Ban, J.O.; Cho, M.C.; Jo, M.; Jung, J.K.; Ahn, B.; Yoon, D.Y.; Han, S.B.; Hong, J.T. 4-O-methylhonokiol inhibits colon tumor growth via p21-mediated suppression of NF-kappaB activity. J. Nutr. Biochem. 2012, 23, 706–715. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Shi, Y.; Xiang, B.; Qu, B.; Su, W.; Zhu, M.; Zhang, M.; Bao, G.; Wang, F.; Zhang, X.; et al. A nuclear function of beta-arrestin1 in GPCR signaling: Regulation of histone acetylation and gene transcription. Cell 2005, 123, 833–847. [Google Scholar] [CrossRef] [PubMed]
- Perez-Luna, M.; Aguasca, M.; Perearnau, A.; Serratosa, J.; Martinez-Balbas, M.; Jesus Pujol, M.; Bachs, O. PCAF regulates the stability of the transcriptional regulator and cyclin-dependent kinase inhibitor p27 Kip1. Nucleic Acids Res. 2012, 40, 6520–6533. [Google Scholar] [CrossRef] [PubMed]
- Khattar, V.; Fried, J.; Xu, B.; Thottassery, J.V. Cks1 proteasomal degradation is induced by inhibiting Hsp90-mediated chaperoning in cancer cells. Cancer Chemother. Pharmacol. 2015, 75, 411–420. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Liao, C.H.; Chien, M.H.; Tsai, T.Y.; Lin, J.K.; Weng, M.S. Induction of p21(Waf1/Cip1) by garcinol via downregulation of p38-MAPK signaling in p53-independent H1299 lung cancer. J. Agric. Food. Chem. 2014, 62, 2085–2095. [Google Scholar] [CrossRef] [PubMed]
- Weng, M.S.; Liao, C.H.; Chen, C.N.; Wu, C.L.; Lin, J.K. Propolin H from Taiwanese propolis induces G1 arrest in human lung carcinoma cells. J. Agric. Food. Chem. 2007, 55, 5289–5298. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.; Li, J.; Atadja, P.; Bhalla, K.; Haura, E.B. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol. Cancer Ther. 2007, 6, 2515–2524. [Google Scholar] [CrossRef] [PubMed]
- Caron, C.; Boyault, C.; Khochbin, S. Regulatory cross-talk between lysine acetylation and ubiquitination: Role in the control of protein stability. BioEssays 2005, 27, 408–415. [Google Scholar] [CrossRef] [PubMed]
- Alao, J.P.; Lam, E.W.; Ali, S.; Buluwela, L.; Bordogna, W.; Lockey, P.; Varshochi, R.; Stavropoulou, A.V.; Coombes, R.C.; Vigushin, D.M. Histone deacetylase inhibitor trichostatin A represses estrogen receptor alpha-dependent transcription and promotes proteasomal degradation of cyclin D1 in human breast carcinoma cell lines. Clin. Cancer Res. 2004, 10, 8094–8104. [Google Scholar] [CrossRef] [PubMed]
- Murphy, P.J.; Morishima, Y.; Kovacs, J.J.; Yao, T.P.; Pratt, W.B. Regulation of the dynamics of hsp90 action on the glucocorticoid receptor by acetylation/deacetylation of the chaperone. J. Biol. Chem. 2005, 280, 33792–33799. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the NBM-T-BBX-OS01 (TBBX) is not available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pai, J.-T.; Hsu, C.-Y.; Hua, K.-T.; Yu, S.-Y.; Huang, C.-Y.; Chen, C.-N.; Liao, C.-H.; Weng, M.-S. NBM-T-BBX-OS01, Semisynthesized from Osthole, Induced G1 Growth Arrest through HDAC6 Inhibition in Lung Cancer Cells. Molecules 2015, 20, 8000-8019. https://doi.org/10.3390/molecules20058000
Pai J-T, Hsu C-Y, Hua K-T, Yu S-Y, Huang C-Y, Chen C-N, Liao C-H, Weng M-S. NBM-T-BBX-OS01, Semisynthesized from Osthole, Induced G1 Growth Arrest through HDAC6 Inhibition in Lung Cancer Cells. Molecules. 2015; 20(5):8000-8019. https://doi.org/10.3390/molecules20058000
Chicago/Turabian StylePai, Jih-Tung, Chia-Yun Hsu, Kuo-Tai Hua, Sheng-Yung Yu, Chung-Yang Huang, Chia-Nan Chen, Chiung-Ho Liao, and Meng-Shih Weng. 2015. "NBM-T-BBX-OS01, Semisynthesized from Osthole, Induced G1 Growth Arrest through HDAC6 Inhibition in Lung Cancer Cells" Molecules 20, no. 5: 8000-8019. https://doi.org/10.3390/molecules20058000