LC-ESI-MS/MS Analysis and Pharmacokinetics of Plantainoside D Isolated from Chirita longgangensis var. hongyao, a Potential Anti-Hypertensive Active Component in Rats


Abstract
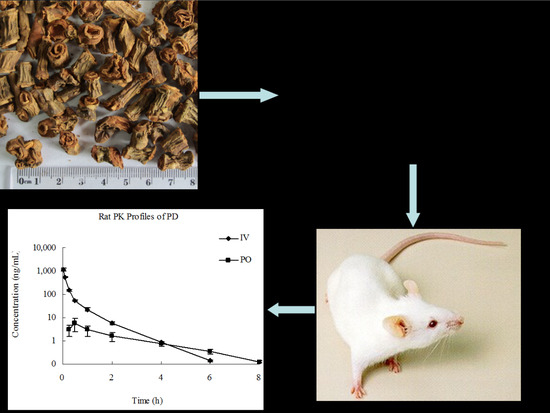
:
1. Introduction

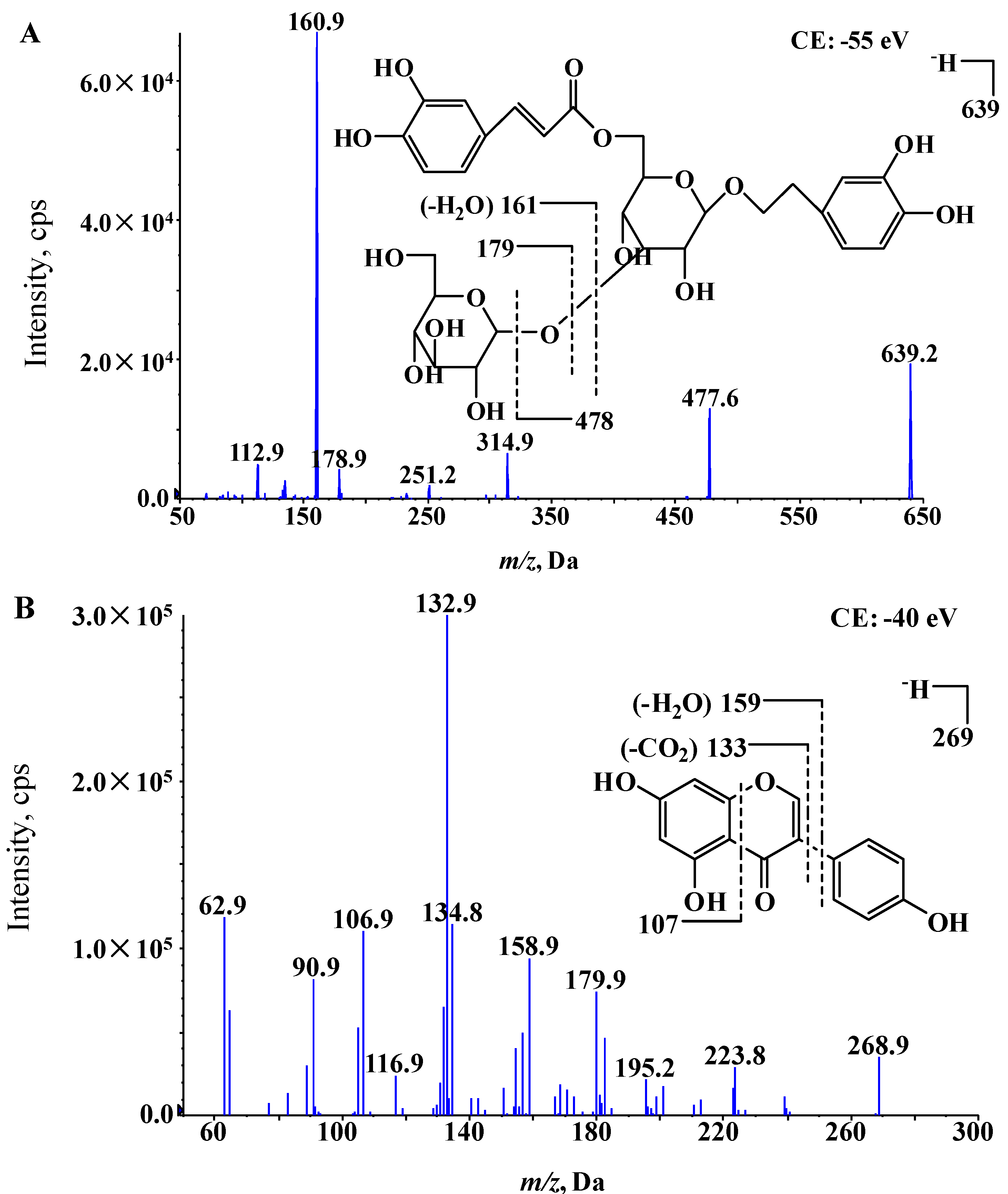
2. Results and Discussion
2.1. Mass Spectrometry and Chromatography
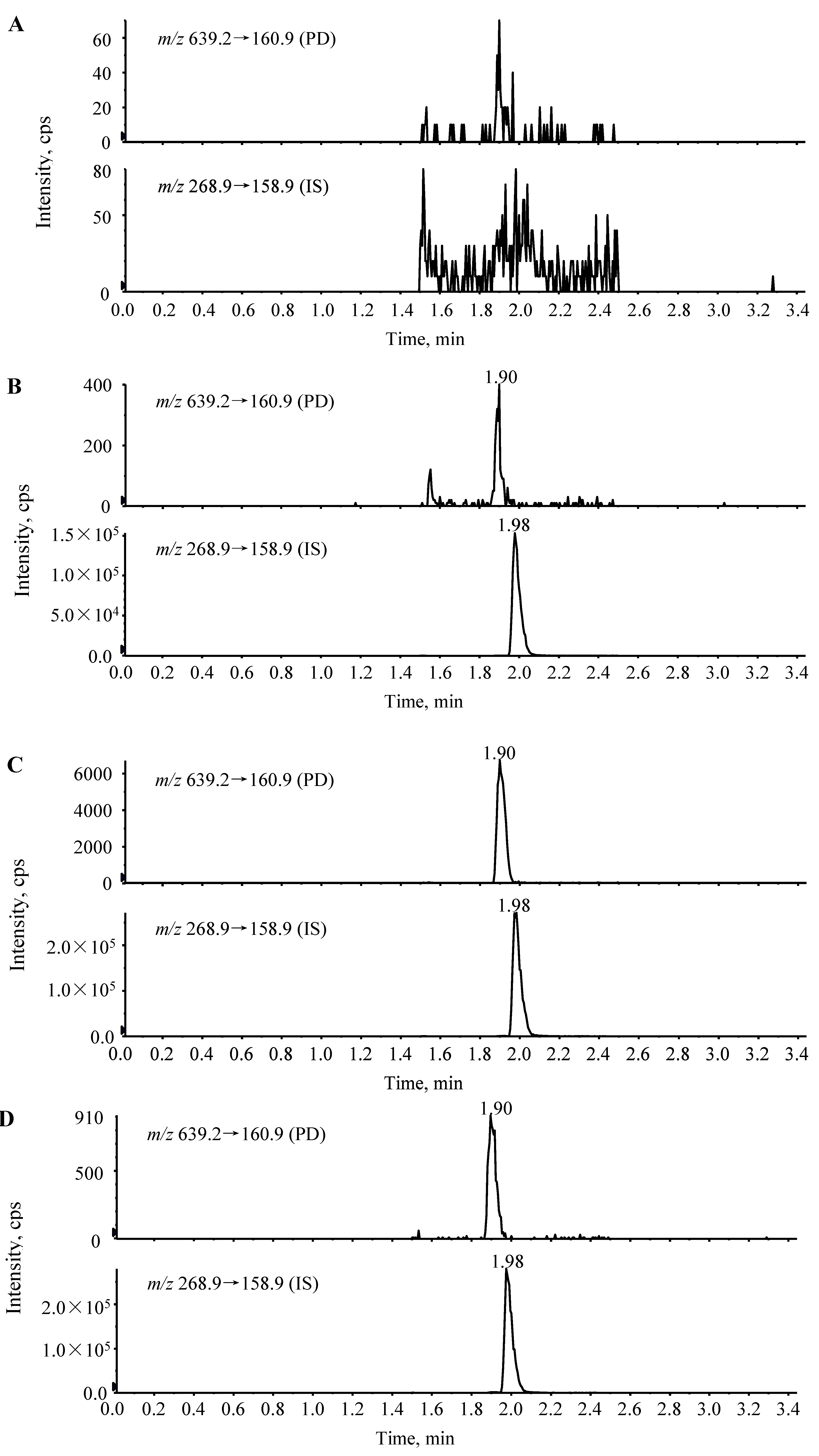
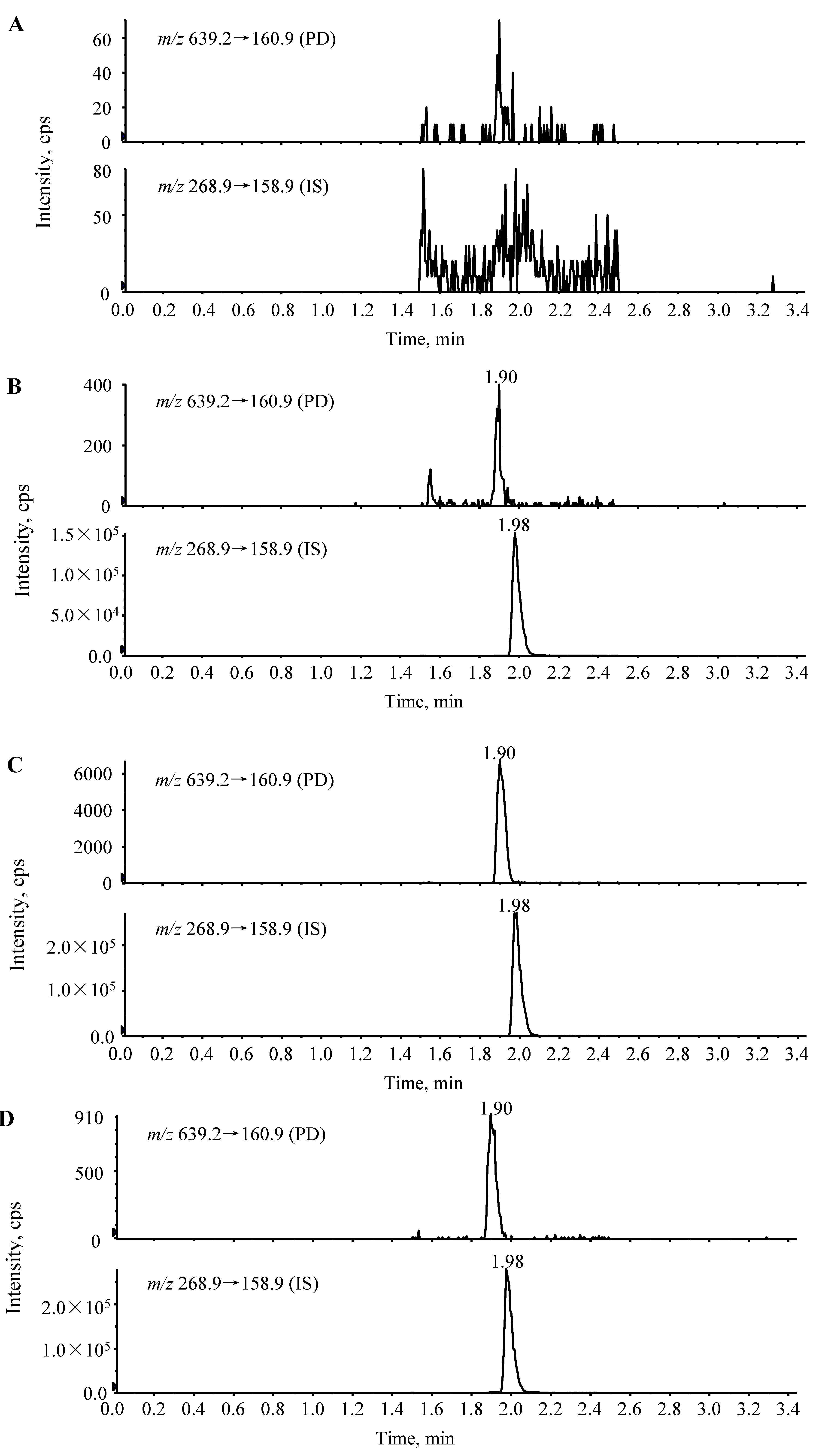
2.2. Linearity, Sensitivity and Detection Limit of the Assay
2.3. Recovery and Matrix Effect

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Spiked Concentration (ng/mL) | Matrix Effect (%) | Mean ± SD (%) | Recovery (%) | RSD (%) |
|---|---|---|---|---|
| 0.20 | 93.1 | 93.0 ± 1.25 | 88.3 | 3.27 |
| 10.0 | 94.2 | 92.0 | 0.58 | |
| 160 | 91.7 | 90.4 | 1.44 |
2.4. Accuracy and Precision of the Assay
| Batch | No. | Low | Medium | High |
|---|---|---|---|---|
| 0.20 ng/mL | 10.0 ng/mL | 160 ng/mL | ||
| Day 1 | Mean ± SD | 0.20 ± 0.02 | 10.1 ± 0.46 | 159 ± 9.04 |
| RSD (%) | 7.75 | 4.61 | 5.68 | |
| Accuracy (%) | 100.0 | 100.7 | 99.5 | |
| Day 2 | Mean ± SD | 0.19 ± 0.01 | 10.3 ± 0.69 | 165 ± 7.04 |
| RSD (%) | 7.44 | 6.64 | 4.27 | |
| Accuracy (%) | 95.0 | 103.2 | 103.1 | |
| Day 3 | Mean ± SD | 0.20 ± 0.02 | 10.2 ± 0.73 | 163 ± 6.34 |
| RSD (%) | 9.59 | 7.14 | 3.88 | |
| Accuracy (%) | 97.5 | 102.0 | 102.0 | |
| Inter-Day | Mean ± SD | 0.20 ± 0.02 | 10.2 ± 0.61 | 162 ± 7.53 |
| RSD (%) | 8.11 | 5.95 | 4.64 | |
| Accuracy (%) | 97.5 | 102.0 | 101.5 |
2.5. Stability
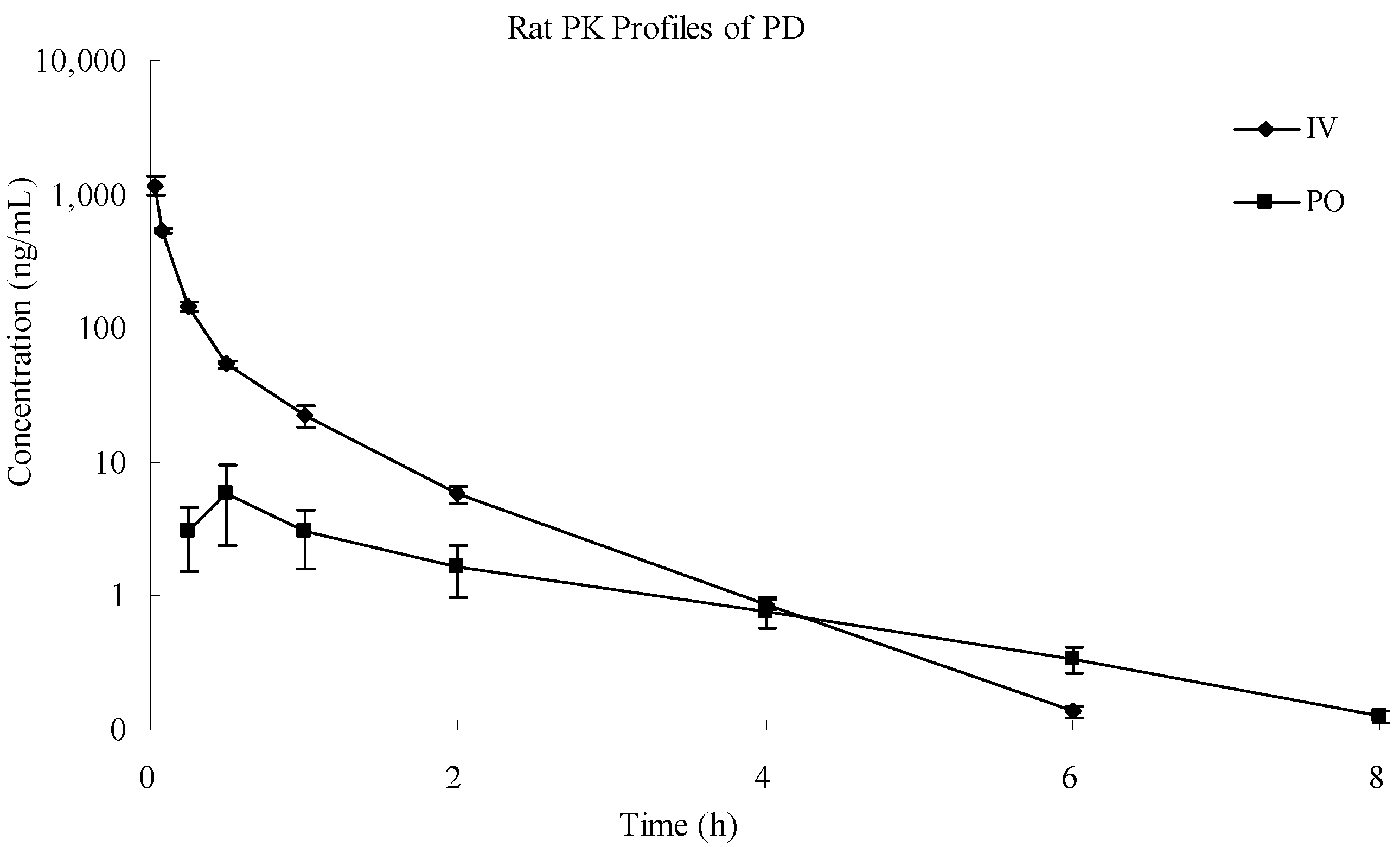
2.6. Application of the Assay Method
| Stability Conditions | Added Conc. | 0.20 ng/mL | 10.0 ng/mL | 160 ng/mL |
|---|---|---|---|---|
| Three freeze–thaw cycles | Mean ± SD | 0.21 ± 0.01 | 9.89 ± 0.45 | 161 ± 6.11 |
| RSD (%) | 4.76 | 4.54 | 3.80 | |
| Recovery (%) | 105.0 | 98.9 | 100.4 | |
| Room temperature for 24 h | Mean ± SD | 0.19 ± 0.02 | 10.5 ± 0.25 | 168 ± 4.73 |
| RSD (%) | 7.90 | 2.39 | 2.81 | |
| Recovery (%) | 96.7 | 105.3 | 105.2 | |
| Storage at −80 °C for 1 month | Mean ± SD | 0.19 ± 0.02 | 9.66 ± 0.08 | 151 ± 6.24 |
| RSD (%) | 7.90 | 0.85 | 4.14 | |
| Recovery (%) | 96.7 | 96.6 | 94.4 |
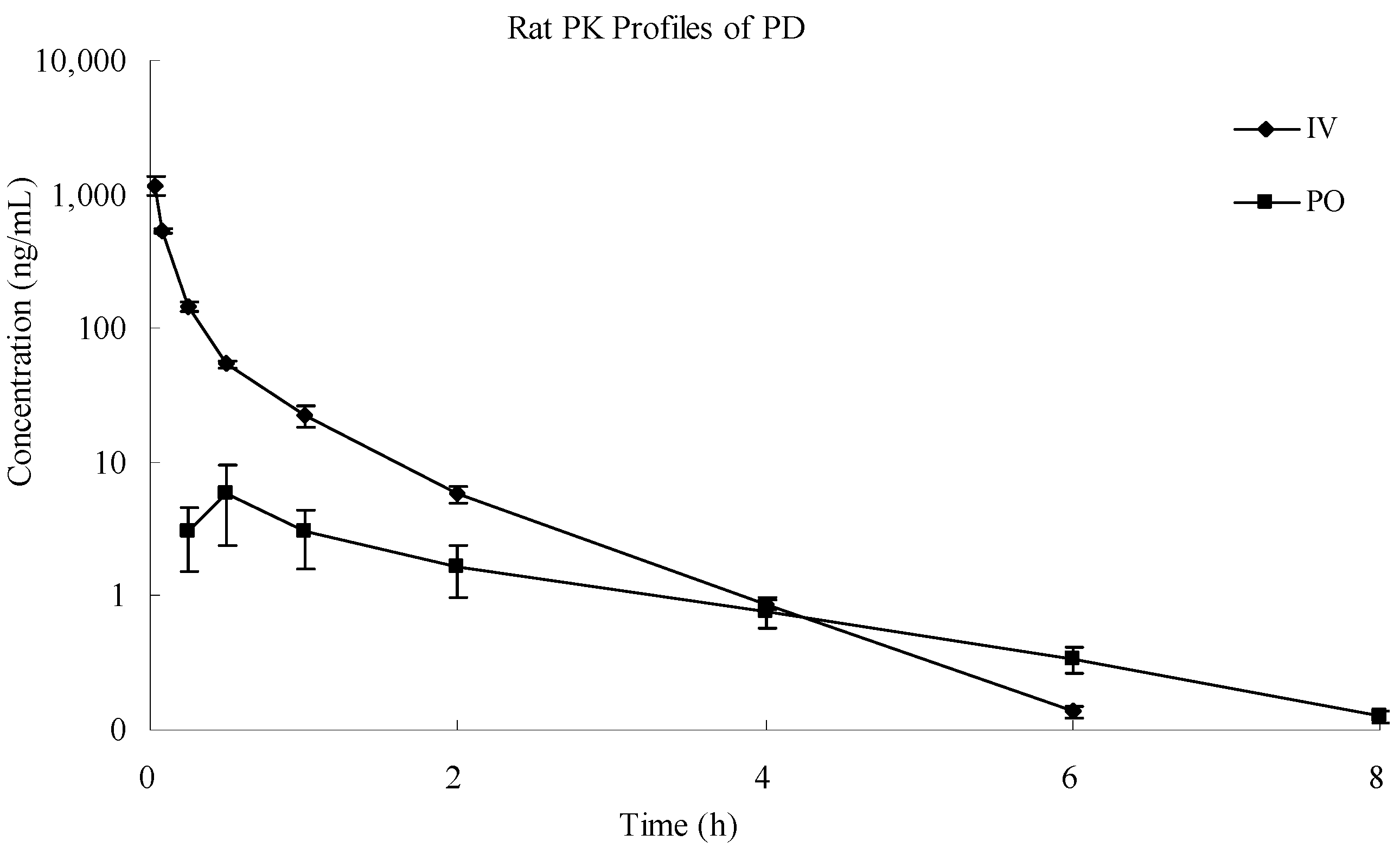
| Parameters | Unit | IV | PO |
|---|---|---|---|
| AUC(0-t) | ng/mL × h | 185 ± 11.7 | 10.1 ± 4.28 |
| AUC(0-∞) | ng/mL × h | 185 ± 11.7 | 10.4 ± 4.28 |
| MRT(0-t) | h | 0.36 ± 0.01 | 2.04 ± 0.32 |
| MRT(0-∞) | h | 0.36 ± 0.01 | 2.31 ± 0.43 |
| t1/2z | h | 0.69 ± 0.06 | 1.63 ± 0.19 |
| Tmax | h | 0.03 ± 0.00 | 0.50 ± 0.00 |
| CLz | L/h/kg | 10.8 ± 0.70 | 1080 ± 436 |
| Vz | L/kg | 10.8 ± 0.96 | 2626 ± 1340 |
| Cmax | ng/mL | 1154 ± 183 | 5.92 ± 3.51 |
| F | % | - | 1.12 ± 0.46 |
3. Experimental Section
3.1. Chemicals and Reagents
3.2. LC–MS/MS
3.3. Preparation of Standards and Calibration Curves
3.4. Sample Preparation
3.5. Assay Validation
3.5.1. Linearity, Accuracy, Precision, and Recovery
3.5.2. Stability
3.5.3. Matrix Effects
3.6. Pharmacokinetic Study
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- National Group Corporation of Traditional & Herbal Medicine. Compendium of Chinese Medical Material Resources China, Science Press: Beijing, China, 1994; 1181.
- Huang, X.C. Compendium of Guangxi National Medicine, 1st ed. Science Press: Beijing, China, 1980; 117.
- Wang, M.Y.; Yang, L.; Tu, Y.Y. Phenylethanoid glycosides from stem of Chiritalonggangensis var. hongyao. Chin. J. Chin. Mater. Med. 2005, 30, 1921–1923. [Google Scholar]
- Wang, M.Y.; Fan, Y.; Zhang, J.; Gong, M.X. Simultaneous determination of plantainoside D and verbascoside from stem of Chirita longgangensis var. Hongyao by RP-HPLC. Chin. J. Chin. Mater. Med. 2010, 35, 3188–3191. [Google Scholar]
- Zou, L.C.; Zhu, T.F.; Xiang, H.; Yu, L.; Yan, Z.H.; Gan, S.C.; Wang, D.C.; Zeng, S.; Deng, X.M. New secoiridoid glycosides from the roots of Picrorhiza scrophulariiflora. Molecules 2008, 13, 2049–2057. [Google Scholar]
- Zou, X.; Liao, X.; Ding, L.S.; Peng, S.L. Phenyl and phenylethyl glycosides from Picrorhiza scrophulariiflora. J. Asian Nat. Prod. Res. 2007, 9, 443–448. [Google Scholar]
- Wang, H.; Ye, W.C.; Xiong, F.; Zhao, S.X. Phenylethanoid glycosides from root of Picrorhiza scrophulariiflora. Chin. J. Chin. Mater. Med. 2004, 29, 531–534. [Google Scholar]
- Budzianowska, A.; Skrzypczak, L.; Budzianowski, J. Phenylethanoid glucosides from in vitro propagated plants and callus cultures of Plantagolanceolata. Planta Med. 2004, 70, 834–840. [Google Scholar]
- Zhou, B.N.; Bahler, B.D.; Hofmann, G.A.; Mattern, M.R.; Johnson, R.K.; Kingston, D.G. Phenylethanoid glycosides from Digitalis purpurea and Penstemon linarioides with PKCalpha-inhibitory activity. J. Nat. Prod. 1998, 61, 1410–1412. [Google Scholar]
- Geng, F.; Yang, L.; Chou, G.; Wang, Z. Bioguided isolation of angiotensin-converting enzyme inhibitors from the seeds of Plantago asiatica L. Phytother. Res. 2010, 24, 1088–1094. [Google Scholar]
- Kim, D.S.; Woo, E.R.; Chae, S.W.; Ha, K.C.; Lee, G.H.; Hong, S.T.; Kwon, D.Y.; Kim, M.S.; Jung, Y.K.; Kim, H.M.; et al. Plantainoside D protects adriamycin-induced apoptosis in H9c2 cardiac muscle cells via the inhibition of ROS generation and NF-kappa Bactivation. Life Sci. 2007, 80, 314–323. [Google Scholar]
- Lee, J.Y.; Woo, E.; Kang, K.W. Screening of new chemopreventive compounds from Digitalis purpurea. Pharmazie 2006, 61, 356–358. [Google Scholar]
- Oh, J.W.; Lee, J.Y.; Han, S.H.; Moon, Y.H.; Kim, Y.G.; Woo, E.R.; Kang, K.W. Effects of phenylethanoid glycosides from Digitalis purpurea L. on the expression of inducible nitric oxide synthase. J. Pharm. Pharmacol. 2005, 57, 903–910. [Google Scholar]
- Wang, H.; Sun, Y.; Ye, W.C.; Xiong, F.; Wu, J.J.; Yang, C.H.; Zhao, S.X. Antioxidativephenylethanoid and phenolic glycosides from Picrorhiza scrophulariiflora. Chem. Pharm. Bull. 2004, 52, 615–617. [Google Scholar]
- US Department of Health and Human Services Food and Drug Administration; Center for Drug Evaluation and Research (CDER). Guidance for Industry; Bioanalytical Method Validation, Center for Veterinary Medicine (CV): Silver Spring, MD, USA, 2001.
- Koufopantelis, P.; Georgakakou, S.; Kazanis, M.; Giaginis, C.; Margeli, A.; Papargiri, S.; Panderi, I. Direct injection liquid chromatography/positive ion electrospray ionization mass spectrometric quantification of methotrexate, folinic acid, folic acid and ondansetron in human serum. J. Chromatogr. B 2009, 877, 3850–3856. [Google Scholar]
- Sample Availability: Samples of plantainoside D are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, M.; Fu, S.; Zhang, X.; Li, J.; Gong, M.; Qiu, F. LC-ESI-MS/MS Analysis and Pharmacokinetics of Plantainoside D Isolated from Chirita longgangensis var. hongyao, a Potential Anti-Hypertensive Active Component in Rats. Molecules 2014, 19, 15103-15115. https://doi.org/10.3390/molecules190915103
Wang M, Fu S, Zhang X, Li J, Gong M, Qiu F. LC-ESI-MS/MS Analysis and Pharmacokinetics of Plantainoside D Isolated from Chirita longgangensis var. hongyao, a Potential Anti-Hypertensive Active Component in Rats. Molecules. 2014; 19(9):15103-15115. https://doi.org/10.3390/molecules190915103
Chicago/Turabian StyleWang, Manyuan, Shujun Fu, Xinshi Zhang, Jing Li, Muxin Gong, and Feng Qiu. 2014. "LC-ESI-MS/MS Analysis and Pharmacokinetics of Plantainoside D Isolated from Chirita longgangensis var. hongyao, a Potential Anti-Hypertensive Active Component in Rats" Molecules 19, no. 9: 15103-15115. https://doi.org/10.3390/molecules190915103