Design and Synthesis of New Cholesterol-Conjugated 5-Fluorouracil: A Novel Potential Delivery System for Cancer Treatment


Abstract
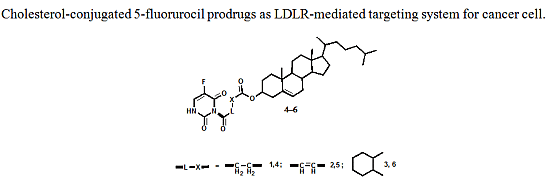
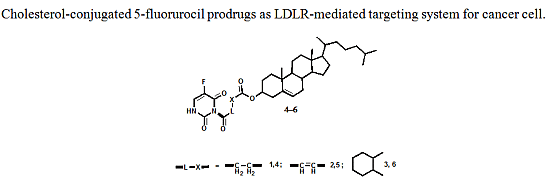
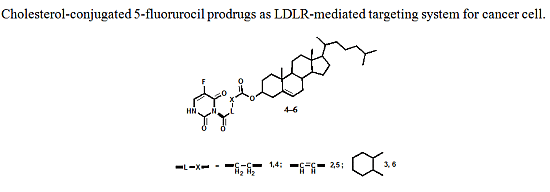
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
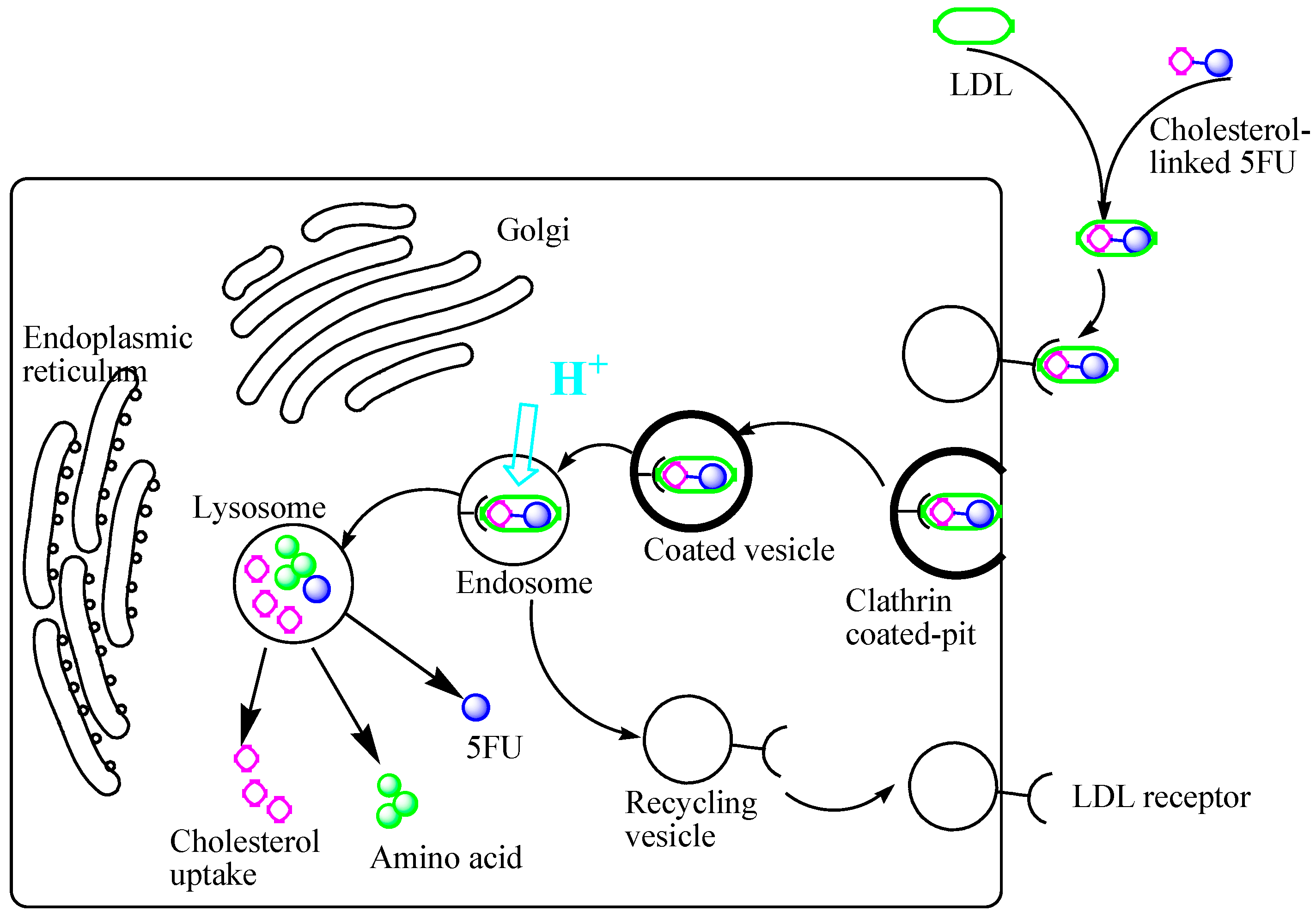
2. Results and Discussion
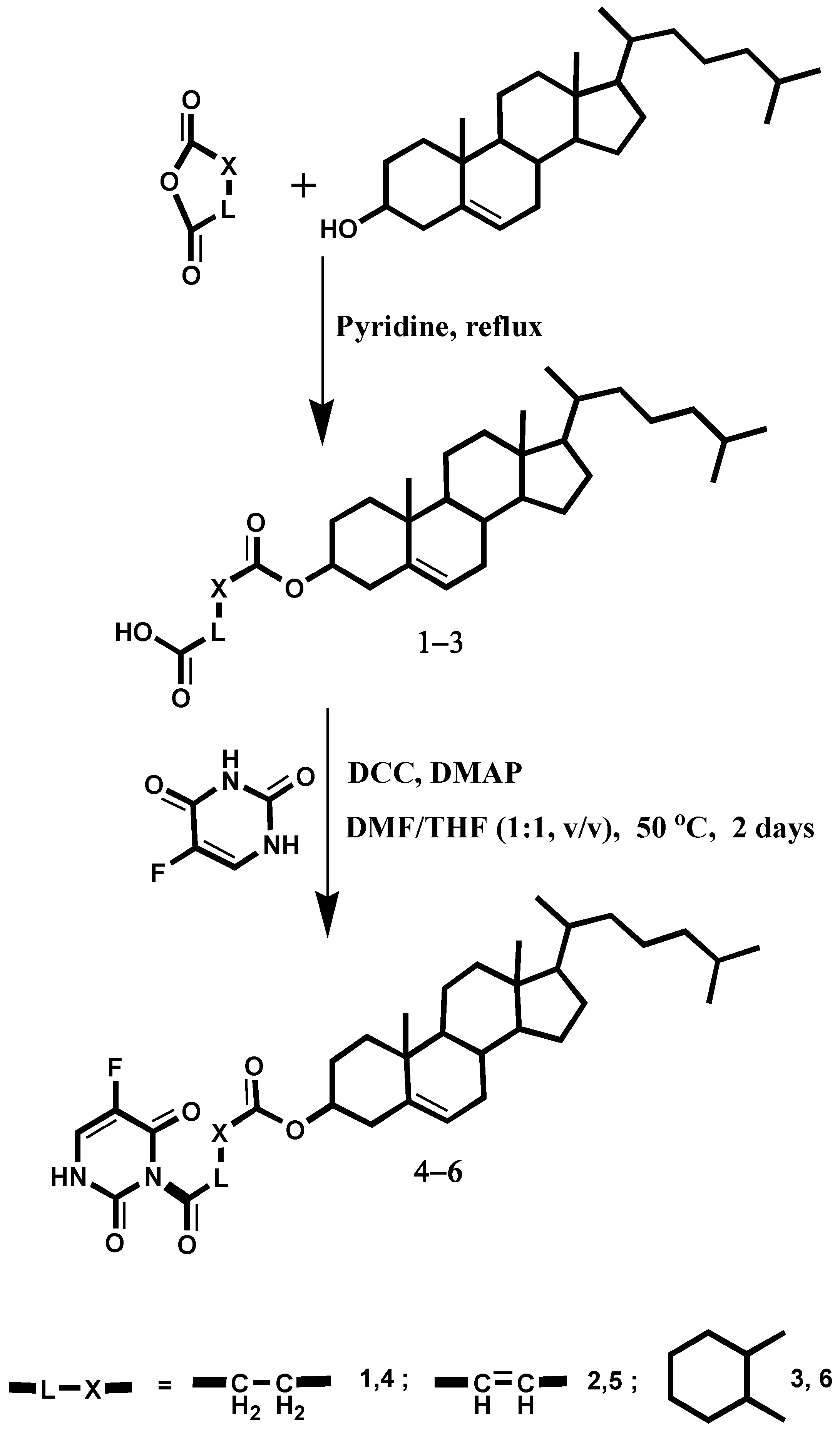
2.1. Chemistry
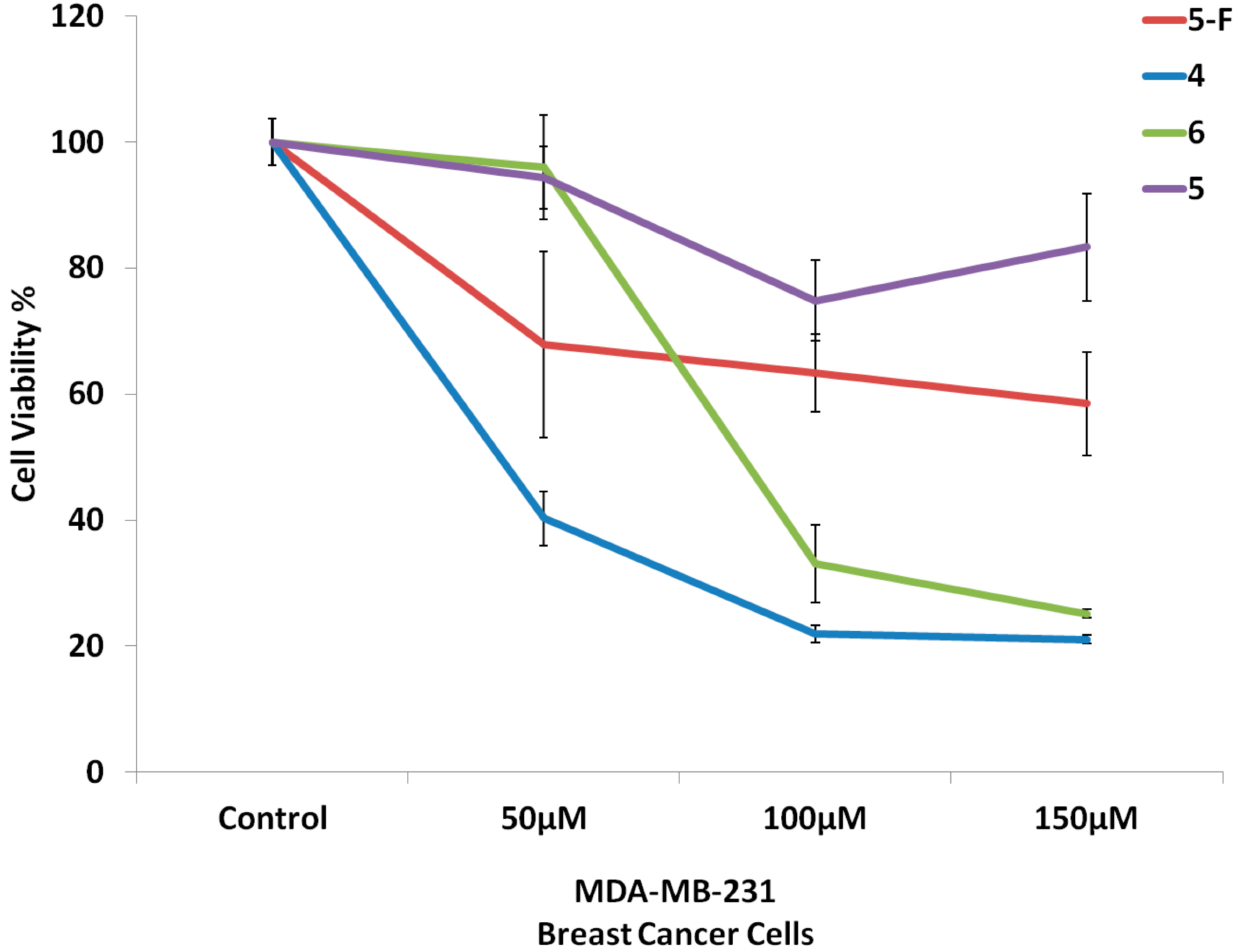
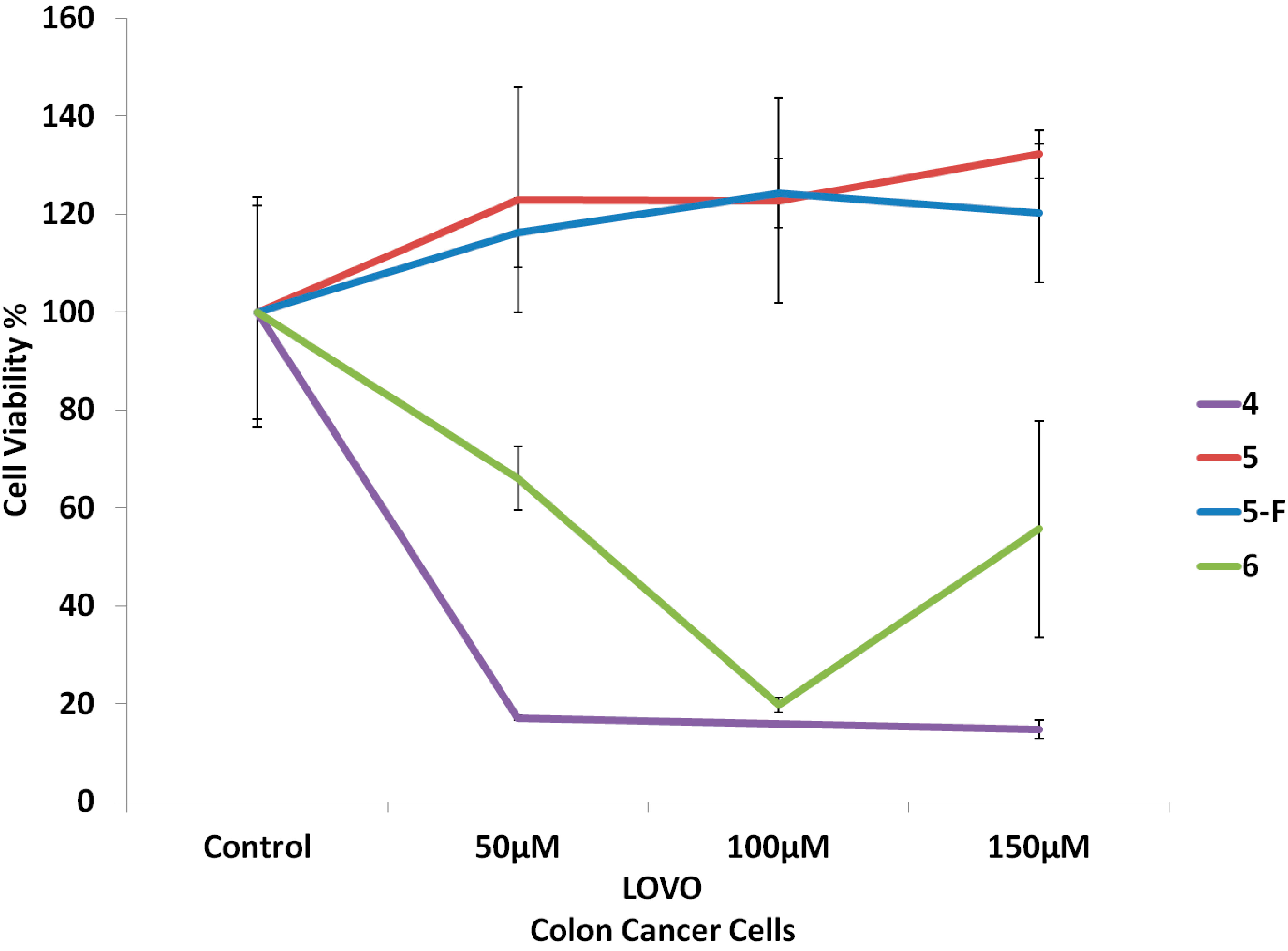
2.2. Biological Screening
2.2.1. Cell Culture and Cytotoxicity Assay
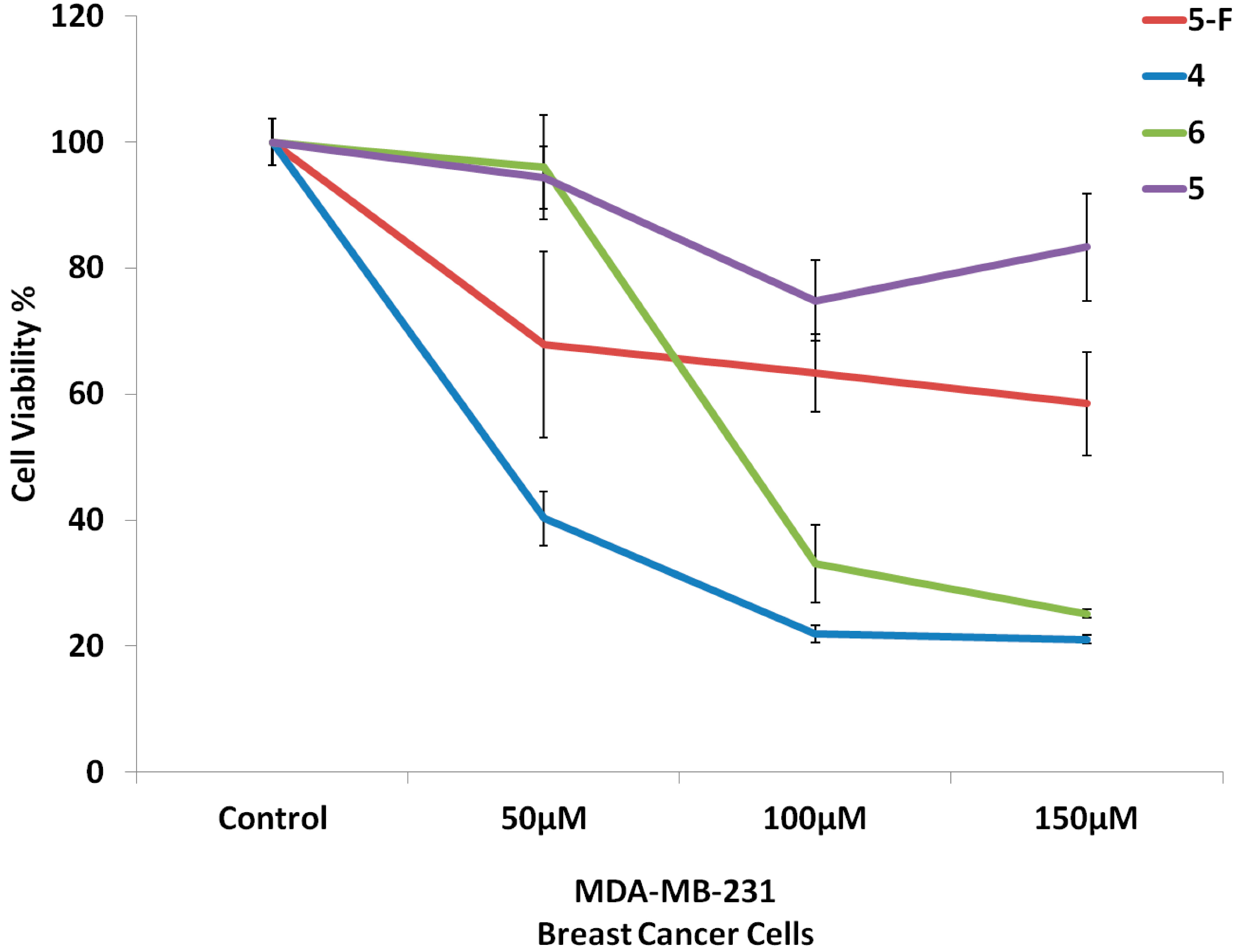
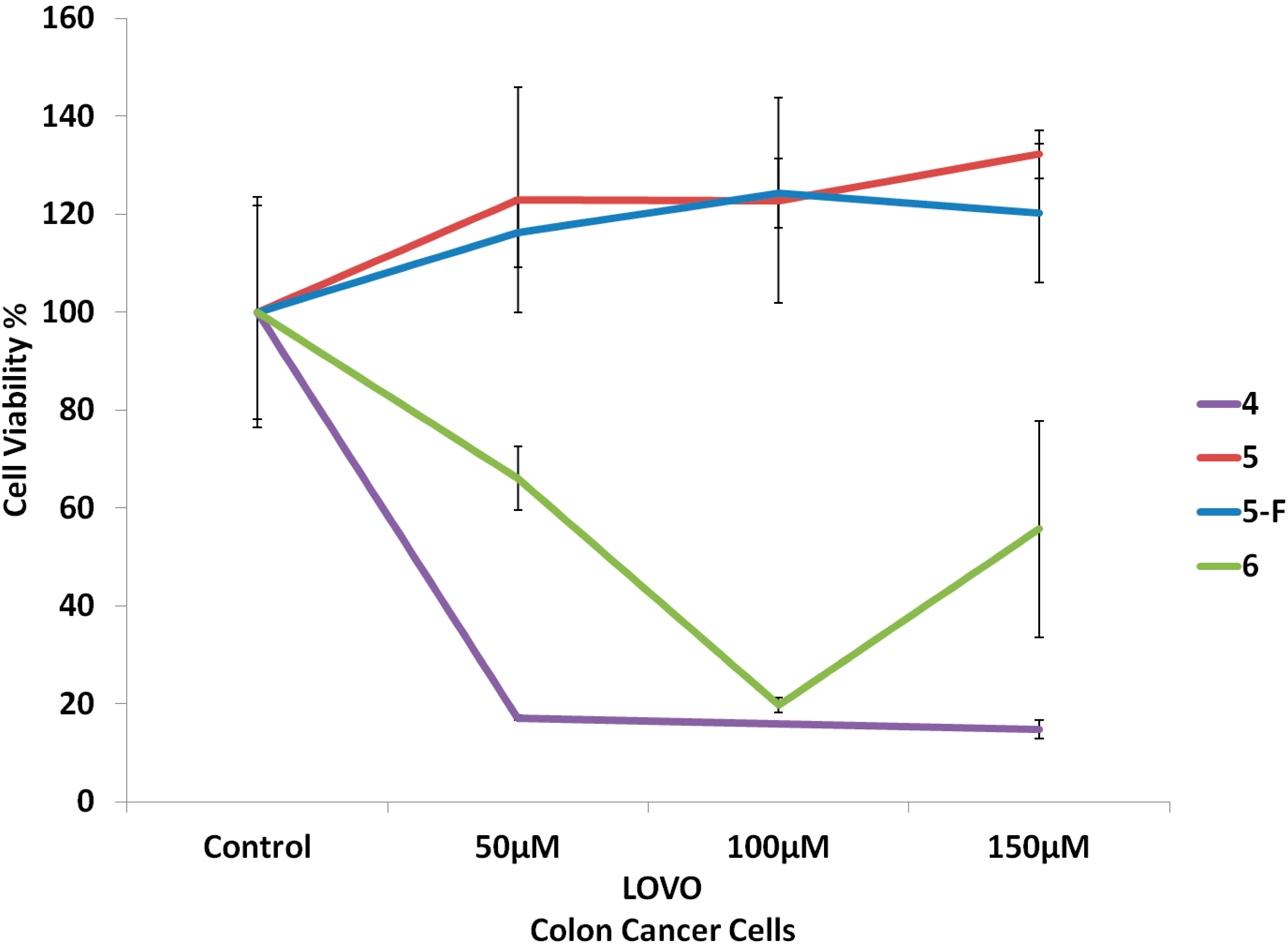
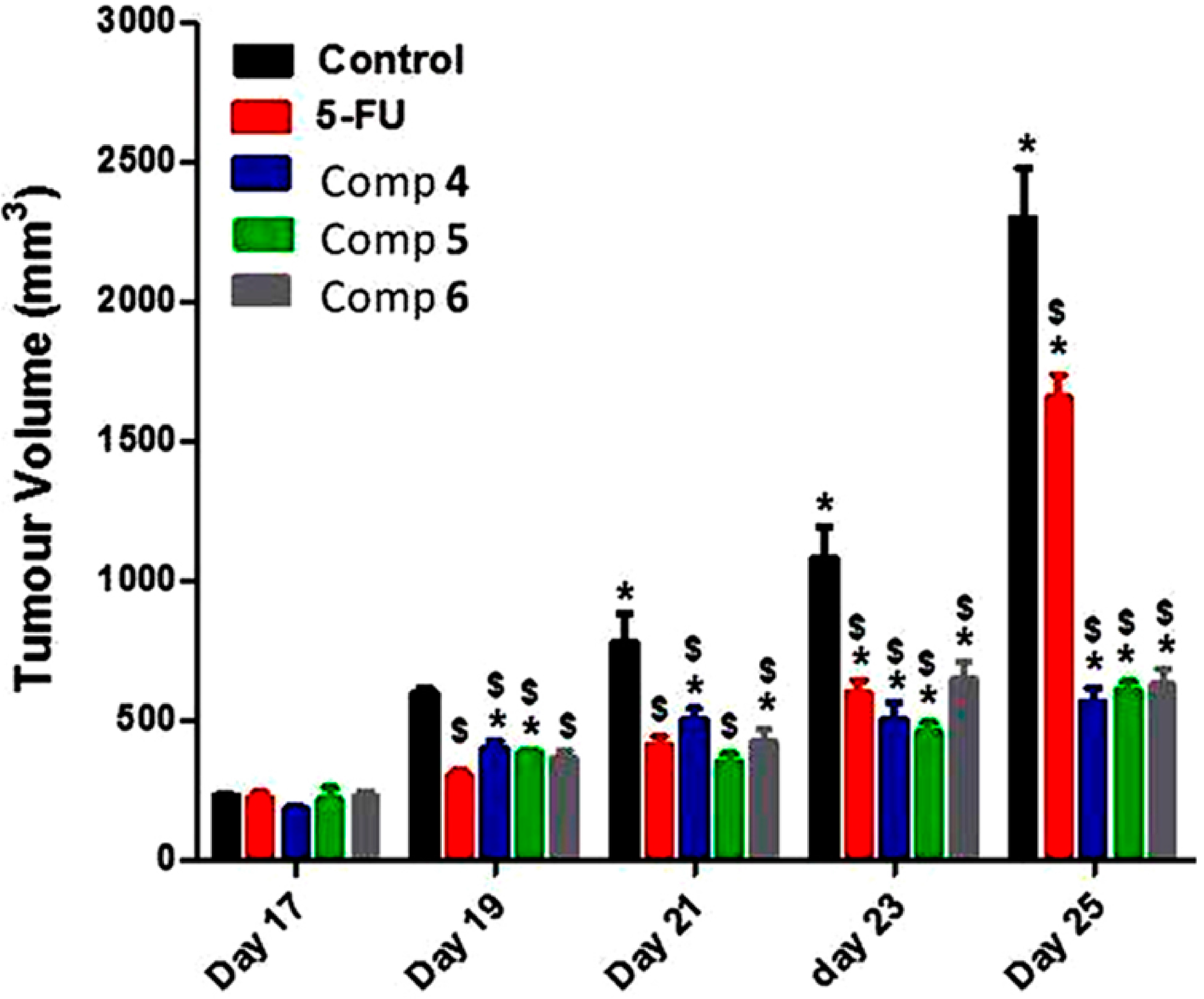
2.2.2. In Vivo Antitumor Activity of Compounds 4–6 on Growth of Solid Ehrlich Carcinoma (SEC) in Mice Model
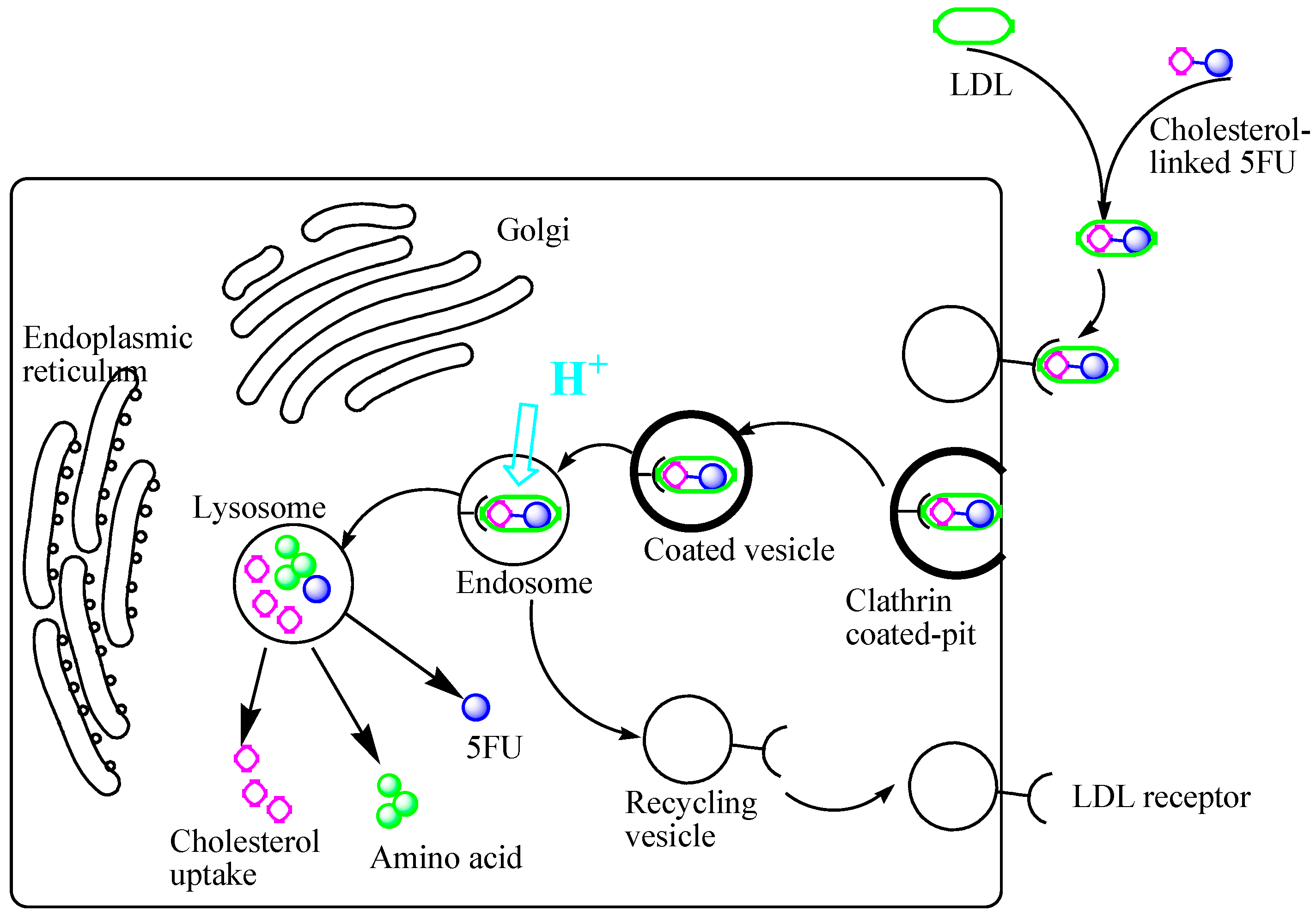
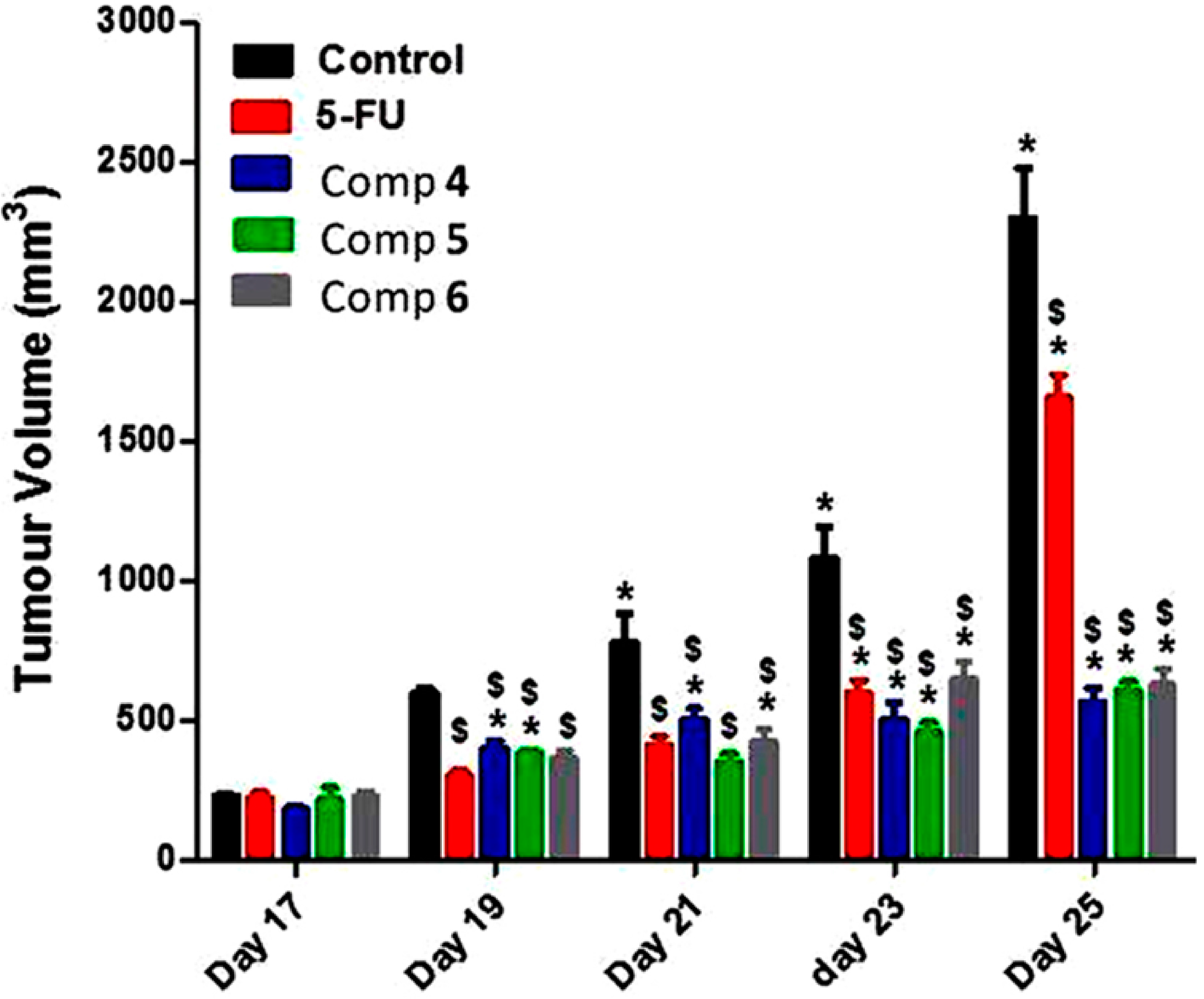
3. Experimental
3.1. Chemistry
3.2. Synthesis of Cholesteryl Hemicarboxylate (1–3)
3.3. Preparation of 5-FU-Cholesterol Conjugates 4–6
3.4. Biological Evaluation
3.4.1. Cell Culture and Cytotoxicity Assay
3.4.2. In Vivo Antitumor Activity in Solid Ehrlich-Carcinoma-Bearing Mice (SEC)
Animals
Antitumor Evaluation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Vitols, S. Uptake of low-density lipoprotein by malignant cells-possible therapeutic applicantions. Cancer Cells 1991, 3, 488–495. [Google Scholar]
- Polo, L.; Valduga, G.; Jori, G.; Reddi, E. Density lipoprotein receptors in the uptake of tumor photosensitizer by human and rat transformed fibroblasts. Int. J. Biochem. Cell Biol. 2002, 34, 10–23. [Google Scholar]
- Sarkar, R.; Halpern, D.S.; Jacobs, S.K.; Lu, D.R. LDL-receptor mediated drug targeting to malignant tumors. In Biomedical Aspects of Drug Targeting; Muzykantov, V., Torchilin, V., Eds.; Kluwer Academic Publisher: Norwell, MA, USA, 2002; pp. 150–168. [Google Scholar]
- Gueddari, N.; Favre, G.; Hachem, H.; Marek, E.; le Gaillard, F.; Soula, G. Evidence for up-regulated low density lipoprotein receptor in human lung adenocarcinoma cell line A549. Biochimie 1993, 75, 811–819. [Google Scholar]
- Firestone, R.A. Low density lipoprotein as a vehicle for targeting antitumor compounds to cancer cells. Bioconjug. Chem. 1994, 5, 105–113. [Google Scholar]
- Yen, C.F.; Kalunta, C.I.; Chen, F.S.; kaptein, J.S.; Lin, C.K.; Lad, P.M. Regulation of low-density lipoprotein receptors and assessment of their functional role in Burkitt’s lymphoma cells. Biochim. Biophys. Acta 1995, 1257, 47–57. [Google Scholar]
- Maletinska, L.; Blakely, E.A.; Bjornstad, K.A.; Deen, D.F.; Knoff, L.J.; Forte, T.M. Human glioblastoma cell lines: Levels of low-density lipoprotein receptor and low-density lipoprotein receptor-related protein. Cancer Res. 2000, 60, 2300–2303. [Google Scholar]
- Chen, Y.; Hughes-Fulford, M. Human prostate cancer cells lack feedback regulation of low-density liprotein receptor and its regulator, SREBP2. Int. J. Cancer 2001, 91, 41–45. [Google Scholar]
- Kostner, G.M.; Laggner, P. Human Plasma Lipoprotein; Walter de Gruyter: Berlin, Germany, 1989; pp. 23–51. [Google Scholar]
- Versluis, A.J.; Rump, E.T. Synthesis of a lipophilic Daunorubicin derivative and its incorporation into lipidic carriers developed for LDL receptor-mediated tumor therapy. Pharm. Res. 1998, 15, 531–537. [Google Scholar]
- Feakes, D.A.; Spinler, J.K.; Harris, F.R. Synthesis of boron-containing cholesterol derivatives for incorporation into unilamellar liposomes and evaluation as potential agents for BNCT. Tetrahedron 1999, 55, 11177–11186. [Google Scholar]
- Ji, B.; Lu, D.R. A Novel Approach to Targeted Drug Delivery for Treatment of Brain Cancer. Ph.D. Thesis, The University of Georgia, Athens, GA, USA, 2001. [Google Scholar]
- Alanazi, F.K.; Haq, N.; Radwan, A.A.; Alsarra, I.A.; Shakeel, F. Development and validation of UHPLC-DAD method for the determination of cholesteryl-hexahydrophthaloyl-5-fluorouracil in lipid nanoemulsion. J. Anal. Chem. 2014, in press. [Google Scholar]
- Sayed-Ahmed, M.M.; Shaarawy, S.; Shouman, S.A.; Osman, A.-M.M. Reversal of Dozorubicin-induced cardiac metabolic damage by L-carnitine. Pharm. Res. 1999, 39, 289–295. [Google Scholar]
- Klein, B.; Kleinman, N.B.; Foreman, J.A. Preparation and evaluation of a water-soluble cholesterol standard. Clin. Chem. 1974, 20, 482–485. [Google Scholar]
- Sample Availability: Samples of the compounds 1–6 are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Radwan, A.A.; Alanazi, F.K. Design and Synthesis of New Cholesterol-Conjugated 5-Fluorouracil: A Novel Potential Delivery System for Cancer Treatment. Molecules 2014, 19, 13177-13187. https://doi.org/10.3390/molecules190913177
Radwan AA, Alanazi FK. Design and Synthesis of New Cholesterol-Conjugated 5-Fluorouracil: A Novel Potential Delivery System for Cancer Treatment. Molecules. 2014; 19(9):13177-13187. https://doi.org/10.3390/molecules190913177
Chicago/Turabian StyleRadwan, Awwad A., and Fares K. Alanazi. 2014. "Design and Synthesis of New Cholesterol-Conjugated 5-Fluorouracil: A Novel Potential Delivery System for Cancer Treatment" Molecules 19, no. 9: 13177-13187. https://doi.org/10.3390/molecules190913177