Recent Advances in Chemoenzymatic Peptide Syntheses

Abstract
:1. Introduction
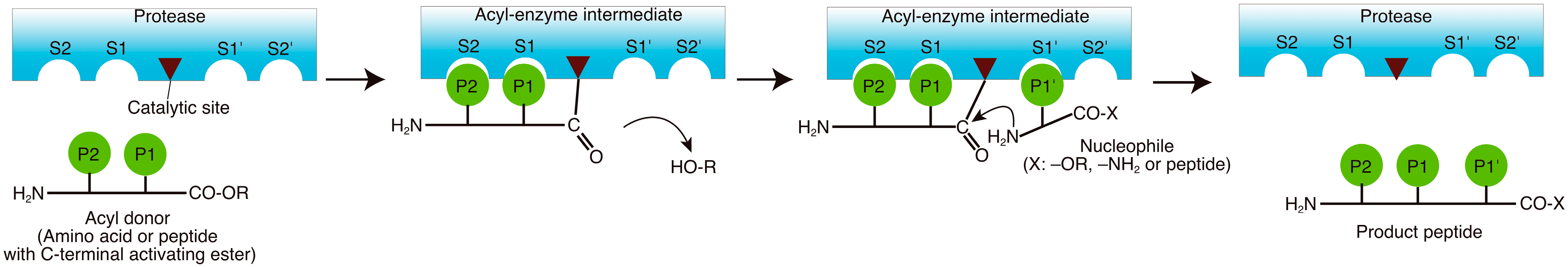
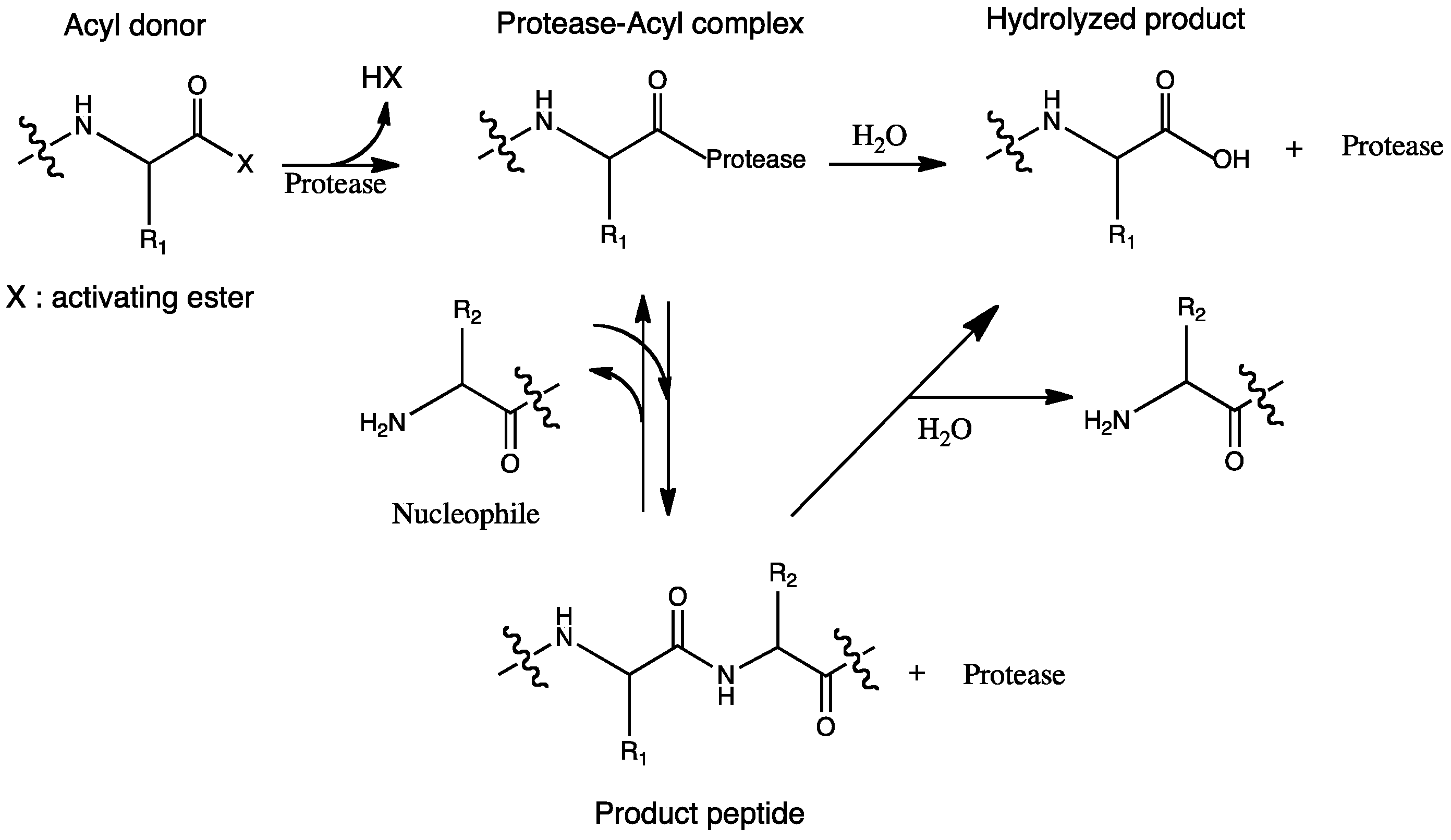
2. Reaction Mechanisms
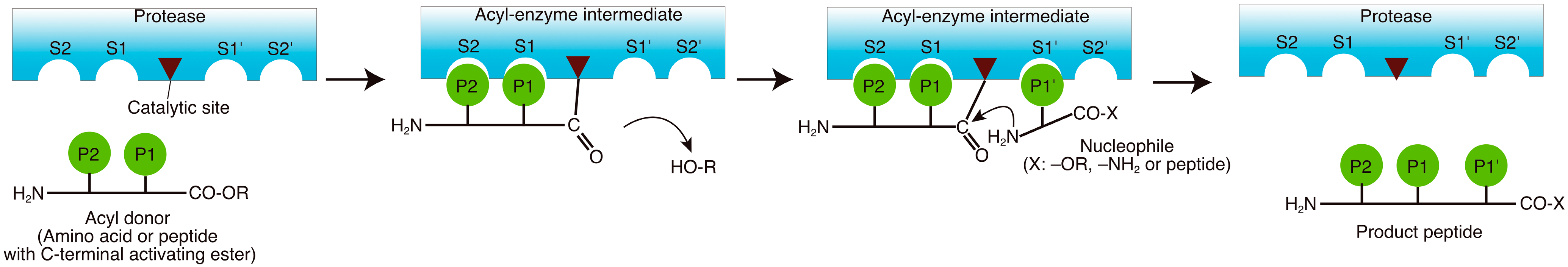
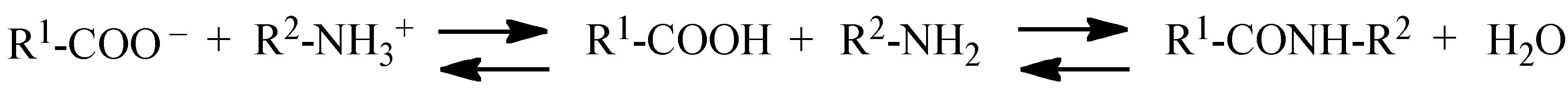
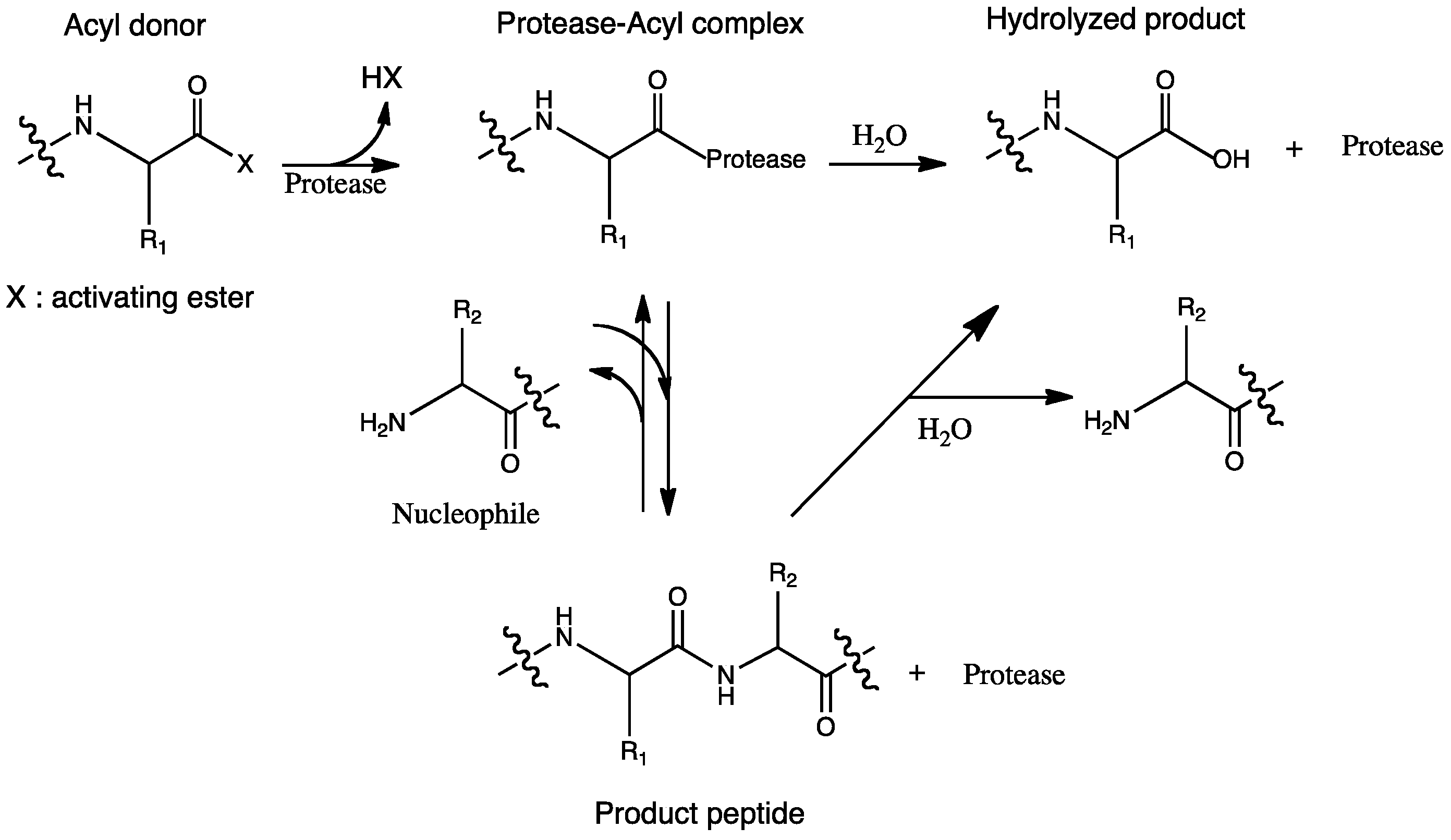
3. Enzymes Used in the Peptide Synthesis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
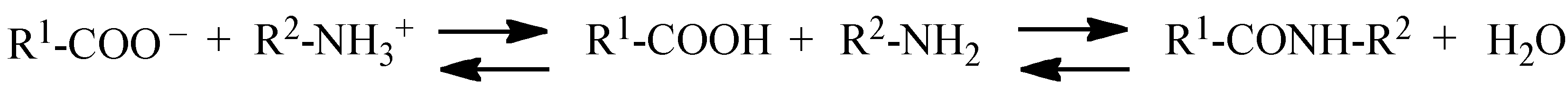{kind=link}
{kind=link}
| Protease | Monomer | pH | Temp. | Substrate | Enzyme | Yield | DPavg | Ref |
|---|---|---|---|---|---|---|---|---|
| papain | Ala-OEt | 11 | 40 °C | 0.7 M | 7.1 mg/mL | 36% | 13.5 (NMR) | [15] |
| papain | Ala-OEt | 7 | 40 °C | 0.7 M | 7.1 mg/mL | 67% | 9.3 (NMR) | [15] |
| papain | Tyr-OEt | 7 | 25 °C | 0.2 M | 20 mg/mL | 64% | 12 (SEC) | [16] |
| papain | Glu-(OEt)2 | 7 | 40 °C | 0.5 M | 40 mg/mL | 60% | 9.5 (NMR) | [17] |
| papain | Glu-(OEt)2Cys-OEt | 8 | 40 °C | 0.2 M,0.3 M | 16 units/mL | 34% | 9.2 (NMR) | [18] |
| papain | Tyr-OEt | 7 | 40 °C | 0.2 M | 12 mg/mL | 80% | 9 (NMR) | [19] |
| papain | Ala-Gly-OEt | 9 | 40 °C | 0.5 M | 20 mg/mL | 48% | 9.5 (NMR) | [20] |
| papain | Leu-OEt | 7 | 40 °C | 1.0 M | 10 mg/mL | 25% | 6–9 (MS) | [21] |
| papain | Tyr-OEtLys-OEt | 9.5 | 40 °C | 0.3 M0.3 M | 7.0 mg/mL | 42% | 8 (NMR) | [22] |
| bromelain | Phe-OEt | 8 | 40 °C | 0.1 M | 18.6 mg/mL | 45% | 8.2 (NMR) | [23] |
| bromelain | Lys-OEt | 7.6 | 40 °C | 0.5 M | 20 mg/mL | 80% | 3.6 (NMR) | [24] |
| α-chymotrypsin | Cys-OEt | 8 | −20 °C | 0.1 M | 20 μM | 85% | 6–11 (MS) | [25] |
| α-chymotrypsin | Lys-Leu-OEt | 8.5 | 40 °C | 0.3 M | 10 mg/mL | - | 4.7 (NMR) | [26] |
| proteinase K | Phe-OEt | 8 | 40 °C | 0.6 M | 1.0 mg/mL | 65% | 12 (NMR) | [27] |
| trypsin | Lys-OEt | 10 | 25 °C | 0.2 M | 10 μM | 50% | 2–8 (MS) | [28] |
| trypsin | Arg-OEt | 10 | 25 °C | 0.5 M | 10 μM | 43% | - | [29] |
| subtilisin | Bz-Arg-OEtGly-NH2 | 10 | 45 °C | 0.05 M0.4 M | - | 83% | - | [30] |
| lipase | Bz-Arg-OEtGly-Asp-Ser-NH2 | 7.5 | 10 °C | 0.05 M0.4 M | 10 mg/mL | 74% | - | [31] |
| lipase | 2-azetidinone | - | 90 °C | 0.3 M | 20 mg/mL | 73% | 8 (NMR) | [32] |
3.1. Cysteine Protease
3.1.1. Papain
3.1.2. Bromelain
3.2. Serine Protease
3.2.1. α-Chymotrypsin
3.2.2. Proteinase K
3.2.3. Trypsin
3.2.4. Subtilisin (Alcalase)
3.3. Lipase
3.4. Engineered Proteases
4. Design of Reaction Conditions
4.1. pH, Temperature, and Substrate and Enzyme Concentrations
4.2. Frozen Aqueous Media
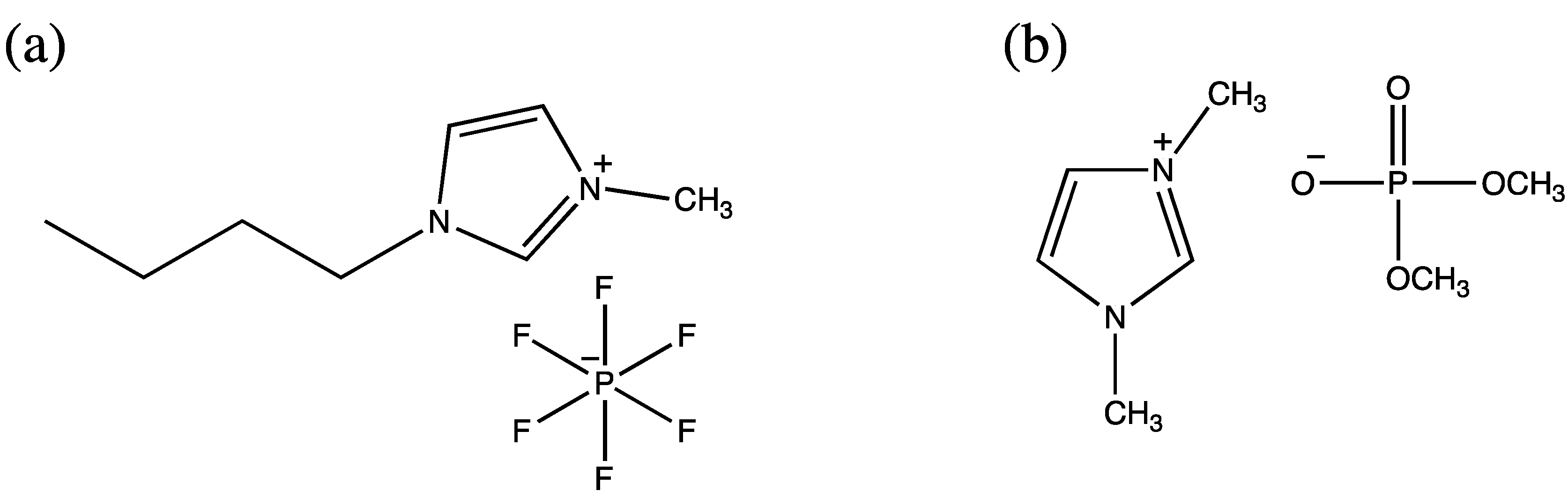
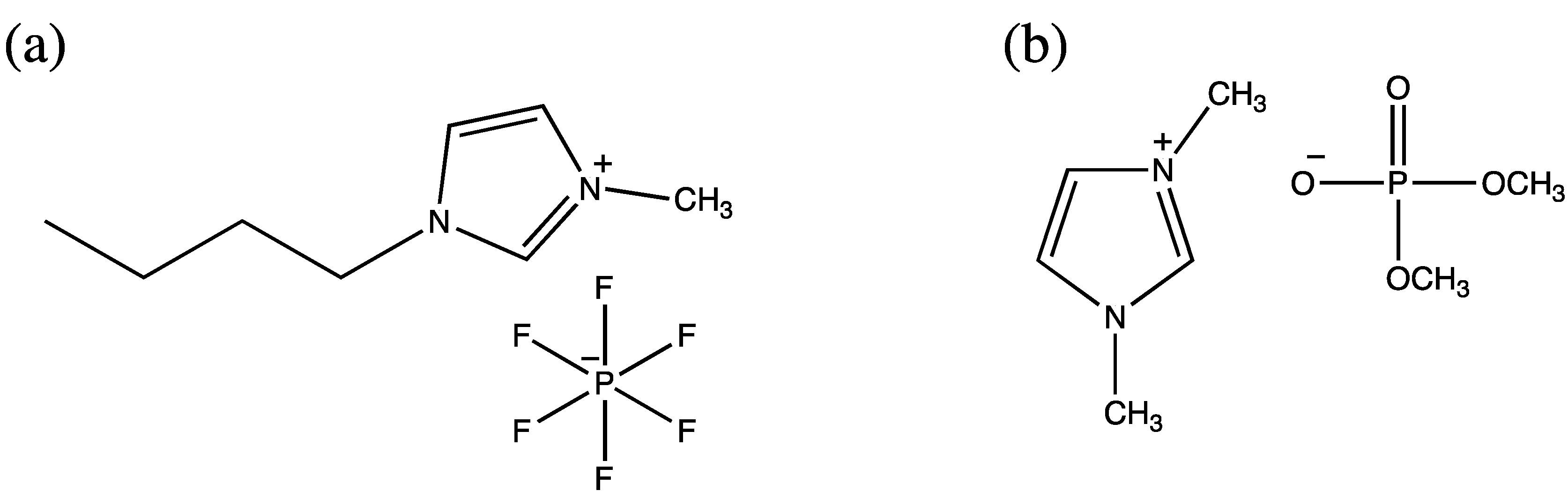
4.3. Ionic Liquids
4.4. Supercritical Carbon Dioxide
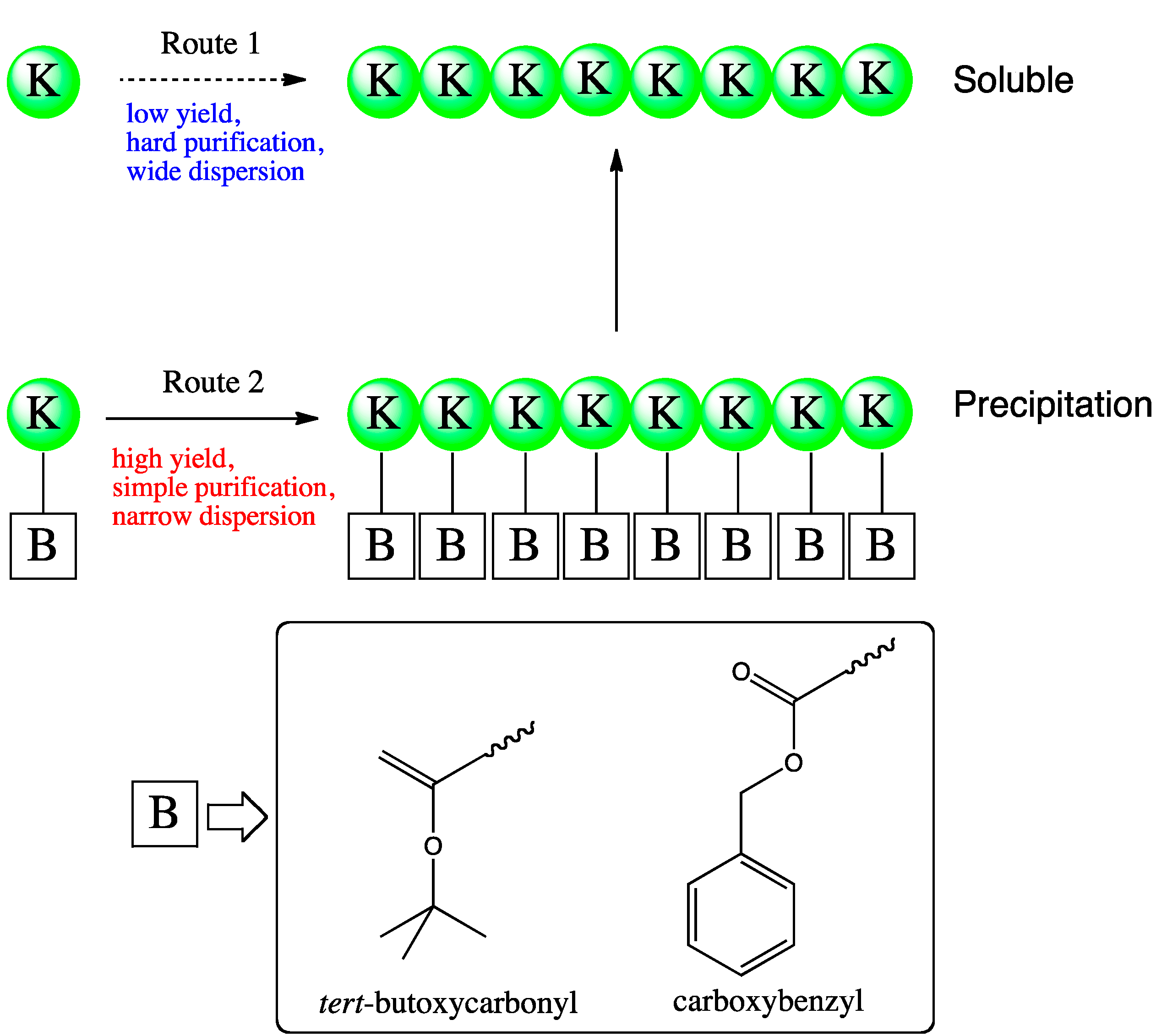
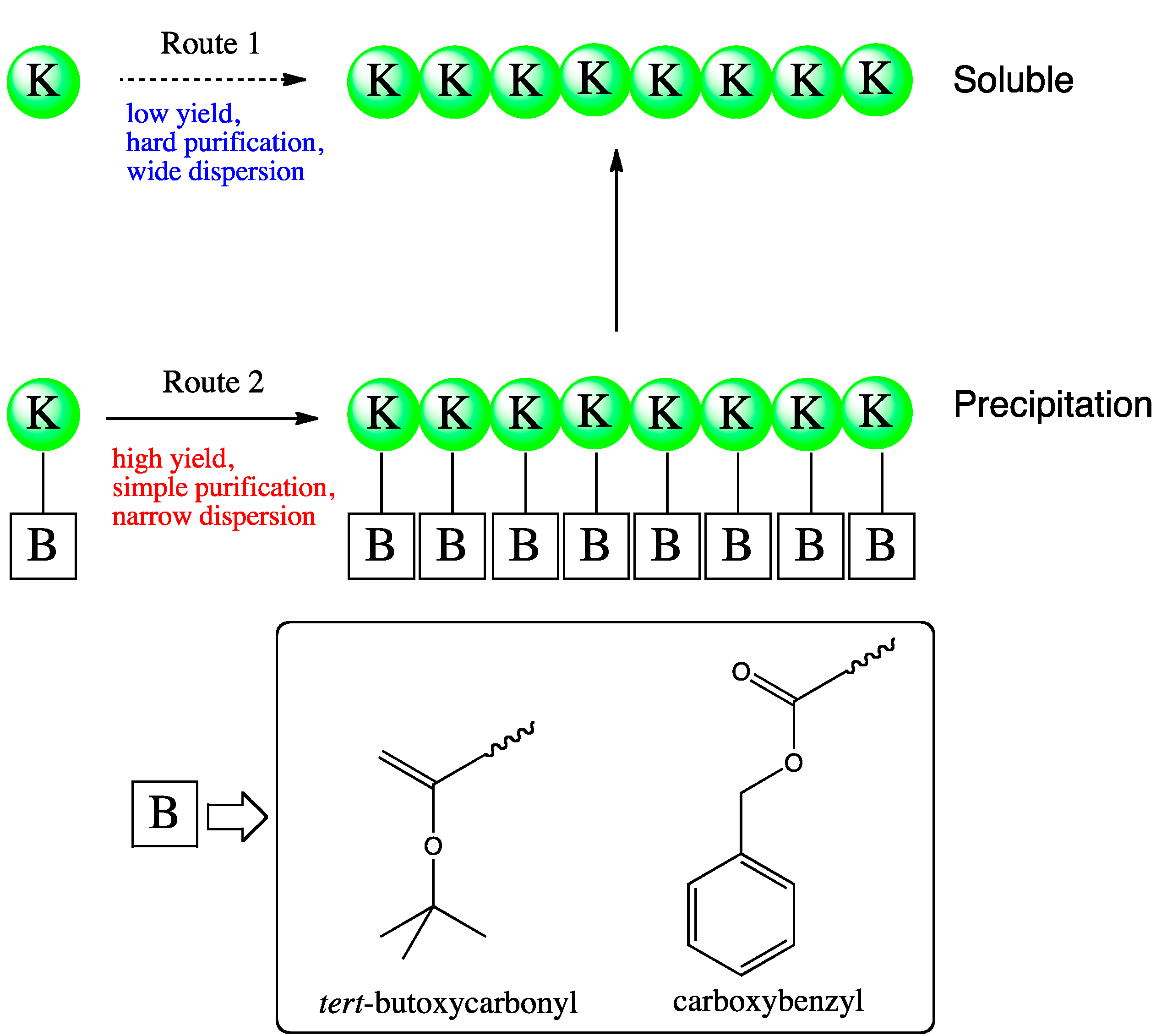
4.5. Immobilization of Substrate or Enzyme
4.6. Cross-Linked Enzyme Aggregate
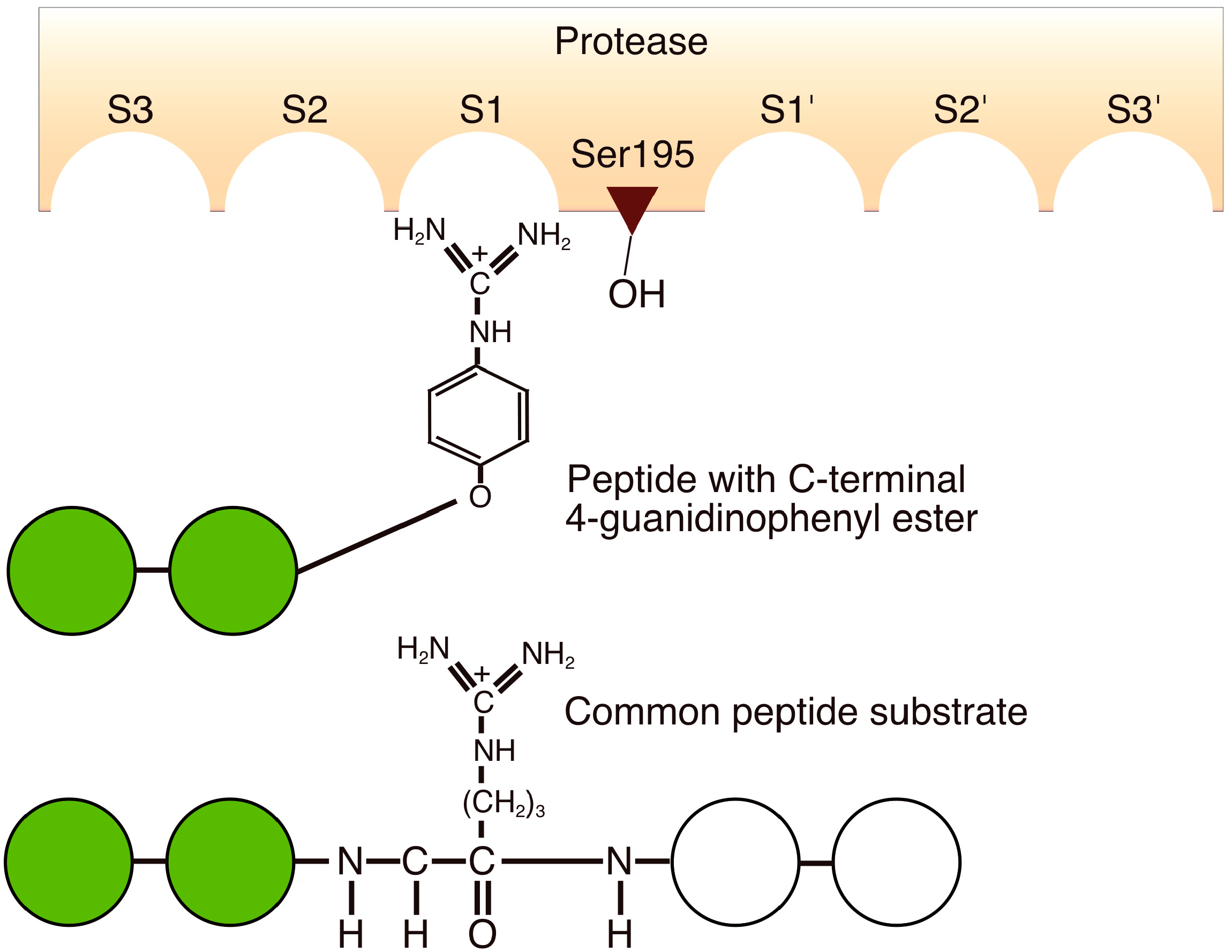
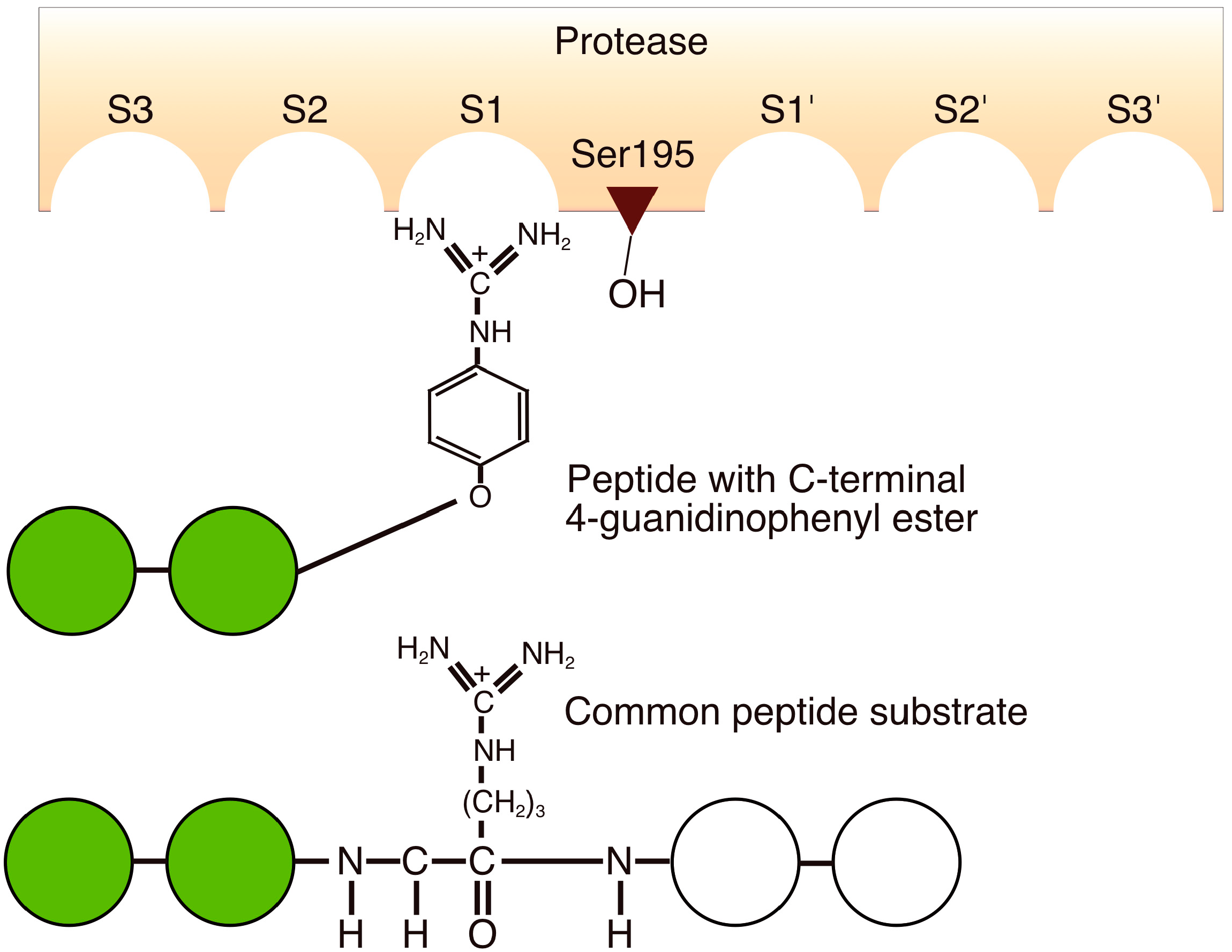
4.7. Substrate Mimetics
5. Various Applications using Chemoenzymatic Peptide Synthesis
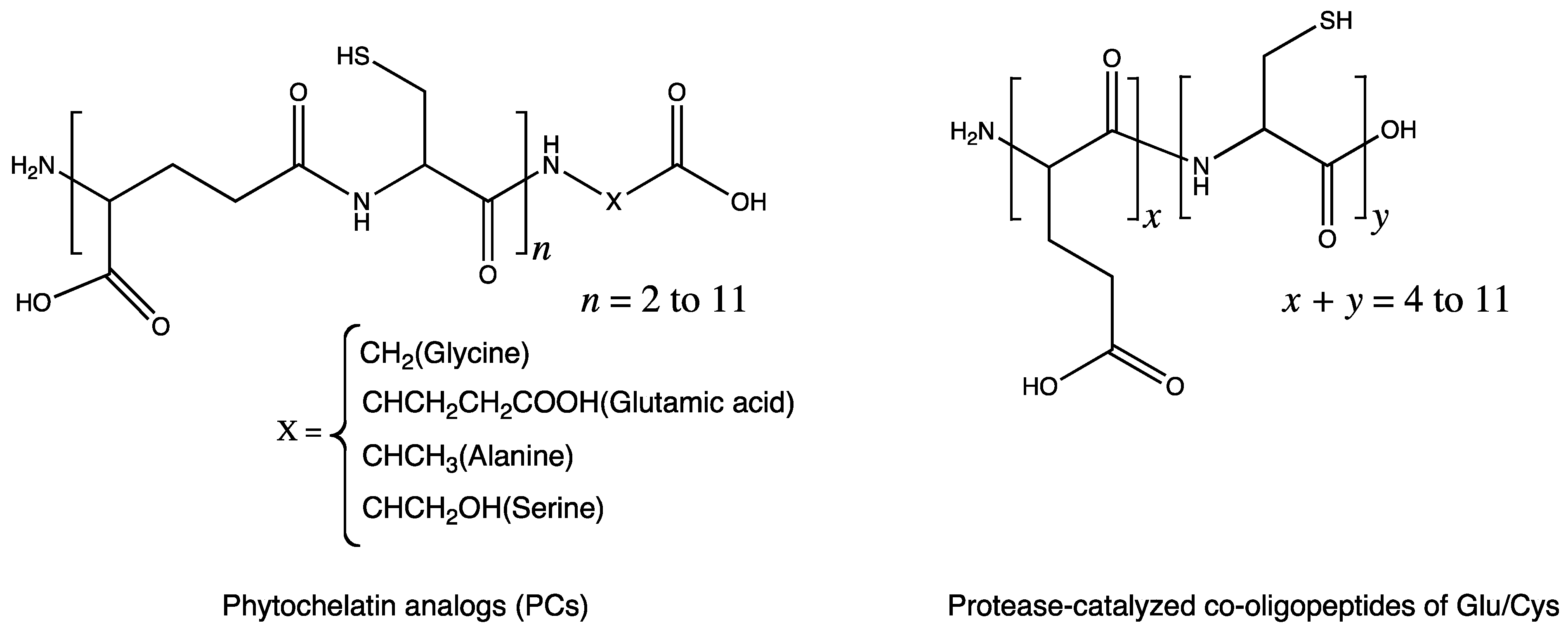
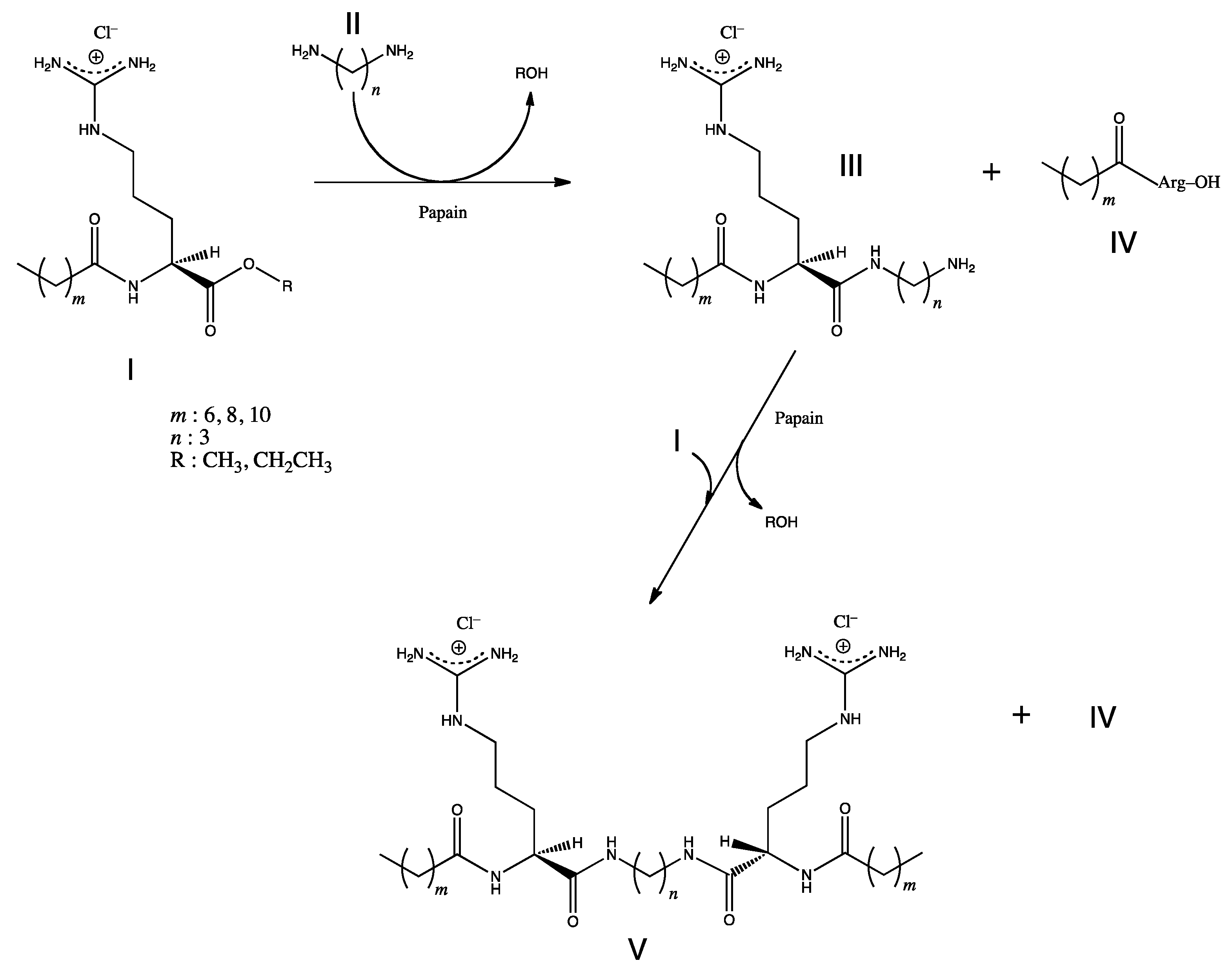
5.1. Metal-Chelating Agents
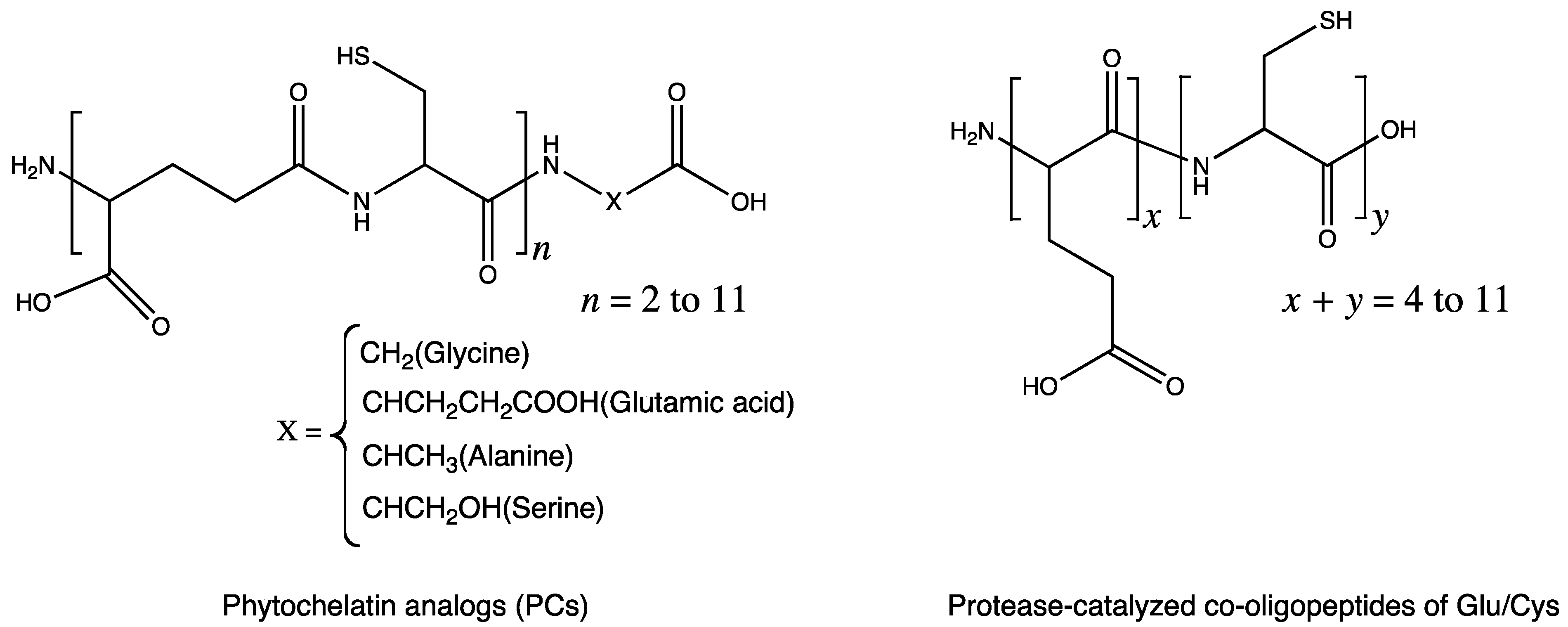
5.2. Antibiotics
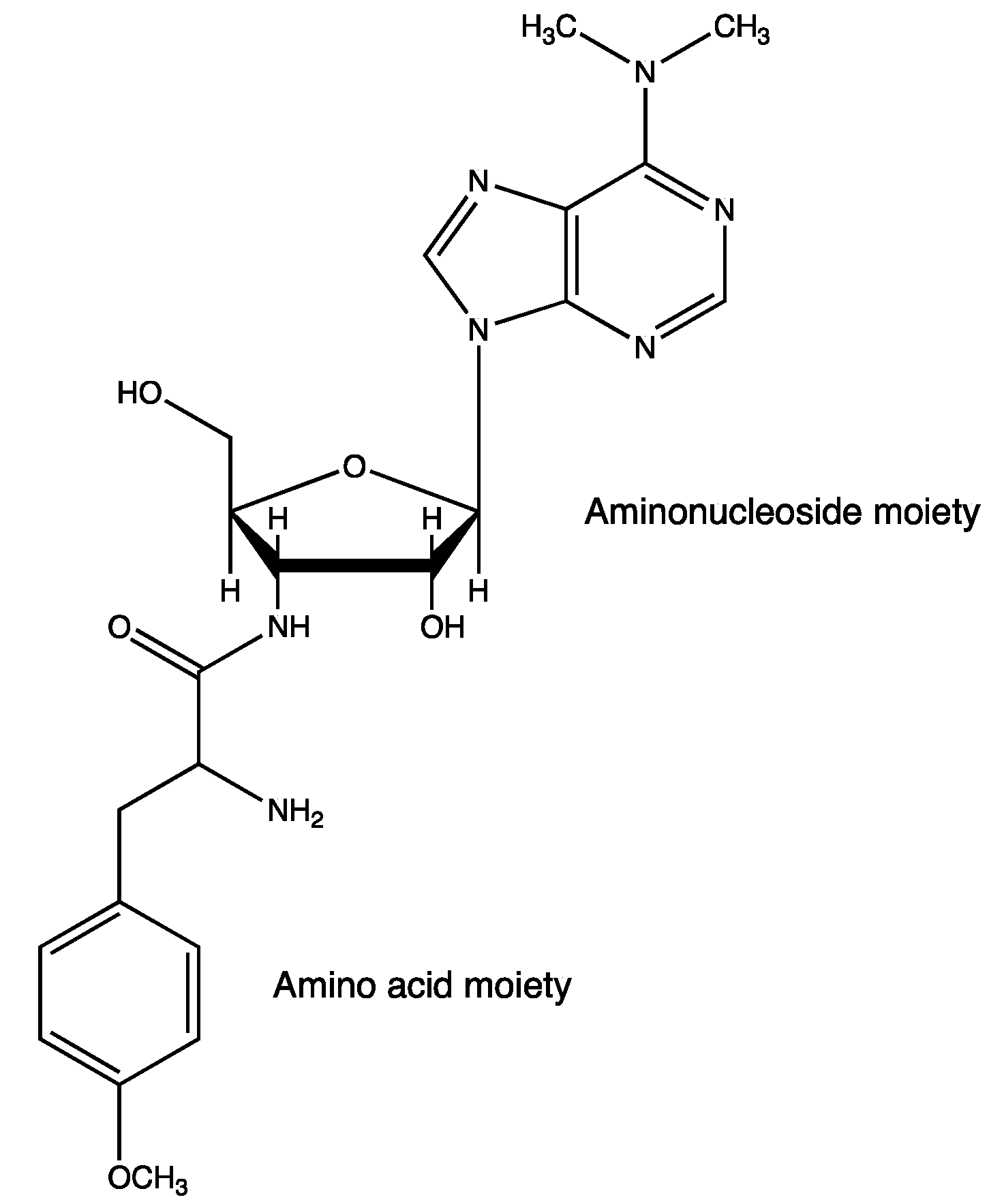
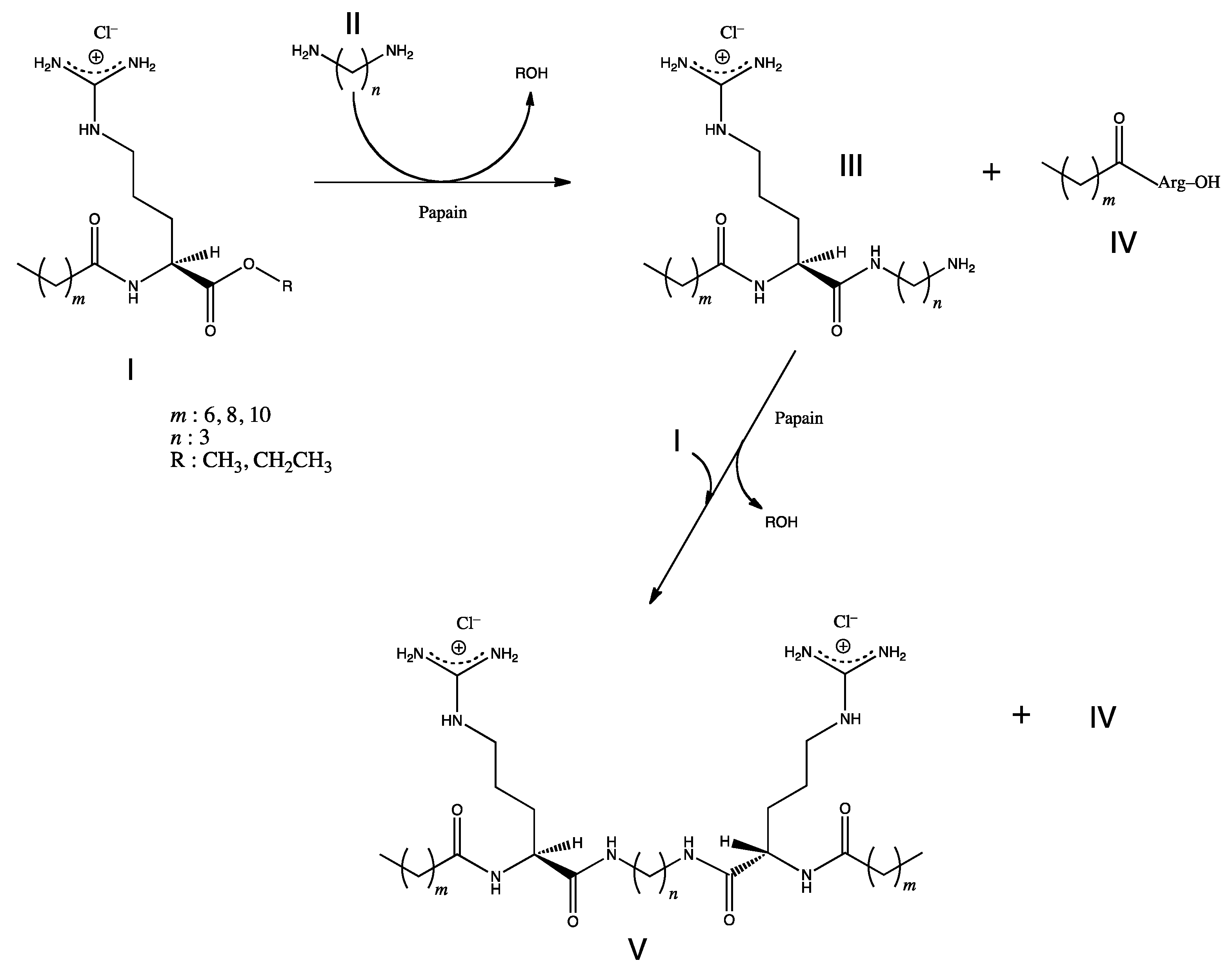
5.3. Surfactants
5.4. Potential Use in Tissue Engineering
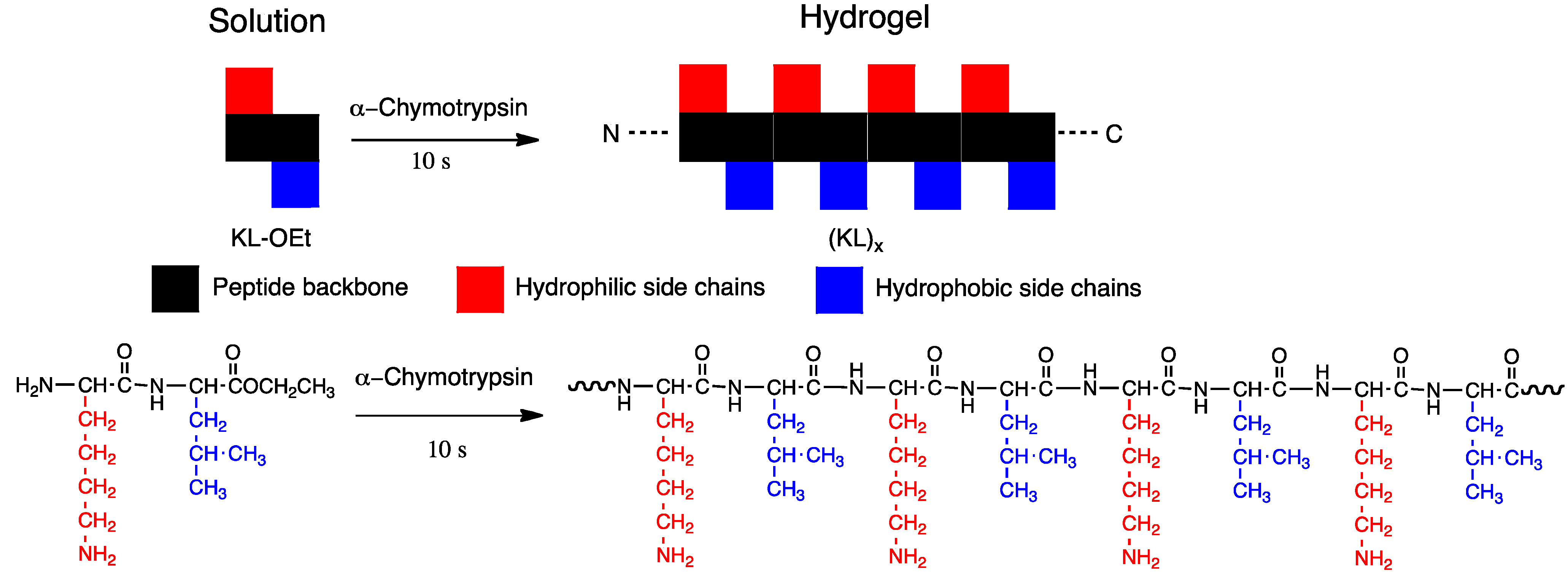
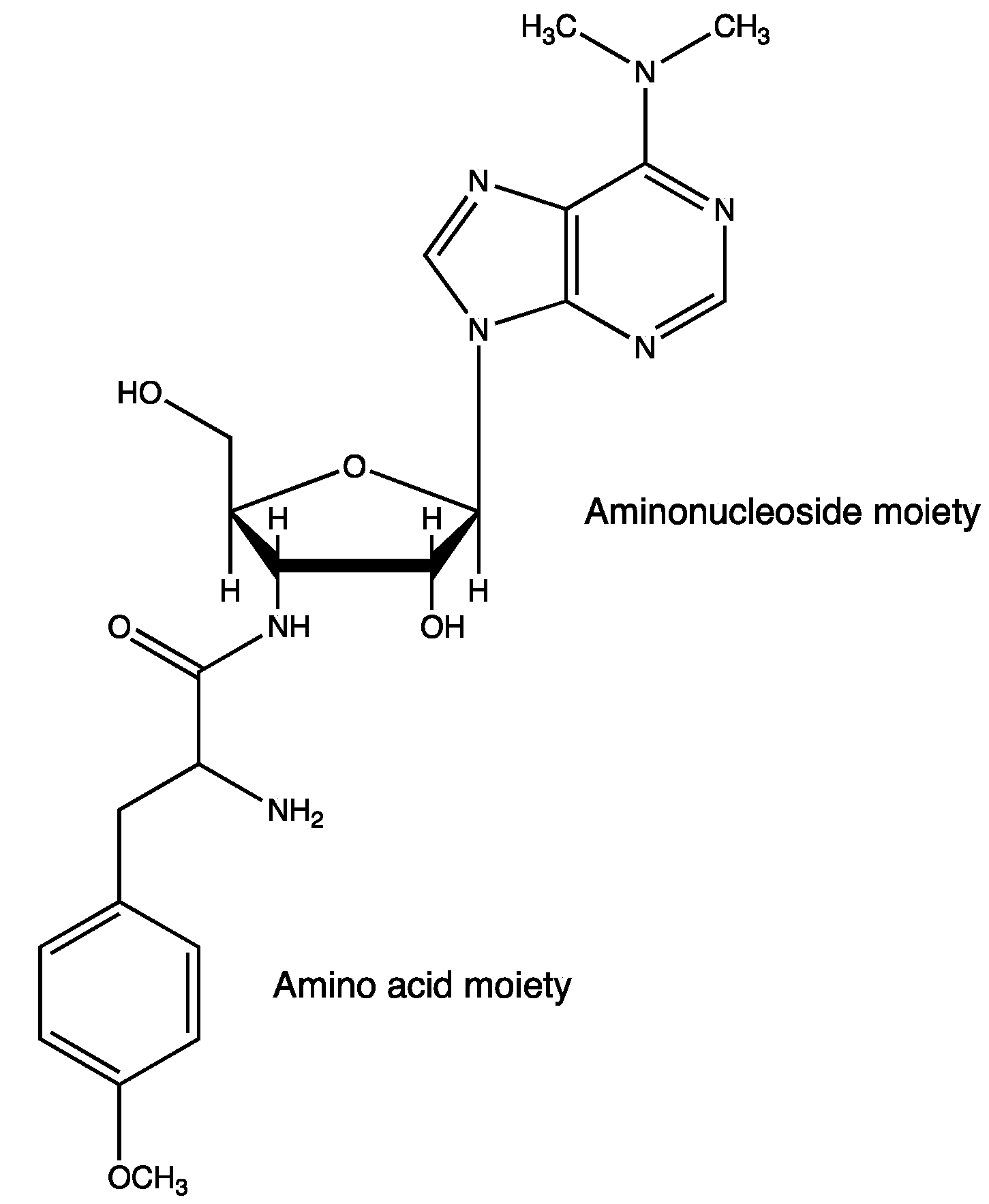
5.5. Analgesics
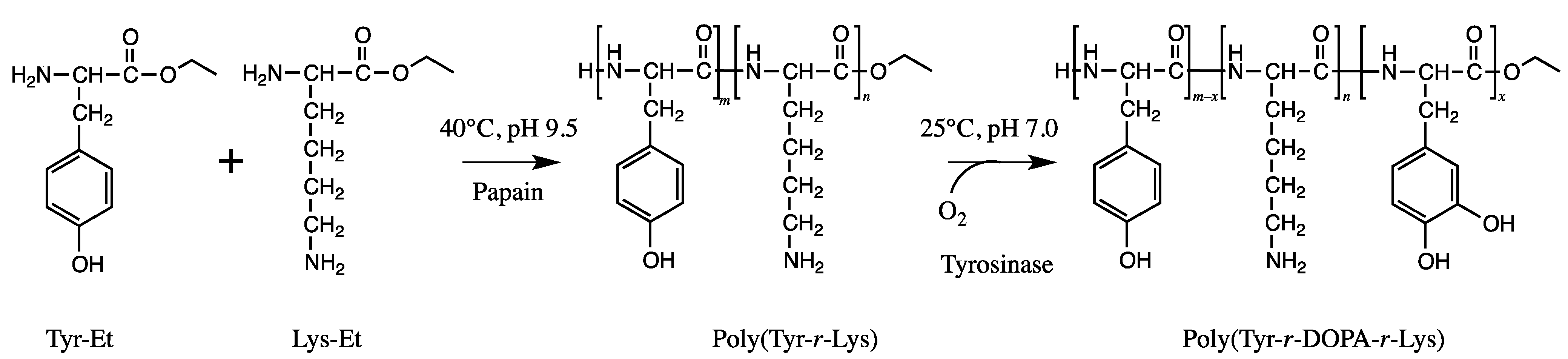
5.6. Adhesive Peptides
6. Conclusions and Future Perspectives
Acknowledgments
Conflicts of Interest
References
- Nuijens, T.; Quaedflieg, P.J.L.M.; Jakubke, H.D. Hydrolysis and synthesis of peptides. In Enzyme Catalysis in Organic Synthesis, 3rd ed.; Drauz, K., Gröger, H., May, O., Eds.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2012; pp. 675–748. [Google Scholar]
- Merrifield, R.B. Solid-phase peptide synthesis. Adv. Enzymol. Relat. Areas Mol. Biol. 1969, 32, 221–296. [Google Scholar]
- Löfblom, J. Bacterial display in combinatorial protein engineering. Biotechnol. J. 2011, 6, 1115–1129. [Google Scholar] [CrossRef]
- Baker, P.J.; Numata, K. Polymerization of peptide polymers for biomaterial applications. In Polymer Science; Yılmaz, F., Ed.; Intech: Rijeka, Croatia, 2013; pp. 229–246. [Google Scholar]
- Bergmann, M.; Niemann, C. Newer biological aspects of protein chemistry. Science 1937, 86, 187–190. [Google Scholar]
- Bergmann, M.; Fruton, J.S. Some synthetic and hydrolytic experiments with chymotrypsin. J. Biol. Chem. 1938, 124, 321–329. [Google Scholar]
- Yu, H.; Chokhawala, H.A.; Huang, S.; Chen, X. One-pot three-enzyme chemoenzymatic approach to the synthesis of sialosides containing natural and non-natural functionalities. Nat. Protoc. 2006, 1, 2485–2492. [Google Scholar] [CrossRef]
- Guzmán, F.; Barberis, S.; Illanes, A. Peptide synthesis: Chemical or enzymatic. Electron. J. Biotechnol. 2007, 10, 279–314. [Google Scholar]
- Schröder, H.; Strohmeier, G.A.; Leypold, M.; Nuijens, T.; Quaedflieg, P.J.L.M.; Breinbauer, R. Racemization-free chemoenzymatic peptide synthesis enabled by the ruthenium-catalyzed synthesis of peptide enol esters via alkyne-addition and subsequent conversion using alcalase-cross-linked enzyme aggregates. Adv. Synth. Catal. 2013, 355, 1799–1807. [Google Scholar]
- Cheng, H.N. Enzyme-catalyzed synthesis of polyamides and polypeptides. In Biocatalysis in Polymer Chemistry; Loos, K., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; pp. 131–141. [Google Scholar]
- Schechter, I.; Berger, A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. [Google Scholar] [CrossRef]
- Bordusa, F. Proteases in organic synthesis. Chem. Rev. 2002, 102, 4817–4867. [Google Scholar] [CrossRef]
- Chen, F.; Zhang, F.; Wang, A.; Li, H.; Wang, Q.; Zeng, Z.; Xie, T. Recent progress in the chemo-enzymatic peptide synthesis. Afr. J. Pharm. Pharmacol. 2010, 4, 721–730. [Google Scholar]
- Yagasaki, M.; Hashimoto, S. Synthesis and application of dipeptides; current status and perspectives. Appl. Microbiol. Biotechnol. 2008, 81, 13–22. [Google Scholar] [CrossRef]
- Baker, P.J.; Numata, K. Chemoenzymatic synthesis of poly(l-alanine) in aqueous environment. Biomacromolecules. 2012, 13, 947–951. [Google Scholar] [CrossRef]
- Fukuoka, T.; Tachibana, Y.; Tonami, H.; Uyama, H.; Kobayashi, S. Enzymatic polymerization of tyrosine derivatives. Peroxidase- and protease-catalyzed synthesis of poly(tyrosine)s with different structures. Biomacromolecules 2002, 3, 768–774. [Google Scholar]
- Uyama, H.; Fukuoka, T.; Komatsu, I.; Watanabe, T.; Kobayashi, S. Protease-catalyzed regioselective polymerization and copolymerization of glutamic acid diethyl ester. Biomacromolecules 2002, 3, 318–323. [Google Scholar] [CrossRef]
- Viswanathan, K.; Schofield, M.H.; Teraoka, I.; Gross, R.A. Surprising metal binding properties of phytochelatin-like peptides prepared by protease-catalysis. Green Chem. 2012, 14, 1020–1029. [Google Scholar] [CrossRef]
- Schwab, L.W.; Kloosterman, W.M.J.; Konieczny, J.; Loos, K. Papain catalyzed (co)oligomerization of α-amino acids. Polymers 2012, 4, 710–740. [Google Scholar] [CrossRef]
- Qin, X.; Khuong, A.C.; Yu, Z.; Du, W.; Decatur, J.; Gross, R.A. Simplifying alternating peptide synthesis by protease-catalyzed dipeptide oligomerization. Chem. Commun. 2013, 49, 385–387. [Google Scholar] [CrossRef]
- Baker, P.J.; Patwardhan, S.V.; Numata, K. Synthesis of homopolypeptides by aminolysis mediated by proteases encapsulated in silica nanospheres. Macromol. Biosci. 2014, in press. [Google Scholar]
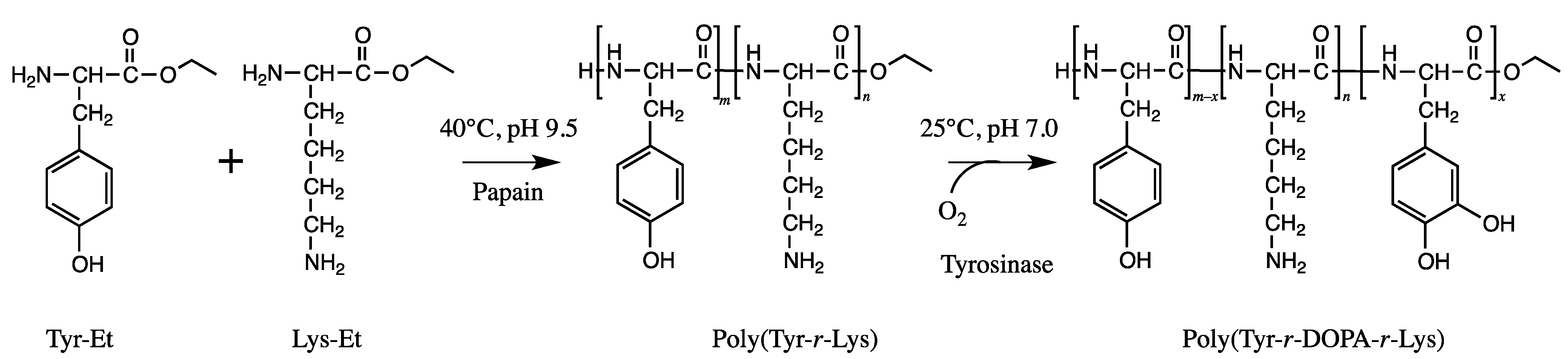
- Numata, K.; Baker, P.J. Synthesis of adhesive peptides similar to those found in blue mussel (Mytilus edulis) using papain and tyrosinase. Biomacromolecules 2014, 15, 3206–3212. [Google Scholar] [CrossRef]
- Viswanathan, K.; Omorebokhae, R.; Li, G.; Gross, R.A. Protease-catalyzed oligomerization of hydrophobic amino acid ethyl esters in homogeneous reaction media using l-phenylalanine as a model system. Biomacromolecules 2010, 11, 2152–2160. [Google Scholar] [CrossRef]
- Qin, X.; Xie, W.; Su, Q.; Du, W.; Gross, R.A. Protease-catalyzed oligomerization of l-lysine ethyl ester in aqueous solution. ACS Catal. 2011, 1, 1022–1034. [Google Scholar] [CrossRef]
- Narai-Kanayama, A.; Hanaishi, T.; Aso, K. α-Chymotrypsin-catalyzed synthesis of poly-l-cysteine in a frozen aqueous solution. J. Biotechnol. 2012, 157, 428–436. [Google Scholar] [CrossRef]
- Qin, X.; Xie, W.; Tian, S.; Cai, J.; Yuan, H.; Yu, Z.; Gross, R.A. Enzyme-triggered hydrogelation via self-assembly of alternating peptides. Chem. Commun. 2013, 49, 4839–4841. [Google Scholar]
- Ageitos, J.M.; Baker, P.J.; Sugahara, M.; Numata, K. Proteinase K-catalyzed synthesis of linear and star oligo-l-phenylalanine conjugates. Biomacromolecules 2013, 14, 3635–3642. [Google Scholar] [CrossRef]
- Aso, K.; Kodaka, H. Trypsin-catalyzed Oligomerization of l-Lysine Esters. Biosci. Biotechnol. Biochem. 1992, 56, 755–758. [Google Scholar] [CrossRef]
- Aso, K.; Kodaka, H.; Fukushi, H.; Lee, H. Trypsin-catalyzed synthesis of the arginyl-arginine dipeptide from l-arginine ethyl ester. Biotechnol. Lett. 1992, 14, 451–454. [Google Scholar] [CrossRef]
- Hou, R.Z.; Yang, Y.; Li, G.; Huang, Y.B.; Wang, H.; Liu, Y.J.; Xu, L.; Zhang, X.Z. Synthesis of a precursor dipeptide of RGDS (Arg-Gly-Asp-Ser) catalysed by the industrial protease alcalase. Biotechnol. Appl. Biochem. 2006, 44, 73–80. [Google Scholar]
- Huang, Y.B.; Cai, Y.; Yang, S.; Wang, H.; Hou, R.Z.; Xu, L.; Zhang, X.Z. Synthesis of tetrapeptide Bz-RGDS-NH2 by a combination of chemical and enzymatic methods. J. Biotechnol. 2006, 125, 311–318. [Google Scholar] [CrossRef]
- Schwab, L.W.; Kroon, R.; Schouten, A.J.; Loos, K. Enzyme-catalyzed ring-opening polymerization of unsubstituted β-Lactam. Macromol. Rapid Commun. 2008, 29, 794–797. [Google Scholar] [CrossRef]
- Ménard, R.; Storer, A.C. Papain. In Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Academic Press: London, UK, 1998; pp. 555–557. [Google Scholar]
- Berger, A.; Schechter, I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos. Trans. R. Soc. Lond. BBiol. Sci. 1970, 257, 249–264. [Google Scholar]
- Kamphuis, I.G.; Drenth, J.; Baker, E.N. Thiol proteases: Comparative studies based on the high-resolution structures of papain and actinidin, and on amino acid sequence information for cathepsins B and H, and stem bromelain. J. Mol. Biol. 1985, 182, 317–329. [Google Scholar] [CrossRef]
- Fagerland, J.; Finne-Wistrand, A.; Numata, K. Short one-pot chemo-enzymatic synthesis of l-lysine and l-alanine diblock co-oligopeptides. Biomacromolecules 2014, 15, 735–743. [Google Scholar] [CrossRef]
- Qin, X.; Xie, W.; Tian, S.; Ali, M.A.; Shirke, A.; Gross, R.A. Influence of Nε-Protecting Groups on the Protease-Catalyzed Oligomerization of l-Lysine Methyl Ester. ACS Catal. 2014, 4, 1783–1792. [Google Scholar] [CrossRef]
- Rowan, A.D. Stem bromelain. In Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Academic Press: London, UK, 1998; pp. 566–567. [Google Scholar]
- Appel, W. Chymotrypsin: Molecular and catalytic properties. Clin. Biochem. 1986, 19, 317–322. [Google Scholar] [CrossRef]
- Beck-Piotraschke, K.; Jakubke, H. Protease-catalysed synthesis of peptides containing histidine and lysine. Tetrahedron Asymmetry 1998, 9, 1505–1518. [Google Scholar]
- Saenger, W.; Proteinase, K. Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Academic Press: London, UK, 1998; pp. 322–325. [Google Scholar]
- Ebeling, W.; Hennrich, N.; Klockow, M.; Metz, H.; Orth, H.D.; Lang, H. Proteinase K from Tritirachium album Limber. Eur. J. Biochem. 1974, 47, 91–97. [Google Scholar] [CrossRef]
- Morihara, K.; Tsuzuki, H. Specificity of proteinase K from Tritirachium album Limber for synthetic peptides. Agric. Biol. Chem. 1975, 39, 1489–1492. [Google Scholar]
- Halfon, S.; Craik, C.S. Trypsin. In Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Academic Press: London, UK, 1998; pp. 12–21. [Google Scholar]
- Manea, M.; Mezo, G.; Hudecz, F.; Przybylski, M. Mass spectrometric identification of the trypsin cleavage pathway in lysyl-proline containing oligotuftsin peptides. J. Pept. Sci. 2007, 13, 227–236. [Google Scholar] [CrossRef]
- Ballinger, M.D.; Wells, J.A. Subtilisin. In Handbook of Proteolytic Enzymes; Barrett, A.J., Rawlings, N.D., Woessner, J.F., Eds.; Academic Press: London, UK, 1998; pp. 289–294. [Google Scholar]
- Nuijens, T.; Schepers, A.H.M.; Cusan, C.; Kruijtzer, J.A.W.; Rijkers, D.T.S.; Liskamp, R.M.J.; Quaedflieg, P.J.L.M. Enzymatic fragment condensation of side chain-protected peptides using subtilisin a in anhydrous organic solvents: A general strategy for industrial peptide synthesis. Adv. Synth. Catal. 2013, 355, 287–293. [Google Scholar]
- Gandhi, N.N.; Patil, N.S.; Sawanta, S.B.; Joshi, J.B.; Wangikar, P.P.; Mukesh, D. Lipase-catalyzed esterification. Catal. Rev. Sci. Eng. 2000, 42, 439–480. [Google Scholar] [CrossRef]
- West, J.B.; Hennen, W.J.; Lalonde, J.L.; Bibbs, J.A.; Zhong, Z.; Meyer, E.F.; Wong, C. Enzymes as Synthetic Catalysts: Mechanistic and Active-Site Considerations of Natural and Modified Chymotrypsin. J. Am. Chem. Soc. 1990, 5320, 5313–5320. [Google Scholar]
- Grünberg, R.; Domgall, I.; Günther, R.; Rall, K.; Hofmann, H.J.; Bordusa, F. Peptide bond formation mediated by substrate mimetics. Eur. J. Biochem. 2000, 267, 7024–7030. [Google Scholar] [CrossRef]
- Chen, K.Q.; Robinson, A.C.; van Dam, M.E.; Martinez, P.; Economou, C.; Arnold, F.H. Enzyme engineering for nonaqueous solvents. II. Additive effects of mutations on the stability and activity of subtilisin E in polar organic media. Biotechnol. Prog. 1991, 7, 125–129. [Google Scholar] [CrossRef]
- Salam, S.M.A.; Kagawa, K.; Kawashiro, K. α-Chymotrypsin-catalyzed peptide synthesis in frozen aqueous solution using N-protected amino acid carbamoylmethyl esters as acyl donors. Tetrahedron Asymmetry 2006, 17, 22–29. [Google Scholar] [CrossRef]
- Salam, S.M.A.; Kagawa, K.; Kawashiro, K. Proteasecatalyzed dipeptide synthesis from N-protected amino acid carbamoylmethyl esters and free amino acids in frozen aqueous solutions. Enzym. Microb. Technol. 2008, 43, 537–543. [Google Scholar] [CrossRef]
- Wehofsky, N.; Wespe, C.; Cerovsky, V.; Pech, A.; Hoess, E.; Rudolph, R.; Bordusa, F. Ionic liquids and proteases: A clean alliance for semisynthesis. ChemBioChem 2008, 9, 1493–1499. [Google Scholar]
- Noritomi, H. Protease-catalyzed synthetic reactions in ionic liquids. In Ionic Liquids: Applications and Perspectives; Kokorin, A., Ed.; Intech: Rijeka, Croatia, 2011; pp. 517–528. [Google Scholar]
- Erbeldinger, M.; Mesiano, A.J.; Russell, A.J. Enzymatic catalysis of formation of Z-aspartame in ionic liquid—An alternative to enzymatic catalysis in organic solvents. Biotechnol. Prog. 2000, 16, 1129–1131. [Google Scholar]
- Mishima, K.; Matsuyama, K.; Baba, M.; Chidori, M. Enzymatic dipeptide synthesis by surfactant-coated α-chymotrypsin complexes in supercritical carbon dioxide. Biotechnol. Prog. 2003, 19, 281–284. [Google Scholar] [CrossRef]
- Noritomi, H.; Miyata, M.; Kato, S.; Nagahama, K. Enzymatic synthesis of peptide in acetonitrile/supercritical carbon dioxide. Biotechnol. Lett. 1995, 17, 1323–1328. [Google Scholar]
- Ulijn, R.V.; Baragaña, B.; Halling, P.J.; Flitsch, S.L. Protease-catalyzed peptide synthesis on solid support. J. Am. Chem. Soc. 2002, 124, 10988–10989. [Google Scholar] [CrossRef]
- Klibanov, A.M. Enzyme stabilization by immobilization. Anal. Biochem. 1979, 93, 1–25. [Google Scholar] [CrossRef]
- Meng, L.; Joshi, R.; Eckstein, H. Application of enzymes for the synthesis of the Cholecystokinin Pentapeptide (CCK-5). Chimica OggiChem. Today 2006, 24, 1–4. [Google Scholar]
- Talekar, S.; Joshi, A.; Joshi, G.; Kamat, P.; Haripurkar, R.; Kambale, S. Parameters in preparation and characterization of cross linked enzyme aggregates (CLEAs). RSC Adv. 2013, 3, 12485–12511. [Google Scholar] [CrossRef]
- Sheldon, R.A. Characteristic features and biotechnological applications of cross-linked enzyme aggregates (CLEAs). Appl. Microbiol. Biotechnol. 2011, 92, 467–477. [Google Scholar] [CrossRef]
- Nuijens, T.; Cusan, C.; Schepers, A.C.H.M.; Kruijtzer, J.A.W.; Rijkers, D.T.S.; Liskamp, R.M.J.; Quaedflieg, P.J.L.M. Enzymatic synthesis of activated esters and their subsequent use in enzyme-based peptide synthesis. J. Mol. Catal. B Enzym. 2011, 71, 79–84. [Google Scholar] [CrossRef]
- Bordusa, F. Nonconventional amide bond formation catalysis: Programming enzyme specificity with substrate mimetics. Braz. J. Med. Biol. Res. 2000, 33, 469–485. [Google Scholar]
- Beer, R.J.; Zarzycka, B.; Mariman, M.; Amatdjais-Groenen, H.I.; Mulders, M.J.; Quaedflieg, P.J.; Delft, F.L.; Nabuurs, S.B.; Rutjes, F.P. Papain-specific activating esters in aqueous dipeptide synthesis. ChemBioChem 2012, 13, 1319–1326. [Google Scholar] [CrossRef]
- Bread, N.S.; Armentrout, S.A.; Weisberger, A.S. Inhibition of mammalian protein synthesis by antibiotics. Pharmacol. Rev. 1969, 21, 213–245. [Google Scholar]
- Nathans, D. Puromycin inhibition of protein synthesis: Incorporation of puromycin into peptide chains. Proc. Natl. Acad. Sci. USA 1964, 51, 585–592. [Google Scholar] [CrossRef]
- Usuki, H.; Yamamoto, Y.; Arima, J.; Iwabuchi, M.; Miyoshi, S.; Nitoda, T.; Hatanaka, T. Peptide bond formation by aminolysin-A catalysis: A simple approach to enzymatic synthesis of diverse short oligopeptides and biologically active puromycins. Org. Biomol. Chem. 2011, 9, 2327–2335. [Google Scholar] [CrossRef]
- Thongyoo, P.; Jaulent, A.M.; Tate, E.W.; Leatherbarrow, R.J. Immobilized protease-assisted synthesis of engineered cysteine-knot microproteins. ChemBioChem 2007, 8, 1107–1109. [Google Scholar] [CrossRef]
- Rosen, M.J. Surfactants and Interfacial Phenomena, 3rd ed.; John Wiley & Sons, Inc.: New York, NY, USA, 2004. [Google Scholar]
- Piera, E.; Infante, M.R.; Clapés, P. Chemo-enzymatic synthesis of arginine-based gemini surfactants. Biotechnol. Bioeng. 2000, 70, 323–331. [Google Scholar] [CrossRef]
- Huang, Y.B.; Cai, Y.; Yang, S.; Wang, H.; Hou, R.Z.; Xu, L.; Zhang, X.Z. Chemo-enzymatic synthesis of precursor tetrapeptide Bz–RGDS–NH2 of cellular adhesion motif in low-water organic media. Enzym. Microb. Technol. 2006, 39, 1159–1165. [Google Scholar] [CrossRef]
- Huang, Y.B.; Xiao, Y.P.; Wang, H.; Hou, R.Z.; Zhang, N.; Wu, X.X.; Xu, L.; Zhang, X.Z. Chemo-enzymatic synthesis of tripeptide RGD diamide in organic solvents. J. Biotechnol. 2005, 116, 51–59. [Google Scholar] [CrossRef]
- Sun, H.; He, B.; Xu, J.; Wu, B.; Ouyang, P. Efficient chemo-enzymatic synthesis of endomorphin-1 using organicsolvent stable proteases to green the synthesis of the peptide. Green Chem. 2011, 13, 1680–1685. [Google Scholar] [CrossRef]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Yazawa, K.; Numata, K. Recent Advances in Chemoenzymatic Peptide Syntheses. Molecules 2014, 19, 13755-13774. https://doi.org/10.3390/molecules190913755
Yazawa K, Numata K. Recent Advances in Chemoenzymatic Peptide Syntheses. Molecules. 2014; 19(9):13755-13774. https://doi.org/10.3390/molecules190913755
Chicago/Turabian StyleYazawa, Kenjiro, and Keiji Numata. 2014. "Recent Advances in Chemoenzymatic Peptide Syntheses" Molecules 19, no. 9: 13755-13774. https://doi.org/10.3390/molecules190913755
APA StyleYazawa, K., & Numata, K. (2014). Recent Advances in Chemoenzymatic Peptide Syntheses. Molecules, 19(9), 13755-13774. https://doi.org/10.3390/molecules190913755