
The Role of Phosphoglycans in the Susceptibility of Leishmania mexicana to the Temporin Family of Anti-Microbial Peptides


Abstract
:1. Introduction
2. Results and Discussion
2.1. The Antileishmanial Properties of Temporin Antimicrobial Peptides

{kind=link}
{kind=link}
| Peptide | Sequence | Empirical Formula | Mass Calculated [M+2H]2+ | Accurate Mass Found [M+2H]2+ |
|---|---|---|---|---|
| Temporin A | FLPLIGRVLSGIL-NH2 | C68H117N17O14 | 698.9561 | 698.9548 |
| Temporin B | LLPIVGNLLKSLL-NH2 | C67H122N16O15 | 696.4716 | 696.4735 |
| Temporin 1Sa | FLSGIVGMLGKLF-NH2 | C67H109N15O14S | 690.9078 | 690.9075 |
| Temporin F | FLPLIGKVLSGIL-NH2 | C68H117N15O14 | 684.9531 | 684.9504 |
| Temporin L | FVQWFSKFLGRIL-NH2 | C83H122N20O15 | 820.9792 | 820.9792 |
| Peptide | ED50 (µM) | |||
|---|---|---|---|---|
| L. mexicana Promastiogte | L. mexicana Amastigote | L. mexicana ∆lpg1 | L. mexicana ∆lpg2 | |
| Temporin A | 8 (6–14) | ~100 | 11 (8–16) | 26 (21–39) |
| Temporin B | 38 (24–64) | >100 | 39 (28–70) | 41 (40–41) |
| Temporin 1Sa | 4 (3–13) | 42 (35–44) | 6 (3–18) | 31 (28–35) |
| Temporin F | 14 (10–27) | >100 | 17 (13–29) | 23 (16–49) |
| Temporin L | 5 (5–6) | 83 (46–93) | 4 (3–6) | 9 (8–12) |
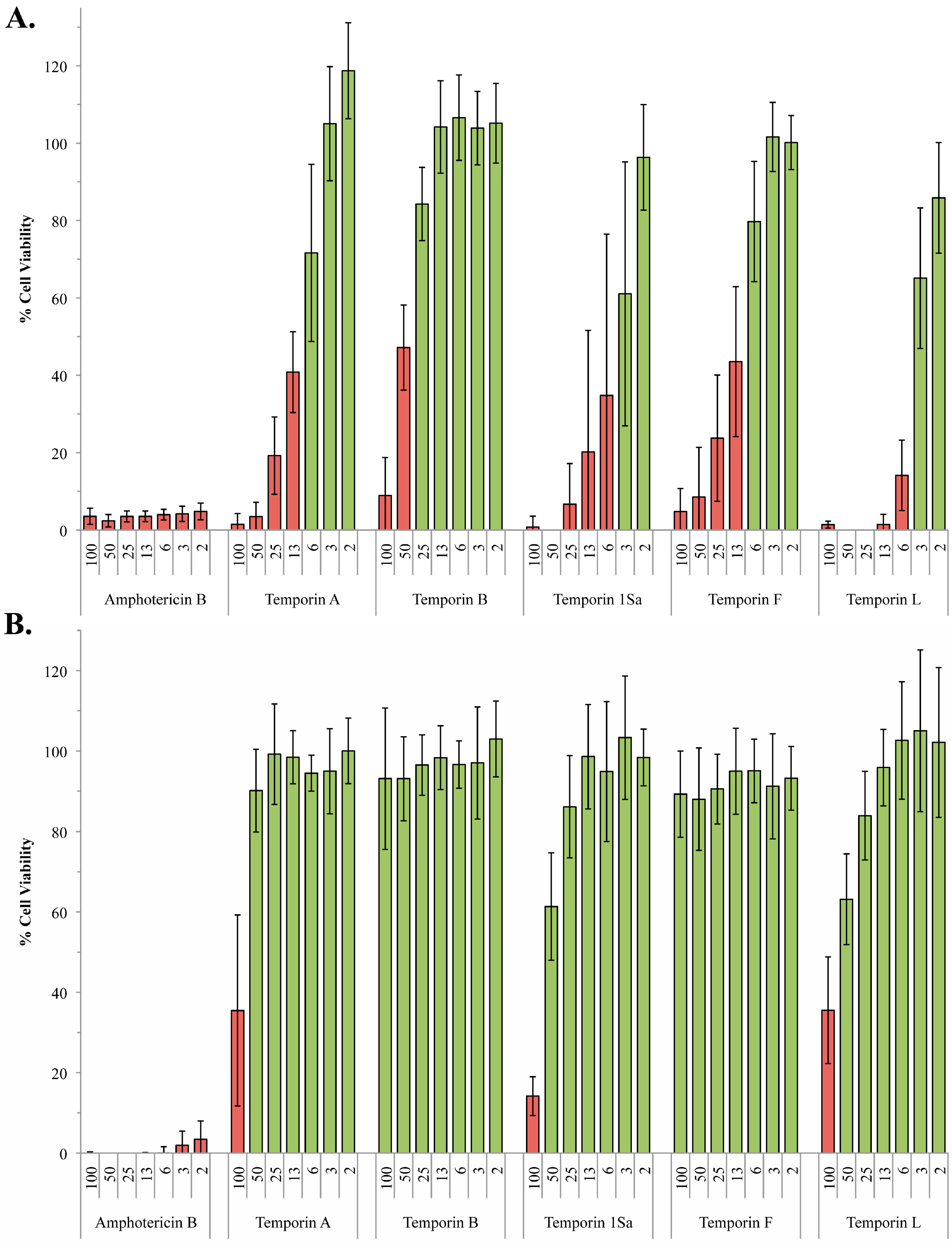
2.2. The Role of Phosphoglycans in the Susceptibility of L. Mexiciana to the Temporin Family of Antimicrobial Peptides

3. Experimental Section
3.1. Materials and Reagents
3.2. Peptide Synthesis
3.3. Antileishmanial Assay
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gurtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croft, S.L.; Coombs, G.H. Leishmaniasis—Current chemotherapy and recent advances in the search for novel drugs. Trends Parasitol. 2003, 19, 502–508. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L.; Sakthianandeswaren, A.; Curtis, J.M.; Andrews, P.C.; Junk, P.C.; Kedzierska, K. Leishmaniasis: Current treatment and prospects for new drugs and vaccines. Curr. Med. Chem. 2009, 16, 599–614. [Google Scholar] [CrossRef] [PubMed]
- Chappuis, F.; Sundar, S.; Hailu, A.; Ghalib, H.; Rijal, S.; Peeling, R.W.; Alvar, J.; Boelaert, M. Visceral leishmaniasis: What are the needs for diagnosis, treatment and control? Nat. Rev. Microbiol. 2007, 5, 873–882. [Google Scholar] [CrossRef] [PubMed]
- Demicheli, C.; Ochoa, R.; da Silva, J.B.; Falcao, C.A.; Rossi-Bergmann, B.; de Melo, A.L.; Sinisterra, R.D.; Frezard, F. Oral delivery of meglumine antimoniate-beta-cyclodextrin complex for treatment of leishmaniasis. Antimicrob. Agents Chemother. 2004, 48, 100–103. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Sundar, S.; Fairlamb, A.H. Drug resistance in leishmaniasis. Clin. Microbiol. Rev. 2006, 19, 111–126. [Google Scholar] [CrossRef] [PubMed]
- Ephros, M.; Waldman, E.; Zilberstein, D. Pentostam induces resistance to antimony and the preservative chlorocresol in Leishmania donovani promastigotes and axenically grown amastigotes. Antimicrob. Agents Chemother. 1997, 41, 1064–1068. [Google Scholar] [PubMed]
- Thakur, C.P.; Singh, R.K.; Hassan, S.M.; Kumar, R.; Narain, S.; Kumar, A. Amphotericin B deoxycholate treatment of visceral leishmaniasis with newer modes of administration and precautions: A study of 938 cases. Trans. R. Soc. Trop. Med. Hyg. 1999, 93, 319–323. [Google Scholar] [CrossRef] [PubMed]
- Bray, P.G.; Barrett, M.P.; Ward, S.A.; de Koning, H.P. Pentamidine uptake and resistance in pathogenic protozoa: past, present and future. Trends Parasitol. 2003, 19, 232–239. [Google Scholar] [CrossRef] [PubMed]
- Di Giorgio, C.; Faraut-Gambarelli, F.; Imbert, A.; Minodier, P.; Gasquet, M.; Dumon, H. Flow cytometric assessment of amphotericin B susceptibility in Leishmania infantum isolates from patients with visceral leishmaniasis. J. Antimicrob. Chemother. 1999, 44, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Cobb, S.L.; Denny, P.W. Antimicrobial peptides for leishmaniasis. Curr. Opin. Investig. Drugs 2010, 11, 868–875. [Google Scholar] [PubMed]
- McGwire, B.S.; Kulkarni, M.M. Interactions of antimicrobial peptides with Leishmania and trypanosomes and their functional role in host parasitism. Exp. Parasitol. 2010, 126, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Ulvatne, H. Antimicrobial peptides: Potential use in skin infections. Am. J. Clin. Dermatol. 2003, 4, 591–595. [Google Scholar] [CrossRef] [PubMed]
- Chadbourne, F.L.; Raleigh, C.; Ali, H.Z.; Denny, P.W.; Cobb, S.L. Studies on the antileishmanial properties of the antimicrobial peptides temporin A, B and 1Sa. J. Pept. Sci. 2011, 17, 751–755. [Google Scholar] [CrossRef] [PubMed]
- Naderer, T.; Vince, J.E.; McConville, M.J. Surface determinants of Leishmania parasites and their role in infectivity in the mammalian host. Curr. Mol. Med. 2004, 4, 649–665. [Google Scholar] [CrossRef] [PubMed]
- Bahr, V.; Stierhof, Y.D.; Ilg, T.; Demar, M.; Quinten, M.; Overath, P. Expression of lipophosphoglycan, high-molecular weight phosphoglycan and glycoprotein 63 in promastigotes and amastigotes of Leishmania mexicana. Mol. Biochem. Parasitol. 1993, 58, 107–121. [Google Scholar] [PubMed]
- Torrent, M.; Pulido, D.; Rivas, L.; Rivas, L.; Andreu, D. Antimicrobial peptide action on parasites. Curr. Drug Targets 2012, 13, 1138–1147. [Google Scholar] [CrossRef] [PubMed]
- Mangoni, M.L.; Saugar, J.M.; Dellisanti, M.; Barra, D.; Simmaco, M.; Rivas, L. Temporins, small antimicrobial peptides with leishmanicidal activity. J. Biol. Chem. 2005, 280, 984–990. [Google Scholar] [CrossRef] [PubMed]
- Abbassi, F.; Oury, B.; Blasco, T.; Sereno, D.; Bolbach, G.; Nicolas, P.; Hani, K.; Amiche, M.; Ladram, A. Isolation, characterization and molecular cloning of new temporins from the skin of the North African ranid Pelophylax saharica. Peptides 2008, 29, 1526–1533. [Google Scholar] [CrossRef] [PubMed]
- Mauricio, I.L.; Stothard, J.R.; Miles, M.A. The strange case of Leishmania chagasi. Parasitol. Today 2000, 16, 188–189. [Google Scholar] [CrossRef] [PubMed]
- Miranda, A.; Samudio, F.; Saldana, A.; Castillo, J.; Brandao, A.; Calzada, J.E. The calmodulin intergenic spacer as molecular target for characterization of Leishmania species. Parasit. Vectors 2014, 7, 35. [Google Scholar] [CrossRef] [PubMed]
- De Assis, R.R.; Ibraim, I.C.; Nogueira, P.M.; Soares, R.P.; Turco, S.J. Glycoconjugates in New World species of Leishmania: Polymorphisms in lipophosphoglycan and glycoinositolphospholipids and interaction with hosts. Biochim. Biophys. Acta 2012, 1820, 1354–1365. [Google Scholar] [CrossRef] [PubMed]
- Ilg, T. Lipophosphoglycan is not required for infection of macrophages or mice by Leishmania mexicana. EMBO J. 2000, 19, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- King, D.L.; Turco, S.J. A ricin agglutinin-resistant clone of Leishmania donovani deficient in lipophosphoglycan. Mol. Biochem. Parasitol. 1988, 28, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Ilg, T.; Demar, M.; Harbecke, D. Phosphoglycan repeat-deficient Leishmania mexicana parasites remain infectious to macrophages and mice. J. Biol. Chem. 2001, 276, 4988–4997. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.L.; Kim, H.K.; Sass, P.M.; He, S.; Geng, J.; Xu, H.; Zhu, B.; Turco, S.J.; Lo, S.K. Structure-function analysis of Leishmania lipophosphoglycan. Distinct domains that mediate binding and inhibition of endothelial cell function. J. Immunol. 1996, 157, 3013–3020. [Google Scholar] [PubMed]
- Ilg, T.; Craik, D.; Currie, G.; Multhaup, G.; Bacic, A. Stage-specific proteophosphoglycan from Leishmania mexicana amastigotes. Structural characterization of novel mono-, di-, and triphosphorylated phosphodiester-linked oligosaccharides. J. Biol. Chem. 1998, 273, 13509–13523. [Google Scholar] [CrossRef] [PubMed]
- Ramos, H.; Saint-Pierre-Chazalet, M.; Bolard, J.; Cohen, B.E. Effect of ketoconazole on lethal action of amphotericin B on Leishmania mexicana promastigotes. Antimicrob. Agents Chemother. 1994, 38, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Bates, P.A. Complete developmental cycle of Leishmania mexicana in axenic culture. Parasitology 1994, 108, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ilg, T. Proteophosphoglycans of Leishmania. Parasitol. Today 2000, 16, 489–497. [Google Scholar] [CrossRef] [PubMed]
- Secundino, N.; Kimblin, N.; Peters, N.C.; Lawyer, P.; Capul, A.A.; Beverley, S.M.; Turco, S.J.; Sacks, D. Proteophosphoglycan confers resistance of Leishmania major to midgut digestive enzymes induced by blood feeding in vector sand flies. Cell. Microbiol. 2010, 12, 906–918. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the temporins discussed are available from the authors on request.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eggimann, G.A.; Sweeney, K.; Bolt, H.L.; Rozatian, N.; Cobb, S.L.; Denny, P.W. The Role of Phosphoglycans in the Susceptibility of Leishmania mexicana to the Temporin Family of Anti-Microbial Peptides. Molecules 2015, 20, 2775-2785. https://doi.org/10.3390/molecules20022775
Eggimann GA, Sweeney K, Bolt HL, Rozatian N, Cobb SL, Denny PW. The Role of Phosphoglycans in the Susceptibility of Leishmania mexicana to the Temporin Family of Anti-Microbial Peptides. Molecules. 2015; 20(2):2775-2785. https://doi.org/10.3390/molecules20022775
Chicago/Turabian StyleEggimann, Gabriela A., Kathryn Sweeney, Hannah L. Bolt, Neshat Rozatian, Steven L. Cobb, and Paul W. Denny. 2015. "The Role of Phosphoglycans in the Susceptibility of Leishmania mexicana to the Temporin Family of Anti-Microbial Peptides" Molecules 20, no. 2: 2775-2785. https://doi.org/10.3390/molecules20022775